

Abstract
Cigarette smoke (CS) is a leading cause of death worldwide. The aryl hydrocarbon receptor (AHR) is partially responsible for tobacco-induced carcinogenesis although the underlying mechanisms involving early effector genes have yet to be determined. Here, we report that adrenomedullin (ADM) significantly contributes to the carcinogenicity of tobacco activated AHR. CS and AHR activating ligands induced ADM in vitro and in vivo but not in AHR-deficient fibroblasts and mice. Ectopic transfection of AHR rescued ADM expression in AHR−/− fibroblasts while AHR blockage with siRNA in wild type cells significantly decreased ADM expression. AHR regulates ADM expression through two intronic xenobiotic response elements located close to the start codon in the ADM gene. Using tissue microarrays we showed that ADM and AHR were coupregulated in lung tumor biopsies from smoker patients. Microarray metaanalysis of 304 independent microarray experiments showed that ADM is elevated in smokers and smokers with cancer. Additionally, ADM coassociated with a subset of AHR responsive genes and efficiently differentiated patients with lung cancer from non-smokers. In a novel preclinical model of CS-induced tumor progression, host exposure to CS extracts significantly elevated tumor ADM while systemic treatment with the ADM antagonist NSC16311 efficiently blocked tobacco-induced tumor growth. In conclusion, ADM significantly contributes the carcinogenic effect of AHR and tobacco combustion products. We suggest that therapeutics targeting the AHR/ADM axis may be of clinical relevance in the treatment of tobacco-induced pulmonary malignancies.
Keywords: cigarette smoke, lung cancer, adrenomedullin, aryl hydrocarbon receptor, microarray meta analysis
Introduction
Cigarette smoking causes 87% of lung cancer fatalities and represents the leading preventable cause of death in developed countries (1, 2). First and second hand exposure to cigarette combustion products promote tumor angiogenesis and cancer in lung and several other anatomical sites including esophagus, bladder, pancreas and cervix (3). While much is known about the epidemiology of tobacco smoke, the underlying cellular and molecular mechanisms responsible for its carcinogenic potential are unclear. Tumor angiogenesis, epigenetic regulation and inflammation have been identified as processes contributing to smoke-related lung cancer (4–6), although we are only beginning to recognize the controlling individual molecular players. The AHR (a basic helix-loop-helix transcription factor) is a main factor of the complex subcellular circuitry linking tobacco smoke and tumor promotion and progression (7). Upon activation by polycyclic aromatic hydrocarbons (PAH) present in CS (8), AHR binds to specific DNA consensus sequences denoted as xenobiotic response elements (XRE) and drives the expression of proinflamatory and oncogenic genes such as COX-2 (9) thereby enhancing tumor growth (10). AHR expression is also elevated in PAH-induced lung carcinomas (11). Constitutive activation of AHR results in spontaneous stomach tumors and promotes hepatocarcinogenesis and lymphoma (12) whereas PAH carcinogenicity is lost in AHR deficient mice (13). Hence, in the context of cancer, AHR can be regarded as a master transcription regulator that controls the expression of a large array of gene clusters some of which display oncogenic properties and can potentially be implicated in lung cancer (14). However, identification of individual AHR-activated effector genes remains elusive.
ADM is a protooncogene which plays a multifaceted role in cancer (15). Hypoxia drives its expression through a HIF1 based mechanism (16). Within the tumor microenvironment ADM supports tumor progression through a variety of mechanisms. It acts as a growth factor for tumor cells (17) and confers resistance to apoptosis through inhibition of proapoptotic factors (18) and upregulation of antiapoptotic factors (19). ADM promotes tumor growth by mediating the crosstalk between tumor and immune cells (20) and induces angiogenesis and lymphangiogenesis in a direct fashion (21, 22) or through induction of angiogenic factors such as vascular endothelial growth factor (23). It is also a migratory factor contributing to enhanced metastasis in ADM producing tumors (24).
The present study was undertaken to determine the role of ADM as an effector gene functionally associated with AHR mediating the oncogenic potential of tobacco in lung cancers.
Materials and Methods
Cell Lines and chemicals
A549, MCF7, Panc1, CaPan, Hep3B2, HepG2, H209 and H1264 (American Type Culture Collection, Manassas, VA, USA) were cultured following manufacturer’s instructions. Immortalized fibroblast bearing or lacking the AHR gene (AHR+/+, AHR−/−) have been previously described (25). 3-methylcholanthrene (3MC) and 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) we obtained from Cambridge Isotopes Laboratories (Andover, MA).
Plasmids and Luciferase Reporter Assays
Three 5′-flanking region of the human ADM gene containing 6, 4 and 2 putative XREs were amplified from human genomic DNA using primers AM2725, AM1754 and AM689 respectively (supplemental Table 1), subcloned into pcDNA3.1 (Invitrogen Carlsbad, CA) and subsequently excised by digestion with MluI and BglII. The resulting DNA fragments were cloned into the promoterless luciferase reporter pGL3Promoter vector (Promega Corp, Madison, WI) to generate plasmids pGL3P-AM2725, pGL3P-AM1754, and pGL3P-AM689. The cloned sequence of all plasmids was confirmed to be 100% identical to the published ADM gene. The XREs sequences present in plasmid pGL3P-AM689 were mutated by site directed mutagenesis (QuikChange®, Stratagene, La Jolla, CA) and primers XRE1 and XRE2 (supplemental Table 1). The expression vectors for mouse and human AHR (pCI-mAHR and pCI-hAHR) were a kind gift from Dr. Frank Gonzalez (NCI, NIH Bethesda, MD). The expression vectors for mouse and human ARNT (pmARNT and phARNT) were a kind gift from Dr. Oliver Hankinson (School of Medicine, University of California, Los Angeles, CA). The XRE-driven reporter plasmid pGudLuc was kindly provided by Professor Michael S. Denison (University of California, Los Angeles, CA).
For the luciferase assays, transfections were carried out in triplicate using FuGene (Roche Diagnostics, Mannheim, Germany) and cells exposed to the indicated treatments. Luciferase activities were determined using the Dual-Luciferase Assay Kit (Promega, Madison, WI, USA) and a M200 Infinite multireader plate scanner (Tecan, Männedorf, Switzerland) and normalized to and internal reference standard of renilla luciferase activity.
RNA extraction, reverse transcription and real time PCR
Total RNA was extracted from cell lines and mouse tissues using the RNeasy Mini Kit (Qiagen, Valencia, CA) and 3.5 μg reverse-transcribed using the SuperScript First-Strand Synthesis system (Invitrogen Carlsbad, CA). Quantitative real-time PCR reactions were run in an Opticon cycler (MJ Research, Waltham, MA) using Sybr Green PCR master mix (Applied Biosystems, Foster City, CA) and primers in supplemental Table 1 as previously described (26).
Preparation of CSE
CSE derived from Kentucky Reference 3R4F research blend cigarettes (University of Kentucky, KY) were prepared as described (27).
RIA of immunoreactive ADM
Concentrations of ADM in culture media of A549 cells were measured by RIA (Phoenix Pharmaceuticals, Burlingame, CA) following manufacturer’s instructions and as previously described (28).
In vivo exposure to benzo[a]pyrene (BP)
Four months old AHR−/− and AHR+/+ mice (29) were treated for 24 h with a single i.p. injection of BP (Sigma Chemical Co, St. Louis, MO, USA, 10 mg/Kg) in 100 μl corn oil, or corn oil only. Animals were euthanized by cervical dislocation and tissues snap frozen in dry ice and stored at −80 °C until used. Two treatments were made for each experimental condition and the experiment was repeated twice. These experiments were done following the guidelines set forth by the Animal Care and Use Committee of the University of Extremadura.
In vivo CS exposure
CS exposures were performed using a nose-only exposure apparatus (Expose, Scireq Inc., Tempe, AZ) daily, 5 days per week, for 6 weeks (20 mice). Two mainstream reference cigarettes (1R1, Kentucky Tobacco Research and Development Center) were lit and fed into a pump programmed to puff for 2 seconds in every 30-seconds period, thus simulating actual smoking inhalation. The CS was mixed with bias-flow air and led to the exposure tower. Mice (C57BL/6) were positioned in net restrainers and allowed to acclimate for 5 minutes before smoking inhalation was initiated. Two cigarettes were used simultaneously to feed smoke to the exposure chamber, and the process was immediately repeated once (4 cigarettes total for 20 mice) per day. Mice were then allowed access to water and chow ad libitum. Studies were conducted under animal protocols approved by the NIEHS/ACUC.
In vivo exposure to CSE
A549 cells (1 × 106 cells/100 μL) were inoculated subcutaneously in the left and right flanks of athymic nude mice. The mice (10 animals/group) were checked daily for tumor formation by palpation and the width and length measured twice a week. Once tumors reached ~25 mm2, mice were injected intraperitoneally with saline, CSE (100 μl; 0.06, 0.12, 0.18 and 0.24 puffs) and/or the ADM antagonist small molecule NSC16311 (100 μl, 10 μM (30)), three times a week for 4 weeks. Tumors were then surgically removed and processed for RNA extraction. This experiment was conducted in a blind fashion under animal protocol approved by the Animal Care and Usage Committee of NCI–Frederick Cancer Research Center.
Tissue microarrays
Tissue microarrays containing a total of 133 non-small cell lung cancers, including primary and secondary (metastatic) cancers, were prepared as described before (11). All tissues were obtained through an NCI/Taiwan MTA and considered IRB exempt by the NCI Patient Care Review Board.
ADM and AHR immunodetection
AHR and ADM immunocytochemistry (ICH) were performed as previously described (31). For immunofluorescence a chicken polyclonal anti-rabbit IgG-FITC (1:100 for cultured cells; Abcam Inc., Cambridge, MA) was used. Tissue microarrays were assessed by two independent researchers.
Microarray meta analysis
Microarray meta analysis included 304 arrays from five human data sets publicly available from Gene Expression Omnibus (supplemental Table 2). All data sets were from Affymetrix GeneChips and the “signal” or “average difference” values after MAS5 normalization were downloaded as supplied from the source. Intensity values for each experiment were log2 transformed and the probe sets from each array were mapped to HUGO gene symbols. When multiple probes/probe sets were mapped to the same gene, the expressions were processed using customized script to rule out combining values with potential splice variants. In short, a Pearson correlation was computed between all probes/probe sets for a given gene and the two profiles with highest correlation were averaged to represent the gene (32).
Z-score normalization was used to adjust the systematic bias of datasets generated by different platforms. The Z-scores were computed according to the formula:
where G is any gene on the microarray and G1...Gn represents aggregate measure of all of the genes. The data were regrouped into 4 sample classes: non-smokers, smokers, smokers with lung cancer, and former smokers for comparative analysis.
An AHR module was compiled from the network of ligand activated AHR-binding targets (14) containing a list of 693 genes. Out of these 693 genes 479 mapped to the genes on the arrays. To identify genes that show significant correlation with the ADM gene, we calculated the pair-wise correlation of ADM gene-expression profile with all genes in the chip. To limit the number of false positives, we used the multivariate permutation test, with 100 permutations at each comparison, to give the P value estimate of significance (33, 34). Genes with P value <= 0.01 were flagged for enrichment analysis. Next, we showed the enrichment of the AHR module among the 4 sample classes using Fisher’s exact test. The probability that the AHR module was significantly enriched among a specified set of genes can be calculated with the following formula:
Where n and nf are the total and flagged number of genes in AHR category and N and Nf are the total and flagged number of genes on the microarray (35). Sweave code documentation for the analysis of microarray data is provided as supplemental file.
Statistical Analysis
Statistical tests were performed using either GraphPad Prism 5 software or R bioconductor resource (36, 37). Chi-square test was used to evaluate correlations of AHR and ADM immunocytochemistry (supplemental Table 3), multivariate permutation tests was used to identify genes co-expressed with ADM module, and Fisher exact test was used for the enrichment analysis of ADM module in non smoker, former smoker, smoker with or without lung cancer groups. Differences were regarded as significant at a value of P<0.05 and noted as follows throughout the text: *, P<0.05; **, P<0.01; ***, P<0.001.
Results
CSE induces ADM in vitro and concomitantly activates AHR
CSE was used to explore the influence of tobacco combustion products on ADM expression in lung cancer cells in vitro. Exposure of A549 cells to CSE resulted in a dose (Fig 1A) and time dependent (Fig 1B) increase in expression of ADM that paralleled the well characterized AHR target gene CYP1A1. To better understand this response, A549 cells expressing a reporter plasmid under the regulation of the full ADM promoter, pGL3P-AM2725, were exposed to CSE. A statistically significant induction in luciferase activity was observed 24 h after exposure to CSE (Fig 1C). Activation of AHR in the same cells was demonstrated by significant luciferase induction of the XRE control reporter plasmid pGudLuc after exposure to CSE (Fig 1C, insert). Further, a significant increase in ADM was detected by RIA in culture media from A549 cells treated with 0.06 puffs/ml of CSE for 48 hours (Fig 1D). Together, these in vitro results support a positive regulation of the ADM gene by CSE, concomitant with activation of the AHR pathway, and encouraged us to study ADM expression in an in vivo model of CS exposure.
Figure 1.

Tobacco smoke induces ADM expression and activates the AHR pathway in lung cancer cells. Exposure of A549 cells to CSE resulted in induction of ADM and CYP1A1 mRNA expression in a dose (A) and time (B) dependent manner. (C) CSE significantly enhanced transactivation of a luciferase reporter under the control of the ADM promoter (pGL3P-AM2575) and a XRE reporter control plasmid (pGudLuc; C insert) in A549 cells. (D) Elevated levels of ADM protein were found in conditioned media of A549 cells exposed to CSE.
ADM and AHR are upregulated in lungs of mice exposed to CS
Further supporting our in vitro results, lung tissue obtained from mice challenged with CS for six weeks showed over two fold increase in ADM mRNA expression when compared to animals exposed to normal air (Fig 2A). No histological differences were appreciable between the lungs of control (normal air exposed) and CS exposed mice (supplemental Fig 1). Immunocytochemical analysis revealed that, ADM overexpression was restricted to the ciliated epithelial cells of the bronchioles (Fig 2 B, C) and the vascular endothelium (Fig 2F). Interestingly, increased AHR protein expression was observed in lungs of mice exposed to CS and colocalized with ADM to the bronchiole’s epithelium (Fig 2 D, E), although no AHR immunostaining was observed in the vascular endothelium in the same mice (Fig 2 G). Both ADM and AHR proteins were localized in the cytoplasm of bronchiolar epithelial cells (Fig 2 C,E). No ADM or AHR immunostaining was detected in the bronchiolar epithelium of mice exposed to normal air (Fig 2 H–K).
Figure 2.

ADM is elevated in lungs of mice exposed to CS. (A) Expression of ADM mRNA is elevated in lungs of mice exposed to CS compared to animals exposed to normal air. Strong ADM (B, C) and AHR (D, E) immunoreactivity colocalized in the epithelium of bronchioles of mice exposed to CS (x100). ADM expression was also elevated in the vascular endothelium of the lung in mice upon exposure to CS (F), although AHR was undetectable in the same vessels (G) (x600). In mice exposed to normal air, ADM immunoreactivity was localized exclusively to some muscle fibers around the bronchioles (H, I) and no AHR staining was detected (J, K) (x100).
AHR regulates ADM expression
The above in vitro and in vivo data, together with recent studies demonstrating high levels of AHR agonists in CS (8), support the involvement of AHR in the regulation of ADM by tobacco smoke. Consistent with this idea, exposure of A549 cells to 3-methylcholanthrene (3MC), a known AHR agonist, resulted in a significant induction of ADM mRNA and intracellular protein levels (Fig 3A). Furthermore, tumor cell lines from different anatomical origins showed an increase in ADM mRNA upon exposure to TCDD (Fig 3B) which peaked 24 hours after the initial insult (Fig 3C). Additionally, in vivo preliminary data suggested that BP triggers ADM expression in liver, kidney, heart, lung and testis in AHR+/+ mice (supplemental Fig 2) while no induction was observed in AHR−/− mice.
Figure 3.

AHR activation results in induction of ADM expression in vitro and in vivo. (A) Increased ADM mRNA and protein was observed in A549 cells after exposure to 3MC for 24h. (B) Exposure of cancer cells from different anatomical origins to TCDD resulted in enhanced ADM mRNA levels, (C) which was found to be time dependent in breast and lung cells lines. (D) AHR null fibroblasts showed no increase in ADM transactivation after exposure to CSE (0.06 puffs/ml for 24h) as opposed to isogenic AHR+/+ cells. A marked increase in basal ADM transactivation was observed between untreated AHR+/+ and AHR−/− cells. (E) Forced co-expression of AHR and ARNT was required in A549 cells to induce upregulation of ADM mRNA. Consistently, transient transfection of two different siRNA for AHR resulted in significant blockage of ADM expression. (F) AHR-ARNT dependent stimulation of ADM expression was shown to be time dependent. (G) Low ADM transactivation driven by the complete ADM promoter (pGL3P-AM2725) in AHR null cells was restored by ectopic expression of AHR in a dose dependent manner.
Several reports in the literature have implicated ADM as part of a generalized physiological response to different stress conditions (38, 39). In order to rule out non-specific effects and to confirm the involvement of AHR as part of a defined molecular response involved in the regulation of ADM expression upon exposure to tobacco smoke, AHR−/− and AHR+/+ fibroblasts were compared for their ability to activate ADM transactivation. A twofold induction in luciferase activity (under the regulation of the complete ADM promoter; pGL3P-AM2725) was observed only in AHR+/+ but not AHR−/− cells exposed to CSE (Fig 3D). Interestingly, a significant difference in the reporter activity was noted between untreated AHR+/+ and AHR−/− cells, suggesting that basal activity of AHR (unrelated to activation by xenobiotic substances) is also relevant in the regulation of ADM expression. As expected, significant differences in transactivation activity were observed when pGudLuc was transfected in AHR+/+ and AHR−/− fibroblasts (supplemental Fig 3).
Forced overexpression of AHR in mice results in the development of spontaneous tumors (40). Following the same rationale in an in vitro system, we artificially expressed AHR in A549 cells and tested levels of ADM. The AHR is a heterodimeric transcription factor which exerts its transcriptional activity upon binding to ARNT (41). Co-expression of AHR and ARNT was needed to trigger a significant increase in ADM transactivation (Fig 3E) that was subsequently shown to be time dependent (Fig 3F). Consistently, blockage of ADM expression was achieved after transfection of siRNA hairpins targeting AHR (Fig 3E and supplemental Fig 4 A,B). A high level of correlation between AHR and ADM mRNA levels was observed in murine normal tissues (supplemental Fig 4 C,D). In a recovery experiment, ectopic reintroduction of AHR in AHR−/− fibroblast restored the transcriptional activation of ADM in a dose dependent manner to the levels in AHR wild type cells (Fig 3G).
Two intronic XREs drive CSE-activated AHR-induced transcriptional transactivation of ADM
XREs contain a 5-nucleotide core sequence (GCGTG) flanked by variable residues (42–44). A search for XREs in the ADM gene revealed the presence of 12 consensus sequences located in the promoter and both intronic and exonic regions (Fig 4A). Deletion of the 5′ region of the ADM promoter containing 4 of 6 XREs located before the start codon, in exon 2 (pGL3P-AM689), had no significant effect on the relative luciferase activity in CSE (0.06 puffs/ml) treated A549 or AHR+/+ fibroblasts (Fig 4B). In contrast, deletion of the region containing the two intronic XREs proximal to the ADM start codon resulted in a significant decrease in luciferase activity in both A549 and AHR+/+ (Fig 4B). No differences were found in luciferase activity when the same plasmids were transfected in AHR−/− fibroblasts (data not shown) demonstrating that the observed differences were mediated through AHR. These experiments suggested that the two XREs located in intron 1 are more relevant to the overall transcriptional activation of the ADM gene by AHR than the ones located upstream of exon 1. Mutation of these XREs individually (pGL3P-AM689Δ-190 and pGL3P-AM689Δ-50) resulted in a moderate reduction in the luciferase activity while mutation of both XREs (pGL3P-AM689Δ-50Δ-190) notably hindered the transcriptional activity of the ADM gene in A549 and AHR+/+ cells (Fig 4C).
Figure 4.

Two intronic XREs are responsible for transactivation of ADM by AHR. (A) 12 putative XREs (orange ovals) were found across the entire genomic sequence of the ADM gene. (B) The two XREs contained in the first intron of the ADM gene are largely responsible for its transactivation in A549 cells and AHR+/+ fibroblasts exposed to CSE (0.06 puffs/ml). (C) Single mutation of the XREs present in this region results in significant diminution of ADM transactivation in both A549 and AHR wild type cells exposed to CSE (0.06 puffs/ml). Mutation of both XREs further impedes ADM transactivation. Exons are indicated green boxes.
ADM mediates tobacco-induced tumor growth in vivo
To study the functional role of ADM in tobacco-induced cancer progression in vivo we developed a new preclinical model in which mice bearing A549 subcutaneous xenograft tumors were exposed to CSE intraperitoneal injections. CSE significantly enhanced tumor growth in a dose dependent manner reaching a maximum at 0.18 mg tar (Fig 5A; no further growth stimulation was observed using 0.24 mg tar injection in a separate experiment; data not shown). Gene expression analysis of tumor tissues at the completion of the experiment revealed a CSE dose dependent increase of ADM expression and of several known AHR target genes including CYP1A1, CYP1B1, APP and AAKT (Fig 5B). To further ascertain the involvement of ADM in the observed CSE enhanced tumor progression, we used a previously identified small molecule inhibitor of ADM (NSC16311, (30)). Treatment with the ADM small molecule inhibitor completely abrogated the enhanced tumor growth induced by CSE further supporting the role of ADM in tobacco induced tumor progression in vivo (Fig 5C).
Figure 5.
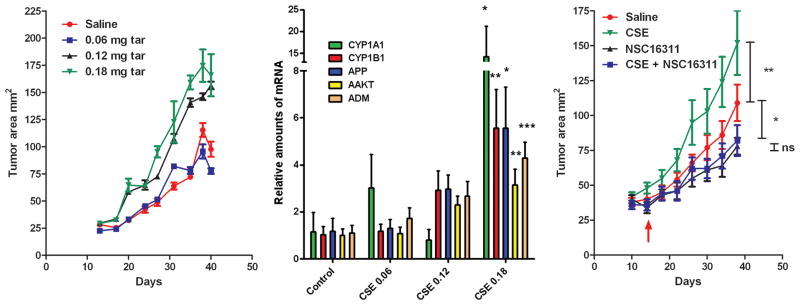
ADM drives tobacco-induced tumor progression. (A) Dose dependent tumor growth response to systemic injections of increasing concentrations of CSE. (B) Dose dependent gene expression of ADM and AHR responsive genes in xenograft tumors exposed to increasing concentrations of CSE (0.06–0.18 mg tar). (C) Blockage of ADM with the small molecule antagonist NSC16311 inhibits CSE-induced tumor growth. Treatments with NSC16311 started two weeks after injection of tumor cells (red arrow).
ADM and AHR are co-upregulated in lung cancer biopsies
The expression of ADM and AHR was analyzed in serial lung sections from 133 patients with adenocarcinoma, squamous cell carcinoma or bronchioloalveolar carcinoma (Fig 6 and supplemental Table 3). A high degree of correlation (70%) between AHR and ADM expression was found independently of the tumor etiology (chi-square test < 0.001 for all groups in supplemental Table 3). Over 56.4% of the biopsies were positive for both AHR and ADM and about 14.3% of them were negative for both markers. 10.5% of the samples were positive for one of the markers but not for the other (18.8% of the spots in the tissue array were missing or did not contain tumor mass and were not included in the analysis). In all matching tumors AHR and ADM colocalized to the same tissue areas (Fig 6 A–F). Illustrating the complexity of this colocalization, the ADM and AHR proteins were localized to either the nucleus and/or the cytoplasm in different biopsies (Fig 6 A,B and C,D). Tumor associated inflammatory cells consistently showed staining for both AHR and ADM (Fig 6 E,F).
Figure 6.

ADM and AHR are co-upregulated in patient lung tumors. A–F, immunodetection of AHR (A,C,E) and ADM (B,D,F) in serial sections of lung cancer biopsies (100x for panoramic images and 600x for inserts).
ADM and AHR target genes are coassociated and efficiently differentiate between smoke-induced cancer and non smoker patients in clinical sample microarrays
To validate the results observed in patient biopsies, we extended the comparison to microarray analysis on metadata consisting of 304 human subjects (supplemental Table 2) stratified into 4 groups of samples: 1) non smoker group, 2) smokers without cancer, 3) smokers with lung cancer, and 4) former smokers. Welsh t-test analysis of unequal variance was implemented to test the statistical differences between non smokers and patients in each of the other 3 groups (Fig 7A). Interestingly a significant difference was observed in smokers without/with cancer groups (p < 0.05), but not in former smoker group (p = 0.197), confirming the results from tissue biopsies. It is proposed that genes co-expressed with any given gene in both parallel and anti-parallel directions are enriched for significant biological functions, processes and/or cellular components (32). Using multivariate analysis we estimated 51,748 pair wise computations (supplemental table 4) and selected genes with correlation p-value of <= 0.01 (after 100 permutations for each computation) in each group for further analysis. In order to understand the relationship between AHR and smoking, we used an AHR module generated as described in the methods section and applied the Fisher’s exact analysis to estimate the level of significance of enrichment (Fig 7B). At a p-value cutoff of 0.05, only smoker groups without/with cancer were found to be significant but not former smoker group. Unsupervised clustering of the 136 AHR target genes (14) co-associated with ADM (pooled list with MPT p-value <=0.05 in any of the groups) efficiently separated the smoker with cancer group from the non-smokers and former smokers (Fig 7C). Smokers with no cancer clustered generally with patients in the cancer group although some smokers were grouped with the non smokers and former smokers groups.
Figure 7.

ADM and AHR target genes are coassociated and efficiently differentiate smoking-induced cancer and non-smokers in clinical sample microarrays. (A) Comparison of ADM gene expression in non smokers, and various smoker groups as indicated. The category smoker includes group of smokers who do not have cancer. P value from t-test analysis between non smokers and each of the smoker groups is indicated on top of each bar. (B) Bar graph of –log10 Fisher test P values depicting the enrichment scores of AHR module (479 genes) in each of the groups. The number of genes co-expressed with ADM gene (percentage enrichment in parentheses) is shown on top of each bar (C). Unsupervised clustering of expression of AHR target genes co-associated with ADM in non smoker (red), smoker without cancer (green), smoker with cancer (brown) and former smoker (blue) groups. Rows represent genes and columns represent sample classes.
Discussion
According to the World Health Organization, cigarette smoking is one of the leading causes of preventable death in the world, accounting for 6 million deaths each year worldwide (45). Despite the relevance of this epidemic, the molecular mechanisms underlying tobacco smoke toxicity and carcinogenic potential remain elusive. The complexity of tobacco smoke, containing over 55 carcinogens, has generated confusion about the mechanisms by which it induces lung cancer (46). Lung cells are equipped with molecular sensors able to detect and trigger the processing of carcinogens present in tobacco smoke. A well known example is the AHR gene which is activated by tumorigenic substances in tobacco smoke and regulates the expression of vast proto-oncogenic gene networks (47). However, the individual effector genes responsible for the carcinogenic effect of tobacco smoke-activated AHR are largely unknown. Here, we provide in vitro, in vivo and clinical data supporting the critical role of ADM as a mediator of the carcinogenic potential of AHR and CS.
Although ADM is elevated in lung cancer (48), little is known about its regulatory expression and functional implication in tobacco smoke related lung malignancies. In this study we have found that CS and prototypical AHR-activating exogenous ligands, such as TCDD, 3MC and BP, activate AHR and stimulate ADM expression both in vitro and in vivo, but not in cells and mice lacking the AHR gene. However, AHR-induced ADM expression in AHR−/− fibroblasts is rescued upon ectopic re-expression of AHR. Using site directed mutagenesis we demonstrate that ADM transactivation is regulated through binding of AHR to two XREs mapped to the intronic region proximal to the translation start codon in the ADM gene. Taken together these data demonstrate that AHR directly regulates ADM expression. This regulation occurs under normal physiological conditions as suggested by a strong correlation of AHR and ADM expression in different organs and lower ADM levels in AHR-/- fibroblast compared to their wild type counterparts, in absence of external stimulus. However, we provide evidence that this regulatory mechanism may also play a role during early stages of tumorigenesis in the lung. Mice inhaling CS for short periods of time (6 weeks) showed overall overexpression of ADM mRNA and significant upregulation of AHR and ADM protein in bronchial epithelial cells prior to any noticeable histological changes. It is known that ADM induces activation of proto-oncogenic early response genes such as c-jun (49). Microarray metaanalysis shows increased ADM levels and significant coassociation with AHR target genes (14) in smokers with no cancer, including c-jun, supporting an early role of the AHR/ADM axis in cigarette exposed non transformed lung cells, prospectively involved in tumor promotion.
Forced overexpression of AHR in mice results in spontaneous tumors in different anatomical sites (40, 50) and abnormal overexpression of AHR has been shown in different tumor types including lung adenocarcinoma (11). In our model, ectopic expression of AHR in the lung adenocarcinoma cell line A549 causes upregulation of ADM. This provides a working model to explain ADM overexpression in patients with lung tumors (48) and to understand the oncogenic and angiogenic properties of AHR in smoke-induced lung cancers (51). Supporting this model, we found paralleled protein overexpression of AHR and ADM in 133 patient lung tumors and associated inflammatory cells and significant coassociation between ADM and AHR targeted genes in microarray data from smokers with cancer. Unsupervised clustering of AHR module subset correlated with ADM gene from 304 sample microarray validation set separated smokers without/with cancer from non smokers and former smoker. Interestingly, while most smokers with no cancer clustered together with the smokers with cancer group, some clustered with non smokers and former smokers. A likely explanation is that smokers without cancer group have a considerable level of heterogeneity in the expression of AHR associated genes potentially reflecting the level of progression towards a malignant phenotype. The prospective prognostic value of ADM/AHR coassociated gene data sets requires further study.
Further functional evidence of the role of ADM in tobacco/AHR enhanced tumor progression comes from a new preclinical model of cancer progression in which lung adenocarcinoma A549 cells are subcutaneously implanted in mice which are systemically treated with CSE. In this model, increasing doses of CSE were directly correlated to tumor growth and expression of tumor ADM and other AHR induced genes such as CYP1A1. More importantly, tobacco driven tumor growth was suppressed by the ADM small molecule antagonist NSC16311 (30) providing a direct indication that ADM mediates tobacco smoke induced tumor progression.
In conclusion, CS drives ADM expression in lung cancer cells through AHR. Our data supports that tobacco induced ADM plays a significant role in lung cancer progression and reveals the AHR/ADM axis as a rational target for drug therapy in the clinical management of cigarette-induced cancers.
Supplementary Material
Acknowledgments
We thank Drs. James Mulshine (Rush University Medical Center, Chicago, Illinois, USA) and Dimitra Bourmpoulia (Extracellular Matrix Pathology Section, Center for Cancer Research, National Cancer Institute, NIH, Gaithersburg, MD, USA) for their critical comments on the manuscript.
Footnotes
The authors have declared that no conflict of interest exists.
References
- 1.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–9. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, et al. Cancer statistics, 2008. CA Cancer J Clin. 2008;58:71–96. doi: 10.3322/CA.2007.0010. [DOI] [PubMed] [Google Scholar]
- 3.Vineis P, Alavanja M, Buffler P, Fontham E, Franceschi S, Gao YT, et al. Tobacco and cancer: recent epidemiological evidence. Journal of the National Cancer Institute. 2004;96:99–106. doi: 10.1093/jnci/djh014. [DOI] [PubMed] [Google Scholar]
- 4.Herbst RS, Onn A, Sandler A. Angiogenesis and lung cancer: prognostic and therapeutic implications. J Clin Oncol. 2005;23:3243–56. doi: 10.1200/JCO.2005.18.853. [DOI] [PubMed] [Google Scholar]
- 5.Pleasance ED, Stephens PJ, O’Meara S, McBride DJ, Meynert A, Jones D, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–90. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Takahashi H, Ogata H, Nishigaki R, Broide DH, Karin M. Tobacco Smoke Promotes Lung Tumorigenesis by Triggering IKKbeta- and JNK1-Dependent Inflammation. Cancer Cell. 2010;17:89–97. doi: 10.1016/j.ccr.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitamura M, Kasai A. Cigarette smoke as a trigger for the dioxin receptor-mediated signaling pathway. Cancer Lett. 2007;252:184–94. doi: 10.1016/j.canlet.2006.11.015. [DOI] [PubMed] [Google Scholar]
- 8.Kasai A, Hiramatsu N, Hayakawa K, Yao J, Maeda S, Kitamura M. High levels of dioxin-like potential in cigarette smoke evidenced by in vitro and in vivo biosensing. Cancer Res. 2006;66:7143–50. doi: 10.1158/0008-5472.CAN-05-4541. [DOI] [PubMed] [Google Scholar]
- 9.Martey CA, Baglole CJ, Gasiewicz TA, Sime PJ, Phipps RP. The aryl hydrocarbon receptor is a regulator of cigarette smoke induction of the cyclooxygenase and prostaglandin pathways in human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2005;289:L391–9. doi: 10.1152/ajplung.00062.2005. [DOI] [PubMed] [Google Scholar]
- 10.Chang JT, Chang H, Chen PH, Lin SL, Lin P. Requirement of aryl hydrocarbon receptor overexpression for CYP1B1 up-regulation and cell growth in human lung adenocarcinomas. Clin Cancer Res. 2007;13:38–45. doi: 10.1158/1078-0432.CCR-06-1166. [DOI] [PubMed] [Google Scholar]
- 11.Lin P, Chang H, Tsai WT, Wu MH, Liao YS, Chen JT, et al. Overexpression of aryl hydrocarbon receptor in human lung carcinomas. Toxicol Pathol. 2003;31:22–30. doi: 10.1080/01926230390173824. [DOI] [PubMed] [Google Scholar]
- 12.Vogel CF, Li W, Sciullo E, Newman J, Hammock B, Reader JR, et al. Pathogenesis of aryl hydrocarbon receptor-mediated development of lymphoma is associated with increased cyclooxygenase-2 expression. Am J Pathol. 2007;171:1538–48. doi: 10.2353/ajpath.2007.070406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, et al. Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor. Proc Natl Acad Sci U S A. 2000;97:779–82. doi: 10.1073/pnas.97.2.779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sartor MA, Schnekenburger M, Marlowe JL, Reichard JF, Wang Y, Fan Y, et al. Genomewide analysis of aryl hydrocarbon receptor binding targets reveals an extensive array of gene clusters that control morphogenetic and developmental programs. Environ Health Perspect. 2009;117:1139–46. doi: 10.1289/ehp.0800485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zudaire E, Martinez A, Cuttitta F. Adrenomedullin and cancer. Regul Pept. 2003;112:175–83. doi: 10.1016/s0167-0115(03)00037-5. [DOI] [PubMed] [Google Scholar]
- 16.Garayoa M, Martinez A, Lee S, Pio R, An WG, Neckers L, et al. Hypoxia-inducible factor-1 (HIF-1) up-regulates adrenomedullin expression in human tumor cell lines during oxygen deprivation: a possible promotion mechanism of carcinogenesis. Mol Endocrinol. 2000;14:848–62. doi: 10.1210/mend.14.6.0473. [DOI] [PubMed] [Google Scholar]
- 17.Ouafik L, Sauze S, Boudouresque F, Chinot O, Delfino C, Fina F, et al. Neutralization of adrenomedullin inhibits the growth of human glioblastoma cell lines in vitro and suppresses tumor xenograft growth in vivo. Am J Pathol. 2002;160:1279–92. doi: 10.1016/S0002-9440(10)62555-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinez A, Vos M, Guedez L, Kaur G, Chen Z, Garayoa M, et al. The effects of adrenomedullin overexpression in breast tumor cells. J Natl Cancer Inst. 2002;94:1226–37. doi: 10.1093/jnci/94.16.1226. [DOI] [PubMed] [Google Scholar]
- 19.Oehler MK, Norbury C, Hague S, Rees MC, Bicknell R. Adrenomedullin inhibits hypoxic cell death by upregulation of Bcl-2 in endometrial cancer cells: a possible promotion mechanism for tumour growth. Oncogene. 2001;20:2937–45. doi: 10.1038/sj.onc.1204422. [DOI] [PubMed] [Google Scholar]
- 20.Zudaire E, Martinez A, Garayoa M, Pio R, Kaur G, Woolhiser MR, et al. Adrenomedullin is a cross-talk molecule that regulates tumor and mast cell function during human carcinogenesis. Am J Pathol. 2006;168:280–91. doi: 10.2353/ajpath.2006.050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kahn ML. Blood is thicker than lymph. J Clin Invest. 2008;118:23–6. doi: 10.1172/JCI34485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nikitenko LL, Fox SB, Kehoe S, Rees MC, Bicknell R. Adrenomedullin and tumour angiogenesis. Br J Cancer. 2006;94:1–7. doi: 10.1038/sj.bjc.6602832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Evans JJ, Chitcholtan K, Dann JM, Guilford P, Harris G, Lewis LK, et al. Adrenomedullin interacts with VEGF in endometrial cancer and has varied modulation in tumours of different grades. Gynecol Oncol. 2012;125:214–9. doi: 10.1016/j.ygyno.2011.12.429. [DOI] [PubMed] [Google Scholar]
- 24.Deng B, Zhang S, Miao Y, Han Z, Zhang X, Wen F, et al. Adrenomedullin expression in epithelial ovarian cancers and promotes HO8910 cell migration associated with upregulating integrin alpha5beta1 and phosphorylating FAK and paxillin. J Exp Clin Cancer Res. 2012;31:19. doi: 10.1186/1756-9966-31-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mulero-Navarro S, Pozo-Guisado E, Perez-Mancera PA, Alvarez-Barrientos A, Catalina-Fernandez I, Hernandez-Nieto E, et al. Immortalized mouse mammary fibroblasts lacking dioxin receptor have impaired tumorigenicity in a subcutaneous mouse xenograft model. J Biol Chem. 2005;280:28731–41. doi: 10.1074/jbc.M504538200. [DOI] [PubMed] [Google Scholar]
- 26.Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–50. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du B, Leung H, Khan KM, Miller CG, Subbaramaiah K, Falcone DJ, et al. Tobacco smoke induces urokinase-type plasminogen activator and cell invasiveness: evidence for an epidermal growth factor receptor dependent mechanism. Cancer Res. 2007;67:8966–72. doi: 10.1158/0008-5472.CAN-07-1388. [DOI] [PubMed] [Google Scholar]
- 28.Martinez A, Elsasser TH, Muro-Cacho C, Moody TW, Miller MJ, Macri CJ, et al. Expression of adrenomedullin and its receptor in normal and malignant human skin: a potential pluripotent role in the integument. Endocrinology. 1997;138:5597–604. doi: 10.1210/endo.138.12.5622. [DOI] [PubMed] [Google Scholar]
- 29.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, et al. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–6. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 30.Martinez A, Julian M, Bregonzio C, Notari L, Moody TW, Cuttitta F. Identification of vasoactive nonpeptidic positive and negative modulators of adrenomedullin using a neutralizing antibody-based screening strategy. Endocrinology. 2004;145:3858–65. doi: 10.1210/en.2003-1251. [DOI] [PubMed] [Google Scholar]
- 31.Martinez A, Miller MJ, Unsworth EJ, Siegfried JM, Cuttitta F. Expression of adrenomedullin in normal human lung and in pulmonary tumors. Endocrinology. 1995;136:4099–105. doi: 10.1210/endo.136.9.7649118. [DOI] [PubMed] [Google Scholar]
- 32.Gustin MP, Paultre CZ, Randon J, Bricca G, Cerutti C. Functional meta-analysis of double connectivity in gene coexpression networks in mammals. Physiol Genomics. 2008;34:34–41. doi: 10.1152/physiolgenomics.00008.2008. [DOI] [PubMed] [Google Scholar]
- 33.Yoder PJ, Blackford JU, Waller NG, Kim G. Enhancing power while controlling family-wise error: an illustration of the issues using electrocortical studies. J Clin Exp Neuropsychol. 2004;26:320–31. doi: 10.1080/13803390490510040. [DOI] [PubMed] [Google Scholar]
- 34.Peter H, Westfall SSY. Resampling-based multiple testing: examples and methods for P-value adjustment. New York: Wiley and Sons; 1993. [Google Scholar]
- 35.Zeeberg BR, Feng W, Wang G, Wang MD, Fojo AT, Sunshine M, et al. GoMiner: a resource for biological interpretation of genomic and proteomic data. Genome Biol. 2003;4:R28. doi: 10.1186/gb-2003-4-4-r28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Team RDC. A language and environment for statistical computing. R Foundation for Statistical Computing; Vienna: 2012. [Google Scholar]
- 37.The R Project for Statistical Computing. 2012 [cited; Available from: http://www.r-project.org/
- 38.Khan S, Michaud D, Moody TW, Anisman H, Merali Z. Effects of acute restraint stress on endogenous adrenomedullin levels. Neuroreport. 1999;10:2829–33. doi: 10.1097/00001756-199909090-00024. [DOI] [PubMed] [Google Scholar]
- 39.Tang F, Wong MP, Hwang IS, Li YY. Ether stress increases adrenomedullin gene expression and levels in the rat adrenal. Horm Metab Res. 2005;37:585–8. doi: 10.1055/s-2005-870534. [DOI] [PubMed] [Google Scholar]
- 40.Andersson P, McGuire J, Rubio C, Gradin K, Whitelaw ML, Pettersson S, et al. A constitutively active dioxin/aryl hydrocarbon receptor induces stomach tumors. Proc Natl Acad Sci U S A. 2002;99:9990–5. doi: 10.1073/pnas.152706299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–40. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 42.Saatcioglu F, Perry DJ, Pasco DS, Fagan JB. Aryl hydrocarbon (Ah) receptor DNA-binding activity. Sequence specificity and Zn2+ requirement. J Biol Chem. 1990;265:9251–8. [PubMed] [Google Scholar]
- 43.Yao EF, Denison MS. DNA sequence determinants for binding of transformed Ah receptor to a dioxin-responsive enhancer. Biochemistry. 1992;31:5060–7. doi: 10.1021/bi00136a019. [DOI] [PubMed] [Google Scholar]
- 44.Swanson HI, Chan WK, Bradfield CA. DNA binding specificities and pairing rules of the Ah receptor, ARNT, and SIM proteins. J Biol Chem. 1995;270:26292–302. doi: 10.1074/jbc.270.44.26292. [DOI] [PubMed] [Google Scholar]
- 45.World Health Organization. Tobacco. 2011 [cited; Available from: http://www.who.int/mediacentre/factsheets/fs339/en/index.html.
- 46.Hecht SS. Tobacco smoke carcinogens and lung cancer. J Natl Cancer Inst. 1999;91:1194–210. doi: 10.1093/jnci/91.14.1194. [DOI] [PubMed] [Google Scholar]
- 47.Dertinger SD, Nazarenko DA, Silverstone AE, Gasiewicz TA. Aryl hydrocarbon receptor signaling plays a significant role in mediating benzo[a]pyrene- and cigarette smoke condensate-induced cytogenetic damage in vivo. Carcinogenesis. 2001;22:171–7. doi: 10.1093/carcin/22.1.171. [DOI] [PubMed] [Google Scholar]
- 48.Beer DG, Kardia SL, Huang CC, Giordano TJ, Levin AM, Misek DE, et al. Gene-expression profiles predict survival of patients with lung adenocarcinoma. Nat Med. 2002;8:816–24. doi: 10.1038/nm733. [DOI] [PubMed] [Google Scholar]
- 49.Ouafik L, Berenguer-Daize C, Berthois Y. Adrenomedullin promotes cell cycle transit and up-regulates cyclin D1 protein level in human glioblastoma cells through the activation of c-Jun/JNK/AP-1 signal transduction pathway. Cell Signal. 2009;21:597–608. doi: 10.1016/j.cellsig.2009.01.001. [DOI] [PubMed] [Google Scholar]
- 50.Moennikes O, Loeppen S, Buchmann A, Andersson P, Ittrich C, Poellinger L, et al. A constitutively active dioxin/aryl hydrocarbon receptor promotes hepatocarcinogenesis in mice. Cancer Res. 2004;64:4707–10. doi: 10.1158/0008-5472.CAN-03-0875. [DOI] [PubMed] [Google Scholar]
- 51.Zhu BQ, Heeschen C, Sievers RE, Karliner JS, Parmley WW, Glantz SA, et al. Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell. 2003;4:191–6. doi: 10.1016/s1535-6108(03)00219-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.