

Abstract
Objectives
This is a pilot study designed to identify gene expression profiles able to stratify head and neck squamous cell carcinoma (HNSCC) tumors that may or may not respond to chemoradiation or radiation therapy.
Study Design
We prospectively evaluated 14 HNSCC specimens, arising from patients undergoing chemoradiotherapy or radiotherapy alone with curative intent. A complete response was assessed by clinical evaluation with no evidence of gross tumor after a 2-year follow-up period.
Methods
Residual biopsy samples from eight complete responders (CR) and six nonresponders (NR) were evaluated by genome-wide gene expression profiling using HG-U133A 2.0 arrays. Univariate parametric t-tests with proportion of false discoveries controlled by multivariate permutation tests were used to identify genes with significantly different gene expression levels between CR and NR cases. Six different prediction algorithms were used to build gene predictor lists. Three representative genes showing 100% crossvalidation support after leave-one-out crossvalidation (LOOCV) were further validated using real-time QRT-PCR.
Results
We identified 167 significant probe sets that discriminate between the two classes, which were used to build gene predictor lists. Thus, 142 probe sets showed an accuracy of prediction ranging from 93% to 100% across all six prediction algorithms. The genes represented by these 142 probe sets were further classified into different functional networks that included cellular development, cellular movement, and cancer.
Conclusions
The results presented herein offer encouraging preliminary data that may provide a basis for a more precise prognosis of HNSCC, as well as a molecular-based therapy decision for the management of these cancers.
Keywords: Head and neck cancer, squamous cell carcinoma, chemoradiation response, microarrays
INTRODUCTION
Head and neck squamous cell carcinomas (HNSCC) may arise in diverse locations but have a common etiologic association with tobacco and/or alcohol exposure. The management of HNSCC often consists of surgical resection, followed by postoperative radiotherapy or chemoradiation therapy. However, cure rates from chemoradiotherapy or radiation alone, with preservation of organ function, have been reported comparable to those achieved by combined surgery and postoperative radiotherapy.1 Unfortunately, a significant percentage of patients treated with chemoradiation or radiation therapy alone do not respond and must subsequently undergo surgical resection, which is associated with high morbidity. This subset of patients might benefit from surgery prior to adjunctive therapy but their identification remains a clinical challenge requiring new testing methods.
To date, most of the studies that define the molecular markers for prediction of radiation response are based on the observation of gene expression using immunostaining, Northern blot, or Western blot analysis of a single or several genes, such as p532 and p16,3 among others. The results vary among different studies, and some results are contradictory. Genomic information, in the form of massive profiles of gene expression within tumor samples, has recently demonstrated the ability to identify characteristics that reflect tumor behavior, disease progression, and outcomes, including cancer recurrence. Several studies have now reported the use of gene expression patterns to classify and sometimes to predict disease outcomes in cancer patients.4,5
Lately, a study was published assessing gene expression by utilizing a gene array technique with a restricted number of candidate genes, and compared a very limited number of tumors to identify genes involved in radiation therapy resistance. In the resulting analysis, investigators reported that 60 genes correlated with radiation therapy resistance of two squamous cell tumors.6 Furthermore, high-density microarrays, including the Affymetrix® GeneChip arrays, have been recently used as a tool for pretherapeutic gene expression profiling for response prediction of rectal adenocarcinomas to pre-operative chemoradiotherapy.7 In the present study, we propose to extend this approach to the prediction of radiation response of human HNSCC tumors, using the Affymetrix® GeneChip platform, through analysis of the expression profiles of a series of tumor samples demonstrating chemoradiation or radiation resistance and chemoradiation or radiation sensitivity.
MATERIALS AND METHODS
Tissue Specimens and RNA Isolation
Specimens for analysis were obtained under a Virginia Commonwealth University institutional review board-approved protocol and anonymized. Fourteen HNSCC tumor biopsies were collected at the time of surgery and transported within 15 minutes to the surgical pathology laboratory for immediate processing. Residual tissue from each surgical pathology specimen was snap-frozen in liquid nitrogen and stored at −80°C until RNA extraction. A complete description of the clinical features and chemoradiation therapy response is listed in Table I. Histopathological scoring of standard features (tumor content, stroma contribution, presence of necrosis, and grade) was performed by a pathologist on hematoxylin and eosin-stained frozen sections adjacent, above, and below the tissue used for RNA isolation. All samples used for this study contained more than 70% tumor. Total RNA was extracted and the quality evaluated using a sample-processing method previously established in our laboratory.8 To extract RNA, multiple 10 μm-thick frozen sections of the tissue samples were placed directly in TRIZOL reagent (InvitrogenTM Life Technologies, Carlsbad, CA). A subsequent cleanup process with RNeasy reagents (QIAGEN Inc., Valencia, CA) was performed according to the manufacturer’s protocol. RNA purity was judged by spectrophotometry at 260, 270, and 280 nm. RNA integrity as well as cDNA and cRNA synthesis products were assessed by running 1 μL of every sample in RNA 6000 Nano LabChips® on the 2100 Bioanalyzer (Agilent Technologies, Foster City, CA).
TABLE I.
HNSCC Patient Demographics and Tumor Location/Staging.
| Responder | Nonresponder | ||
|---|---|---|---|
| Age at biopsy (in years) | Mean | 54 | 62 |
| Range | 39–70 | 45–79 | |
| Gender | Male | 8 | 3 |
| Female | 0 | 3 | |
| Stage | III | 3 | 4 |
| IV A | 4 | 2 | |
| IV B | 1 | 0 | |
| Chemotherapy | Yes | 6 | 4 |
| No | 2 | 2 | |
| Location | Lip, oral cavity | 2 | 0 |
| Larynx | 1 | 5 | |
| Pharynx | 5 | 1 |
HNSCC = head and neck squamous cell carinoma.
Microarray Analysis
The Affymetrix® protocol has been previously described.8,9 Briefly, starting from 5 μg of total RNA, we performed a single-strand cDNA synthesis primed with a T7-(dT24) oligonucleotide and a second-strand cDNA synthesis with the Escherichia coli DNA Polymerase I, using the GeneChip® One-Cycle cDNA Synthesis Kit (Manufactured by Invitrogen for Affymetrix). The resulting cDNA was purified using the cDNA cleanup reagents from the GeneChip Sample Cleanup Module (developed and manufactured by QIAGEN® especifically for GeneChip arrays). Biotinylation of the cRNA was achieved by an in vitro transcription (IVT) reaction using the GeneChip IVT Labeling Kit. After a 37°C incubation for 16 hours, the labeled cRNA was purified using the cRNA cleanup reagents from the GeneChip Sample Cleanup Module and fragmented in fragmentation buffer (40 mM Tris-Acetate, pH 8.1, 100 mM KOAc, 30 mM MgOAc) for 35 minutes at 94°C. Ten micrograms of fragmented cRNA were hybridized on the GeneChip® Human Genome U133A 2.0 (HG-U133A 2.0) array for 16 hours at 60 rpm in a 45°C hybridization oven. The HG-U133A 2.0 array provides comprehensive coverage of the transcribed human genome by including 22,277 probe sets that analyze the expression level of over 17,000 human transcripts. The arrays were washed and stained with streptavidin phycoerythrin (SAPE; Molecular Probes, Eugene, OR) in the Affymetrix fluidics workstation. To amplify the fluorescent signal, SAPE solution was added twice with an antistreptavidin biotinylated antibody (Vector Laboratories, Burlingame, CA) staining step in between. Every chip was scanned at a high resolution, with pixelations ranging from 2.5 μm down to 0.51 μm, by the Affymetrix GeneChip® Scanner 3000 according to the GeneChip® Expression Analysis Technical Manual procedures (Affymetrix, Santa Clara, CA). After scanning, the raw intensities for every probe were stored in electronic files (in .DAT and .CEL formats) by the GeneChip® Operating Software (GCOS) (Affymetrix). The overall quality of each array was assessed by monitoring the 3′/5′ ratios for a housekeeping gene (GAPDH) and the percentage of “Present” genes (%P); where arrays exhibiting GAPDH 3′/5′ <3.0 and %P >40% were considered good quality arrays. Moreover, whisker boxplots were used to assess and compare the intensity distribution across all the arrays included in this study, using the boxplot function from the Bioconductor affy package, run on R 2.4.0.10
Statistical Analysis
Continuous clinical variables were analyzed with t-tests, whereas the significance of categorical variables was assessed by examining the resulting P-value from the Fisher’s exact test.
For the microarray data analysis, background correction, normalization, and estimation of probe set expression summaries was performed using the log-scale Robust Multiarray Analysis (RMA) method.11 Filtering and analyses were performed with the BRB-ArrayTools v3.1.0 (Biometric Research Branch, National Cancer Institute), an Excel add-in that collates microarray data with sample annotations. The resulting expression dataset was filtered to eliminate genes that are presumably not expressed in any of the samples. The latter was assessed by filtering out probe sets with expression summaries lower than the mean value for the limit of quantification (1.5 pM) assessed with the lowest hybridization control gene, AFFX-BioB ± 1.96 × standard deviation (SD). Using this reduced dataset, we performed t-tests for each probe, and assessed statistical significance for multivariate analysis by performing permutations to identify those genes that significantly distinguish between complete responder (CR) and nonresponder (NR) HNSCC tumor samples. In addition to adjusting the P-values with permutation analyses, we estimated the q-values to assess probe set specific false discovery rates (FDR) using the Bioconductor q-value package.12
Prediction analyses were performed by applying a leave-one-out crossvalidation (LOOCV) approach. For this, six prediction algorithms were used: compound covariate predictor, diagonal linear discriminant analysis, 1-nearest neighbor, 3-nearest neighbors, nearest centroid, support vector machines. We performed 2,000 permutations, which resulted in P <.002 for all prediction algorithms. The LOOCV method takes into account the sample size and is suitable for small sample numbers.13
Functional Analysis of Response Predicting Genes
A data set containing Affymetrix® probe set IDs as gene identifiers and corresponding fold changes in expression levels and their associated significance (P-value) was uploaded into in the Ingenuity Pathways Analysis (Ingenuity® Systems, www.ingenuity.com) application. Each gene identifier was mapped to its corresponding gene object in the Ingenuity Pathways Knowledge Base. These genes, called Focus Genes, were overlaid onto a global molecular network developed from information contained in the Ingenuity Pathways Knowledge Base. Networks of these Focus Genes were then algorithmically generated based on their connectivity, or interactions between one another. Biological networks were ranked by score, where the score corresponds to the likelihood of a set of genes being found in the networks due to random chance; that is, a score of 3 indicates that there is a 1/1,000 chance that the focus genes are in a network due to random chance. A score of 3 was used as the cutoff for identifying gene networks significantly involved in chemoradiation or radiation therapy response in HNSCC tumors.
QRT-PCR
Real-time, quantitative reverse transcriptase polymerase chain reaction (QRT-PCR) was used to assess gene expression levels of selected genes using TaqMan® chemistry. Hence, probes and primer sets for detection of TNC, PTHLH, and LTB transcripts were obtained from inventoried assays (Applied Bio-systems, Foster City, CA). Thus, gene-specific probes labeled in the 5′ end with FAM (6-carboxyfluorescein) and in the 3′ end with a dark quencher were used for all the target genes of interest. For all the samples, cyclophilin A (PPIA) from the Predeveloped TaqMan® Assay Reagents (Applied Biosystems) was used as endogenous control. The experiments were performed in the ABI Prism 7500 Sequence Detection System (Applied Biosystems) using the TaqMan® One-Step PCR Master Mix Reagents Kit. All the samples were tested in triplicate. The cycling conditions were 48°C for 30 minutes, 95°C for 10 minutes, 40 cycles of 95°C for 15 seconds, and 60°C for 1 minute. The 2−ΔΔCt method was used to calculate fold changes in the expression levels of the genes of interest.14 The efficiency of amplification for each gene was calculated by running 10-fold serial dilutions of template samples.15
Immunohistochemical Analysis
Genes that satisfied the following criteria were studied further by immunohistochemical (IHC) analysis of HNSCC formalin-fixed paraffin-embedded (FFPE) sections: 1) gene expression in NR tumors was significantly higher than in CR tumors by the two different methods, QRT-PCR and GeneChip (Affymetrix) analysis, and 2) the relative gene expressions determined by the two methods were significantly correlated with each other. Thus, we further validated the protein levels of tenascin C (TNC) and parathyroid hormone-related peptide (PTHrP).
The TNC IHC was performed using a rabbit affinity isolated anti-TNC antibody (Sigma-Aldrich, St. Louis, MO) at a dilution of 1:150; and PTHrP immunohistochemical analysis was performed using a rabbit polyclonal anti-PTHrP (1–34) antibody (Bachem Americas, Inc., Torrance, CA) at a dilution of 1:200.
Slides were deparaffinized through three changes of Xylene for 5 minutes each and rehydrated through two changes of 100% reagent alcohol for 3 minutes each, one change of 95% reagent alcohol for 3 minutes, and one change of 80% reagent alcohol for 3 minutes. Endogenous peroxidase activity was blocked by incubation in 0.3% hydrogen peroxide at room temperature for 5 minutes. Both IHC stains were performed using Heat Induced Epitope Retrieval (HIER), consisting of a 20-minute incubation in Target Retrieval solution, pH = 6.0 (Dako Corporation, Carpinteria, CA) and a 20-minute cooling period.
Slides were placed on an Autostainer Plus™ (Dako Corporation), on which all subsequent incubations were performed at room temperature and all washes consisted of a rinse with Wash Buffer (Dako Corporation) and an aspiration. Nonspecific binding sites were blocked with Serum Free Universal Protein Block (Dako Corporation) for 5 minutes. Slides were then washed and incubated with the primary antibody at the above-mentioned dilutions in Antibody Diluent with Background Reducing Agents (Dako Corporation) for 1 hour. Then, we performed an incubation with biotinylated goat antirabbit secondary antibody (Santa Cruz Biotechnology, Inc., Santa Cruz, CA), dilution 1:500 in previously noted antibody diluent, a wash, and subsequent incubation in Envision+™ Dual Link (Dako Corporation) for 30 minutes. Slides were washed again and incubated for 10 minutes in DAB+ (diaminobenzidine; Dako Corporation).
Slides were removed from the Autostainer Plus™ and were counterstained with Gills III Hematoxylin (Poly Scientific, Bay Shore, NY), blued in tap water, rinsed in deionized water, dehydrated through one change of 80% reagent alcohol for 3 minutes, one change of 95% reagent alcohol for 3 minutes, and three changes of 100% reagent alcohol for 3 minutes each. The dehydration was followed with three clearing changes of Xylene for 5 minutes each and coverslipped using Tissue Tek Glas Mounting Media (Sakura, Torrance, CA) and 24 × 55 #1 cover-glasses (Cardinal Health, McGaw Park, IL). Images of the immunohistochemical-stained tissue sections were digitized using the Nikon ACT-1, version 2.63 system.
RESULTS
Patients, RNA, and Microarray Quality
Characteristics of the 14 HNSCC patients and tumor biopsies are presented in Table I. No associations were found between chemoradiotherapy response and age at biopsy (P= .205), gender (P = .110), stage (III vs. IV A or higher) (P = .767), or tumor location (lip, oral cavity vs. larynx and pharynx (P = .615). Chemoradiotherapy response was assessed by complete clinical evaluation and CR was determined when there was no evidence of gross tumor after a 2-year follow-up period.
Capillary electrophoresis was performed to assess the quality of the total RNA preparations from all the tissue samples processed in this study. Specifically, we aimed at assessing the degradation level, if any, in every sample. No signs of degradation were evident in any of the samples analyzed in this study as revealed by RNA integrity number (RIN) >7.0, suggesting that good quality RNA material was systematically obtained from these samples (Table II). Moreover, the hybridized arrays did not show any significant difference in performance (Table II); therefore, no sample was excluded in subsequent analyses for failing to pass quality control (QC) standards. Likewise, boxplots for the RMA expression summaries across the 14 GeneChips (Fig. 1) revealed similar intensity distributions, as expected.
TABLE II.
RNA and Microarray Quality Control Parameters.
| Microarray Alias | RIN | 3′/5′ GAPDH | %P |
|---|---|---|---|
| VLA1 | 9.9 | 0.96 | 59.10 |
| VLA5 | 8.9 | 1.04 | 56.50 |
| VLA6 | 9.3 | 1.04 | 55.70 |
| VLA9 | 8.3 | 1.02 | 49.60 |
| VLA10 | 8.8 | 1.08 | 54.20 |
| VLA11 | 8.2 | 1.37 | 56.40 |
| VLA15 | 8.3 | 1.06 | 58.00 |
| VLA16 | 7.0 | 1.57 | 53.90 |
| VLOO4 | 10.0 | 0.93 | 52.00 |
| VLOO6 | 8.4 | 0.96 | 57.30 |
| VLOO7 | 8.5 | 0.94 | 55.60 |
| VLOO8 | 8.3 | 1.04 | 49.30 |
| VLOO9 | 8.0 | 0.94 | 52.70 |
| VLOO13 | 8.5 | 1.10 | 57.50 |
RIN = RNA integrity number; %P = percent present.
Fig. 1.
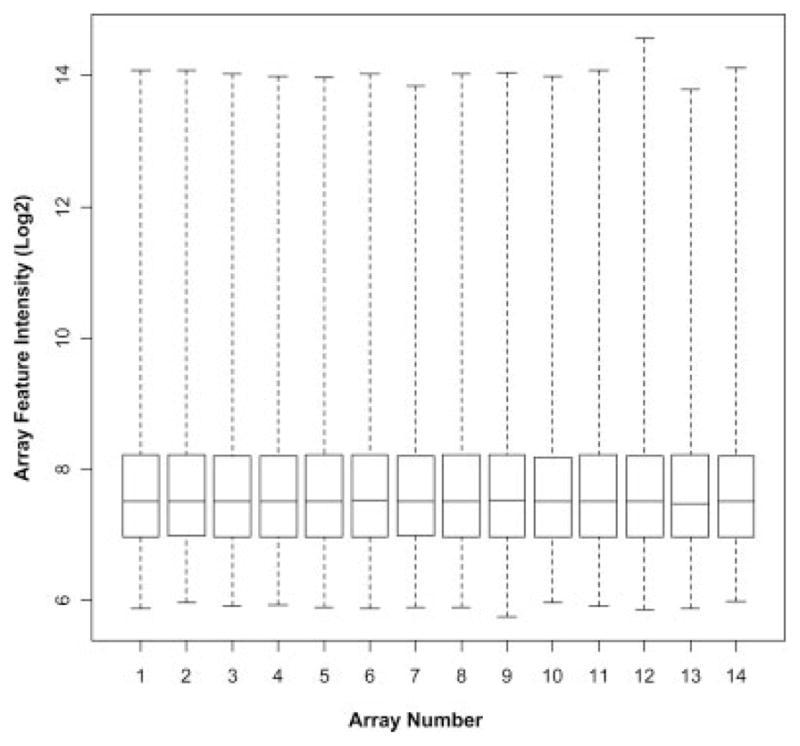
Feature intensity distribution on HG-U133A 2.0 arrays. Box plot representation of the array feature intensities after RMA normalization of all 14 arrays. Boxes represent the interquartile range, 75th percentile at the top and 25th percentile at the bottom. The line in the box represents the 50th percentile, or median. Whiskers represent the rest of the distribution of the log2-transformed feature intensity values.
Differential Gene Expression Between CR and NR HNSCC Tumors
We evaluated 14 HNSCC tumor biopsies, comprising eight chemoradiotherapy or radiation therapy responders (CR) and six NRs, by genome-wide gene expression profiling using HG-U133A 2.0 arrays. After background correction, normalization, and probe set expression summaries using RMA, 16,592 probe sets (above AFFX-BioB ± 1.96 SD) were retained for further analysis.
Unsupervised hierarchical cluster analyses based on these 16,592 probe sets failed to reveal a clear association between samples from same location or discriminate between CR and NR tumor samples (Fig. 2), suggesting that a small subset of the genes may be involved in the genetics of these tumors’ response to chemoradiotherapy or radiation therapy.
Fig. 2.

Unsupervised hierarchical cluster analysis. Unsupervised cluster dendrogram using Euclidian distance and average linkage, based on 16,592 probe sets for the 14-array dataset. Tumor biopsy locations are indicated as follows: PH, pharynx; LA, larynx; and LOC, lip, oral cavity. The response status for each sample is also indicated as complete responder (CR) or nonresponder (NR).
Univariate parametric t-tests with proportion of false discoveries controlled by multivariate permutation tests were used to identify genes with significantly different gene expression levels between CR and NR cases. Thus, we identified 167 significant probe sets (univariate P < .001, multivariate P = .008, based on 10,000 permutations) that discriminate between the two classes (Fig. 3). Among them, we identified probe sets corresponding to genes involved in cell adhesion and motility (such as: ADAM9, ARHGAP5, ICAM3, LAMA3, MYH10, MYO5A, TNC, and TSPAN7, among others); and genes involved in regulation of transcription (such as: TIEG, MEF2A, MYCPBP, SMAD5, and TCF4, among others). Also, we found genes corresponding to cell proliferation (including: CDC37L1, PCTK2, PTHLH, and SEPT11) and genes related to apoptosis (comprising: TOSO, BAG5, FOSL2, SULF1, and LTB) were differentially expressed in these samples. Interestingly, we also found that the ankyrin repeat domain 17 (ANKRD17) gene, involved in the MutS/MutL/MutH mismatch repair pathway, was upregulated in NR HNSCC tumor biopsy samples.
Fig. 3.
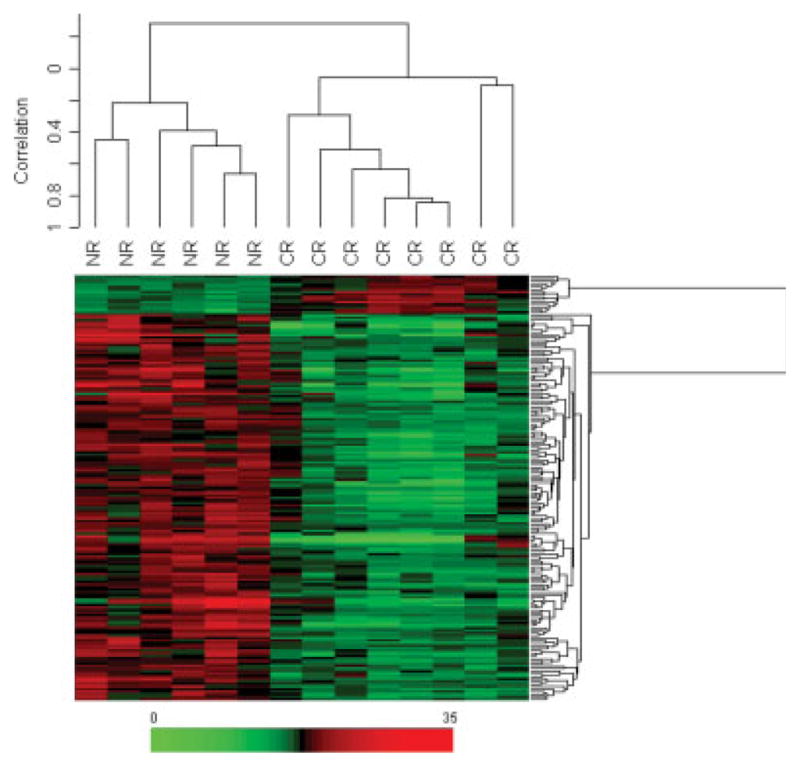
Supervised samples and genes cluster analysis. Two-dimensional hierarchical clustering of samples and genes using Pearson (centered) correlation and average linkage, based on the 167 probe sets that were significantly (P <.001) different between complete responders (CR) and nonresponder (NR) HNSCC tumors. Each of the 167 rows in the heat map located beneath the dendrogram shows the relative expression for that specific gene in the separate 14 cases (columns). The relative gene expression levels (0-fold increase to 35-fold increase) are plotted according to the color scale at the bottom of the heat map, where red and green areas correspond to genes overexpressed and underexpressed, respectively, compared to the median intensity across the 14 samples.
Chemoradiation Response Prediction Analysis
To build a list of chemoradiation or radiation therapy response predictor genes, six different prediction algorithms were used in this study: Compound Covariate Predictor, Diagonal Linear Discriminant Analysis, 1-Nearest Neighbor, 3-Nearest Neighbors, Nearest Centroid and Support Vector Machines. The average success rate and significance of each algorithm are listed in Table III. Of the 167 significant probe sets, 142 probe sets, corresponding to 120 genes, showed an accuracy of prediction ranging from 93% to 100%, depending on the prediction algorithm. In addition, the 142-probe set prediction signature was then applied to an independent cohort of new HNSCC cases, corresponding to five NR tumors: two tumors from the larynx (one stage II and one stage III tumor) and three tumors from a lip, oral cavity location (one stage III and two stage IV B tumors). Thus, we found that this signature was able to correctly assign a response status to 100% of the samples when using five of the six prediction algorithms, whereas the 1-Nearest Neighbor predictor showed 80% accuracy by correctly classifying 4/5 samples. From the 142-probe set predictor, 46 probe sets significantly different, between the CR and NR HNSCC tumors, at an α-level of 0.001, with less than 8% FDR, showed 100% support of the LOOCV algorithms (Table IV).
TABLE III.
Performance of the Predictor Algorithms.
| Microarray Alias | Class | Number of Probe Sets in Classifier | Compound Covariate Predictor Correct? | Diagonal Linear Discriminant Analysis Correct? | 1-Nearest Neighbor Correct? | 3-Nearest Neighbors Correct? | Nearest Centroid Correct? | Support Vector Machines Correct? |
|---|---|---|---|---|---|---|---|---|
| VLA9 | CR | 79 | YES | YES | YES | YES | YES | YES |
| VLA10 | CR | 110 | YES | YES | YES | YES | YES | YES |
| VLOO4 | CR | 107 | YES | YES | YES | YES | YES | YES |
| VLOO6 | CR | 83 | YES | YES | YES | YES | YES | YES |
| VLOO7 | CR | 87 | YES | YES | YES | YES | YES | YES |
| VLOO8 | CR | 101 | YES | YES | YES | YES | YES | YES |
| VLOO9 | CR | 114 | YES | YES | YES | YES | YES | YES |
| VLOO13 | CR | 138 | YES | NO | YES | YES | YES | YES |
| VLA1 | NR | 94 | YES | YES | YES | YES | YES | YES |
| VLA5 | NR | 78 | YES | YES | YES | YES | YES | YES |
| VLA6 | NR | 90 | YES | YES | YES | YES | YES | YES |
| VLA11 | NR | 112 | YES | YES | YES | YES | YES | YES |
| VLA15 | NR | 85 | YES | YES | YES | YES | YES | YES |
| VLA16 | NR | 94 | YES | YES | YES | YES | YES | YES |
| % Correctly classified: | 100 | 93 | 100 | 100 | 100 | 100 | ||
| P-value | < 5e-04 | 0.002 | < 5e-04 | < 5e-04 | < 5e-04 | < 5e-04 |
CR = complete responder; NR = non-responder.
TABLE IV.
Genes Significantly Different (α = 0.001) between CR and NR Samples, Which Support 100% LOOCV.
| Probe Set | Gene Symbol | Fold Change | Parametric P-Value | Q-Value |
|---|---|---|---|---|
| 211756_at | PTHLH | 16.2 | .00027 | 0.080 |
| 206300_s_at | PTHLH | 12.3 | .00015 | 0.080 |
| 210355_at | PTHLH | 8.8 | .00015 | 0.080 |
| 201645_at | TNC | 7.9 | .00045 | 0.080 |
| 203789_s_at | SEMA3C | 7.8 | .00035 | 0.080 |
| 222108_at | AMIGO2 | 6.1 | .00034 | 0.080 |
| 212353_at | SULF1 | 6.0 | .00032 | 0.080 |
| 220253_s_at | LRP12 | 5.8 | .00002 | 0.080 |
| 202600_s_at | NRIP1 | 5.5 | .00034 | 0.080 |
| 202599_s_at | NRIP1 | 5.5 | .00013 | 0.080 |
| 212354_at | SULF1 | 5.0 | .00033 | 0.080 |
| 219926_at | POPDC3 | 4.6 | .00005 | 0.080 |
| 217196_s_at | KIAA1078 | 4.3 | .00022 | 0.080 |
| 203355_s_at | PSD3 | 4.2 | .00008 | 0.080 |
| 212344_at | SULF1 | 4.1 | .00039 | 0.080 |
| 212977_at | CMKOR1 | 4.0 | .00033 | 0.080 |
| 205809_s_at | WASL | 3.9 | 2.50E-06 | 0.054 |
| 202242_at | TM4SF2 | 3.9 | .00012 | 0.080 |
| 218880_at | FOSL2 | 3.8 | .00020 | 0.080 |
| 202363_at | SPOCK | 3.8 | .00004 | 0.080 |
| 203810_at | DNAJB4 | 3.7 | .00006 | 0.080 |
| 208370_s_at | DSCR1 | 3.3 | .00010 | 0.080 |
| 204567_s_at | ABCG1 | 3.3 | .00014 | 0.080 |
| 212887_at | SEC23A | 3.3 | .00013 | 0.080 |
| 222209_s_at | FLJ22104 | 3.1 | .00009 | 0.080 |
| 208663_s_at | TTC3 | 3.0 | .00030 | 0.080 |
| 212794_s_at | KIAA1033 | 3.0 | .00015 | 0.080 |
| 204527_at | MYO5A | 3.0 | .00021 | 0.080 |
| 218613_at | PSD3 | 2.9 | .00022 | 0.080 |
| 212417_at | SCAMP1 | 2.8 | .00016 | 0.080 |
| 202393_s_at | TIEG | 2.8 | .00025 | 0.080 |
| 214449_s_at | RHOQ | 2.8 | .00007 | 0.080 |
| 211760_s_at | VAMP4 | 2.6 | .00026 | 0.080 |
| 212163_at | KIDINS220 | 2.5 | .00010 | 0.080 |
| 212220_at | PSME4 | 2.4 | .00014 | 0.080 |
| 204944_at | PTPRG | 2.4 | .00008 | 0.080 |
| 202739_s_at | PHKB | 2.3 | .00013 | 0.080 |
| 217904_s_at | BACE1 | 2.3 | 1.07E-05 | 0.054 |
| 212492_s_at | JMJD2B | 2.2 | .00012 | 0.080 |
| 203354_s_at | PSD3 | 2.1 | .00010 | 0.080 |
| 212979_s_at | KIAA0738 | 2.1 | 1.40E-06 | 0.005 |
| 221787_at | PHF10 | 2.1 | .00007 | 0.080 |
| 212122_at | RHOQ | 2.0 | .00009 | 0.080 |
| 204840_s_at | EEA1 | 2.0 | .00007 | 0.080 |
| 204220_at | GMFG | −2.2 | .00022 | 0.080 |
| 207339_s_at | LTB | −3.0 | .00019 | 0.080 |
Functional Analysis of the Differential Gene Expression Between CR and NR
The 142 predictor probe sets were overlaid onto a global molecular network developed from information contained in the IPKB. Networks of these genes were then algorithmically generated based on their connectivity, or interactions between one another. Significant biological networks were ranked by score (Table V), identifying Cellular Development, Cellular Movement, Cancer, and Cell Death as the most relevant functional networks. Interconnections within the most significant functional network corresponding to Cellular Development and Cellular Movement (P < 10−50) are shown in Figure 4.
TABLE V.
Significant Functional Networks.
| Network ID | Genes in Network | Score | Focus Genes | Top Functions |
|---|---|---|---|---|
| 1 | ABCG1, ADM, Akt, ARHGAP5, CD2AP, CFLAR, CXCR7, FOSL2, GJA9, IL1R1, INSIG1, Insulin, KLF10, LAMA3, LDL, LTB, Mapk, MCFD2, NF1, Nfat, NFkB, PALLD*, Pdgf, PDGF BB, Pkc(s), PTHLH*, PTPN11, RCAN1, RNF111, SMAD5, SMURF2, SULF1*, TCR, Tgf beta, TNC | 50 | 24 | Cellular development, cellular movement, cell-to-cell signaling and interaction |
| 2 | ACVR1, ARHGAP5, BAG5, CASD1, CDC37L1, COPB1, COPG, CREB1, CYP19A1, EGF, ENTPD7, ETS2, GABPA, GALNT1, GH1, GNAI2, GNB5, GTP, Hsp70, IRAK2, KPNB1, MAP3K12, MAPK8, MBIP, MYH10, ODC1, PCTK2, PTEN, PTHLH*, RGS2, SCG2, SEC23A*, SEC23IP, TP63, VDR | 27 | 15 | Cellular development, cancer, developmental disorder |
| 3 | ADAM9, Ap1, BAZ1B, CDKN1A, CHGN, CHI3L1, CXCL11, CYP19A1, dihydrotestosterone, FOSL2, GMFG, HAND1, ID2, IL11, IL1B, INHBB, IRAK2, IRAK4, KLF10, ME1, MEF2A, MYO1D, MYOD1, ORM1, PCNA, PELI2, PRNP, progesterone, PTHLH*, SHH, SLC4A7, SOX9, VAMP4, VCAN, YTHDC1 (includes EG:91746) | 24 | 14 | Cellular development, cancer, connective tissue development and function |
| 4 | ARHGAP5, ARRB1, ARRB2, BACE1, CAND1, CASP1, CASP3, CD48, CDC42, CDC42BPA, CDC42EP3, COL2A1, DSTN, EDNRA, EIF4G3, HOXD3, IL11, IL13, KIDINS220, LIMK2, MAPK1, MYC, NFKBIA, PPP1CB, PTPRG, RSU1, SCAMP1, SEPT11, SPINT2, SPOCK1, SRC, TGFB1, TNC, TPM1, TSPAN7 | 24 | 14 | Cell death, cellular development, cellular movement |
| 5 | ABL2, ACSL1*, AEBP1, ASPH, CNP, EDEM3*, EIF2S1, glutathione, hydrogen peroxide, IL11, IL1R1, IMPACT, Jnk, MPO, NR1H2, NRIP1*, NRP1, ODC1, PDS5A, QKI (includes EG:9444), RAD21, RBPMS, RHOQ*, SEMA3C*, SIRT1, SLC27A1, SLC7A11*, SMC3, STAG1, STAG2, STAR, TM4SF1, TNF, TRA@, TRAF4 | 22 | 13 | Cell death, lipid metabolism, small molecule biochemistry |
| 6 | CA12, Calmodulin, CAMK2D, CARM1, CDH1, DDX5, DENND4A, DLG1, DNAJB4, EEA1, ETS1, FMR1, GAST, HELZ, Histone h3, ICAM3, IQGAP1 (includes EG:8826), MEIS1, MN1, MTMR2*, MYO5A, NCOA3, NXF1, OGT, PHKB (includes EG:5257)*, PIK3CB, PURA, RNA polymerase I, SBF1, SMYD3, STAU1, TTC3, VDR, WASL, WNT1 | 22 | 13 | Gene expression, RNA trafficking, molecular transport |
Gene symbols in boldface type belong to the 142 probe set predictor list.
Corresponds to genes represented by multiple probe sets in the predictor list.
Fig. 4.

Functional networks. Interconnection of significant functional networks, where gene nodes in different shades of red and green or white depending on being upregulated and downregulated or no change, respectively, in nonresponder (NR) HNSCC samples, as well as the fold change and significance are indicated. The most significant functional network corresponding to Cellular Development, Cellular Movement, and Cell-to-Cell Signaling and Interaction (P < 10−50). The meaning of the node shapes is also indicated.
Validation of Selected Predictor Genes
QRT-PCR assays were used to examine expression levels of representative differentially expressed genes involved in significant biological functions, such as TNC, PTHLH, and LTB to further validate differences in expression seen in the Affymetrix® microarray studies. The 2−ΔΔCt method used to calculate fold changes in the expression levels of the genes of interest compared to one of the tumor samples, assumes that the efficiencies for the endogenous control amplicon (PPIA) and the gene of interest amplicon are the same. Amplification efficiencies were determined for the TNC, PTHLH, LTB, and PPIA amplicons 1:10 dilution series. Thus, we obtained 100.0%, 95.3%, 96.2%, and 92.3% efficiency for the TNC, PTHLH, LTB, and PPIA amplicons, respectively, allowing us to conclude that all amplicons amplify with similar efficiencies. Very good correlations, with Pearson’s r = 0.96 (P < .0001); r = 0.95 (P < .0001), and r = 0.92 (P < .0001) were observed in the fold changes measured by QRT-PCR and those observed in the micro-array analyses for TNC, PTHLH, and LTB, respectively (Fig. 5). These results further validate the microarray data obtained in this study. Furthermore, we also validated the overexpression of TNC and PTHLH genes at the protein level, by performing IHC analyses on representative NR and CR HNSCC cases using FFPE specimens that matched our cohort of frozen samples. Thus, we observed a strong staining of TNC and PTHrP in tumor cells on NR cases, whereas no substantial immunostaining was detected in the CR cases (Fig. 6).
Fig. 5.

QRT-PCR gene expression validation. Relative gene expression of the TNC, PTHLH, and LTB genes in the 14 HNSCC samples, measured by Affymetrix gene expression arrays (bar chart) and by TaqMan chemistry (line chart). The PPIA-normalized fold changes (Log2) were calculated against an arbitrarily chosen sample from this cohort.
Fig. 6.
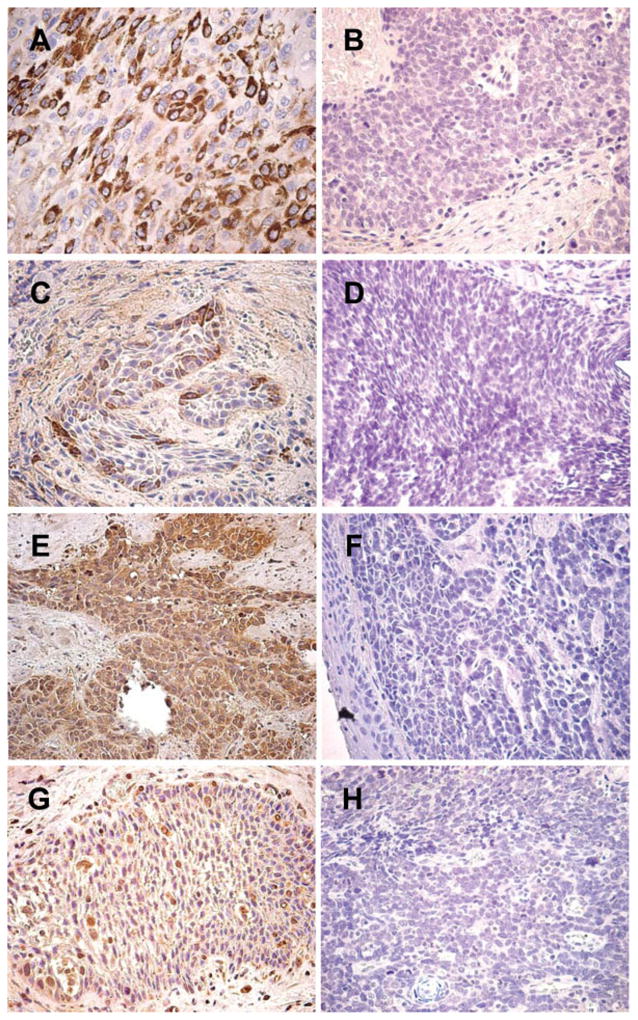
Immunohistochemical analyses of HNSCC FFPE sections. TNC (A, B, C, and D) and PTHrP (E, F, G, and H) IHC staining on NR cases (A, C, E, and G) and CR cases (B, D, F, and H). Both TNC and PTHrP antibodies gave strong cytoplasmic positive staining in NR cases, whereas CR cases showed no substantial reactivity with these antibodies. Magnification, ×400.
DISCUSSION
Concomitant chemoradiotherapy and definitive radiation therapy have become a popular and promising treatment for head and neck cancer, yielding relatively high tumor response rates while achieving organ preservation. However, not all tumors respond to this treatment, and resistance has been observed in a great number of HNSCC cases. Nonresponsive tumors could be spared from definitive chemoradiotherapy or radiation therapy and might be better managed with combined surgical and nonsurgical therapy. Hence, improving the metrics for response prediction is becoming increasingly important in HNSCC tumor management. In this pilot study, we aimed to identify gene expression profiles that could differentiate and further predict chemoradiation or radiation response status in HNSCC tumor biopsy samples. Because tissue handling and RNA degradation could lead to erroneous results, quality assessment of RNA samples as well as microarray data is crucial to overriding technical variability and ensuring robust data sets. Thus, all the samples processed in this study passed rigorous QC criteria previously established in our laboratory8 and the distribution of feature intensities per array was highly similar across all the arrays analyzed in this study, suggesting good-quality microarray data that would ensure reliable results.
For the pilot study presented here, we prospectively collected diagnostic biopsies from 14 HNSCC patients who were treated with definitive chemoradiotherapy or radiation therapy followed by tumor resection in case of nonresponse. Our sample size was taken into account when calculating the significance of the predictor algorithms.
Thus, after identifying 167 probe sets that were significantly (P < .001) different between CR and NR HNSCC tumors, we were able to obtain between 93% to 100% of average classification success rate, depending on the algorithm, with a 142-probe set prediction signature. Interestingly, when applying the 142-probe set predictor to five new HNSCC samples, we found that this signature was able to stratify CR form NR tumors with 100% (5/5) accuracy when using Compound Covariate Predictor, Diagonal Linear Discriminant Analysis, 3-Nearest Neighbors, Nearest Centroid and Support Vector Machines methods, and with 80% (4/5) accuracy when using the 1-Nearest Neighbor algorithm.
Among the 142 probe sets, we identified genes significantly involved in Cellular Development, Cellular Movement, and Cancer, such as TNC, and PTHLH. These genes were significantly overexpressed in NR HNSCC tumors compared to the CRs, both at the mRNA and the protein level, correlating with a more aggressive phenotype in NR tumors. In particular, TNC has been found to facilitate a strong proliferative response when cells are coexposed to TGFβ1, via TGFβIIR-Integrins interactions.16 In addition, adhesion receptors of the integrin family are further associated with tetraspanins, which regulate integrin-dependent cell migration.17 In that respect, we found that the Integrin Signaling Pathway was the most significant canonical pathway across the entire dataset (P = .0226), including the TSPAN7 gene, which was significantly overexpressed in NR tumors.
Additionally, we found that PTHLH was overexpressed in NR HNSCC tumors. It has been proven that PTHrP, encoded by the PTHLH gene, suppresses cell maturation and stimulates cell proliferation.18 Also, it has been reported that PTHrP is produced by oral squamous cell carcinoma, and is involved in malignant conversion by increasing cell proliferation, survival, adhesion, migration, and invasion.19 In addition, PTHrP may have intracrine functions that seem to be connected to cell adhesiveness and motility through integrin expression and actin reorganization in breast cancer cells.20 Interestingly, the localization of β-actin mRNA is dependent on myosin II-B interaction, encoded by the MYH10 gene,21 which we found to be overexpressed in NR samples.
Moreover, regulation of apoptosis seems to play an important role in chemoradiation response in these samples. Our results showed that several differentially expressed genes in this cohort of HNSCC cases were involved in regulation of cell death by apoptosis. Interestingly, TOSO and BAG5, which are antiapopotic genes, were upregulated in NR samples, whereas LTB, which, in cell culture models induces cell death through LTβR signaling,22 was upregulated in CR HNSCC tumor biopsy samples. Also, the fact that ANKRD17, encoding for a protein that interacts selectively with double-stranded DNA containing one or more mismatches, was upregulated in NR HNSCC tumor biopsy samples, suggests that the mismatch repair properties of this protein maybe involved in chemoradiation resistance in HNSCC tumors.
Altogether, our findings strongly suggest that NR HNSCC tumors exhibit a gene expression profile that correlates with a more aggressive tumor growth phenotype, with a deregulation of apoptotic pathways, which may impact radiation and chemoradiation treatment outcome.
CONCLUSIONS
The results presented herein offer encouraging preliminary data that may provide a basis for a more precise prognosis for patients with HNSCC, as well as molecular-based therapeutic management of these cancers. Nonetheless, the implementation of gene expression profiles for treatment stratification and clinical management of cancer patients requires validation in large, independent studies, which are now under evaluation at our institution. These preliminary findings have the potential to shed light on the underlying biology of these tumors and additional studies using an increased number of tumor samples will allow us to further elucidate the mechanisms of chemoradiotherapy and radiation therapy resistance.
Acknowledgments
This work was supported by a grant from the Richmond Eye & Ear Foundation and the Virginia Tobacco Settlement Fund.
We thank Julia Schaum, Carrie Glassman, and Tana Blevins for technical assistance in the tissue acquisition and handling process, as well as Irene Gonzalez for clinical data management. Also, we specially thank Travis Mullan for his expertise on immunohistochemical staining of the HNSCC cases. Human tissues, patient consents, and clinical data were obtained through collaboration with by the Tissue and Data Acquisition and Analysis Core (TDAAC)—Massey Cancer Center and Department of Pathology, VCU, in support of the Tissue Acquisition System to Support Cancer Research (TASSCR) protocol.
BIBLIOGRAPHY
- 1.Adelstein DJ, Li Y, Adams GL, et al. An intergroup phase III comparison of standard radiation therapy and two schedules of concurrent chemoradiotherapy in patients with unresectable squamous cell head and neck cancer. J Clin Oncol. 2003;21:92–98. doi: 10.1200/JCO.2003.01.008. [DOI] [PubMed] [Google Scholar]
- 2.Osman I, Sherman E, Singh B, et al. Alteration of p53 pathway in squamous cell carcinoma of the head and neck: impact on treatment outcome in patients treated with larynx preservation intent. J Clin Oncol. 2002;20:2980–2987. doi: 10.1200/JCO.2002.06.161. [DOI] [PubMed] [Google Scholar]
- 3.Yuen PW, Man M, Lam KY, Kwong YL. Clinicopathological significance of p16 gene expression in the surgical treatment of head and neck squamous cell carcinomas. J Clin Pathol. 2002;55:58–60. doi: 10.1136/jcp.55.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Golub TR, Slonim DK, Tamayo P, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science. 1999;286:531–537. doi: 10.1126/science.286.5439.531. [DOI] [PubMed] [Google Scholar]
- 5.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 6.Hanna E, Shrieve DC, Ratanatharathorn V, et al. A novel alternative approach for prediction of radiation response of squamous cell carcinoma of head and neck. Cancer Res. 2001;61:2376–2380. [PubMed] [Google Scholar]
- 7.Ghadimi BM, Grade M, Difilippantonio MJ, et al. Effectiveness of gene expression profiling for response prediction of rectal adenocarcinomas to preoperative chemoradiotherapy. J Clin Oncol. 2005;23:1826–1838. doi: 10.1200/JCO.2005.00.406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dumur CI, Nasim S, Best AM, et al. Evaluation of quality-control criteria for microarray gene expression analysis. Clin Chem. 2004;50:1994–2002. doi: 10.1373/clinchem.2004.033225. [DOI] [PubMed] [Google Scholar]
- 9.Sugita M, Geraci M, Gao B, et al. Combined use of oligonucleotide and tissue microarrays identifies cancer/testis antigens as biomarkers in lung carcinoma. Cancer Res. 2002;62:3971–3979. [PubMed] [Google Scholar]
- 10.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Irizarry RA, Bolstad BM, Collin F, et al. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storey JD. A direct approach to false discovery rates. J R Stat Soc B. 2002;64:479–498. [Google Scholar]
- 13.Radmacher MD, McShane LM, Simon R. A paradigm for class prediction using gene expression profiles. J Comput Biol. 2002;9:505–511. doi: 10.1089/106652702760138592. [DOI] [PubMed] [Google Scholar]
- 14.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 15.Dumur CI, Dechsukhum C, Wilkinson DS, Garrett CT, et al. Analytical validation of a real-time reverse transcription-polymerase chain reaction quantitation of different transcripts of the Wilms’ tumor suppressor gene (WT1) Anal Biochem. 2002;309:127–136. doi: 10.1016/s0003-2697(02)00265-8. [DOI] [PubMed] [Google Scholar]
- 16.Scaffidi AK, Petrovic N, Moodley YP, et al. αν β3 integrin interacts with the transforming growth factor β (TGFβ) type II receptor to potentiate the proliferative effects of TGFβ1 in living human lung fibroblasts. J Biol Chem. 2004;279:37726–37733. doi: 10.1074/jbc.M403010200. [DOI] [PubMed] [Google Scholar]
- 17.Stipp CS, Hemler ME. Transmembrane-4-superfamily proteins CD151 and CD81 associate with alpha 3 beta 1 integrin, and selectively contribute to alpha 3 beta 1-dependent neurite outgrowth. J Cell Sci. 2000;113:1871–1882. doi: 10.1242/jcs.113.11.1871. [DOI] [PubMed] [Google Scholar]
- 18.Ionescu AM, Schwarz EM, Vinson C, et al. PTHrP modulates chondrocyte differentiation through AP-1 and CREB signaling. J Biol Chem. 2001;276:11639–11647. doi: 10.1074/jbc.M006564200. [DOI] [PubMed] [Google Scholar]
- 19.Yamada T, Tsuda M, Ohba Y, Kawaguchi H, Totsuka Y, Shindoh M. PTHrP promotes malignancy of human oral cancer cell downstream of the EGFR signaling. Biochem Biophys Res Commun. 2008;368:575–581. doi: 10.1016/j.bbrc.2008.01.121. [DOI] [PubMed] [Google Scholar]
- 20.Shen X, Qian L, Falzon M. PTH-related protein enhances MCF-7 breast cancer cell adhesion, migration, and invasion via an intracrine pathway. Exp Cell Res. 2004;294:420–433. doi: 10.1016/j.yexcr.2003.11.028. [DOI] [PubMed] [Google Scholar]
- 21.Latham VM, Yu EH, Tullio AN, Adelstein RS, Singer RH. A Rho-dependent signaling pathway operating through myosin localizes beta-actin mRNA in fibroblasts. Curr Biol. 2001;11:1010–1016. doi: 10.1016/s0960-9822(01)00291-3. [DOI] [PubMed] [Google Scholar]
- 22.Force WR, Glass AA, Benedict CA, et al. Discrete signaling regions in the lymphotoxin-beta receptor for tumor necrosis factor receptor-associated factor binding, subcellular localization, and activation of cell death and NF-kappaB pathways. J Biol Chem. 2000;275:11121–11129. doi: 10.1074/jbc.275.15.11121. [DOI] [PubMed] [Google Scholar]