

Abstract
Tissue fibrosis is a major cause of death in developed countries. It commonly occurs after either acute or chronic injury and affects diverse organs, including the heart, liver, lung, and kidney. Using the renal ablation model of chronic kidney disease, we previously found that the development of progressive renal fibrosis was dependent on p21WAF1/Cip1 expression; the genetic knockout of the p21 gene greatly alleviated this disease. In the present study, we expanded on this observation and report that fibrosis induced by two different acute injuries to the kidney is also dependent on p21. In addition, when p21 expression was restricted only to the proximal tubule, fibrosis after injury was induced in the whole organ. One molecular fibrogenic switch we describe is transforming growth factor-β induction, which occurred in vivo and in cultured kidney cells exposed to adenovirus expressing p21. Our data suggests that fibrosis is p21 dependent and that preventing p21 induction after stress could be a novel therapeutic target.
Keywords: p21, proximal tubule, transforming growth factor-β, fibrosis, cyclin-dependent kinase inhibitor 1A
fibroproliferative diseases contribute to almost half of all deaths in developed countries (53). Progressive kidney fibrosis resulting in chronic kidney disease (CKD) is a prominent member of this disorder. Recently, interest has focused on CKD developing after an acute kidney injury (AKI) (5). Tubular injury initiates regeneration, which is accompanied by cell cycle activation and differentiation to support recovery of the nephron. This same process can also initiate profibrotic pathways. Two fundamental unanswered questions in understanding the pathogenesis of tissue fibrosis are how the fibrotic signal occurs and, especially after kidney injury, which cell type(s) initiates the signal.
Cell cycle activation concomitantly activates genes for cyclin-dependent kinase (Cdk) inhibitory proteins that regulate checkpoint controls (19). We previously described that one such cell cycle regulatory protein, p21WAF1,CIP1, was expressed in all renal injuries examined (33). In the present study, we report that in a model of renal fibrosis caused by unilateral ureteral obstruction (UUO), the genetic knockout of p21 is protective. Furthermore, when we induced the p21 gene specifically in kidney proximal tubule epithelium in mice with a p21 knockout background, UUO resulted in fibrotic changes in the kidney similar to those observed in wild-type mice. These findings indicate that p21 expression can induce the molecular factor(s) needed for fibrosis development, and its expression only in proximal tubules is sufficient to initiate fibrosis throughout the kidney. This fibrotic factor(s) acts through paracrine mechanisms since when only kidney proximal tubule cells were capable of producing p21, the entire kidney was affected and, furthermore, the area of highest proximal tubular density was relatively less fibrotic.
The transforming growth factor-β (TGF-β) signaling pathway plays a major role in all fibrotic diseases, and according to our data, the fibrogenic cytokine TGFβ-1 was not only induced in cultured kidney proximal tubule cells in vitro after p21 expression but also its induction was greater when p21 was overexpressed than after treatment with aristolochic acid (AA), a known inducer of renal fibrosis. TGF-β exposure is known to induce p21 mRNA, but this is the first demonstration that increased p21 expression can similarly result in increased TGF-β mRNA.
METHODS
Proximal tubule cell culture.
Mouse kidney proximal tubule cells (TKPTS cells) (10) were grown at 37°C with 5% CO2 in DMEM-Ham's F-12 medium supplemented with 50 μU/ml insulin and 7% FBS. Cells were grown for 30 h after being split, and we added adenoviruses where indicated to a final multiplicity of infection of 100. Purvalanol, a chemical cell cycle inhibitor, was dissolved in DMSO and added to a final concentration of 9 μM. AA is known to cause progressive renal fibrosis (40, 57) and was added to the culture medium at 0.5 μg/ml. We harvested cells and isolated total RNA using TRIzol reagent (Life Technologies, Grand Island, NY). RNA samples were used for quantitative RT-PCR to analyze mRNA as previously described (25).
For the analysis of TGF-β proteins secreted from cells, we cultured TKPTS cells as described above in complete medium for 24 h, changed the medium to serum-free medium, cultured cells for an additional 3 h, and added adenovirus as indicated. Cells were cultured for 48 h, and medium (10 ml from 4–5 × 106 cells) was concentrated by acetone precipitation. Proteins were redissolved in PBS, resolved by 15% SDS-PAGE, and transferred onto a polyvinylidene difluorid membrane using a Bio-Rad mini-blot apparatus. Membranes were stained with Ponceau to detect protein, and later the stain was removed by washing (data not shown). Membranes were blocked in 5% nonfat milk solution and then incubated with primary antibodies against TGF-β1 (dilution 1:1,000, catalog no. sc-146, Santa Cruz Biotechnology, Santa Cruz, CA) and TGF-β3 (sc-82, Santa Cruz Biotechnology) overnight at 4°C. Membranes were washed extensively, incubated with horseradish peroxidase-linked anti-rabbit antibody (GE Healthcare Bio-Sciences, Pittsburgh, PA), and then washed again. Detection was performed with ECL Western blotting detection reagent (GE Healthcare). After detection, membranes were stripped of antibodies and redeveloped using TGF-β2 antibody (sc-90, Santa Cruz Biotechnology).
For quantification of the rate of decay of TGF-β1 mRNA, TKPTS cells were cultured for 24 h after the addition of p21 adenovirus. Transcription was then inhibited by the addition of the RNA polymerase II-specific inhibitor 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB; Sigma- Aldrich, St. Louis, MO) to a final concentration of 50 μM. At various times after the addition of DRB, total RNA was isolated as described above. The rate of mRNA degradation was subsequently determined using real-time RT-PCR as previously described (47).
Adenoviruses.
Recombinant viruses were generated by homologous recombination using the AdEasy (14) vector system (supplied by Dr. B. Vogelstein). Human p21WAF cDNA (9) was subcloned into pBluescript SK (+) plasmid (Agilent Technologies, Santa Clara, CA). Adenoviruses expressing fusion proteins of green fluorescent protein (GFP) and p21 full-length protein or truncations (NH2-terminal, amino acids 1–91; COOH-terminal, amino acids 83–164) were constructed as previously described (56). Briefly, pEGFP-N3 plasmid (Clontech, Mountain View, CA) was used to fuse p21 in frame with the NH2-terminal end of GFP. The cassette with these genes was excised with HindIII and HpaI (New England Biolabs) and cloned between the HindIII/HpaI sites of pAdTrack-CMV. The resultant plasmids were linearized by digestion with PmeI and subsequently cotransformed into Escherichia coli BJ5183 cells (Agilent) with the pAdEasy-1 adenoviral backbone plasmid. Recombinants were selected for kanamycin resistance, and recombination was confirmed by restriction endonuclease analysis. Recombinant plasmids were linearized with PacI and transfected into Ad-293 cells. Human Cdk2 wild-type and Cdk2 dominant negative (49) (D145N, DN-Cdk2) cDNA were obtained from Dr. Sander van den Heuvel and Dr. Ed Harlow (Massachusetts General Hospital), and the corresponding adenoviruses were constructed as previously described (56). Recombinant adenoviruses were amplified in human embryonic kidney-293 cells, purified by CsCl banding, and stored at −20°C. Adenoviral infections were performed on TKPTS cells at a multiplicity of infection of 100.
Animals.
Mice carrying a deletion of a large portion of the p21 gene in which neither p21 mRNA nor p21 protein is expressed were obtained from Dr. Philip Leder (Harvard Medical School, Cambridge, MA). Mice homozygous for the p21 gene deletion were selected from the offspring of heterozygous matings using PCR analysis of tail DNA as previously described (Tyler Jacks laboratory protocol). Wild-type littermates were used as controls.
Full-length human p21WAF1 cDNA or DN-Cdk2 was fused to GFP as described above. The p21-GFP construct was excised with BglII and NotI and inserted between the BglII and NotI sites in pKAP2 (22). The DN-Cdk2-GFP construct was excised with EcoRV and NotI and inserted between the BglII/Klenow and NotI sites in pKAP2. Both transgenes were constructed to contain a translation termination site immediately after GFP. The resulting constructions contained the transgene-GFP fusion downstream of the testosterone-inducible, kidney proximal tubule-specific kidney androgen-regulated promoter (KAP) and upstream of KAP gene open reading frames 2–5 and termination/polyadenylation signals. The plasmids were linearized with SpeI/NdeI and used to generate transgenic mice. Mice were screened for the presence of the transgenes by PCR amplification of tail DNA using primers specific for the KAP promoter (5′-CCAACTGTGGAAAACCACCT-3′) and GFP (5′-GCGGCCGCTTTACTTGTACAGCTCG-3′), generating fragments of 1,751 or 1,333 bases for DN-Cdk2 or p21 transgenics, respectively. After positive mice had been bred, additional confirmation of the integrity of the transgenes was obtained by sequencing the cDNA-GFP inserts.
Transgenic and p21 knockout mice were crossed to generate strains of mice containing both the p21 gene deletion and either KAP2-p21 or KAP2-DN-Cdk2 transgenes. All mice were bred into a background of 129S1/SvImJ (Jackson Laboratory, Bar Harbor, ME) for at least 10 generations.
Animals were housed at the Veterinary Medical Unit at the Central Arkansas Veterans Healthcare System (Little Rock, AR). When appropriate, animals were painlessly euthanized according to methods of euthanasia approved by the Panel on Euthanasia of the American Veterinary Medical Association. Our animal study protocols were in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee of the Central Arkansas Veterans Healthcare System.
In vivo models of renal fibrosis.
UUO and release were performed similar to previously described techniques (6, 43). Transgene expression was induced in 2-mo-old female mice using either 5-mg 21-day release or 12.5-mg 60-day release subcutaneous testosterone pellets (Innovative Research of America, Sarasota, FL) for 2 wk. The left kidney was exposed through a midline incision under sterile conditions; the ureter was dissected and securely clamped using a micro-clamp (B-1, Fine Science Tools, Foster City, CA) for 3 days and then followed by removal of the clamp. Volume depletion was prevented by the administration of 0.1 ml saline into the peritoneal cavity during both surgeries. The midline incision was closed, and mice were returned to their cages and allowed free access to food and water. As control, sham surgery was performed without clamp of the ureter. Mice were euthanized after 2 wk, and left kidneys were collected for protein, RNA isolation, and histological evaluation. Animals with 3 days of UUO were euthanized without release of the clamp; kidneys were analyzed similarly to the UUO/release samples. Obstruction and reversal of obstruction were confirmed visually by the presence and absence of hydronephrosis (43).
Ischemia was induced (33) in anesthetized animals [pentobarbital sodium (50 mg/kg)] by exposure of the left kidney under sterile conditions through a midline incision. The kidney was decapsulated, and the renal hilum was clamped with a small arterial clamp for 35 min and then released. During the course of the surgery, volume depletion in the mice was prevented by the administration of 0.3 ml saline into the peritoneal cavity. After surgery, animals were returned to their cages and allowed free access to food and water. Forty-two days after reflow, as indicated, animals were euthanized, and left kidneys were prepared for protein isolation.
Quantitative PCR.
TGF-β1 and GAPDH mRNA levels were determined by quantitative RT-PCR. Total RNA was extracted from cells or mouse kidney tissue and treated with RNase-free DNase before reverse transcriptase reaction. Real-time PCR was carried out using the StepOnePlus real-time PCR system (Invitrogen, Foster City, CA) with iTaqSYBR Green Supermix with Rox (Bio-Rad, Hercules, CA). In each experiment, triplicates of 50 ng cDNA (total RNA equivalent) samples were amplified in a 20-μl reaction. Specificity of the amplified product was confirmed by melting curve analysis and agarose gel electrophoresis. For relative quantification, a standard curve was generated from a six-step cDNA dilution series. Samples were amplified with primers for TGF-β1, GAPDH, and 18S rRNA. The relative expression of genes was calculated from the standard curve. Relative quantity was calculated by the ratio of gene-specific and appropriate 18S rRNA expression. The primer sequences in the RT-PCR were as follows: TGF-β1, forward 5′-CGAGGCGGTGCTCGCTTTGT-3′ and reverse 5′-CATAGATGGCGTTGTTGCGGTCCA-3′; and 18S rRNA, forward 5′-AGGAGTGGGCCTGCGGCTTA-3′ and reverse 5′-AACGGCCATGCACCACCACC-3′.
In situ determination and quantitative analysis of fibrotic markers in kidney tissues.
Formalin-fixed, paraffin-embedded 5-μm kidney sections from sham and UUO kidneys were stained with picrosirius red for 1 h. Positive collagen staining in the interstitium was detected by light microscopy using circularly polarized light. Photographs from the entire kidney cross-section were analyzed by ImageJ software (http://rsbweb.nih.gov/ij/download.html) to quantify collagen accumulation. Sections from the same kidneys were stained with periodic acid-Schiff reagent or Masson's trichrome and used to evaluate morphological changes by light microscopy.
Both p21 and Cdk2 were fused in frame with COOH-terminal GFP. These fusion proteins retained the biological activity of the native proteins (42, 56). GFP, which indicated the presence of the transgene, was localized using 5-μm paraffin-embedded kidney sections from transgenic and nontransgenic mice. Sections were deparaffinized, endogenous autofluorescence was quenched by treatment with glycine, and sections were mounted with medium containing 4′,6-diamidino-2-phenylindole. GFP was detected using a Nikon E-800 fluorescent microscope.
Statistical analysis.
Results are presented as means ± SE. Statistical analyses were performed using an unpaired Student's t-test for independent samples. P values of ≤0.02 were considered to be statistically significant.
RESULTS
p21-dependent fibrosis in the mouse kidney.
Using a model of 3-day UUO and 14-day release, we found that induced kidney fibrosis was dependent on p21 expression. As shown in Fig. 1, p21 knockout mice were protected from fibrosis compared with wild-type mice.
Fig. 1.

Detection of interstitial fibrosis in the mouse kidney. Fibrosis was detected by Masson's trichrome stain in mouse kidney sections in p21+/+ (A and C) and p21−/− (B and D) mice harvested from untreated mice (A and B) and mice 2 wk after 3-day unilateral ureteral obstruction (UUO) was released (C and D). Magnification: ×122.
Identification of cells signaling fibrotic changes.
There are conflicting evidences to identify the cell population that eventually differentiates into a fibrotic phenotype. Similarly, the cellular source of the molecular signal for fibrosis has also not been described. We have developed strains of mice in which either p21 or DN-Cdk2 can be induced specifically in kidney proximal tubules (Fig. 2) using testosterone-inducible KAP (22). This promoter confers kidney proximal tubule-specific and androgen-responsive expression on several transgenes (8, 22, 34). Since both p21 and Cdk2 are endogenous proteins, to confirm protein expression of the transgenes, they were fused in frame with COOH-terminal GFP. The activity of the fused protein is unaffected in the transgene (56). As shown in Fig. 2, GFP localization indicated the colocalization of the GFP-fused transgene. Induction of the transgene by testosterone was confirmed by RT-PCR with primers representing the 5′-end of p21 and the 3′-end of GFP (data not shown). Double staining for expressed transgene and proximal tubule-specific protein expression (l-aromatic amino acid decarboxylase) showed colocalization of the transgene with proximal tubule cells (data not shown).
Fig. 2.

Expression of induced transgenes in the mouse kidney. A–C: the kidney cortex (A and C) and corticomedularry junction (B) in nontransgenic (A) and testosterone-treated transgenic (B and C) mice. Green fluorescence shows the localization of p21-green fluorescent protein (GFP) or dominant negative (DN)-cyclin-dependent kinase 2 (Cdk2)-GFP fusion protein transgenes. Magnification: ×244.
To determine the extent of fibrosis in kidney after 3-day UUO and 14-day release, we stained kidney sections from mice with picrosirius red, analyzed the entire kidney cross-section (Fig. 3, representative sections), and quantified fibrosis (Fig. 4, fibrosis bar graphs) using ImageJ software. Compared with kidneys from control mice (Figs. 3A and 4A, fibrosis), there was a small increase in fibrosis in p21 knockout mice that underwent UUO/release (1.0 ± 0.21 vs 2.26 ± 0.42, P = 0.02; Figs. 3B and 4B, fibrosis). The differences in fibrosis after UUO/release were more apparent when p21 knockout mice were compared with mice in which the p21 gene was intact (wild-type: Figs. 3C and 4C fibrosis, 2.26 ± 0.42 vs 7.13 ± 1.00, P = 0.0013; wild-type p21/KAP2-p21 transgenic: Figs. 3D and 4D fibrosis, 2.26 ± 0.42 vs. 5.97 ± 0.48, P = 0.0008). Induction of either p21 or DN-Cdk2 in kidney proximal tubules in mice from a p21 knockout background, which would otherwise be minimally fibrotic after UUO/release, showed a high induction of fibrosis relative to the same surgery in p21 knockout mice (testosterone-treated KAP2-p21 in p21 knockout: Figs. 3E and 4E fibrosis, 2.26 ± 0.42 vs. 7.19 ± 0.41, P = 0.0001; testosterone-treated KAP2-DN-Cdk2 in p21 knockout: Figs. 3F and 4F fibrosis, 2.26 ± 0.42 vs. 8.54 ± 0.66, P < 0.0001).
Fig. 3.

Fibrosis in picrosirius-stained mouse kidney sections. Representative sections of mouse kidneys were stained with picrosirius and analyzed for fibrosis using ImageJ. Kidney sections are representative images from untreated control mice (A) or 2 wk after the release of 3-day UUO (B–F). Mouse genotypes were either p21 wild-type (A, C, and D) or p21 knockout (B, E, and F) mice. Transgenics were treated with testosterone to induce p21-GFP (D and E) or DN-Cdk2-GFP (F). Sections represent areas of the kidney cortex, cortico-medullary (C-M) junction, inner stripe of the outer medulla (ISOM), and medulla. Magnification: ×122.
Fig. 4.

Fibrosis in the kidney. Mouse kidneys were harvested 2 wk after the release of 3-day UUO and processed either for histological picrosirius red staining or RNA isolation. Fibrosis was quantified in picrosirius red-stained sections from the entire cross-section using ImageJ. Mouse genotypes were either p21 wild-type (A, C, and D), p21 knockout (B, E, and F), or transgenic treated with testosterone (D–F) to induce the transgene. Transgenes were either p21 (D and E) or DN-Cdk2 (F). Transgenic mice were either wild-type (D) or p21 knockout (E and F). Mice were either sham treated (A) or underwent UUO/release surgery (B–F). Bar graphs are means ± SE. TGF, transforming growth factor.
The localization of fibrosis observed with picrosirius staining was primarily in the inner stripe of the outer medulla and secondarily in the corticomedullary junction (Fig. 3, C–F). At the same time, very little and patchy fibrosis was observed in the kidney cortex.
Induction of TGF-β in response to p21 expression.
We found that a fibrotic signal was generated by p21 competent kidney proximal tubule cells in vivo. We next wanted to see whether TKPTS cells, which were originally derived from mouse proximal tubule cells (10), were able to generate similar signals in vitro. We used AA exposure as a positive control, since it is known to cause progressive renal fibrosis (40, 57). TGF-β1 mRNA expressed in untreated cells was induced in TKPTS cells after 24-h AA exposure (Fig. 5A, lanes A and B, 1.014 ± 0.016 and 1.813 ± 0.103, P < 0.0001). Also, 48 h after transduction with p21-expressing adenovirus, TGF-β1 mRNA was induced to an even higher level than AA (Fig. 5A, lane C, 4.968 ± 0.493, P < 0.0001 compared with control cells or AA-treated cells). Furthermore, mRNA was induced and TGF-β1 protein was secreted into the medium (Fig. 5B). Similarly, other TGF-β family members were induced and secreted after p21 treatment. We have previously used expression of NH2-terminal and COOH-terminal fragments of p21 protein to differentiate between several possible functions of this protein, and expression of the NH2-terminal fragment increased TGF-β1 mRNA relative to control (Fig. 5A, lane D, 1.488 ± 0.0883, P < 0.0001), but the level of expression was significantly lower than that with full-length p21. Expression of the COOH-terminal fragment did not induce higher TGF-β1 mRNA relative to control (Fig. 5A, lane E, 0.999 ± 0.112, P = 0.81). This indicates that p21 protein could be acting as a Cdk inhibitor to control TGF-β1 mRNA expression, since the binding moiety of Cdk is in this fragment. To explore this possibility, we transduced TKPTS cells with adenovirus expressing DN-Cdk2 or treated with a Cdk-inhibitory drug, purvalanol. Both DN-Cdk2 expression (Fig. 5A, lane F) and purvalanol exposure (Fig. 5A, lane G) increased TGF-β1 mRNA compared with control (1.652 ± 0.2675, P = 0.0004, and 1.686 ± 0.0964, P < 0.0001, respectively). Compared with AA, the increases of TGF-β1 mRNA caused by DN-Cdk2 and purvalanol were statistically equivalent (P = 0.56 and P = 0.39, respectively). p21-GFP primarily localized to the nucleus, whereas DN-Cdk2 primarily localized to both the nucleus and cytoplasm (Fig. 6). The level of induction of TGF-β1 mRNA by p21 compared with DN-Cdk2 could have been influenced by this difference in intracellular localization, as will be discussed below.
Fig. 5.

A: TGF-β mRNA induction in cultured mouse kidney proximal tubule cells (TKPTS cells). TKPTS cells were cultured and either not treated (A) or exposed to aristolochic acid (B), transduced with adenovirus (C–F), or exposed to purvalanol (G). After either 24 h of aristolochic acid or 48 h of virus, RNA was isolated and mRNA induction was determined by real-time PCR. Adenoviruses were expression vectors for either full-length mouse p21 (C), NH2-terminal p21 (D), COOH-terminal p21 (E), or DN-Cdk2 (F). Bar graphs are means ± SE. B: TGF-β proteins in TKPTS cells. TKPTS cells were cultured in complete medium for 24 h, medium was changed into serum-free medium, and cells were infected with adenovirus. Cells were incubated for an additional 48 h before collection. Medium was concentrated by acetone precipitation, and proteins were resolved by 15% PAGE. Western blots were developed using TGF-β1 antibody, TGF-β2 antibody, and TGF-β3 antibody. Samples represent uninfected TKPTS cells (A) and p21 adenovirus-transduced TKPTS cells (B).
Fig. 6.
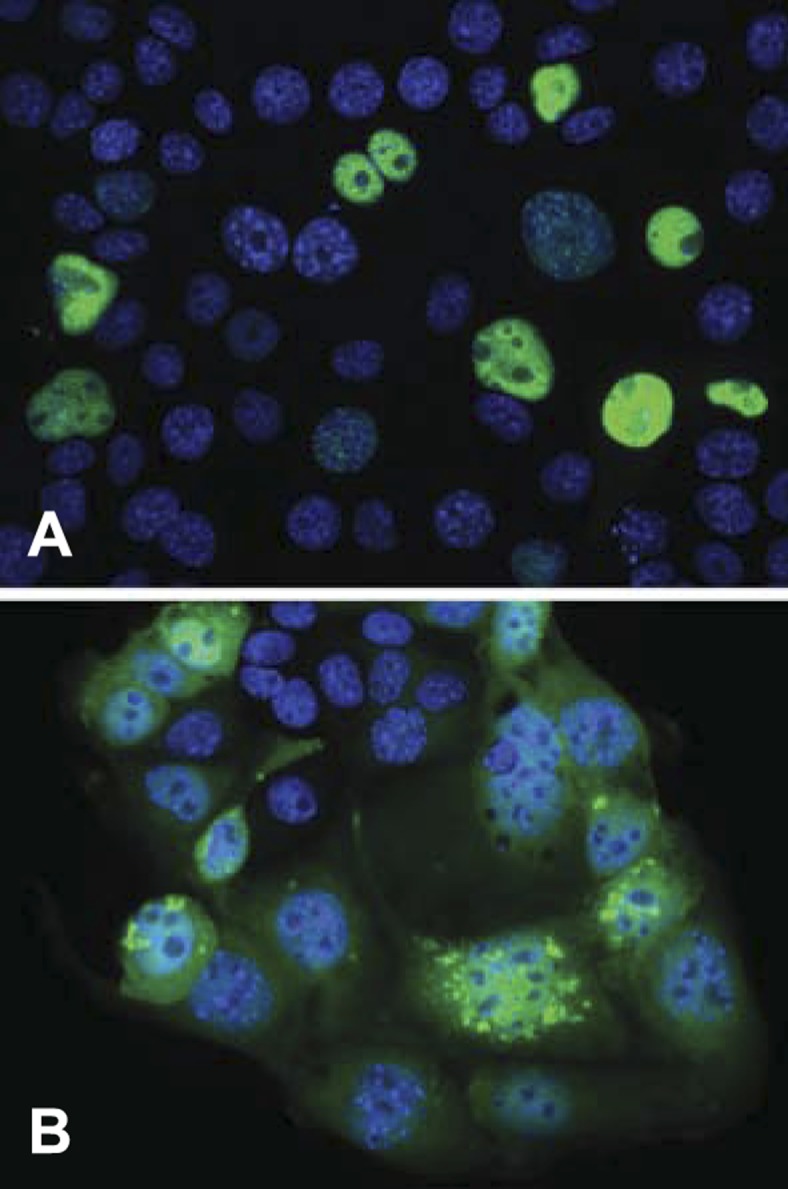
Localization of p21-GFP and DN-Cdk2-GFP in TKPTS cells. TKPTS cells were transduced with adenoviruses expressing either p21-GFP (A) or DN-Cdk2-GFP (B) and photographed after 24 h. Cells were stained with 4′,6-diamidino-2-phenylindole to visualize the nucleus, and the images merged.
To determine whether TGF-β1 expression in vivo after different renal injuries was dependent on p21, we measured mRNA levels after 3 days of UUO (Fig. 7A) and protein expression 42 days after renal ischemia (Fig. 7B). Compared with control, TGF-β1 mRNA was elevated in wild-type mouse kidneys (Fig. 7A, columns A and B, 1.13 ± 0.13 and 2.09 ± 0.07, P = 0.0017) but not elevated in p21 knockout compared with control (Fig. 7A, column C, 1.22 ± 0.06, P = 0.65). Induction of the p21-GFP transgene in kidney proximal tubules in a p21 knockout background also elevated TGF-β1 mRNA relative to control (Fig. 7A, column D, 1.94 ± 0.18, P = 0.085), which was not statistically different from the elevation in wild-type mouse kidneys after UUO (P = 0.49). After renal ischemia, TGF-β1 protein compared with control (Fig. 7B, lane A) was elevated in all p21 knockout mice expressing either p21 or DN-Cdk2 transgene in proximal tubules (Fig. 7B, lanes B and C or lanes D and E, respectively) or in wild-type mice (Fig. 7B, lanes F and G). TGF-β1 protein expression in kidneys from p21 knockout mice after ischemia (Fig. 7B, lanes H and I) was not elevated compared with control. Similarly, TGF-β1 mRNA levels were measured after 3-day UUO/2-wk release (Fig. 4, TGF-β mRNA bar graphs). Compared with kidneys from control mice (Fig. 4A, TGF-β mRNA), there was no difference in the amount of TGF-β1 mRNA in p21 knockout mice that underwent UUO/release (Fig. 4B, TGF-β mRNA, 4.3 ± 0.17 vs 5.4 ± 0.57, P = 0.19). There were minor differences in the amount of TGF-β1 mRNA when control mice were compared with wild-type, UUO/release (Fig. 4C, TGF-β mRNA, 7.5 ± 1.54, P = 0.05), or testosterone-treated KAP2-DN-Cdk2 in p21 knockout (Fig. 4F, TGF-β mRNA, 7.0 ± 1.02, P = 0.04). The greatest statistical differences in the amount of TGF-β1 mRNA expression was when we compared control with either wild-type p21/KAP2-p21 transgenics (Fig. 4D, TGF-β mRNA, 8.15 ± 1.03, P = 0.0074) or p21 knockout/KAP2-p21 transgenics (Fig. 4E, TGF-β mRNA, 7.38 ± 1.09, P = 0.0083). There were no statistical differences when we compared any of the mouse populations from mice that underwent UUO/release.
Fig. 7.

A: TGF-β mRNA induction in the mouse kidney after 3-day UUO. Mouse kidneys were harvested from untreated mice (A) or after 3 days of UUO without release (B–D) and processed for RNA isolation. TGF-β mRNA was quantified by real-time PCR. Mouse genotypes were either wild-type (B), p21 knockout (C), or p21 knockout containing kidney androgen-regulated promoter (KAP)2-p21 transgene (D). Mice with transgenes were treated with testosterone to induce the transgene. The control genotype was wild-type. Bar graphs are means ± SE. B: TGF-β1 protein induction in the mouse kidney after unilateral ischemia. Mouse kidneys were harvested from untreated mice (A) or 42 days after 35 min of unilateral ischemia-reperfusion (B–I) and processed for protein isolation. Proteins were resolved by 15% PAGE, and Western blots were developed using TGF-β1 antibody. Mouse genotypes were either p21 knockout containing KAP2-p21 transgene (B and C), p21 knockout containing KAP2-DN-Cdk2 transgene (D and E), wild-type (F and G), or p21 knockout (H and I). Mice with transgenes were treated with testosterone to induce the transgene. The control genotype was wild-type. Tubulin detection is included as a loading control.
Mechanism of TGF-β1 mRNA induction by p21.
The induction of TGF-β1 mRNA by p21 was significantly higher using full-length protein (Fig. 5A, lane C) than either the NH2-terminal fragment (Fig. 5A, lane D), the COOH-terminal fragment (Fig. 5A, lane E), or both fragments coexpressed together (data not shown). mRNA stability assays (Fig. 8) showed that during the first 8 h after transcription was terminated, the half-life of GAPDH mRNA (∼8 h) was unaffected by p21. In contrast, the half-life of TGF-β1 mRNA increased from ∼10 to ∼30 h after p21 exposure, which would explain its accumulation after p21 adenovirus transduction. This interpretation is supported by our inability to demonstrate any effect on the mouse TGF-β1 promoter (nt −1799 to +59) in a luciferase-linked plasmid by p21 expression (data not shown). Similar conclusions of mRNA stabilization were reached for TGF-β1 mRNA induction by 12-O-tetradecanoylphorbol-13-acetate (51) and by TNF-α induction of TGF-β1 mRNA (47).
Fig. 8.

mRNA stability assay. For quantification of the rate of decay of TGF-β mRNA, TKPTS cells were transduced with p21 adenovirus for 24 h. Transcription was then inhibited by the addition of 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB). At various times after the addition of DRB, medium was removed and total RNA was isolated. The rate of mRNA degradation was subsequently determined using real-time RT-PCR.
DISCUSSION
Studies on AKI using animals models (3, 12, 46) and recent epidemiological studies (2, 17, 28) have showed that AKI significantly contributes to the progression of CKD. At present, the relative importance of contributions from either tubular or interstitial processes to fibrosis and CKD is unclear (18). In this work, we report that p21-depleted kidney cells were protected from UUO-initiated fibrosis (Figs. 1, 3, and 4). Furthermore, in this background, expression of p21 only in proximal tubule cells was sufficient to completely restore fibrosis development throughout the kidney (Figs. 3 and 4). Although it is not possible to determine at this time whether other kidney cells could also generate a fibrotic signal, it is apparent that tubular epithelial cells are sufficient to induce fibrosis after an acute injury. From this present work, we cannot demonstrate the differentiation of cells into myofibroblasts, but it is apparent that the fibrotic signal can be initiated from damaged p21-competent proximal tubules even in a genetic background (p21 knockout) that is resistant to fibrosis.
There is general agreement that interstitial processes of fibrosis are started by paracrine factors. Venkatachalam et al. (50) postulated that signaling events in regenerating tubules that fail to regenerate can produce paracrine factors resulting in inflammation and fibrosis. Here, we show (Fig. 3, E and F) that the fibrotic signal can be initiated solely in proximal tubules, which are concentrated primarily in the kidney cortex and are minimally present in the inner stripe of the kidney. However, induction of p21 in proximal tubules resulted in little fibrosis in the kidney cortex, and similar to the wild-type mouse kidney, was mostly confined to the inner stripe. This observation supports the idea that the signal for fibrosis is most likely a paracrine factor. There is little knowledge with respect to how cellular stress controls the production of profibrotic paracrine molecules. One main factor is the production of TGF-β (11, 20, 21, 23, 25, 26, 44, 45). The TGF-β signaling pathway plays a major role in all fibrotic diseases. Previously published results have demonstrated the importance of TGF-β signaling for myofibroblast transdifferentiation (1) and blockade of integrins thought to activate latent TGF-β reduced collagen staining in kidneys of ureteral obstructed mice by 50% (15). TGF-β stimulates fibrosis not only via its direct effects on fibroblasts but additionally by inducing the production of other molecules like Notch (4), connective tissue growth factor (36), or PDGF-β (11, 48, 50), which also stimulate fibrosis (38). TGF-β receptor-2 (TGFβR-2) is located on the cell membrane and forms heterocomplexes in reaction to ligand binding. TGFβR-2 phosphorylates TGF-β type 1 receptors, which then propagate the signal (52). Secreted TGF-β is arguably the major profibrotic cytokine and a central mediator of fibrosis in multiple organs. It is secreted as a latent complex, and much of its functional regulation in tissues is based on extracellular activation of this latent complex (13, 35). In many epithelial cells, TGF-β induces both p15Ink4b and p21WAF1/Cip1, which in growing cells inhibit Cdk4/6 and Cdk2/1, respectively, although the particular Cdk inhibitors in this response depend on the cell type (29). Expression of p21 and/or p16Ink4a have been shown to be associated with the induction of senescence in a variety of cell types (16), but an examination of several nonproliferative states, including reversible and replicative quiescence and terminal differentiation, showed that mitotic reactivation could be accomplished most effectively by inhibition of p21 expression (37).
Cell cycle arrest or dysregulation has recently been linked to fibrosis. We first described that genetic deletion of the p21 gene in the fibroproliferative model of CKD induced by 5/6 nephrectomy (31) resulted in a hyperplastic rather than hypertrophic response in kidney tubular cells. In contrast with wild-type mice, p21 knockout mice were protected from functional changes, such as severely decreased kidney inulin clearance and elevated blood pressure, and from histological changes, including glomerulosclerosis and interstitial fibrosis. We reported that these differences were apparent from 6 to 16 wk after surgery, but interstitial fibrosis in p21 knockout mice was not evident even 52 wk after renal ablation (data not reported). This model of chronic renal fibrosis is induced by severe decrease of kidney parenchyma and does not represent the progression of chronic disease from an acute form of renal injury. We previously showed that p21 mRNA was induced in the mouse kidney after UUO (33), an acute injury that progresses to chronic fibrosis (6, 24, 25, 27, 39, 43, 44). Using the model of UUO, in which the obstruction was released 3 days after initial surgery (43), we show (Fig. 1) that p21 knockout mice were protected from fibrosis, as visualized with Masson's trichrome stain, compared with wild-type mice. Yang et al. (55), using five in vitro models of AKI, demonstrated the development of fibrosis and production of profibrotic cytokines that was correlated with cell cycle arrest of cultured proximal tubule epithelial cells. We now show that expression of p21, which we found to be elevated in all models of kidney injury (31, 33), directly induced the accumulation of TGF-β mRNA and secretion of TGF-β protein from cultured renal proximal tubule cells (see Fig. 5, A and B). Also, we show that in two in vivo models of acute injury to the kidney (Figs. 4 and 7, A and B), expression of either p21 or DN-Cdk2 only in proximal tubules was sufficient to induce fibrotic changes in the kidney interstitium similar to those observed in wild-type animals. These data solidify the observations that fibrotic factors are paracrine and that kidney proximal tubules play a major role in kidney fibrosis. The acute injury models of UUO and unilateral ischemia-reperfusion extend our previous observation (31) in the 5/6 nephrectomy model of end-stage kidney disease to provide a mechanistic explanation of how proximal tubular injury induces fibrosis. It remains to be seen whether fibrotic cytokines accumulate because of the activity of p21 to inhibit Cdk2 or Cdk1 or affect other proteins. The model of UUO/release also illustrates that p21 expression after 3-day UUO, either by endogenous gene expression in wild-type mice or by induced expression in p21-GFP transgenics, was sufficient to induce TGF-β1 mRNA and lead to fibrotic changes visible 2 wk after release of UUO. In this model, in which the stress of UUO is relieved after 3 days, TGF-β1 mRNA amounts, although still elevated, approach control levels after UUO release (Fig. 4, TGF-β mRNA).
The finding that p21 induction is deleterious in models of chronic renal failure but is beneficial in acute models (31, 32, 41) points toward possible different protein interaction domains of the molecule (56). In addition, it is noteworthy that the nuclear localization of the Cdk-inhibitory domain of p21 seems to be necessary for TGF-β induction since induction was limited using the NH2-terminal half of the protein, which contains the Cdk domain but lacks the nuclear localization signal. This could be the reason why induction by p21 was significantly higher than that by DN-Cdk2, since only the p21 protein localized exclusively to the nucleus (Fig. 6). The COOH-terminal half of p21 was incapable of inducing TGF-β mRNA accumulation (Fig. 5A).
There is evidence of a feedback loop involving TGF-β transcription, p15, Cdk4, and Cdk2 (30). The canonical signaling pathway is that after TGF-β signals and activates its receptors, Smad 2 and Smad 3 then mediate the transcription of TGF-β-responsive genes, including the antiproliferative genes p15 and p21. G1 cyclin-dependent kinases (Cdk4 and Cdk2), which are inhibited by p15 and p21, respectively, can phosphorylate Smad 2 and Smad 3, decreasing their transcriptional activity. The data we show in Figs. 5 and 7 suggest that p21 induction precedes TGF-β signaling. This does not conflict with known pathways of TGF-β signaling, but it suggests that induction of TGF-β in response to kidney injury is dependent on prior expression of p21. It had been pointed out by Cochrane et al. (6) that 6 wk after release in the UUO/release model, there was significant structural and functional repair in the injured kidney. The level of p21 was not measured in this study, but it is possible that decreased stress will reduce p21 induction, which could downregulate TGF-β and resolve fibrosis. Even without relieving stress, it may also be possible to reduce p21 induction therapeutically. Genetic ablation of the p21 gene has little if any known consequences (7), so that it is likely that therapy targeting p21 expression would be beneficial in fibroproliferative diseases.
GRANTS
This work was supported in part by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-54471 (to P. M. Price) and DK-75976 (to D. Portilla) and by a Veterans' Affairs Merit Review (to P. M. Price and D. Portilla) and with resources and the use of facilities at the John L. McClellan Memorial Veterans' Hospital (Little Rock, AR).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
Author contributions: J.M. and P.M.P. conception and design of research; J.M., A.T., S.L., R.H., N.S.H.L.S., D.P., and P.M.P. performed experiments; J.M., D.P., and P.M.P. analyzed data; J.M. and P.M.P. interpreted results of experiments; J.M. and P.M.P. prepared figures; J.M. and P.M.P. drafted manuscript; J.M., A.T., S.L., R.H., N.S.H.L.S., D.P., and P.M.P. edited and revised manuscript; J.M., A.T., S.L., R.H., N.S.H.L.S., D.P., and P.M.P. approved final version of manuscript.
ACKNOWLEDGMENTS
The authors thank Dr. Bert Vogelstein (Johns Hopkins University) for supplying the AdEasy adenovirus cloning vectors and p21 cDNA. The authors also thank Dr. Sander van den Heuvel and Dr. Ed Harlow (Massachusetts General Hospital) for supplying human Cdk2 wild-type and Cdk2 dominant negative cDNAs. The authors also thank Dr. Philip Leder (Harvard Medical School, Cambridge, MA) for p21 knockout mice.
Present address of R. Hodeify: Dept. of Physiology and Biophysics, Weill Cornell Medical College in Qatar, Doha, Qatar.
Present address of D. Portilla: Div. of Nephrology, Univ. of Virginia, Box 800133, Charlottesville, VA 22908.
REFERENCES
- 1.Akhmetshina A, Palumbo K, Dees C, Bergmann C, Venalis P, Zerr P, Horn A, Kireva T, Beyer C, Zwerina J, Schneider H, Sadowski A, Riener MO, MacDougald OA, Distler O, Schett G, Distler JH. Activation of canonical Wnt signalling is required for TGF-β-mediated fibrosis. Nat Commun 3: 1–12, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amdur RL, Chawal LS, Amodeo S, Kimmel PL, Palant C. Outcomes following diagnosis of acute renal failure in U. S veterans: focus on acute tubular necrosis. Kidney Int 76: 1089–1097, 2009. [DOI] [PubMed] [Google Scholar]
- 3.Basile DP, Donohoe D, Roethe K, Osborn JL. Renal ischemic injury results in permanent damage to peritubular capillaries and influences long-term function. Am J Physiol Renal Physiol 281: F887–F899, 2001. [DOI] [PubMed] [Google Scholar]
- 4.Bielesz B, Sirin Y, Si H, Niranjan T, Gruenwald A, Ahn S, Kato H, Pullman J, Gessler M, Haase V, Susztak K. Epithelial notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest 120: 4040–4054, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Canaud G, Bonventre JV. Cell cycle arrest and the evolution of chronic kidney disease from acute kidney injury. Nephrol Dial Transplant; doi: 10.1093/ndt/gfu230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cochrane AL, Kett MM, Samuel CS, Campanale NV, Anderson WP, Hume DA, Little MH, Bertram JF, Ricardo SD. Renal structural and functional repair in a mouse model of reversal of ureteral obstruction. J Am Soc Nephrol 16: 3623–3630, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Deng C, Zhang P, Harper JW, Elledge SJ, Leder P. Mice lacking p21CIP1/WAF1 undergo normal development, but are defective in G1 checkpoint control. Cell 82: 675–684, 1995. [DOI] [PubMed] [Google Scholar]
- 8.Ding Y, Davisson RL, Hardy DO, Zhu LJ, Merrill DC, Catterall JF, Sigmund CD. The kidney androgen-regulated protein promoter confers renal proximal tubule cell-specific and highly androgen-responsive expression on the human angiotensinogen gene in transgenic mice. J Biol Chem 272: 28142–28148, 1997. [DOI] [PubMed] [Google Scholar]
- 9.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF-1, a potential mediator of p53 tumor suppression. Cell 75: 817–825, 1993. [DOI] [PubMed] [Google Scholar]
- 10.Ernest S, Bello-Reuss E. Expression and function of P-glycoprotein in a mouse kidney cell line. Am J Physiol Cell Physiol 269: C323–C333, 1995. [DOI] [PubMed] [Google Scholar]
- 11.Geng H, Lan R, Singha P, Gilchrist A, Weinreb P, Violette S, Weinberg J, Saikumar P, Venkatachalam K. Lysophosphatidic acid increases proximal tubule cell secretion of profibrotic cytokines PDGF-B and CTGF through LPA2- and Gαq-mediated Rho and αvβ6 integrin-dependent activation of TGF-β. Am J Pathol 181: 1236–1249, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geng H, Lan R, Wang G, Siddiqi AR, Naski MC, Brooks AI, Barnes JL, Saikur P, Weinberg JM, Venkatachalam MA. Inhibition of autoregulated TGFβ signaling simultaneously enhances proliferation and differentiation of kidney epithelium and promotes repair following renal ischemia. Am J Pathol 174: 1291–1308, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gleizes PE, Munger JS, Nunes I, Harpel JG, Mazzieri R, Noguera I, Rifkin DB. TGF-β latency: biological significance and mechanisms of activation. Stem Cells 15: 190–197, 1997. [DOI] [PubMed] [Google Scholar]
- 14.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci USA 95: 2509–2514, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Henderson NC, Arnold TD, Katamura Y, Giacomini MM, Rodriguez JD, McCarty JH, Pellicoro A, Raschperger E, Betsholtz C, Ruminski PG, Griggs DW, Prinsen MJ, Maher JJ, Iredale JP, Lacy-Hulbert A, Adams RH, Sheppard D. Targeting of αv integrin identifies a core molecular pathway that regulates fibrosis in several organs. Nat Med 19: 1617–1624, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Herbig U, Sedivy JM. Regulation of growth arrest in senescence: telomere damage is not the end of the story. Mech Ageing Dev 127: 16–24, 2006. [DOI] [PubMed] [Google Scholar]
- 17.Ishani A, Xue JL, Himmelfarb J, Eggers PW, Kimmel PL, Molitoris BA, Collins AJ. Acute kidney injury increases risk of ESRD among elderly. J Am Soc Nephrol 20: 223–228, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kaissling B, LeHir M, Kriz W. Renal epithelial injury and fibrosis. Biochim Biophys Acta 1832: 931–939, 2013. [DOI] [PubMed] [Google Scholar]
- 19.Kastan MB, Bartek J. Cell cycle checkpoints and cancer. Nature 432: 316–323, 2004. [DOI] [PubMed] [Google Scholar]
- 20.Koesters R, Kaissling B, Le Hir M, Picard N, Theilig F, Gebhardt R, Glick A, Hähnel B, Hosser H, Gröne H, Kriz W. Tubular overexpression of transforming growth factor-β1 induces autophagy and fibrosis but not mesenchymal transition of renal epithelial cells. Am J Pathol 177: 632–643, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lan R, Geng H, Polichnowski A, Singha P, Saikumar P, McEwen D, Griffin K, Koesters R, Weinberg J, Bidani A, Kriz W, Venkatachalam K. PTEN loss defines a TGF-β induced tubule phenotype of failed differentiation and JNK signaling during renal fibrosis. Am J Physiol Renal Physiol 302: F1210–F1223, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lavoie JL, Lake-Bruse KD, Sigmund CD. Increased blood pressure in transgenic mice expressing both human renin and angiotensinogen in the renal proximal tubule. Am J Physiol Renal Physiol 286: F965–F971, 2004. [DOI] [PubMed] [Google Scholar]
- 23.Leask A, Abraham DJ. TGF-β signaling and the fibrotic response. FASEB J 18: 816–827, 2004. [DOI] [PubMed] [Google Scholar]
- 24.Li L, Zepeda-Orozco D, Black R, Lin F. Autophagy is a component of epithelial cell fate in obstructive uropathy. Am J Pathol 176: 1767–1778, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Mariappan N, Megyesi J, Shank B, Kannan K, Theus S, Price PM, Duffield JS, Portilla D. Proximal tubule PPARα attenuates renal fibrosis and inflammation caused by unilateral ureteral obstruction. Am J Physiol Renal Physiol 305: F618–F627, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lin H. Transforming growth factor β and the kidney revisited: introduction. Semin Nephrol 32: 225–227, 2012. [DOI] [PubMed] [Google Scholar]
- 27.Lin SL, Kisseleva T, Brenner DA, Duffield JS. Pericytes and perivascular fibroblasts are the primary source of collagen-producing cells in obstructive fibrosis of the kidney. Am J Pathol 173: 1617–1627, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mammen C, Al Abbas A, Skippen P, Nadel H, Levine D, Collet JP, Matsell DG. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis 59: 523–530, 2012. [DOI] [PubMed] [Google Scholar]
- 29.Massagué J. TGFβ in cancer. Cell 134: 215–230, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Matsuura I, Denissova NG, Wang G, He D, Long J, Liu F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 430: 226–231, 2004. [DOI] [PubMed] [Google Scholar]
- 31.Megyesi J, Price PM, Tamayo E, Safirstein RL. The lack of a functional p21WAF1/CIP1 gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci USA 96: 10830–10835, 1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest 101: 777–782, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Megyesi J, Udvarhelyi N, Safirstein RL, Price PM. The p53-independent activation of transcription of p21WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol Renal Fluid Electrolyte Physiol 271: F1211–F1216, 1996. [DOI] [PubMed] [Google Scholar]
- 34.Meseguer A, Catterall JF. Mouse kidney androgen-regulated protein messenger ribonucleic acid is expressed in the proximal convoluted tubules. Mol Endocrinol 1: 535–541, 1987. [DOI] [PubMed] [Google Scholar]
- 35.Munger JS, Harpel JG, Gleizes PE, Mazzieri R, Nunes I, Rifkin DB. Latent transforming growth factor-β structural features and mechanisms of activation. Kidney Int 51: 1376–1382, 1997. [DOI] [PubMed] [Google Scholar]
- 36.Okada H, Kikuta T, Kobayashi T, Inoue T, Kanno Y, Takigawa M, Sugaya T, Kopp J, Suzuki H. Connective tissue growth factor expressed in tubular epithelium plays a pivotal role in renal fibrosis. J Am Soc Nephrol 16: 133–143, 2005. [DOI] [PubMed] [Google Scholar]
- 37.Pajalunga D, Mazzola A, Salzano AM, Biferi MG, De Luca G, Crescenzi M. Critical requirement for cell cycle inhibitors in sustaining nonproliferative states. J Cell Biol 176: 807–818, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Phanish MK, Winn SK, Dockrell ME. Connective tissue growth factor-(CTGF, CCN2)–a marker, mediator and therapeutic target for renal fibrosis. Nephron Exp Nephrol 114: e83-e92, 2009. [DOI] [PubMed] [Google Scholar]
- 39.Picard N, Baum O, Vogetseder A, Kaissling B, Le Hir M. Origin of renal myofibroblasts in the model of unilateral ureter obstruction in the rat. Histochem Cell Biol 130: 141–155, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pozdzik AA, Salmon IJ, Debelle FD, Decaestecker C, Van den Branden C, Verbeelen D, Deschodt-Lanckman MM, Vanherweghem JL, Nortier JL. Aristolochic acid induces proximal tubule apoptosis and epithelial to mesenchymal transformation. Kidney Int 73: 595–607, 2008. [DOI] [PubMed] [Google Scholar]
- 41.Price PM, Safirstein RL, Megyesi J. The cell cycle and acute kidney injury. Kidney Int 76: 604–613, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Price PM, Yu F, Kaldis P, Aleem E, Nowak G, Safirstein RL, Megyesi J. Dependence of cisplatin-induced cell death in vitro and in vivo on cyclin-dependent kinase 2. J Am Soc Nephrol 17: 2434–2442, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Puri TS, Shakaib MI, Chang A, Mathew L, Olayinka O, Minto AWM, Sarav M, Hack BK, Quigg RJ. Chronic kidney disease induced in mice by reversible unilateral ureteral obstruction is dependent on genetic background. Am J Physiol Renal Physiol 298: F1024–F1032, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-β1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest 112: 1486–1494, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spurgeon K, Donohoe D, Basile D. Transforming growth factor-β in acute renal failure: receptor expression, effects on proliferation, cellularity, and vascularization after recovery from injury. Am J Physiol Renal Physiol 288: F568–F577, 2005. [DOI] [PubMed] [Google Scholar]
- 46.Spurgeon-Pechman KR, Donohue DL, Mattson DL, Lund H, James L, Basile DP. Recovery from acute renal failure predisposes hypertension and secondary renal disease in response to elevated sodium. Am J Physiol Renal Physiol 293: F269–F278, 2007. [DOI] [PubMed] [Google Scholar]
- 47.Sullivan DE, Ferris MB, Poclask D, Brody AR. Tumor necrosis factor-α induces transforming growth factor-β1 expression in lung fibroblasts through the extracellular signal-regulated kinase pathway. Am J Respir Cell Mol Biol 32: 342–349, 2005. [DOI] [PubMed] [Google Scholar]
- 48.Suzuki T, Kimura M, Asano M, Fujigaki Y, Hishida A. Role of atrophic tubules in development of interstitial fibrosis in microembolism-induced renal failure in rat. Am J Pathol 158: 75–85, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Van den Heuvel S, Harlow E. Distinct roles for cyclin-dependent kinases in cell cycle control. Science 262: 2050–2054, 1993. [DOI] [PubMed] [Google Scholar]
- 50.Venkatachalam M, Griffin K, Lan R, Geng H, Saikumar P, Bidani A. Acute kidney injury: a springboard for progression in chronic kidney disease. Am J Physiol Renal Physiol 298: F1078–F1094, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wager RE, Assoian RK. A phorbol ester-regulated ribonuclease system controlling transforming growth factor β1 gene expression in hematopoietic cells. Mol Cell Biol 10: 5983–5990, 1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wrana JL, Attisano L, Wieser R, Ventura F, Massagué J. Mechanism of activation of the TGF-β receptor. Nature 370: 341–347, 1994. [DOI] [PubMed] [Google Scholar]
- 53.Wynn TA. Common and unique mechanisms regulate fibrosis in various fibroproliferative diseases. J Clin Invest 117: 524–529, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Xu J, Fan X, Langworthy MM, Zhang MZ, Harris RC. Characterization of a putative intrarenal serotonergic system. Am J Physiol Renal Physiol 293: F1468–F1475, 2007. [DOI] [PubMed] [Google Scholar]
- 55.Yang L, Besschetnova T, Brooks C, Shah J, Bonventre J. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med 16: 535–543, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yu F, Megyesi J, Safirstein RL, Price PM. Identification of the functional domain of p21WAF1/Cip1 that protects from cisplatin cytotoxicity. Am J Physiol Renal Physiol 289: F514–F520, 2005. [DOI] [PubMed] [Google Scholar]
- 57.Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN, Lan HY. Mechanism of chronic aristolochic acid nephropathy: role of smad3. Am J Physiol Renal Physiol 298: F1006–F1017, 2010. [DOI] [PubMed] [Google Scholar]