

Abstract
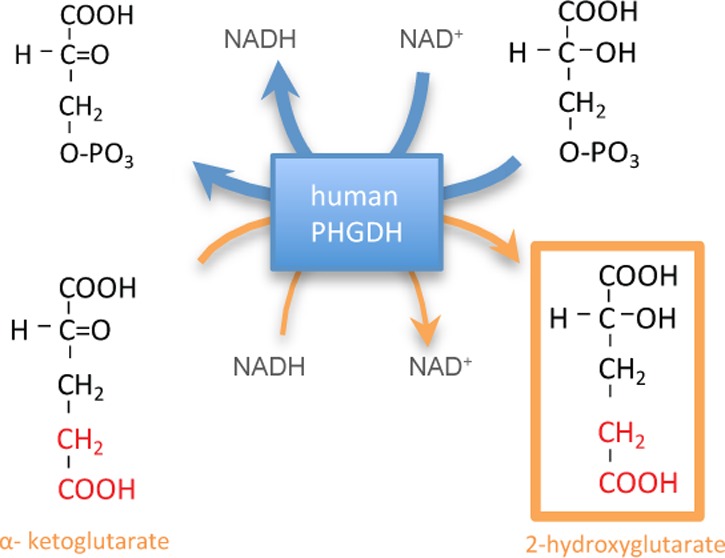
Human d-3-phosphoglycerate dehydrogenase (PHGDH), the first enzyme in the serine biosynthetic pathway, is genomically amplified in tumors including breast cancer and melanoma. In PHGDH-amplified cancer cells, knockdown of PHGDH is not fully rescued by exogenous serine, suggesting possible additional growth-promoting roles for the enzyme. Here we show that, in addition to catalyzing oxidation of 3-phosphoglycerate, PHGDH catalyzes NADH-dependent reduction of α-ketoglutarate (AKG) to the oncometabolite d-2-hydroxyglutarate (d-2HG). Knockdown of PHGDH decreased cellular 2HG by approximately 50% in the PHGDH-amplified breast cancer cell lines MDA-MB-468 (normal concentration 93 μM) and BT-20 (normal concentration 35 μM) and overexpression of PHGDH increased cellular 2HG by over 2-fold in non-PHGDH-amplified MDA-MB-231 breast cancer cells, which normally display very low PHGDH expression. The reduced 2HG level in PHGDH knockdown cell lines can be rescued by PHGDH re-expression, but not by a catalytically inactive PHGDH mutant. The initial connection between cancer and d-2HG involved production of high levels of d-2HG by mutant isocitrate dehydrogenase. More recently, however, elevated d-2HG has been observed in breast cancer tumors without isocitrate dehydrogenase mutation. Our results suggest that PHGDH is one source of this d-2HG.
Introduction
Cancer genome analysis has identified a variety of alterations that affect metabolic enzymes, including point mutations in the gene encoding the TCA-cycle enzyme isocitrate dehydrogenase (IDH)1−3 and amplifications of the gene encoding the serine pathway enzyme phosphoglycerate dehydrogenase (PHGDH).4,5 Active site IDH1 and IDH2 mutations are associated with brain cancer and acute myeloid leukemia,1−3 with the mutant enzyme producing the metabolic error product d-2HG.6,7d-2HG is a competitive inhibitor of AKG-dependent enzymes, including DNA and histone demethylases8−11 and is sufficient to promote leukemic transformation, while l-2HG is not able to induce such transformation.12 More recently, elevated d-2HG has been found in human breast cancers with wild type IDH.13
The committed enzyme of serine biosynthesis, phosphoglycerate dehydrogenase (PHGDH), is involved in a genomic region of copy number gain found across human cancers, and amplifications involving PHGDH occur with highest frequency in subsets of breast cancers and melanomas. When not genomically amplified, PHGDH is often overexpressed.4,5,14 PHDGH catalyzes the first reaction of de novo serine biosynthesis, producing 3-phosphohydroxypyruvate by NAD+-coupled oxidation of 3-phosphoglycerate (3PG). The PHGDH reaction is reversible and, under standard conditions, thermodynamically favors the direction from 3-phosphohydroxypyruvate to 3PG. In cells engaged in serine synthesis, the reaction is driven toward 3-phosphohydroxypyruvate due to its consumption by downstream pathway steps.
Genomic amplification of PHGDH enables cells to grow in serine free media.5 Suppression of PHGDH inhibits growth of PHGDH-amplified cells both in vitro and in vivo.4,5 However, the mechanism by which PHGDH amplification supports cancer growth is not yet fully understood. The simplest hypothesis is that it increases de novo serine synthesis. Serine is an important building block of proteins and lipid head groups, and a major feedstock for one-carbon metabolism, with both glycine and folate species derived from serine.14,15 Serine demand, however, does not completely explain PHGDH dependence, at least in vitro where a standard tissue culture medium contains copious serine and glycine. Even in PHGDH-amplified cell lines, e.g., the breast cancer cell lines MDA-MB-468, less than 20% of serine is generated from de novo synthesis in standard tissue culture conditions.16 Consistent with this, PHGDH knockdown does not significantly reduce the intracellular concentration of serine, and the growth inhibition caused by PHGDH knockdown cannot be rescued by the addition of exogenous serine or serine ester.5
This led us to explore the possibility that PHGDH has an additional enzymatic activity beyond 3PG/3-phosphohydroxypyruvate oxidation/reduction. PHGDH belongs to the d-isomer-specific 2-hydroxyacid dehydrogenase family. The thermodynamically favored PHGDH substrate, 3-phosphohydroxypyruvate, is structurally similar to the more abundant metabolite AKG. Here, we show that PHGDH also catalyzes the reduction of AKG to d-2HG. Thus, PHGDH can also produce d-2HG. Such production could contribute to elevated d-2HG observed in breast cancers without IDH mutations.
Result and Discussion
Promiscuity of Human PHGDH
The favored PHGDH substrate, 3-phosphohydroxypyruvate, is structurally similar to other central metabolites, including pyruvate (lacks the phosphate of 3-phosphohydroxypyruvate), oxaloacetate (OAA, substitutes phosphate with carboxylic acid), and AKG (substitutes phosphate with acetate; Figure 1A). Previous studies have shown that the E. coli homologue of PHGDH, SerA, produces d-2HG from AKG.17 Rat PHGDH, however, was found to lack such activity,18 leading to the assumption that mammalian PHGDH does not produce d- or l-2HG, without the human enzyme being tested. We examined whether recombinant purified human PHGDH (one version His-tagged and another version Streptavidin-tagged) catalyzes the NADH-driven reduction of pyruvate, OAA, or AKG. At pH 7.6 and 37 °C, NADH oxidation was observed in the presence of OAA or AKG, but not pyruvate (Figure 1B). Minimal activity was observed with NADPH in place of NADH. Analysis by LC-MS revealed that the reaction produces malate and 2HG, respectively, and each of the two different tagged versions of PHGDH were able to generate these products (Figure 1C). To confirm the observed activities are indeed due to human PHGDH, rather than from a contaminating activity resulting from the purification process, the same reaction was run with PHGDH replaced by an unrelated enzyme (human galactokinase), which was expressed and purified in an identical manner as the His-tagged PHGDH. No malate or 2HG was produced from OAA or AKG using this negative control enzyme (Figure 1C). While slow NADH oxidation was observed with the control enzyme (consistent with the generally labile nature of NADH), this rate of spontaneous NADH oxidation was much smaller than that observed in the presence of PHGDH (Supplementary Figure 1; all data in the main text are corrected for this background rate). These results demonstrate that OAA and AKG reduction activity is specifically due to PHGDH. Consistent with these findings, throughout the PHGDH reaction time course, 2HG and NAD+ were produced stoichiometrically (Figure 1D).
Figure 1.

Human PHGDH catalyzes NADH-driven reduction of OAA and AKG into malate and d-2HG. (A) Structures of the primary PHGDH substrate/product (box) and various possible alternative endogenous substrates/products. (B) PHGDH-catalyzed NADH-driven reduction of OAA and AKG but not pyruvate. A total of 5 mM of each α-ketoacid was incubated with 0.5 mM NADH and PHGDH (red) or no enzyme control (blue). The reaction was monitored by a decrease in NADH absorbance. For AKG reduction, activity with NADPH was also tested (PHGDH, green; no enzyme control, purple). (C) LC-MS analysis of the AKG reduction product 2HG (left) and the OAA reduction product malate (right). His-tagged PHGDH or strep-tagged PHGDH was incubated with 0.5 mM NADH and 5 mM α-keoglutarate or OAA. The assays included two negative controls: incubation of the same amount of PHGDH without substrate or incubation of the same substrates with human galactokinase (expressed and purified as for His-PHGDH). (D) PHGDH-catalyzed AKG reduction produces NAD+ and 2HG stoichiometrically. (E) PHGDH produces the d- enantiomer of 2HG. GC-MS traces for the l-2HG standard, d-2HG standard, the product of PHGDH-driven AKG reduction, and a mixture of the l- and d-2HG standards. (F) PHGDH oxidizes d-2HG but not l-2HG. PHGDH was incubated with 1 mM NAD+ and 10 mM l-2HG or d-2HG. NADH production, monitored by changes in absorbance at 340 nm, is shown on top, and AKG production analyzed by LC-MS is shown on the bottom.
Because the LC-MS method does not distinguish l-2HG and d-2HG, we also analyzed the reaction mixture by GC-MS following a two step derivatization previously demonstrated to separate the two enantiomers,19 with the PHGDH-derived 2HG coeluting with the d-2HG but not l-2HG standard (Figure 1E). The reverse reaction was observed when d-2HG (but not the corresponding l-enantiomer) was mixed with NAD+ and PHGDH (Figure 1F). Thus, in addition to catalyzing 3PG oxidation, human PHGDH is capable of catalyzing the NADH-driven reduction of OAA into malate, and more importantly AKG into d-2HG.
Kinetics of AKG Reduction Relative to 3PG Oxidation
To test how these additional activities compare to 3PG oxidation, we measured the kinetic parameters for PHGDH-catalyzed 3PG oxidation, OAA reduction, and AKG reduction (Table 1) by varying concentration of one substrate in the presence of saturating concentrations of the other substrate. The results were fitted to Michaelis–Menten kinetics (Supplementary Figure 2). The enzyme has similar kcat for 3PG oxidation and OAA or AKG reduction, with OAA reduction slightly faster than 3PG oxidation. Among these substrates, the highest affinity binder was 3PG (Km = 260 μM), consistent with its being the primary physiological substrate. OAA and AKG both bound similarly, with 20–40-fold higher km than 3PG. NAD(H) binding was high affinity in all cases (Km ≤ 25 μM). The kinetics of NAD+ reduction with malate or 2HG were substantially slower and thus not quantified.
Table 1. Michaelis–Menten Parameters of the Different Reactions Catalyzed by PHGDH, Compared to Intracellular Concentrations of the Corresponding Substrates and Products in Different Cell Lines.
| 3-phosphoglycerate
→ 3-phosphohydroxypyravate |
oxaloacetate
→ malate |
α-ketoglutarate
→ 2-hydroxyglutarate |
||||||
|---|---|---|---|---|---|---|---|---|
| Kcat (1/min) | 4.5 ± 0.6 | Kcat (1/min) | 10.6 ± 1.6 | Kcat (1/min) | 4.7 ± 0.9 | |||
| Km, 3-phosphoglycerate (mM) | 0.26 ± 0.034 | Km, oxaloacetate (mM) | 6.5 ± 1.3 | Km, α-ketoglutarate (mM) | 10.1 ± 1.8 | |||
| [3-phosphoglycerate] (mM) | MDA-MB-468 | 0.18 ± 0.01 | [oxaloacetate] (mM) | MDA-MB-468 | not detectable | [α-ketoglutarate] (mM) | MDA-MB-468 | 0.55 ± 0.02 |
| BT20 | 0.11 ± 0.01 | BT20 | not detectable | BT20 | 0.47 ± 0.02 | |||
| [3-phosphohydroxypyruvate] (mM) | MDA-MB-468 | not detectable | [malate] (mM) | MDA-MB-468 | 3.3 ± 0.08 | [2-hydroxyglutarate] (mM) | MDA-MB-468 | 0. 093 ± 0.009 |
| BT20 | not detectable | BT20 | 0.99 ± 0.07 | BT20 | 0.035 ± 0.002 | |||
| Km, NAD+ (mM) | 0.022 ± 0.003 | Km, NADH (mM) | 0.004 + 0.001 | Km, NADH (mM) | 0.004 ± 0.001 | |||
| [NAD+] (mM) | MDA-MB-468 | 0.99 ± 0.01 | [NADH] (mM) | MDA-MB-468 | 0.37 ± 0.02 | [NADH] (mM) | MDA-MB-468 | 0.37 ± 0.02 |
| BT20 | 0.77 ± 0.02 | BT20 | 0.186 ± 0.001 | BT20 | 0.186 ± 0.001 | |||
| [NADH] (mM) | MDA-MB-468 | 0.37 ± 0.02 | [NAD+] (mM) | MDA-MB-468 | 0.99 ± 0.01 | [NAD+] (mM) | MDA-MB-468 | 0.99 ± 0.01 |
| BT20 | 0.186 ± 0.001 | BT20 | 0.77 ± 0.02 | BT20 | 0.77 ± 0.02 | |||
To put the enzyme kinetic parameters into a physiological context, we determined the absolute intracellular concentrations of the relevant substrates (Table 1). In MDA-MB-468 cells, NAD+ and NADH were both >300 μM, and thus saturating in all cases. Among the other substrates, the most abundant was AKG (550 μM), which was ∼3 fold higher than 3PG. 3-Phosphohydroxypyruvate and OAA, consistent with prior literature, were not detectable. Isotopic labeling experiments showed that the pathways producing these two compounds are active; the absence of a measurable signal by LC-MS indicates that their intracellular concentrations fall below the limit of detection (LOD). For OAA, the LOD is 60 μM; for 3-phosphohydroxypyruvate, the LOD cannot be directly determined due to the lack of a commercially available standard but is likely in the vicinity of 10 μM based on the structurally similar compound 3PG. Thus, the likely main physiological reactions are 3PG oxidation to 3-phosphohydroxypyruvate and AKG reduction to 2HG, with the competition with other potential in vitro substrates likely minor. 3PG and AKG compete for one binding site on PHGDH, and NAD+ and NADH for another. We therefore plugged the observed values into a Michaelis–Menten-type equation that approximates competition for these two active sites, e.g., for the 3PG oxidation reaction:
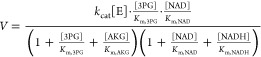 |
The results predicted cellular 2HG production fluxes in MDA-MB-468 and BT-20, respectively, as 17% and 15% of the 3PG oxidation (i.e., serine biosynthetic flux).
Contribution of PHGDH to the Cellular 2HG Pool
To test whether the production of 2HG by PHGDH is significant in the cell, we knocked down PHGDH using shRNA in MDA-MB-468 and BT-20, two breast cancer lines with genomically amplified PHGDH (successful knockdown of PHGDH was obtained for two shRNA sequences in MDA-MB-468 and one in BT-20). We then analyzed metabolites in control and knockdown cell lines (Figure 2A). Consistent with effective knockdown of PHGDH, a large decrease in the unique serine biosynthetic pathway intermediate, 3-phosphoserine, was observed in MDA-MB-468 cells. (3-Phosphoserine was below our limit of detection in BT-20 cells.) In addition to 3-phosphoserine, 2HG levels were decreased by both shRNA knockdowns in the MDA-MB-468 cells, and 2HG was also decreased in the BT-20 cells (Figure 2A,B). Because the 2HG levels in the cell extract were too low to quantitate by GC-MS, especially given background signals from other cellular metabolites, we could not determine stereospecificity and instead report total levels as measured by LC-MS. Serine and malate concentrations were not significantly affected by PHGDH knockdown, and there was a minimal effect on AKG (Figure 2), which can be produced when glutamate transaminates the PHGDH product 3-phosphohydroxypyruvate.4,5 Thus, in these cell lines and culture conditions, PHGDH influences 2HG levels the most of any metabolite that was consistently measured.
Figure 2.

PHGDH knockdown in MDA-MB-468 and BT-20 cells depletes 2HG. (A) Validation of PHGDH knockdown by western (top) and heat map of metabolite levels revealing depletion of 2HG and 3-phosphoserine (bottom). Metabolite levels were averaged and normalized to cell number/volume (which were equivalent as cell size did not change with PHGDH knockdown), then normalized to shNT of the corresponding cell line. Results are log2 transformed and clustered. (B) Relative concentrations of key metabolites in two PHGDH-amplified breast cancer cell lines with shPHGDH or control knockdown.
To determine if the impact of PHGDH on intracellular 2HG is dependent on its catalytic activity, wildtype protein or a catalytically inactive mutant (R236E) of PHGDH was overexpressed in a non-PHGDH-amplified breast cancer cell line, MDA-MB-231. PHGDH expression in these cells increases the intracellular 2HG level by over 2-fold, while overexpression of catalytic dead enzymes does not cause a change in 2HG level; similar, but less profound, changes were also observed in αAKG levels in these cells (Figure 3A). We further re-expressed shRNA-resistant PHGDH or its catalytic dead mutant in control and PHGDH knockdown MDA-MB-468 cells. Re-expression of wild-type PHGDH, but not R263E mutant, rescues the 2HG level in shPHGDH knockdown cells. Additionally, overexpression of wild type, but not R263E, PHGDH increases the 2HG level in cells without PHGDH knockdown (shGFP). No significant changes were observed in the intracellular AKG level in these cells (Figure 3B). Collectively, these data suggest that PHGDH controls the intracellular 2HG concentration by directly catalyzing 2HG synthesis.
Figure 3.
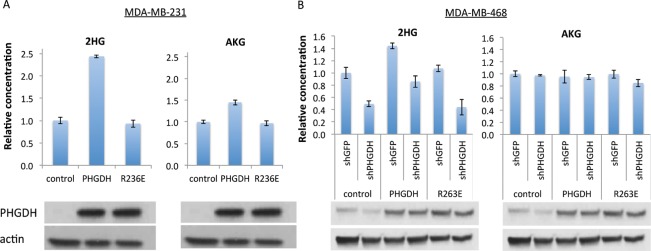
Dependence of cellular 2HG concentration on PHGDH catalytic activity. (A) Relative concentrations of 2-HG and AKG in the non-PHGDH-amplified breast cancer cell line, MDA-MB-231, transfected with control vector, PHGDH, or catalytically inactive PHGDH (R236E). PHGDH expression by western is shown on the bottom. (B) Relative concentrations of 2-HG and AKG in MDA-MB-468 cells, with or without PHGDH knockdown, with catalytically active or inactive PHGDH re-expression. PHGDH expression by western is shown on the bottom. Mean ± SD, N = 3.
Discussion Regarding Biological Significance
2HG impacts protein and DNA covalent modification, including methylation, by competitive inhibition of AKG-dependent dioxygenase enzymes. l-2HG is an error product of malate dehydrogenase and can accumulate to toxic levels in individuals deficient in l-2HG-dehydrogenase activity.20d-2HG is produced in large amounts by IDH active site mutants found in glioma and acute myeloid leukemia (AML). Unlike l-2HG, d-2HG is sufficient to induce leukemic transformation.9 Here, we demonstrate that another cancer-associated core metabolic enzyme, PHGDH, also produces d-2HG, albeit at lower levels than those observed in IDH mutant cancers.
An important question is whether the concentrations of d-2HG produced by PHGDH are biologically significant. PHGDH knockdown reduces the total cellular 2HG concentration by about 50% in the tested cell lines; since the measured cellular 2HG is the sum of the d- and l-stereoisomers, the effect on d-2HG is presumably larger. In another cancer cell line with wild-type IDH, HCC70, we observed higher concentrations of 2HG that were not reduced by PHGDH knockdown, suggesting that other enzymes can produce significant amounts of 2HG (J.F. and X.T., unpublished results). Previously, millimolar concentrations of d-2HG have been shown to directly impact demethylase activity in vitro.8,10 Lower concentrations, however, may inhibit certain enzymes or otherwise impact cellular state. For instance, d-2HG has been shown to inhibit histone demethylase JMJD2A and JMJD2C at an IC50 of 24 μM and 79 μM.11
A recent study found that 2HG accumulates in a subset of breast cancers, and this accumulation is associated with poor prognosis. In breast cancer, PHGDH amplification and overexpression occurs frequently. Interestingly, 2HG accumulation was found to be associated with the activity of the oncogene Myc,13 which positively regulates expression of a large number of genes including PHGDH.
In addition to previously proposed mechanisms by which PHGDH amplification could promote tumor growth, including supplying serine for protein synthesis and one carbon metabolism, promoting TCA cycle anapleurosis,5 and nonenzymatic functions including FOXM1 binding,21 our results raise the possibility that PHGDH amplification could potentially influence cell physiology by overproduction of the oncometabolite d-2HG. Further work is required to elucidate the functional significance of PHGDH’s multiple activities in different physiological and pathological settings.
Methods
Enzyme Activity
To test the activity of human phosphoglycerate dehydrogenase (PHGDH) on its natural substrate, 3-phosphoglycerate, and the potential alternative substrates pyruvate, AKG and OAA, two types of recombinant human PHGDH, were used: His-tagged PHGDH (BPS Biosciences) and Streptavidin-tagged PHGDH (a gift from Dr. Olszewski). Similar AKG and OAA reduction activities were found in both preparations of the enzyme. The kinetic parameters were measured using the His-tagged enzyme in 200 mM Tris buffer (pH = 7.6) at 37 °C. All substrates were purchased from Sigma. The reaction rate was monitored as NADH production or consumption, measured by absorbance at 340 nm using a plate reader (BioTek, Synergy HT). To test the 3PG oxidation reaction, 200 mM of hydrazine was added to the buffer to drive the reaction forward. Kinetic parameters were measured by varying concentration of the substrate of interest, with saturating concentration of the other substrate. To control for non-PHGDH dependent background NADH oxidation, reactions were also run substituting PHGDH with human galactokinase, which was expressed and purified with the same protocol as PHGDH, as a control. When measuring the kinetic parameters the galactokinase data were subtracted from the observed PHGDH raw data. All kinetic parameters were fit to the Michaelis–Menten equation by nonlinear least-squares fitting (MATLAB). The products of PHGDH acting on alternative substrates were analyzed by GC-MS or LC-MS. For MS-based analysis, reactions were quenched with four volumes of methanol and centrifuged to remove protein. Supernatant was further diluted or derivatized before MS-based measurement.
Separation of l- and d-2-Hydroxyglutarate
To determine the chirality of 2-hydroxyglutarate (2HG), samples were derivatized according to a protocol modified from a previous report.19 Samples were dried down under a N2 flow, then resuspended in (R)-2-butanol with 1 M HCl and heated for 2 h at 100 °C. The product was dried down under a nitrogen gas flow and resuspended in 1:1 pyridine/acetic anhydride and heated for 0.5 h at 100 °C. Standards of l- and d-2-hydroxyglutarate and all reagents were purchased from Sigma.
After drying the final product, samples were dissolved in chloroform and analyzed by an Aglient 7890A GC system with Aux EPC column (30 m × 0.25 mm, 0.25 um film thickness) coupled with a TOF mass spectrometer in negative mode. The temperature gradient used in this analysis was 100 °C for 3 min, 4 °C per min from 100 to 230 °C, 15 °C per min from 230 to 300 °C, and 300 °C for 5 min.
Cell Lines and Culture Conditions
Two PHGDH-amplified breast cancer cell lines, MDA-MB-468 and BT-20, and a non-PHGDH-amplified breast cancer cell line, MDA-MB-231, were purchased from ATCC. All cell lines were grown in Dulbecco’s modified eagle media (DMEM) without pyruvate (CELLGRO), supplemented with 10% dialyzed fetal bovine serum (Invitrogen) in a 5% CO2 incubator at 37 °C. Growth medium was replaced every 2 days.
PHGDH Knockdown
Knockdowns were performed by infection with lentivirus expressing the PHGDH shRNA (#1, TRCN0000233031; #2, TRCN0000028532, Sigma) and puromycin selection. To obtain the shRNA-expressing virus, shRNA vectors (Sigma-Aldrich) were cotransfected with lentivirus packaging plasmids into HEK293FT cells using the X-tremeGENE HP DNA transfection reagent (Roche, Catlog # 6366236001). Viral supernatants were collected every 24 h for 3 days. Target cells were infected by a viral supernatant (diluted 1:1 with fresh DMEM). Such treatment was repeated three times, followed by selection with 2 μg/mL puromycin initiated at day 4 and allowed to proceed for 2–3 days. Thereafter, cells were maintained in DMEM with 2 μg/mL puromycin. Knockdown was verified by Western blot using anti-PHGDH antibody (Sigma).
Overexpression was performed by retroviral infection with the following vectors: (1) pLHCX empty vector, (2) PHGDH cDNA coloned into pLHCX with HindIII and ClaI sites with a Kozak sequence, (3) enzymatically dead PHGDH R236E. Mutation of the analogous residue in the E. coli protein destroys enzyme function.22 The mutated site is underlined in the following sequence: tggtgaactgtgccgagggagggatcgtgga. Both PHGDH-containing constructs also included mutations, cccaaagggaccatccaagttatcacacagggaacatccc, designed to confer resistance to the shRNA targeting sequence cttagcaaagaggagctgata. The PHGDH expressing virus was obtained by cotransfecting the overexpression vectors with retrovirus packing plasmids into HEK293T cells using the X-tremeGENE DNA transfection reagent. Target cells were infected every day by virus containing media (with 8 μg/mL Polybrene) which were collected every 24 h for 3 days. Infected cells were selected by 200 μg/mL and 700 μg/mL hygromycin for MDA-MB-468 and MDA-MB-231 cells, respectively. The overexpression cells were then infected by lentiviral shRNA vector.
Measurement of Intracellular Metabolite Concentrations
To measure the effect of PHGDH knockdown on intracellular metabolites levels, cells were harvested at ∼80% confluency. Fresh medium was replaced 2 h before metabolome harvesting. Metabolism was quenched and metabolites extracted by aspirating media and immediately adding −80 °C 80:20 methanol/water (v/v). Supernatants from two rounds of methanol/water extraction were combined, dried under a nitrogen gas flow, and resuspended in HPLC water for analysis.
The LC-MS method involved reversed-phase ion-pairing chromatography coupled by negative mode electrospray ionization to a stand-alone orbitrap mass spectrometer (Thermo Scientific) scanning from m/z 85–1000 at 1 Hz at 100 000 resolution with LC separation on a Synergy Hydro-RP column (100 mm × 2 mm, 2.5 μm particle size, Phenomenex, Torrance, CA) using a gradient of solvent A (97:3 H2O/MeOH with 10 mM tributylamine and 15 mM acetic acid) and solvent B (100% MeOH).23−25 Data were analyzed using the MAVEN software suite.26 Absolute concentrations were quantified by feeding cells U–13C-glucose and U–13C-glutamine to label intracellular metabolites and comparing the signal of isotope-labeled intracellular compound to signal of unlabeled internal standard as described previously.27
Acknowledgments
We thank K. Olszewski for generously sharing the strep-tagged enzyme and helpful discussions and S. Zhang for help with GC-MS analysis. This work was supported by NIH grants 1R01CA163591 and P50GM071508. We also acknowledge support from NIH P30CA14051, NSF, AACR, and the Burroughs Wellcome Fund. J. Fan was supported by a Howard Hughes Medical Institute (HHMI) international student research fellowship.
Supporting Information Available
Supplementary Figures 1 and 2. This material is available free of charge via the Internet at http://pubs.acs.org
Author Contributions
▽ These authors contributed equally.
The authors declare the following competing financial interest(s): J.D.R is a co-founder of Raze Pharmaceuticals and consultant to Kadmon Pharmaceuticals. M.V.H. is a consultant to Agios Pharmaceuticals.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Balss J.; Meyer J.; Mueller W.; Korshunov A.; Hartmann C.; von Deimling A. (2008) Analysis of the IDH1 codon 132 mutation in brain tumors. Acta Neuropathol. 116, 597–602. [DOI] [PubMed] [Google Scholar]
- Yan H.; Parsons D. W.; Jin G.; McLendon R.; Rasheed B. A.; Yuan W.; Kos I.; Batinic-Haberle I.; Jones S.; Riggins G. J.; Friedman H.; Friedman A.; Reardon D.; Herndon J.; Kinzler K. W.; Velculescu V. E.; Vogelstein B.; Bigner D. D. (2009) IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 360, 765–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardis E. R.; Ding L.; Dooling D. J.; Larson D. E.; McLellan M. D.; Chen K.; Koboldt D. C.; Fulton R. S.; Delehaunty K. D.; McGrath S. D.; Fulton L. A.; Locke D. P.; Magrini V. J.; Abbott R. M.; Vickery T. L.; Reed J. S.; Robinson J. S.; Wylie T.; Smith S. M.; Carmichael L.; Eldred J. M.; Harris C. C.; Walker J.; Peck J. B.; Du F.; Dukes A. F.; Sanderson G. E.; Brummett A. M.; Clark E.; McMichael J. F.; Meyer R. J.; Schindler J. K.; Pohl C. S.; Wallis J. W.; Shi X.; Lin L.; Schmidt H.; Tang Y.; Haipek C.; Wiechert M. E.; Ivy J. V.; Kalicki J.; Elliott G.; Ries R. E.; Payton J. E.; Westervelt P.; Tomasson M. H.; Watson M. A.; Baty J.; Heath S.; Shannon W. D.; Nagarajan R.; Link D. C.; Walter M. J.; Graubert T. A.; DiPersio J. F.; Wilson R. K.; Ley T. J. (2009) Recurring mutations found by sequencing an acute myeloid leukemia genome. N. Engl. J. Med. 361, 1058–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale J. W.; Grassian A. R.; Melman T.; Lyssiotis C. A.; Mattaini K. R.; Bass A. J.; Heffron G.; Metallo C. M.; Muranen T.; Sharfi H.; Sasaki A. T.; Anastasiou D.; Mullarky E.; Vokes N. I.; Sasaki M.; Beroukhim R.; Stephanopoulos G.; Ligon A. H.; Meyerson M.; Richardson A. L.; Chin L.; Wagner G.; Asara J. M.; Brugge J. S.; Cantley L. C.; Vander Heiden M. G. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat. Genet. 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R.; Marks K. M.; Shaul Y. D.; Pacold M. E.; Kim D.; Birsoy K.; Sethumadhavan S.; Woo H. K.; Jang H. G.; Jha A. K.; Chen W. W.; Barrett F. G.; Stransky N.; Tsun Z. Y.; Cowley G. S.; Barretina J.; Kalaany N. Y.; Hsu P. P.; Ottina K.; Chan A. M.; Yuan B.; Garraway L. A.; Root D. E.; Mino-Kenudson M.; Brachtel E. F.; Driggers E. M.; Sabatini D. M. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L.; White D. W.; Gross S.; Bennett B. D.; Bittinger M. A.; Driggers E. M.; Fantin V. R.; Jang H. G.; Jin S.; Keenan M. C.; Marks K. M.; Prins R. M.; Ward P. S.; Yen K. E.; Liau L. M.; Rabinowitz J. D.; Cantley L. C.; Thompson C. B.; Vander Heiden M. G.; Su S. M. (2009) Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462, 739–744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward P. S.; Patel J.; Wise D. R.; Abdel-Wahab O.; Bennett B. D.; Coller H. A.; Cross J. R.; Fantin V. R.; Hedvat C. V.; Perl A. E.; Rabinowitz J. D.; Carroll M.; Su S. M.; Sharp K. A.; Levine R. L.; Thompson C. B. (2010) The common feature of leukemia-associated IDH1 and IDH2 mutations is a neomorphic enzyme activity converting alpha-ketoglutarate to 2-hydroxyglutarate. Cancer Cell 17, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W.; Yang H.; Liu Y.; Yang Y.; Wang P.; Kim S. H.; Ito S.; Yang C.; Xiao M. T.; Liu L. X.; Jiang W. Q.; Liu J.; Zhang J. Y.; Wang B.; Frye S.; Zhang Y.; Xu Y. H.; Lei Q. Y.; Guan K. L.; Zhao S. M.; Xiong Y. (2011) Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell 19, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koivunen P.; Lee S.; Duncan C. G.; Lopez G.; Lu G.; Ramkissoon S.; Losman J. A.; Joensuu P.; Bergmann U.; Gross S.; Travins J.; Weiss S.; Looper R.; Ligon K. L.; Verhaak R. G.; Yan H.; Kaelin W. G. Jr. (2012) Transformation by the (R)-enantiomer of 2-hydroxyglutarate linked to EGLN activation. Nature 483, 484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Ward P. S.; Kapoor G. S.; Rohle D.; Turcan S.; Abdel-Wahab O.; Edwards C. R.; Khanin R.; Figueroa M. E.; Melnick A.; Wellen K. E.; O’Rourke D. M.; Berger S. L.; Chan T. A.; Levine R. L.; Mellinghoff I. K.; Thompson C. B. (2012) IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature 483, 474–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury R.; Yeoh K. K.; Tian Y. M.; Hillringhaus L.; Bagg E. A.; Rose N. R.; Leung I. K.; Li X. S.; Woon E. C.; Yang M.; McDonough M. A.; King O. N.; Clifton I. J.; Klose R. J.; Claridge T. D.; Ratcliffe P. J.; Schofield C. J.; Kawamura A. (2011) The oncometabolite 2-hydroxyglutarate inhibits histone lysine demethylases. EMBO Rep. 12, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losman J. A.; Looper R. E.; Koivunen P.; Lee S.; Schneider R. K.; McMahon C.; Cowley G. S.; Root D. E.; Ebert B. L.; Kaelin W. G. Jr. (2013) (R)-2-hydroxyglutarate is sufficient to promote leukemogenesis and its effects are reversible. Science 339, 1621–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma A.; Putluri N.; Mishra P.; Mathe E. A.; Dorsey T. H.; Yi M.; Wallace T. A.; Issaq H. J.; Zhou M.; Killian J. K.; Stevenson H. S.; Karoly E. D.; Chan K.; Samanta S.; Prieto D.; Hsu T. Y.; Kurley S. J.; Putluri V.; Sonavane R.; Edelman D. C.; Wulff J.; Starks A. M.; Yang Y.; Kittles R. A.; Yfantis H. G.; Lee D. H.; Ioffe O. B.; Schiff R.; Stephens R. M.; Meltzer P. S.; Veenstra T. D.; Westbrook T. F.; Sreekumar A.; Ambs S. (2014) MYC-driven accumulation of 2-hydroxyglutarate is associated with breast cancer prognosis. J. Clin. Invest. 124, 398–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullarky E.; Mattaini K. R.; Vander Heiden M. G.; Cantley L. C.; Locasale J. W. (2011) PHGDH amplification and altered glucose metabolism in human melanoma. Pigment Cell Melanoma Res. 24, 1112–1115. [DOI] [PubMed] [Google Scholar]
- Locasale J. W. (2013) Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat. Rev. Cancer 13, 572–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J.; Chung F.; Yang G.; Pu M.; Gao H.; Jiang W.; Yin H.; Capka V.; Kasibhatla S.; Laffitte B.; Jaeger S.; Pagliarini R.; Chen Y.; Zhou W. (2013) Phosphoglycerate dehydrogenase is dispensable for breast tumor maintenance and growth. Oncotarget 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao G.; Winkler M. E. (1996) A novel alpha-ketoglutarate reductase activity of the serA-encoded 3-phosphoglycerate dehydrogenase of Escherichia coli K-12 and its possible implications for human 2-hydroxyglutaric aciduria. J. Bacteriol. 178, 232–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Achouri Y.; Rider M. H.; Schaftingen E. V.; Robbi M. (1997) Cloning, sequencing and expression of rat liver 3-phosphoglycerate dehydrogenase. Biochem. J. 323(Pt 2), 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamerling J. P.; Duran M.; Gerwig G. J.; Ketting D.; Bruinvis L.; Vliegenthart J. F.; Wadman S. K. (1981) Determination of the absolute configuration of some biologically important urinary 2-hydroxydicarboxylic acids by capillary gas--liquid chromatography. J. Chromatogr. 222, 276–283. [DOI] [PubMed] [Google Scholar]
- Van Schaftingen E.; Rzem R.; Veiga-da-Cunha M. (2009) L: −2-Hydroxyglutaric aciduria, a disorder of metabolite repair. J. Inherit. Metab. Dis. 32, 135–142. [DOI] [PubMed] [Google Scholar]
- Liu J.; Guo S.; Li Q.; Yang L.; Xia Z.; Zhang L.; Huang Z.; Zhang N. (2013) Phosphoglycerate dehydrogenase induces glioma cells proliferation and invasion by stabilizing forkhead box M1. J. Neurooncol. 111, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant G. A.; Kim S. J.; Xu X. L.; Hu Z. (1999) The contribution of adjacent subunits to the active sites of d-3-phosphoglycerate dehydrogenase. J. Biol. Chem. 274, 5357–5361. [DOI] [PubMed] [Google Scholar]
- Munger J.; Bennett B. D.; Parikh A.; Feng X. J.; McArdle J.; Rabitz H. A.; Shenk T.; Rabinowitz J. D. (2008) Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 26, 1179–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemons J. M.; Feng X. J.; Bennett B. D.; Legesse-Miller A.; Johnson E. L.; Raitman I.; Pollina E. A.; Rabitz H. A.; Rabinowitz J. D.; Coller H. A. (2010) Quiescent fibroblasts exhibit high metabolic activity. PLoS Biol. 8, e1000514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W.; Clasquin M. F.; Melamud E.; Amador-Noguez D.; Caudy A. A.; Rabinowitz J. D. (2010) Metabolomic analysis via reversed-phase ion-pairing liquid chromatography coupled to a stand alone orbitrap mass spectrometer. Anal. Chem. 82, 3212–3221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melamud E.; Vastag L.; Rabinowitz J. D. (2010) Metabolomic analysis and visualization engine for LC-MS data. Anal. Chem. 82, 9818–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett B. D.; Yuan J.; Kimball E. H.; Rabinowitz J. D. (2008) Absolute quantitation of intracellular metabolite concentrations by an isotope ratio-based approach. Nat. Protoc. 3, 1299–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.