

Abstract
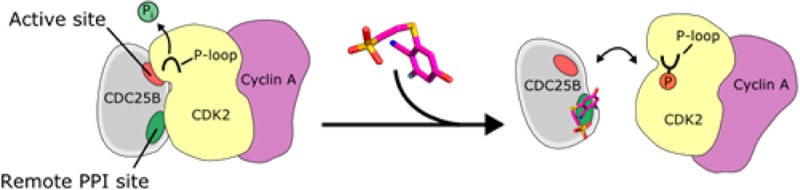
CDC25 phosphatases are key cell cycle regulators and represent very attractive but challenging targets for anticancer drug discovery. Here, we explored whether fragment-based screening represents a valid approach to identify inhibitors of CDC25B. This resulted in identification of 2-fluoro-4-hydroxybenzonitrile, which directly binds to the catalytic domain of CDC25B. Interestingly, NMR data and the crystal structure demonstrate that this compound binds to the pocket distant from the active site and adjacent to the protein–protein interaction interface with CDK2/Cyclin A substrate. Furthermore, we developed a more potent analogue that disrupts CDC25B interaction with CDK2/Cyclin A and inhibits dephosphorylation of CDK2. Based on these studies, we provide a proof of concept that targeting CDC25 phosphatases by inhibiting their protein–protein interactions with CDK2/Cyclin A substrate represents a novel, viable opportunity to target this important class of enzymes.
The CDC25 family of dual-specificity protein phosphatases plays an important role in cell cycle regulation by activating the cyclin-dependent kinases (CDKs) through the removal of inhibitory phosphorylations.1 CDC25 family member CDC25B regulates the G2/M phase transition by removing two inhibitory phosphate groups from the ATP binding loop of the CDK2 kinase.2,3 CDC25B is often overexpressed in various cancers, leading to excessive CDK2/Cyclin A activation and aberrant cell cycle progression resulting in poor clinical outcomes.4−6 Genetic studies have shown the essential role of CDC25B in cancer for tumor cells growth, supporting that CDC25B is an attractive therapeutic target for inhibition by small molecules.7−9 Indeed, the CDC25 phosphatases have been actively pursued as cancer drug targets for over 20 years.10,11 To date, all efforts to inhibit CDC25 phosphatases were focused on targeting the catalytic sites of these enzymes,10,12 which are unusually small and shallow without well-defined binding pockets, making CDC25s somewhat recalcitrant to drug discovery efforts.13 Furthermore, the presence of highly reactive catalytic cysteine in the active sites of CDC25s hampers screening and drug design efforts due to covalent binding and irreversible inhibition by diverse classes of small molecules.10 Indeed, the majority of well-studied and the most potent inhibitors of CDC25s discovered to date, including quinone and Vitamin K3 derivatives, are known to covalently modify cysteines in CDC25s,10,14 raising the question about their potential toxicity and limiting their therapeutic applications.15 Furthermore, no biophysical or structural characterization of known CDC25 inhibitors has been reported to date, leaving the mechanism of their binding largely unknown.
Results and Discussion
To assess whether small molecule compounds binding to CDC25B can be identified, we employed fragment-based screening approach. An in-house library of fragment-like compounds consisting of approximately 1500 chemically diverse small molecules was screened by NMR spectroscopy through the observation of 1H and 15N chemical shift perturbations on 1H–15N HSQC NMR spectra for uniformly 15N labeled CDC25B catalytic domain. Through this screen, we found 2-fluoro-4-hydroxybenzonitrile, (compound 1), as the only compound that binds to CDC25B (Figure 1A). To map the binding site of 1 on CDC25B, we analyzed chemical shift perturbations using previously determined backbone assignment.16 Interestingly, we found that 1 does not bind to the active site but rather perturbs a set of residues in a distal site on CDC25B.
Figure 1.

Identification and characterization of compound 1 as a novel CDC25B ligand. (A) A portion of the 1H–15N HSQC spectrum for the CDC25B catalytic domain in the presence (red) and absence (black) of 2 mM 1. (B) Crystal structure of 1 bound to CDC25B. Dark gray surface denotes the enzymatic active site. Two arginine residues involved in interaction with CDK2/Cyclin A substrate are labeled and shown in red. The distance between the catalytic cysteine and 1 is shown. (C) Molecular details of the interaction of 1 with CDC25B binding pocket. 1 binds in two equally populated orientations with symmetry along CN, OH axis. Distance between position 6 of 1 and the sulfate ion is given (PDB ID: 4WH7). The hydrogen bond network between the hydroxyl of 1 and four waters in the binding pocket is also shown. (D) AlphaLISA signal due to the protein–protein interaction between CDC25B and the CDK2/Cyclin A complex. CDC25B WT is shown in black, and the hotspot mutation R492L is shown in red.
To accurately establish the binding mode of this compound we determined a high-resolution crystal structure of 1 bound to the CDC25B (Figure 1B, Supporting Information Figure 1A). The structure revealed that 1 binds to a relatively small but well-defined pocket on CDC25B located approximately 15 Å away from the active site in agreement with the chemical shift perturbations. This binding pocket is primarily comprised of the Phe386, Leu398, Cys484, Arg488, and Met505 side chains. The phenyl ring of 1 inserts between the side chains of Leu398 and Arg488, forming a hydrophobic and cation-π interactions, respectively (Figure 1C). The nitrile is partly solvent exposed, and the nitrile nitrogen replaces a well-defined water molecule found in the apo-structure near the backbone of Arg485. We also found in the crystal structure that 1 binds in two equally populated orientations with fluorine pointing either toward Cys484 or to the solvent (Figure 1C, Supporting Information Figure 1A and B). An interesting feature of this binding site is the presence of numerous well ordered water molecules that form a network of hydrogen bonds. These waters are hydrogen bonded to backbone carbonyl and amide of Met505 and amides of Lys399 and Gly380 (Figure 1C). Binding of 1 replaces water molecule present in the structure of CDC25B and the hydroxyl group of 1 participates in a network of hydrogen bonds with remaining waters (Figure 1C).
Interestingly, several residues directly adjacent to the binding site of 1 were shown previously to be involved in the protein–protein interaction between CDC25B and its substrate CDK2.3,17 Based on mutagenesis studies, Arg488 and Arg492 in CDC25B (Figure 1B) have been shown to be required for CDK2 substrate recruitment through ionic interactions with Asp206.17 To further validate the importance of this site for interactions with CDK2/Cyclin A substrate, we introduced the R492L mutation. We assessed the ability of the R492L mutant and wild-type CDC25B to interact with the substrate using an AlphaLISA-based protein–protein interaction assay. Consistent with the previous report,17 we found that CDC25B R492L mutant is unable to interact with CDK2/Cyclin A substrate (Figure 1D). This result strongly supports our finding that compound 1 binds to a functionally important site on CDC25B that is involved in the protein–protein interactions with the substrate.
NMR experiments indicated that 1 binds to CDC25B with a relatively low affinity, which is typical for fragment-like compounds.18 We subsequently explored which functional groups in 1 are essential for interactions with CDC25B. We found that removing either hydroxyl or nitrile groups was detrimental for binding to CDC25B (not shown). Then, we tested several commercially available analogs of 1, and to rank their binding to CDC25B, we measured the sum of chemical shift changes for eight of the most perturbed amide resonances. Elimination of the fluorine at position R1 (compound 2) significantly decreased binding, while exchanging it for chlorine (3) had no effect (Figure 2A). Addition of a second fluorine, either at position R2 (5) or R3 (6) enhanced binding, with a preference for the R2 position.
Figure 2.
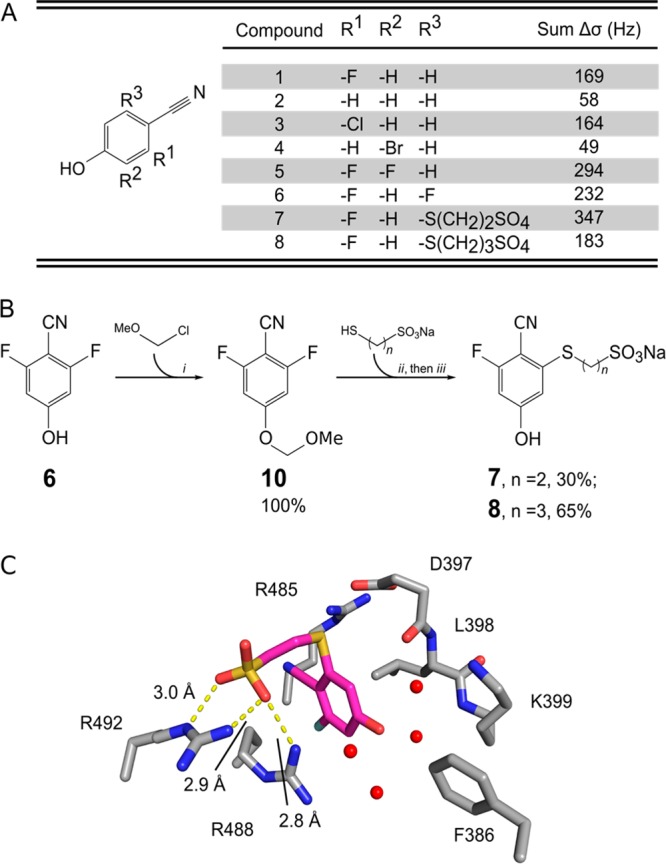
Structure activity relationship (SAR) for 2-fluoro-4-hydroxybenzonitrile analogs. (A) Structures of compounds tested for binding to CDC25B. Sum Δσ was calculated as a sum of the chemical shift perturbations for eight of the most significantly perturbed amide resonances (in Hz) at 2 mM compound concentration. (B) Schematics of the synthesis of compounds 7 and 8. (C) Crystal structure showing the details of the interaction of 7 with CDC25B (PDB ID: 4WH9). Distances between the sulfate oxygens and the side-chain nitrogens of Arg488 and Arg492 are given.
We then explored the possibility to improve 1 by linking to an adjacent sulfate ion that was found in the CDC25B-1 crystal structure and interacts with the side chains of Arg488 and Arg492 (Figure 1C). Based on the structural data we designed and synthesized two compounds, with a thio-ether at R3 position and either two- or three-carbon linker to the sulfate moiety (Figure 2B). Compound 8 containing a three-carbon linker showed limited improvement over 1, while 7 with a two-carbon linker showed more pronounced binding as judged by more extensive chemical shift perturbations (Figure 2A). Subsequently, we determined the crystal structure of 7 in complex with CDC25B (Supporting Information Figure 1B) and found that indeed additional interactions are formed between the sulfate group of 7 and Arg488 and Arg492, which contribute to the enhanced binding of this compound (Figure 2C). This finding further demonstrates the feasibility of developing more potent analogs of 1.
We have selected compound 7, as the most potent and bulky compound, to assess the ability of a small molecule to inhibit the interaction of CDC25B with the CDK2/Cyclin A substrate. For this purpose, we employed the AlphaLISA-based protein–protein interaction assay. A dose-dependent decrease in the AlphaLISA signal was observed upon titration with 7, demonstrating disruption of the CDC25B-CDK2/Cyclin A protein–protein interaction with the IC50 value around 1 mM (Figure 3A). Contrary, much weaker activity has been observed for compound 1, which binds CDC25B with lower affinity. In addition, only 7 forms hydrogen bonds with two arginines (Arg488 and Arg492) that are required for binding of CDK2/CyclinA substrate,19 which may result in higher activity of this compound when compared to 1. Though the activity of 7 is relatively modest, this result provides a proof of principle that ligands that bind to this newly identified site on CDC25B can prevent CDC25B from interacting with the substrate.
Figure 3.
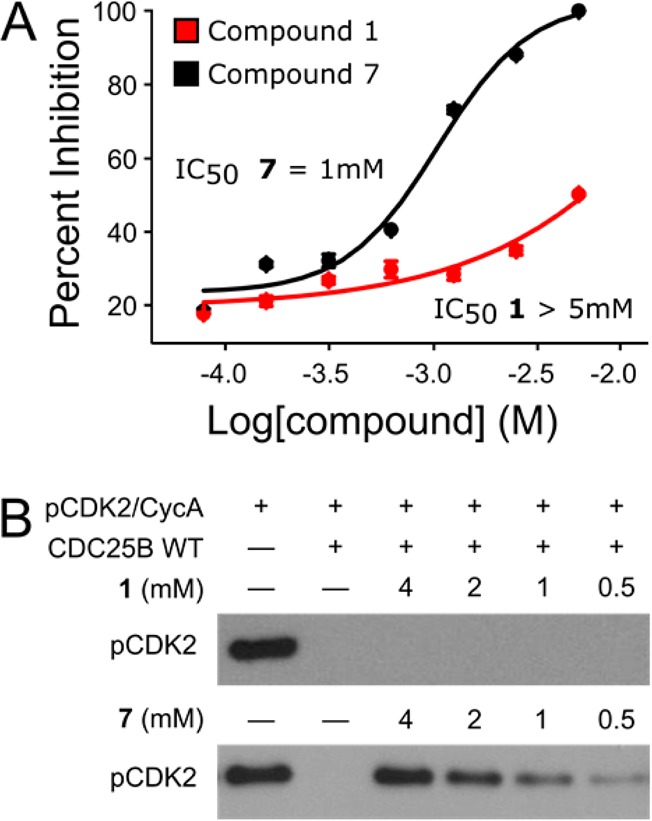
Small molecule ligand binding to the protein–protein interaction site inhibits CDC25B activity. (A) Activity of compound 1 and 7 in an AlphaLISA-based protein–protein interaction assay. (B) In vitro phosphatase assay utilizing phosphorylated CDK2/Cyclin A as a substrate for CDC25B in the presence or absence of 1 or 7. Remaining phosphorylated CDK2/Cyclin A is shown as detected by Western blot.
Finally, to determine whether disruption of the CDC25B-CDK2/Cyclin A protein–protein interaction by a small molecule results in inhibition of CDC25B activity, we assessed the effect of 7 in an in vitro phosphatase assay. We found that incubation with 7 results in a dose-dependent inhibition of CDC25B phosphatase activity toward p-CDK2/Cyclin A substrate (Figure 3B). At the highest concentration of 7, CDC25B activity is completely inhibited and it continues to show inhibitory effect down to 500 μM, with an apparent IC50 between 1 and 2 mM. Importantly, no inhibitory effect of 1 has been observed emphasizing that activity in phosphatase assay is correlated with binding affinity to CDC25B (Figure 3B). These results strongly validate that a small molecule that binds to the protein–protein interaction pocket distal from the active site disrupts the interaction of CDC25B phosphatase with CDK2/Cyclin A substrate and inhibits its phosphatase activity.
We have also assessed whether compounds 1 and 7 bind to CDC25A, a close homologue of CDC25B. We found that 1 and 7 binds to CDC25A, although the chemical shift perturbations are less pronounced than for CDC25B (Supporting Information Figure 2). In addition, compound 7 inhibits the interaction of CDC25A with CDK2/CyclinA with IC50 = 2.2 mM (Supporting Information Figure 3). The binding pockets in CDC25A and CDC25B are very similar, and it is likely that selectivity can be achieved for more elaborated analogs.
In this study, through a fragment-based screening approach, we have identified a small molecule binding site on CDC25B that is distant from the active site and is located adjacent to the residues mediating substrate recognition. Although it has been previously speculated that this pocket might represent a suitable site for inhibitor development,13 our study demonstrates for the first time that (i) it is possible to identify small molecules that can bind to this site; (ii) 2-fluoro-4-hydroxybenzonitrile (compound 1) represents an attractive small molecule scaffold which binds to this site and can be further optimized into more potent ligands; (iii) compounds that bind to this site can disrupt the protein–protein interaction of CDC25B with CDK2/Cyclin A substrate and inhibit the phosphatase activity of CDC25B.
Despite extensive efforts to develop inhibitors of CDC25 phosphatases through targeting the active site,10−12 no structural information for the phosphatase catalytic domain with bound inhibitor has been reported to date. Here, we describe the structure of CDC25B catalytic domain with two small molecule ligands that bind to a pocket in the vicinity of a protein–protein interaction hot-spot. Interestingly, this pocket is relatively polar and fits a network of hydrogen bonded water molecules. Ligands identified in our study can be likely improved through further modifications to replace these water molecules and optimize contacts with the protein. For example, the extensive network of intermolecular hydrogen bonds between biotin and streptavidin is known to contribute to a very high affinity interaction.20 In summary, we propose a novel approach to block activity of CDC25 phosphatases via inhibiting their protein–protein interactions with the substrate as an attractive strategy to develop inhibitors for this important class of enzymes.
Methods
Expression and Purification of Recombinant Proteins
The CDC25B catalytic domain (372–551) and variants were expressed in E. coli BL21 (DE3) with an N-terminal GST tag. The CDC25B cDNA with an N-terminal TEV cleavage site was purchased from Genscript and cloned into a pGST-21a vector with NcoI/XhoI. Cells were grown to in either LB or labeled M9 medium. After 16 h induction with 0.5 mM IPTG at 18 °C, E. coil cells were lysed in a buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM TCEP, and 0.5 mM PMSF. The soluble fraction of the cell lysate was purified using glutathione resin and eluted with lysis buffer containing 50 mM l-glutathione. The eluate was proteolytically cleaved with TEV protease, followed by S-75 size exclusion chromatography in buffer containing 50 mM Tris (pH 8.0), 50 mM NaCl, and 1 mM TCEP. Pure fractions were pooled and frozen at −80 °C. The wild type CDC25A catalytic domain (331–524) and N-terminally Flag tagged CDC25A with the C431S active site mutation were expressed and purified as described above.
pCDK2/Cyclin A was expressed in E. coli BL21 (DE3) by coexpression of three proteins: full-length human CDK2 with a TEV cleavable N-terminal His tag in a pET24 vector, residues 173–432 of human Cyclin A with an N-terminal His-Smt3 tag in a pET24 vector (generously gifted by Dr. Matthew Young, University of Michigan), and full length Xenopus Myt1 kinase in the pGS21a vector (gifted from Dr. Sally Kornbluth, Duke University School of Medicine). Unphosphorylated CDK2/Cyclin A was expressed as above, but without the addition of Myt1. After 16 h induction with 0.1 mM IPTG at 18 °C, E. coil cells were lysed in a buffer containing 50 mM Tris (pH 8.0), 500 mM NaCl, 1 mM β-mercaptoethanol, and 0.5 mM PMSF. The soluble cell lysate was purified using Ni-affinity chromatography. The eluted fractions were pooled and proteolytically cleaved with TEV and ULP1 proteases, followed by S-75 size exclusion chromatography in buffer containing 50 mM Tris (pH 8.0), 150 mM NaCl, and 1 mM TCEP. Pure fractions were pooled and frozen at −80 °C.
NMR-Based Fragment Screen
The fragment library used for screening was a combination of commercially available fragments and compounds synthesized in-house. Samples for fragment screening were made with 80 μM 15N-labeled CDC25B C473S prepared in a buffer containing 50 mM Tris (pH 7.0), 50 mM NaCl, 1 mM TCEP, and 5% D2O. Fragments were screened in mixtures of 20 compounds per sample at 250 μM final concentration, in 5% DMSO. 1H–15N HQSC spectra were acquired at 30 °C on a 600 MHz Bruker Avance III spectrometer equipped with cryoprobe, running Topspin version 2.1. Processing and spectral visualization was performed using NMRPipe21 and Sparky.221H–15N HQSC peak assignments were determined previously.16
Crystallization and Structure Determination
Crystals of apo-CDC25B C473S ΔC (372–551) were produced as previously published.3 For compound 1, crystals were transferred to the mother liquor solution to containing 50 mM 1 in 5% DMSO to soak for 30 min and were then transferred to the mother liquor solution containing 50 mM 1, 5% DMSO, and 20% glycerol for cryoprotection prior to freezing in liquid nitrogen. For compound 7, crystals were transferred directly to the same cryoprotection solution in the presence of 2 mM 7 and 5% DMSO for 2 min prior to freezing. Diffraction data was collected for the CDC25B-1 crystals using an X-ray difractometer at the Center for Structural Biology at the University of Michigan. Diffraction data for the CDC25B-7 complex was collected at the 21-ID-F beamline at the Life Sciences Collaborative Access Team at the Advanced Photon Source. The data was integrated and scaled using HKL-300023 for 1 and Mosflm24 for 7 and both structures were solved by molecular replacement with MOLREP25 using the known apo-CDC25B C473S structure for the search model (PDB code: 2A2K).3 Refinement for both structures was performed using REFMAC,26 COOT,27 and the CCP4 program suite28 (Table 1). Refinement of anisotropic B-factors was done in the late stages of refinement. The structure was validated using the MOLPROBITY29 server.
Table 1. Crystallographic Data Collection and Refinement Statistics.
| CDC25B-1 | CDC25B-7 | |
|---|---|---|
| PDB code | 4WH7 | 4WH9 |
| Data Collection | ||
| space group | P212121 | P212121 |
| cell dimensions a, b, c (Å) | 51.2, 71.4, 73.5 | 51.7, 71.5, 73.9 |
| resolution (Å) | 1.62 (1.65–1.62) | 1.50 (1.58–1.50) |
| unique reflections | 35426 (1746) | 44620 (6450) |
| Rsym | 0.154 (0.467) | 0.081 (0.538) |
| I/σI | 28.6 (2.3) | 14.3 (3.8) |
| completeness (%) | 90.7 (90.2) | 100.0 (100.0) |
| redundancy | 2.3 (2.2) | 7.3 (7.3) |
| Refinement | ||
| Rwork/Rfree (%) | 15.7/19.8 | 12.8/15.2 |
| No. atoms | ||
| protein | 1511 | 1528 |
| water | 287 | 296 |
| mean B-factors (Å2) | 24.4 | 20.3 |
| rms deviations | ||
| bond lengths (Å) | 0.011 | 0.012 |
| bond angles (deg) | 1.187 | 1.928 |
| Ramachandran plot | ||
| most favored regions (%) | 97.2 | 97.3 |
| additional allowed regions (%) | 2.8 | 2.7 |
Protein–Protein Interaction Assay
C-terminally 6xHis tagged CDK2/Cyclin A complex and N-terminally Flag tagged CDC25B C473S (372–566) were expressed and purified as stated above. Proteins were incubated together at a final concentration of 10 nM each for 1 h prior to incubation with compound for 1 h, followed by addition of Ni-chelate AlphaScreen donor beads (PerkinElmer) and Anti-Flag AlphaLISA acceptor beads (PerkinElmer) at a final dilution of 1:1000 for 1 h. Protein–protein interaction assays were quantified using a PheraStar plate reader with excitation at 680 nm wavelength and emission at 615 nm in 20 uL volumes in an uncoated, white, low-volume, 384-well plate (Corning). Assays were performed in a buffer containing 50 mM MOPS (pH 7.25), 50 mM NaCl, 10 mM MgCl2, 1 mM TCEP, with addition of 1 mM ATP, 0.01% BSA, and 0.01% Tween-20 immediately prior to the start of the assay. The protein–protein interaction assay for the interaction between CDK2/Cyclin A and N-terminally Flag tagged CDC25A C431S (331–524) was performed as above.
CDC25B Phosphatase Activity Assay
Wild-type CDC25B (372–566) at 250 nM was incubated with 7 for 1 h in buffer containing 50 mM Tris (pH 8.0), 50 mM NaCl, 1 mM TCEP, and 5% DMSO, followed by the addition of 500 nM pCDK2/Cyclin A. After 50 min at RT, the reaction was quenched by adding 1:1 equiv of 2× SDS-PAGE loading buffer. Reaction samples were separated on a 4–20% SDS gel 170 V for 45 min. Proteins were transferred to nitrocellulose with a wet electrotransfer system (BioRad) for 60 min at 25 V. Subsequently, the membrane was blocked with 5% BSA in Tris-buffered saline with 0.1% Tween-20 (TBS-T), and incubated overnight at 4 °C with anti-pT15-cdc2 monoclonal antibody (#9111, Cell Signaling Technology, 1:2000). After washing, the membrane was incubated with an HRP conjugated antirabbit antibody (1:10 000) and visualized.
Acknowledgments
This work was supported by NIH R01 CA181185 to T.C. and University of Michigan Gastrointestinal (GI) Specialized Programs of Research Excellence (SPORE) Career Development Award to T.C. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the LS-CAT Sector 21 was supported by the Michigan Economic Development Corporation and the Michigan Technology Tri-Corridor (Grant 085P1000817).
Supporting Information Available
Electron density for compounds 1 and 7 before and after refinement; interaction of compounds 1 and 7 with CDC25A; inhibition of protein–protein interactions by compounds 1 and 7; compound synthesis and characterization. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nilsson I.; Hoffmann I. (2000) Cell cycle regulation by the Cdc25 phosphatase family. Prog. Cell Cycle Res. 4, 107–114. [DOI] [PubMed] [Google Scholar]
- Sebastian B.; Kakizuka A.; Hunter T. (1993) Cdc25M2 activation of cyclin-dependent kinases by dephosphorylation of threonine-14 and tyrosine-15. Proc. Natl. Acad. Sci. U.S.A. 90, 3521–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J.; Buhrman G.; Rudolph J. (2007) Kinetic and structural studies of specific protein–protein interactions in substrate catalysis by Cdc25B phosphatase. Biochemistry 46, 807–818. [DOI] [PubMed] [Google Scholar]
- Nishioka K.; Doki Y.; Shiozaki H.; Yamamoto H.; Tamura S.; Yasuda T.; Fujiwara Y.; Yano M.; Miyata H.; Kishi K.; Nakagawa H.; Shamma A.; Monden M. (2001) Clinical significance of CDC25A and CDC25B expression in squamous cell carcinomas of the oesophagus. Br. J. Cancer 85, 412–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y.; Yoshida H.; Tomoda C.; Uruno T.; Takamura Y.; Miya A.; Kobayashi K.; Matsuzuka F.; Kuma K.; Nakamura Y.; Kakudo K.; Miyauchi A. (2005) Expression of cdc25B and cdc25A in medullary thyroid carcinoma: Cdc25B expression level predicts a poor prognosis. Cancer Lett. 229, 291–297. [DOI] [PubMed] [Google Scholar]
- Broggini M., Buraggi G., Brenna A., Riva L., Codegoni A. M., Torri V., Lissoni A. A., Mangioni C., D’Incalci M.. Cell cycle-related phosphatases CDC25A and B expression correlates with survival in ovarian cancer patients. Anticancer Res. 20, 4835–4840. [PubMed] [Google Scholar]
- Yan X.; Chua M.; He J.; So S. (2008) Small interfering RNA targeting CDC25B inhibits liver tumor growth in vitro and in vivo. Mol. Cancer 7, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liffers S.-T.; Munding J. B.; Vogt M.; Kuhlmann J. D.; Verdoodt B.; Nambiar S.; Maghnouj A.; Mirmohammadsadegh A.; Hahn S. A.; Tannapfel A. (2011) MicroRNA-148a is down-regulated in human pancreatic ductal adenocarcinomas and regulates cell survival by targeting CDC25B. Lab. Invest. 91, 1472–1479. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Zhang G.; Kong C. (2014) High expression of Cdc25B and low expression of 14-3-3σ is associated with the development and poor prognosis in urothelial carcinoma of bladder. Tumour Biol. 35, 2503–2512. [DOI] [PubMed] [Google Scholar]
- Lavecchia A.; Di Giovanni C.; Novellino E. (2010) Inhibitors of Cdc25 phosphatases as anticancer agents: a patent review. Expert Opin. Ther. Pat. 20, 405–425. [DOI] [PubMed] [Google Scholar]
- Lavecchia A.; Di Giovanni C.; Novellino E. (2012) CDC25 phosphatase inhibitors: An update. Mini Rev. Med. Chem. 12, 62–73. [DOI] [PubMed] [Google Scholar]
- Lyon M. A.; Ducruet A. P.; Wipf P.; Lazo J. S. (2002) Dual-specificity phosphatases as targets for antineoplastic agents. Nat. Rev. Drug Discovery 1, 961–976. [DOI] [PubMed] [Google Scholar]
- Rudolph J. (2007) Inhibiting transient protein–protein interactions: Lessons from the Cdc25 protein tyrosine phosphatases. Nat. Rev. Cancer 7, 202–211. [DOI] [PubMed] [Google Scholar]
- Kristjánsdóttir K.; Rudolph J. (2004) Cdc25 phosphatases and cancer. Chem. Biol. 11, 1043–1051. [DOI] [PubMed] [Google Scholar]
- Bolton J. L.; Trush M. A.; Penning T. M.; Dryhurst G.; Monks T. J. (2000) Role of quinones in toxicology. Chem. Res. Toxicol. 13, 135–160. [DOI] [PubMed] [Google Scholar]
- Lund G.; Cierpicki T. (2014) Solution NMR studies reveal no global flexibility in the catalytic domain of CDC25B. Proteins 82, 2889–2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn J.; Parks J.; Buhrman G. (2005) Experimental validation of the docking orientation of Cdc25 with its Cdk2-CycA protein substrate. Biochemistry 44, 16563–16573. [DOI] [PubMed] [Google Scholar]
- Scott D. E.; Coyne A. G.; Hudson S. A.; Abell C. (2012) Fragment-based approaches in drug discovery and chemical biology. Biochemistry 51, 4990–5003. [DOI] [PubMed] [Google Scholar]
- Sohn J.; Rudolph J. (2008) The energetic network of hotspot residues between Cdc25B phosphatase and its protein substrate. J. Mol. Biol. 22, 4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber P.; Ohlendorf D.; Wendoloski J.; Salemme F. (1989) Structural origins of high-affinity biotin binding to streptavidin. Science 243, 85–88. [DOI] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
- Goddard T. G., and Kneller D. G.. SPARKY 3. University of California, San Francisco. [Google Scholar]
- Minor W.; Cymborowski M.; Otwinowski Z.; Chruszcz M. (2006) HKL-3000: The integration of data reduction and structure solution—From diffraction images to an initial model in minutes. Acta Crystallogr., Sect. D: Biol. Crystallogr. 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Read R. J., and Sussman J. L. (Eds.) (2007) Evolving Methods for Macromolecular Crystallography; Springer, Netherlands, Dordrecht. [Google Scholar]
- Vagin A.; Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66, 22–25. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr. 53, 240–255. [DOI] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (1994) The CCP4 suite: Programs for protein crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 50, 760–763. [DOI] [PubMed] [Google Scholar]
- Davis I. W.; Leaver-Fay A.; Chen V. B.; Block J. N.; Kapral G. J.; Wang X.; Murray L. W.; Arendall W. B.; Snoeyink J.; Richardson J. S.; Richardson D. C. (2007) MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.