

Abstract
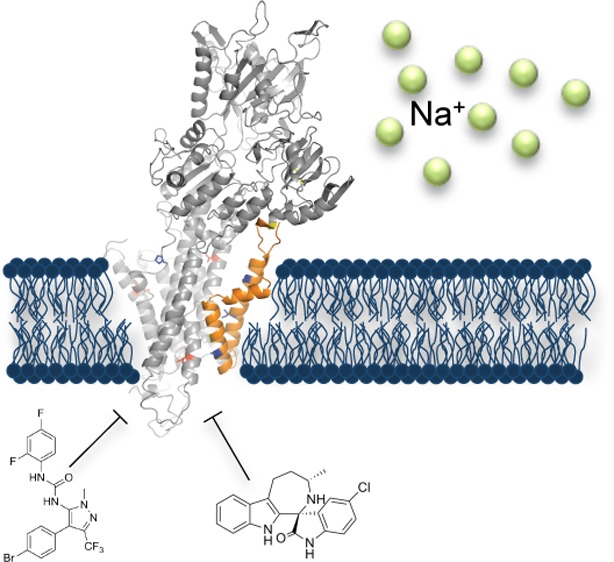
Aminopyrazoles are a new class of antimalarial compounds identified in a cellular antiparasitic screen with potent activity against Plasmodium falciparum asexual and sexual stage parasites. To investigate their unknown mechanism of action and thus identify their target, we cultured parasites in the presence of a representative member of the aminopyrazole series, GNF-Pf4492, to select for resistance. Whole genome sequencing of three resistant lines showed that each had acquired independent mutations in a P-type cation-transporter ATPase, PfATP4 (PF3D7_1211900), a protein implicated as the novel Plasmodium spp. target of another, structurally unrelated, class of antimalarials called the spiroindolones and characterized as an important sodium transporter of the cell. Similarly to the spiroindolones, GNF-Pf4492 blocks parasite transmission to mosquitoes and disrupts intracellular sodium homeostasis. Our data demonstrate that PfATP4 plays a critical role in cellular processes, can be inhibited by two distinct antimalarial pharmacophores, and supports the recent observations that PfATP4 is a critical antimalarial target.
The need to identify new drugs to combat malaria has resulted in high-throughput cellular screening campaigns that have revealed thousands of small molecule inhibitors with antimalarial activity;1−3 several of which are currently in clinical trials.4−8 Although a successful method of drug discovery,9,10 a remaining challenge with cellular screening is in identifying the targets of lead compounds. Target identification is not essential for drug development, but nevertheless improves medicinal chemistry efforts and allows for the design of target-based high-throughput assays that may yield additional potent inhibitors of a target of interest.
The current number of chemically validated targets in Plasmodium spp. remains small11,12 and includes the cytochrome bc1 complex,13 inhibition of hemozoin formation,14 phosphatidylinositol-4-OH kinase,6 dihydroorotate dehydrogenase,15 dihydrofolate reductase,16 and the P-type ATPase, PfATP4.5 As with other antimalarial targets, P-type ATPases are important druggable targets in humans with several clinically relevant inhibitors.17−19 Specifically, PfATP4 belongs to a subfamily of these proteins (family IID) that function to extrude monovalent cations (sodium, lithium, and potassium) from inside the cell. Their presence is limited to lower eukaryotes (fungi, protozoan, and bryophytes) making them an attractive drug target.20,21 PfATP4 is the target of the spiroindolones, a novel antimalarial chemical class discovered in a cellular screen.5,22,23 In a Phase II clinical trial, this class was shown to induce faster parasite clearance times than the current standard of care, artemisinin, and was shown to be active against artemisinin resistant parasites.24
In this study, we use a chemical genomics approach25 to identify PfATP4 as the target of another novel class of inhibitors with antimalarial activity, the aminopyrazoles, which are structurally distinct from the spiroindolones. We further show that the phenotypes of spiroindolone- and aminopyrazole-treated parasites are similar. We additionally identify a third chemotype that interacts with PfATP4. Convergence on this target by multiple chemophores highlights the critical function of this protein in the parasite. Overall, these data suggest that the number of druggable targets in P. falciparum may be smaller than first hypothesized.
Results and Discussion
In Vitro Evolution in the Presence of GNF-Pf4492 Identifies Resistance-Conferring Mutations in P. falciparum pfatp4
Aminopyrazoles were identified in a high-throughput cellular screen against P. falciparum asexual blood stages.1 A representative compound from the aminopyrazole series, GNF-Pf4492 (Figure 1a) (N-[4-(4-bromophenyl)-1-methyl-3-(trifluoromethyl)-1H-pyrazol-5-yl]-N′-(2,4-difluorophenyl)urea, has a mean half maximal (50%) inhibitory concentration (IC50) of 184.1 nM (95% confidence interval (CI) 141.3–239.9) against asexual stages of the multidrug resistant P. falciparum strain, Dd2, and demonstrates no cytotoxicity against the human heptoma cell line Huh7 (>30 μM). Given that the aminopyrazoles have chemical structures that are distinct from currently recommended antimalarials and scaffolds in development, we sought to identify the target of the aminopyrazoles by evolving resistant parasites and examining their genomes for changes that contribute to the resistance phenotype.25
Figure 1.
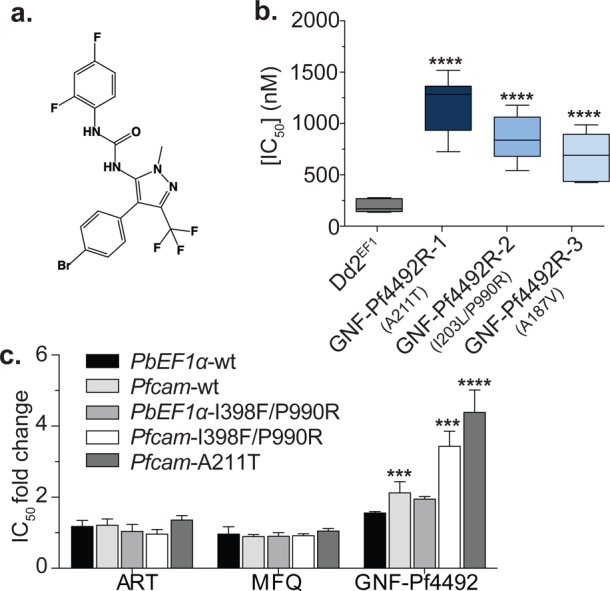
Evolution of P. falciparum GNF-Pf4492-resistant parasite lines. (a) Chemical structure of GNF-Pf4492, a representative compound from the aminopyrazole series. (b) In vitro drug sensitivities of the clonal GNF-Pf4492 evolved resistant lines and the Dd2EF1 parent to GNF-Pf4492 were determined using a SYBR Green I-based cell proliferation assay. Bars represent mean IC50 from a minimum of 3 experiments conducted in duplicate. Error bars = 95% confidence interval. (c) IC50 fold change in transgenic parasite lines harboring additional wild-type pfatp4 copies (PbEF1α-wt and Pfcam-wt) or mutated pfatp4 (PbEF1α-I398F/P990R, Pfcam-I398F/P990R or Pfcam-A211T) under the control of the P. berghei EF1α promoter or the stronger P. falciparum camodulin promoter compared to Dd2attB (contains only the isogenic recombination site). Significance values were determined using one-way ANOVA followed by Dunnett’s multiple comparison post-test to test for a difference in mean log(IC50) value between each strain and the parent: ****p < 0.0001; ***p < 0.001; **p < 0.01.
GNF-Pf4492 drug-resistant lines (GNF-Pf4492R) were selected for in three independent cultures of the multidrug resistant, clonal P. falciparum Dd2 strain (Dd2EF1) by exposure to sublethal concentrations of GNF-Pf4492 for 70 days (SI Figure S1). Each of the resistant clones exhibited mean IC50 values that were significantly greater than the Dd2EF1 parental line (Figure 1b, SI Table S1) (p < 0.0001) and were 1170 nM (95% CI 920.5–1483), 811.0 nM (631.0–1040), and 631.0 nM (458.1–867.0) for GNF-Pf4492R-1, −2 and −3, respectively. None of the mutants appeared to endure any fitness cost as they grew at the same rate as the Dd2EF1 parent. GNF-Pf4492 resistance was stable in these lines; that is, when grown without selective pressure for several months GNF-Pf4492 resistance was still observed. To identify genetic changes that contribute to the GNF-Pf4492 resistance phenotype we compared the genomic sequence of each resistant clone with the sequence of the Dd2EF1 parent (Table 1).
Table 1. Whole Genome Sequencing Identifies SNVs in pfatp4 in All GNF-Pf4492 Resistant Lines.
| GNF-Pf4492 resistant lines |
|||
|---|---|---|---|
| GNF-Pf4492R-1 | GNF-Pf4492R-2 | GNF-Pf4492R-3 | |
| genome coverage (x) | 41 | 55 | 55 |
| % covered by 15 or more reads | 80.3 | 78.1 | 86.5 |
| SNVs identified | |||
| rawa | 63 206 | 62 951 | 67 348 |
| qualityb | 15 868 | 15 983 | 16 367 |
| uniquec | 28 | 22 | 24 |
| intergenic | 21 | 16 | 20 |
| intronic | 4 | 4 | 2 |
| synonymous | 0 | 0 | 0 |
| nonsynonymous | 2 | 2 | 2 |
| gene (mutation) | PF10_0182: conserved protein (K309N), PFL0590c: PfATP4 (A211T) | PFL0590c: PfATP4 (I203L), PFL0590c: PfATP4 (P990R) | PF10_0366: ADP/ATP transporter (I301N), PFL0590c: PfATP4 (A187V) |
After alignment to P. falciparum 3D7 reference genome.
Quality filters based on parameters defined in Methods.
Compared to Dd2 parent. No., number; SNVs, single-nucleotide polymorphisms; PfATP4, P-type cation ATPase 4.
Single-nucleotide variants (SNVs), as well as copy number variants (CNVs), contribute to drug resistance in Plasmodium spp.; therefore, we looked for both types of genetic changes in our resistant lines.5,26,27 We did not observe any unique CNVs in the GNF-Pf4492R lines compared to the Dd2EF1 parent (Figure S2) but did identify SNVs that were unique to the resistant lines, and therefore arose during selective pressure with GNF-Pf4492. A total of 15 868, 15 983, and 16 367 SNVs were confidently identified in GNF-Pf4492R-1, GNF-Pf4492R-2, and GNF-Pf4492R-3, respectively, compared to the 3D7 reference genome (Table 1). After comparative analysis with the Dd2EF1 parental line, 74 SNVs were found to be unique: 28 in GNF-Pf4492R-1, 22 in GNF-Pf4492R-2, and 24 in GNF-Pf4492R-3 (Table 1 and SI Table S2). Of these, six resulted in a nonsynonymous amino acid substitution in an exon of an open reading frame (two in each line) (Table 1) and remarkably, all three lines contained SNVs in PF3D7_1211900 (formerly PFL0590c), the gene that encodes the P-type cation-transporting ATPase, PfATP4. GNF-Pf4492R-1 and GNF-Pf4492R-3 each harbored one mutation in pfatp4 (Ala211Thr and Ala187Val, respectively) while GNF-4492R-2 bore two mutations (Ile203Leu and Pro990Arg) (Table 1). These two mutations were the only nonsynonymous substitutions identified in the entire GNF-Pf4492R-2 genome. Mutations in all resistant clones were covered by 76 to 93 reads (SI Table S2), lending high confidence to the base calls, and were further confirmed by Sanger sequencing.
In addition to the mutations identified in pfatp4, two additional candidate nonsynonymous coding SNVs were identified in GNF-Pf4492R-1 and -3. A nonsynonymous mutation in PF3D7_1018900 (formerly PF10_0182), a gene encoding a conserved protein of unknown function, was identified in GNF-Pf4492R-1. Sanger sequencing showed the expected parental sequence of PF3D7_1018900 in GNF-Pf4492R-1. It was noted that this SNV occurs in a homopolymer tract, which causes difficulty for the read alignment program, therefore causing an isolated false positive call (SI Figure S3a). In GNF-Pf4492R-3, a third nonsynonymous coding SNV (encoding the mutation Ile301Asn) was identified in the last amino acid of the polypeptide encoded by PF3D7_1037300 (formerly PF10_0366). This gene encodes an ADP/ATP transporter, a protein that enables ATP and ADP to traverse the inner mitochondrial membrane.28 The human ADP/ATP transporter, which is critical for cellular respiration, can be chemically inhibited and is the target of the poisons, bongkrekic acid and atractyloside.29 Interestingly, we observed a K544N mutation in pfcdpk5 (Pf3D7_1337800) in all three resistant lines (SI Figure S3b). Although this SNV is not present in the published Dd2 sequence, it was found in our Dd2EF1 line. We thus believe that this SNV was not acquired during GNF-Pf4492 selective pressure.
Because resistance to aminopyrazoles could be conferred by mutations in pfatp4, one of the noncoding genetic changes, the mutation in the ADP/ATP transporter, or a yet unidentified locus, we evaluated a transgenic P. falciparum Dd2 line containing an attB (Dd2attB) site integrated at the cg6 locus30 expressing one extra gene copy of wild-type pfatp4 under the control of the P. berghei elongation factor-1 α (EF1α) promoter5 or the stronger, P. falciparum (camodulin) cam promoter.5,30 Both stable overexpression transgenic lines showed a > 1.5× shift in resistance to GNF-Pf4492 compared to the Dd2attB parent (Figure 1c, SI Table S1). We observed no change in the IC50 of control compounds and Western blot analysis with antibodies to PfATP4 confirmed low levels of overexpression (SI Figure S4a) relative to the Dd2attB parent. Because resistance in the GNF-Pf4492R-lines was caused by mutant alleles and not overexpression (SI Figure S4b), we also evaluated Dd2attB strains expressing mutated copies of pfatp4. The Pfcam-A211T strain harbors the mutated pfatp4 observed in the GNF-Pf4492R-1 line. Two additional pre-existing transgenic lines bearing pfatp4 with two mutations (Ile398Phe and Pro990Arg) under control of the P. berghei EF1α promoter or the P. falciparum cam promotor were also evaluated for cross-resistance.5 We saw a 2-fold increase in resistance in the PbEF1α-I398F/P990R line. Significantly, we observed a 3.6 and 4.4 fold shift in resistance in the Pfcam-I398F/P990R line and Pfcam-A211T line respectively, but no shift for other antimalarials. This increase in resistance in the mutant pfatp4 expressing strains is likely smaller than expected due to concurrent expression of both wild-type and mutant PfATP4. Western Blot analysis showed that this does not appear to be due only to overexpression of the protein (SI Figure S4a). These data show that mutations in pfatp4 alone confer resistance to aminopyrazoles.
Aminopyrazole-Resistant Parasite Lines Exhibit Cross-Resistance with the Spiroindolone Drug Series
Mutations in pfatp4, whose protein product is implicated in parasite sodium tolerance,21 also confer resistance to spiroindolones, a novel class of antimalarial that are currently in clinical trials.5,23 The pfatp4 mutations associated with spiroindolone resistance (SI Table S3) were previously identified using the same methodology, yet with small molecule inhibitors from the spiroindolone class, either KAE609 (formerly NITD609) or KAE678 (formerly NITD678) (Figure 2a,b).5 The pfatp4 SNVs detected in all aminopyrazole- and spiroindolone resistant lines are present at or near the predicted transmembrane (TM) domain of the protein, where cations are translocated, suggesting a shared structure–function relationship (Figure 2c). All GNF-Pf4492-associated mutations, except Pro990Arg, occurred in TM helices 1 and 2 (model backbone colored orange) and corresponded at or near reported KAE678 resistance-conferring mutations: Ala184Ser (yellow), Ile203Met (yellow; identical location of GNF-Pf4492 mutation), and Gly223Arg (yellow) (Figure 2c). The Pro990Arg mutation is located 5 residues before the predicted start of TM helix 7 and, thus, resides near the TM region. Curiously, as does GNF-Pf4492R-3, KAE678R-2 also possessed a nonsynonymous mutation in the ADP/ATP antiporter (Ile119Ser), suggesting that this may function as a compensatory mutation or possibly an additional target.28
Figure 2.
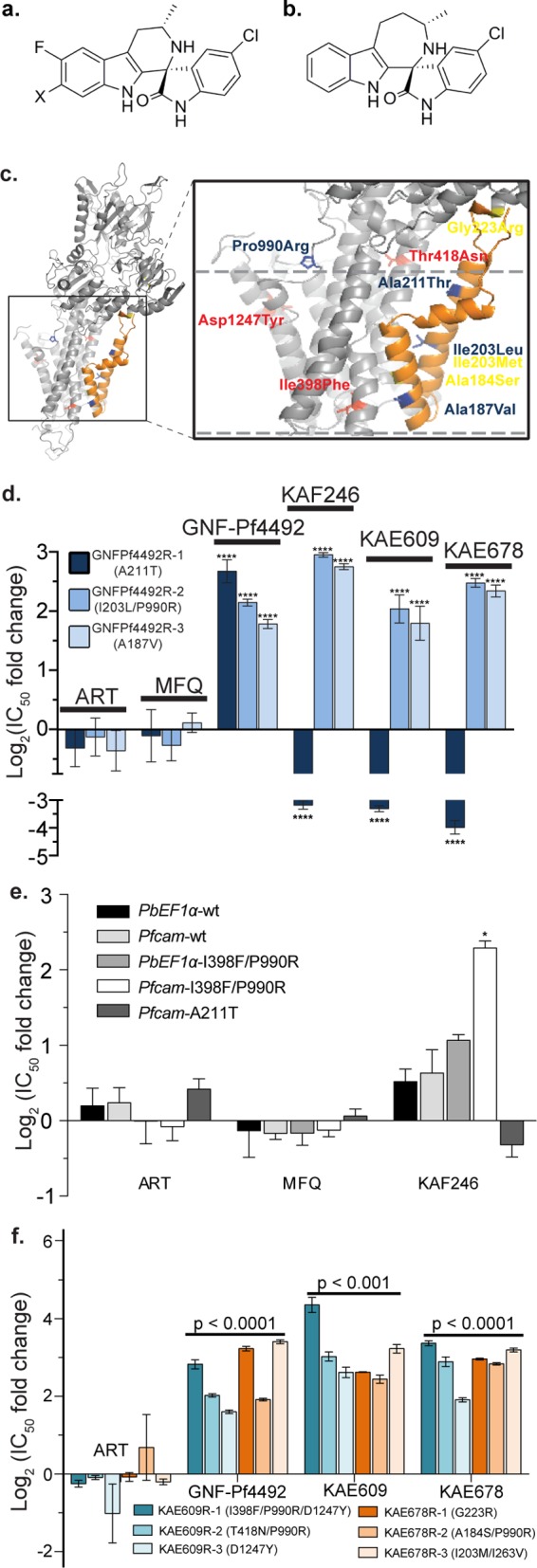
Mutations in pfatp4 confer cross-resistance between GNF-Pf4492 and the spiroindolone class. Chemical structures of representative spiroindolones (a) KAE609 when X = Cl and KAF246 when X = F. (b) KAE678 is distinguished by a 7-membered ring in the tricyclic system. (c) A PfATP4 homology model shows the location of resistance-conferring mutations specific to the aminopyrazole GNF-Pf4492 (blue) and the spiroindolones KAE609 (red) and KAE678 (yellow). Nearly all resistance-conferring mutations occur within or near PfATP4 transmembrane domains (approximated by dashed lines). Transmembranes 1 and 2 are in orange. (d) The three GNF-Pf4492-resistant lines—GNF-Pf4492R-1, GNF-Pf4492R-2, and GNF-Pf4492R-3—were tested for cross-resistance against a panel of spiroindolones (KAE609, KAE678, and KAF246). The IC50 shift is relative to the GNF-Pf4492-sensitive Dd2EF1. Artemisinin (ART) and mefloquine (MFQ) were used as controls. (e) IC50 log2 fold change in transgenic lines harboring either wild-type pfatp4 (PbEF1α-wt and Pfcam-wt) or mutated pfatp4 (PbEF1α-I398F/P990R, Pfcam-I398F/P990R or Pfcam-A211T). (f) Resistant lines (three each) were independently evolved to spiroindolone analogs KAE609 and KAE678. These lines were tested for cross-resistance against GNF-Pf4492. ART and MFQ were used as controls. Significance values were determined using one-way ANOVA followed by Dunnett’s multiple comparison post-test to test for a difference in mean log(IC50) value between each strain and the parent: ****p < 0.0001; *p < 0.01.
Because the spiroindolone chemotype is structurally different from the aminopyrazole class, we did not expect resistance to both chemical classes would be conferred by mutations in the same gene. We characterized the extent of cross-resistance using three closely related members of the spiroindolone series, KAE609, KAE678, and KAF246 (formerly NITD24621) and found GNF-Pf4492R-2 and -3 were 3- to 8-fold more resistant, to the spiroindolones than the Dd2EF1 parent (Figure 2d, SI Table S1). Surprisingly, GNF-Pf4492R-1, the line most resistant to the aminopyrazole, showed an increased sensitivity to the spiroindolones (Figure 2d). This line was 7- to 20-fold more sensitive to all of the spiroindolone compounds tested compared to the Dd2EF1 parent. To further investigate this curious observation, we tested the Pfcam-A211T transgenic line for cross resistance with KAF246. The Pfcam-A211T showed some signs of sensitivity while the Pfcam I398F/P990R was >2-fold more resistant to KAF246 (Figure 2e). Failure to see more sensitivity introduced with the Pfcam-211T mutation is again most likely due to the presence of both wild-type and mutant PfATP4. Similarly, the six independently evolved spiroindolone-resistant lines were also resistant to GNF-Pf4492, exhibiting 3–15 fold higher IC50 values compared to the parent (Figure 2f, SI Table S1). No significant change was observed in IC50 for the controls artemisinin and mefloquine, demonstrating this effect is specific and not due to a multidrug efflux mechanism.
To determine if any other publicly available compounds would have activity against PfATP4, we screened several inhibitors from the “malaria box,” a set of compounds previously identified in cellular screens with activity against blood-stage P. falciparum, for cross-resistance with the GNF-Pf4492R lines.32 Two of these compounds, MMV666124 (Figure 3a) and MMV020660 (Figure 3b), showed cross-resistance with the GNF-Pf4492R lines (Figure 3c). As with the phenotype observed with the spiroindolones, GNF-Pf4492R-1 did not exhibit cross-resistance with MMV020660 but rather was more sensitive than the Dd2EF1 parent. Interestingly, the structure of MMV020660 shows some similarity to GNF-Pf4492 (Figure 3b).
Figure 3.
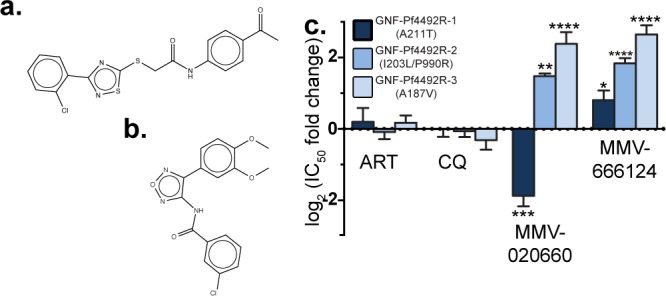
GNF-Pf4492-R lines show cross-resistance to the malaria box inhibitors MMV666124 and MMV020660. Chemical structure of compounds known to have blood-stage antimalarial activity (a) MMV666124 and (b) MMV020660. (c) Log2 fold change in IC50 for GNF-Pf4492R lines compared to the Dd2EF1 parent. Bars represent mean log2(IC50 fold change) from a minimum of 3 experiments conducted in duplicate. Error bars = SEM; Significance values were determined using one-way ANOVA followed by Dunnett’s multiple comparison post-test to test for a difference in mean log2(IC50) between each strain and the Dd2EF1 parent; *p < 0.05, **p < 0.01.
Aminopyrazoles and Spiroindolones Induce Similar Phenotypes In Vitro
Given that mutated PfATP4 mediates aminopyrazole resistance, we hypothesized that the aminopyrazoles and spiroindolones share a mechanism of action. Therefore, we sought to determine whether parasites treated with inhibitors from either class produced the same phenotype. Synchronized cultures were treated with 10× IC50 GNF-Pf4492 or KAF246 to observe when the compounds act during the parasite life cycle. Neither culture treated with inhibitor advanced past the early trophozoite stage, arresting in the ring or early trophozoite stages (Figure 4a). Furthermore, similar to the spiroindolones, GNF-Pf4492 diminished protein synthesis activity in the trophozoite stage of the Dd2 parent at 10× and 100× IC50 concentrations as measured by incorporation of radiolabeled methionine and cysteine (SI Figure S5a). Conversely, protein synthesis progressed unabated in the KAE609R-1 clone after addition of either 100× KAF246 or GNF-Pf4492 (SI Figure S5b).
Figure 4.
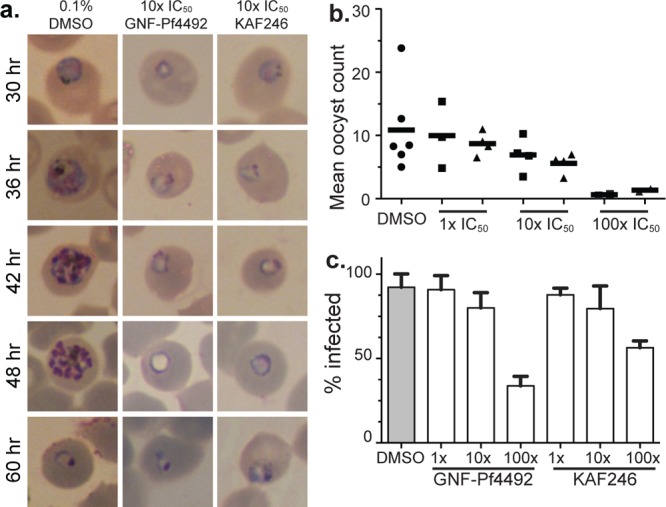
Phenotypic characterizations of parasites treated with aminopyrazoles or spiroindolones. (a) The lifecycle stage of action of GNF-Pf4492 and KAF246 were determined by addition of either 10× IC50 of GNF-Pf4492 or KAF246 and observation over a 60 h period. (b, c) Standard membrane feeding with P. falciparum to determine inhibitor action on transmission. Ten to forty mosquitoes per feeding were dissected. (b) Geometric mean number of oocysts counted per feeding. Bars represent median. Squares, GNF-Pf4492 treated; triangles, KAF246 treated. (c) The percent of mosquitoes that had one or more oocysts per the total number of mosquitoes dissected.
While phenocopy was observed for drug-treated parasites in the asexual blood stages, we continued characterization of these inhibitors in the liver and sexual blood stages. Addition of GNF-Pf4492 or KAF246 to P. yoelii sporozoite-invaded HepG2 cells4 resulted in no detectable reduction in parasite growth at 10 μM, the highest concentration tested (SI Table S4). Van Pelt Koops et al. demonstrated that the spiroindolones are capable of blocking transmission to mosquitoes.33 Therefore, we used a standard membrane-feeding assay to assess the effectiveness of GNF-Pf4492 in preventing P. falciparum oocyst development in the mosquito34 GNF-Pf4492 or KAF246 inhibitors were added at three different concentrations directly to a parasite blood meal. This resulted in a dose-dependent decrease in the number of oocysts formed in the mosquito midguts 8 days after feeding (Figure 4b). The strongest effect was observed for the 100× IC50 concentration for both GNF-Pf4492 and KAF246 (mean oocyst count of 0.63 and 1.33, respectively). At these concentrations, 30 nM KAF246 and 15 μM GNF-Pf4492, a respective reduction of 60% and 38% of the control was observed. Additionally, as compound concentration was increased, the percentage of mosquitoes infected also decreased (Figure 4c).
GNF-Pf4492 Causes an Increase in Intracellular Sodium Concentration in P. falciparum
Recent studies show that PfATP4 functions to transport sodium against a concentration gradient, out of the cell, maintaining a low intracellular sodium concentration in the presence of the high sodium environment of the infected red blood cell.21,35,36 Therefore, we sought to determine whether both compounds would affect sodium concentration within the parasite cytoplasm. When 100× IC50 GNF-Pf4492 (Figure 5a) or KAF246 (Figure 5b) was added, a rapid, steady, and comparable increase in intracellular sodium concentration ([Na+]i) was observed over a 30 min period and this response was demonstrated to be dose-dependent in the presence of GNF-Pf4492. For GNF-Pf4492R-1 and -2, when treated with compound these lines maintained the same [Na+]i over the observation period (Figure 5a, SI Figure S6a). While a dose-dependent response was observed for GNF-Pf4492R-3, [Na+]i concentrations did not reach those of the Dd2EF1 after 30 min of observation after addition of the inhibitor (SI Figure S6a). In the presence of KAF246, in the GNF-Pf4492R-1 mutant, which is hyper-sensitive to KAF246, a similar pattern of increase in [Na+]i was observed as the parent (Figure 5b). For the highest concentration of KAF246 used (100× IC50), increases in [Na+]i were similar for the GNF-Pf4492R-2 and -3 mutants, although the final [Na+]i remained suppressed below that observed in the parental line (SI Figure S6b). The resting [Na+]i concentrations for GNF-Pf4492R-1 and -2 were consistently higher than that of the parent (SI Table S5).
Figure 5.
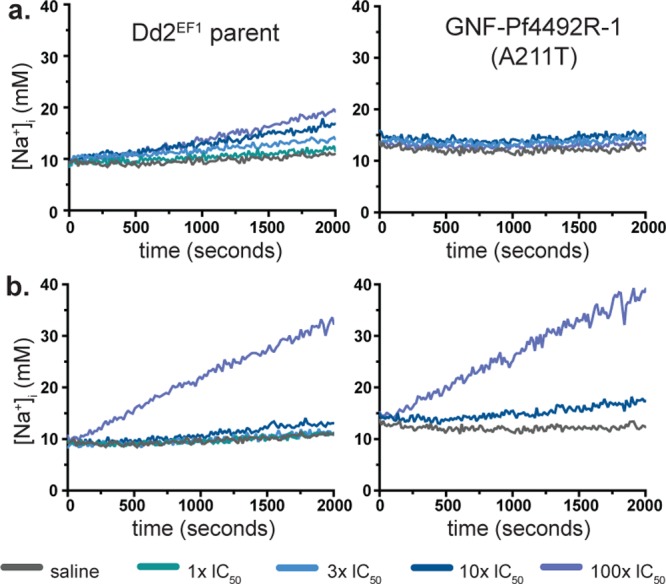
Structure–function relationship between PfATP4 mutations and compound treatment. Effects of (a) aminopyrazole and (b) spiroindolone inhibitors on intracellular sodium concentration [Na+]i in the Dd2EF1 and GNF-Pf4492R-1 strains. [Na+]i traces for extracted trophozoite-stage parasites treated with saline or inhibitor immediately prior to fluorescence acquisition. Inhibitors were added at 1 × , 3×, 10×, or 100× the IC50 for the Dd2EF1 parent. Traces are representative of those obtained from a minimum of four independent experiments performed in duplicate. Calibration curves were generated for each strain and each experimental replicate to determine [Na+]i.
Conclusions
New phenotypic screening efforts have unveiled novel antimalarial chemotypes with correspondingly novel mechanisms of action.4,5,37 Inherently, the identification of targets of compound hits originating from cellular screens requires deconvolution. Target identification for one of these compounds, the aminopyrazole GNF-Pf4492,1 reveals it phenocopies the spiroindolone series, a chemical class shown in Phase II clinical trials to induce faster parasite clearance times than artemisinin.5,24 We demonstrate that cross-resistance between aminopyrazole and spiroindolone series is mediated by mutations in pfatp4, a gene encoding a P-type ATPase that functions as a Na+/H+ pump at the parasite plasma membrane.21 A third chemotype associated with resistance-conferring mutations in pfatp4, dihydroisoquinolones, (http://www.mmv.org/research-development/rd-portfolio),38 has progressed to preclinical evaluation as well. The convergence on the same target by inhibitors of unrelated compound classes is not uncommon for antimalarials and does not preclude their importance as good drug targets. In the case of cytochrome bc1, the validated mitochondrial protein target of atovaquone,39,40 although several other inhibitors target it,27,41,42 atovaquone is used clinically with wide success for prophylaxis.43 Identification of mutations in pfatp4 that mediate resistance to multiple compound classes suggests its importance as a drug target, although the novel mechanism of action is unclear.
Insight into the mechanism of action of PfATP4 targeting inhibitors is gleaned from the location of amino acid changes conferring resistance. The pfatp4 mutations we identify localize to the transmembrane domain suggesting inhibitors bind in the clefts within transmembrane helices. Clinically relevant p-type ATPase inhibitors bind to transmembrane helices leading to poor cation recognition or unfavorable enzymatic conformations.18 We speculate that PfATP4 targeting compounds disrupt sodium export in an analogous manner to other P-type ATPase targeting drugs and that there is most likely a small structural element common to the two classes that accounts for both compound classes binding to the same druggable pocket. Resistance conferring PfATP4 mutations disrupt binding of the inhibitor to this pocket and such mutations could be expected to alter the function of the enzyme, which may be reflected in the slightly elevated resting sodium concentrations observed in two of the resistant lines. In the case of the mutations observed in the GNF-Pf4492R-1 line, a single mutation renders the strain resistant to the aminopyrazole yet sensitive to the spiroindolone (as phenomena observed with other P-type ATPase inhibitors44), suggesting the binding sites of each inhibitor are overlapping, yet unique.
Although our data suggests PfATP4 is the target of the spiroindolone and aminopyrazole compound series, alternative explanations exist, including a different biological function for PfATP4 (i.e., as a nonselective drug transporter), mutation of PfATP4 as a secondary, compensatory mutation in response to an as-of-yet unidentified target and last, PfATP4 function is sodium dependent, thus acting downstream of the true target. We evaluated the activity of other antimalarials (mefloquine and artemisinin) against aminopyrazole resistant lines and noted a low-incidence of cross-resistance, a finding inconsistent with the function of PfATP4 as a nonselective drug transporter. Furthermore, if PfATP4 is only involved in inhibitor efflux, we also might expect different chemical phenotypes, which we do not observe—aminopyrazoles and spiroindolones behave similarly throughout the lifecycle. The expression of these mutants alone was sufficient to confer aminopyrazole resistance, suggesting these are not compensatory mutations nor that PfATP4 is downstream of the true target and that PfATP4 plays a direct role in the mechanism of action of aminopyrazole antimalarials.
Other reports have used in silico modeling to predict that the pyrazole series might act by disrupting the protein–protein interaction between the P. falciparum myosin motor component myosin A and the myosin tail interacting protein (MTIP), a complex necessary for parasite reinvasion of red blood cells.45 Despite showing P. falciparum blood-stage activity, docking studies were done in silico thus not directly validating the interaction between MTIP and aminopyrazoles. In addition, we show aminopyrazoles are active throughout the asexual life cycle and not just during cell invasion or gliding motility, which requires the MTIP-myosin A interaction.46,47
One open question is whether PfATP4 is the target of or a gene involved in resistance to both the aminopyrazoles and the spiroindolones. While the underlying mechanism by which PfATP4 inhibitors affect sodium levels in the cell remains to be elucidated, that mutations in PfATP4 are responsible for resistance to two (and possibly three) structurally unrelated compounds has important implications for future drug discovery efforts. It is difficult to imagine that convergence on PfATP4 is coincidental, given that both these drug series were identified by large chemical library screens and their relationship to PfATP4 identified by unbiased genomic sequencing of independently generated resistant lines. Because these screens produced antimalarial compounds acting through a variety of targets, we do not think the libraries are biased for PfATP4 inhibitors.1−3 Therefore, there must be other reasons for target convergence. One possible explanation is that PfATP4 may represent a particularly strong drug target, perhaps owing to its molecular structure of physiologic importance. Although current Phase II clinical trials of the spiroindolones are very positive, they may have high production costs due to their chiral centers.22 Investigation into other chemical classes, such as the aminopyrazoles, that interact with PfATP4 may represent an important alternative for combination therapies. Another possible reason for target convergence, may be that the number of suitable drug targets in Plasmodium is smaller than anticipated. Either way the role of PfATP4 as an important, novel antimalarial target warrants further study.
Methods
See detailed version in SI Materials and Methods.
Evolution of Compound-Resistant Lines, Sensitivity Testing and Construction of Transgenic Parasite Lines
The P. falciparum multidrug resistant strain Dd2EF1 was cultured in triplicate in the presence of increasing concentrations of GNF-Pf4492 to generate resistant mutants as previously described.5 After ∼70 days of selection, parasites were cloned in 96-well plates by limiting dilution.48 The half maximal (50%) inhibitory concentration (IC50) of each compound against blood-stage P. falciparum Dd2EF1 was determined in dose–response format using a SYBR Green I-based cell proliferation assay as previously described.1 Liver-stage IC50 was determined as previously described.4 Transgenic parasite lines were created as previously described.5 Briefly, P. falciparum Dd2 containing the attB recombination site was transfected with the wild-type or mutant pfatp4 transgene under the control of either the P. berghei elongation factor-1α 5′ UTR (PbEF1α) or the stronger P. falciparum calmodulin 5′ UTR (Pbcam).
Whole Genome Sequencing
DNA libraries of each sample were prepared using the Illumina TruSeq version 3 protocol (Illumina, Inc., San Diego, CA) of fragmentation, end-repair, and adapter ligation. Libraries were clustered and run on an Illumina HiSeq 2000 according to manufacturer’s instructions. Fifty base pair, single-end reads were analyzed and aligned to the P. falciparum 3D7 reference genome (PlasmoDB v. 9.0) as previously described.49 Sequence alignments were put through a custom quality control procedure.49 SNVs were initially detected using the Genome Analysis Toolkit (GATK v1.6) and then filtered using the Plasmodium Type Uncovering Software, an integrated pipeline developed in our lab that can also call CNVs.49 All nonsynonymous SNVs were confirmed using Sanger sequencing.
Phenotypic Assays for Parasite Characterization
An E. coli expression vector was constructed to express pure recombinant protein and mice were immunized to generate an antibody to the N-domain of PFATP4. Western blots were performed with 10 or 50 μg protein lysate and hybridized with the PfATP4 N-domain antibody to quantify protein levels in the cells. A previously generated homology model of PfATP45 was used to map the resistance-conferring mutations identified in the GNF-Pf4492-resistant lines. The PyMOL Molecular Graphics System (version 1.2r2, Schrödinger, LLC, Portland, OR) was used to render the model and prepare the figure. Compound stage of action and protein synthesis inhibition studies were conducted as previously described.5 The standard membrane feeding assay was performed as previously described.50P. falciparum NF54 was maintained in vitro in continuous cultivation with daily media changes and without the addition of fresh red blood cells to stimulate gametocytogenesis. Day 13, 15, and 17 gametocyte cultures that exhibited exflagellation were combined to form a mosquito bloodmeal supplemented with GNF-Pf4492 or KAF246 at 1×, 10×, or 100× the mean IC50 value for blood-stage parasites. Intracellular sodium concentration ([Na+]i) within the parasite was determined as previously described.21 50 μL of dye-loaded cells (2 × 109 cells/mL) were seeded into a black 384-well assay plate fluorescence was measured on an EnVision Multilabel reader (PerkinElmer, Waltham, MA).
Acknowledgments
This work partially supported by National Institutes of Health (NIH) (F32AI102567 to E.L. Flannery, 1R01AI0358-01A1 and 5R01AI090141-03 to E.A. Winzeler), the Bill and Melinda Gates Foundation (BMGF) (OPP1054480), and the Wellcome Trust (WT078285 and WT096157). We acknowledge the Medicines for Malaria Venture for providing the Malaria Box for screening and J. Walker for sequencing of the resistant parasite lines.
Supporting Information Available
Detailed methods. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. (2008) In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 105, 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamo F. J.; Sanz L. M.; Vidal J.; de Cozar C.; Alvarez E.; Lavandera J. L.; Vanderwall D. E.; Green D. V.; Kumar V.; Hasan S.; Brown J. R.; Peishoff C. E.; Cardon L. R.; Garcia-Bustos J. F. (2010) Thousands of chemical starting points for antimalarial lead identification. Nature 465, 305–310. [DOI] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Bouck D.; Duffy S.; Crowther G. J.; Davis P. H.; Smithson D. C.; Connelly M.; Clark J.; Zhu F.; Jimenez-Diaz M. B.; Martinez M. S.; Wilson E. B.; Tripathi A. K.; Gut J.; Sharlow E. R.; Bathurst I.; El Mazouni F.; Fowble J. W.; Forquer I.; McGinley P. L.; Castro S.; Angulo-Barturen I.; Ferrer S.; Rosenthal P. J.; Derisi J. L.; Sullivan D. J.; Lazo J. S.; Roos D. S.; Riscoe M. K.; Phillips M. A.; Rathod P. K.; Van Voorhis W. C.; Avery V. M.; Guy R. K. (2010) Chemical genetics of Plasmodium falciparum. Nature 465, 311–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister S.; Plouffe D. M.; Kuhen K. L.; Bonamy G. M.; Wu T.; Barnes S. W.; Bopp S. E.; Borboa R.; Bright A. T.; Che J.; Cohen S.; Dharia N. V.; Gagaring K.; Gettayacamin M.; Gordon P.; Groessl T.; Kato N.; Lee M. C.; McNamara C. W.; Fidock D. A.; Nagle A.; Nam T. G.; Richmond W.; Roland J.; Rottmann M.; Zhou B.; Froissard P.; Glynne R. J.; Mazier D.; Sattabongkot J.; Schultz P. G.; Tuntland T.; Walker J. R.; Zhou Y.; Chatterjee A.; Diagana T. T.; Winzeler E. A. (2011) Imaging of Plasmodium liver stages to drive next-generation antimalarial drug discovery. Science 334, 1372–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottmann M.; McNamara C.; Yeung B. K.; Lee M. C.; Zou B.; Russell B.; Seitz P.; Plouffe D. M.; Dharia N. V.; Tan J.; Cohen S. B.; Spencer K. R.; Gonzalez-Paez G. E.; Lakshminarayana S. B.; Goh A.; Suwanarusk R.; Jegla T.; Schmitt E. K.; Beck H. P.; Brun R.; Nosten F.; Renia L.; Dartois V.; Keller T. H.; Fidock D. A.; Winzeler E. A.; Diagana T. T. (2010) Spiroindolones, a potent compound class for the treatment of malaria. Science 329, 1175–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara C. W.; Lee M. C.; Lim C. S.; Lim S. H.; Roland J.; Nagle A.; Simon O.; Yeung B. K.; Chatterjee A. K.; McCormack S. L.; Manary M. J.; Zeeman A. M.; Dechering K. J.; Kumar T. R.; Henrich P. P.; Gagaring K.; Ibanez M.; Kato N.; Kuhen K. L.; Fischli C.; Rottmann M.; Plouffe D. M.; Bursulaya B.; Meister S.; Rameh L.; Trappe J.; Haasen D.; Timmerman M.; Sauerwein R. W.; Suwanarusk R.; Russell B.; Renia L.; Nosten F.; Tully D. C.; Kocken C. H.; Glynne R. J.; Bodenreider C.; Fidock D. A.; Diagana T. T.; Winzeler E. A. (2013) Targeting Plasmodium PI(4)K to eliminate malaria. Nature 504, 248–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coteron J. M.; Marco M.; Esquivias J.; Deng X.; White K. L.; White J.; Koltun M.; El Mazouni F.; Kokkonda S.; Katneni K.; Bhamidipati R.; Shackleford D. M.; Angulo-Barturen I.; Ferrer S. B.; Jimenez-Diaz M. B.; Gamo F. J.; Goldsmith E. J.; Charman W. N.; Bathurst I.; Floyd D.; Matthews D.; Burrows J. N.; Rathod P. K.; Charman S. A.; Phillips M. A. (2011) Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 54, 5540–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis Y.; Douelle F.; Feng T. S.; Gonzalez Cabrera D.; Le Manach C.; Nchinda A. T.; Duffy S.; White K. L.; Shackleford D. M.; Morizzi J.; Mannila J.; Katneni K.; Bhamidipati R.; Zabiulla K. M.; Joseph J. T.; Bashyam S.; Waterson D.; Witty M. J.; Hardick D.; Wittlin S.; Avery V.; Charman S. A.; Chibale K. (2012) 3,5-Diaryl-2-aminopyridines as a novel class of orally active antimalarials demonstrating single dose cure in mice and clinical candidate potential. J. Med. Chem. 55, 3479–3487. [DOI] [PubMed] [Google Scholar]
- Swinney D. C.; Anthony J. (2011) How were new medicines discovered?. Nat. Rev. Drug Discovery 10, 507–519. [DOI] [PubMed] [Google Scholar]
- Payne D. J.; Gwynn M. N.; Holmes D. J.; Pompliano D. L. (2007) Drugs for bad bugs: Confronting the challenges of antibacterial discovery. Nat. Rev. Drug Discovery 6, 29–40. [DOI] [PubMed] [Google Scholar]
- Flannery E. L.; Chatterjee A. K.; Winzeler E. A. (2013) Antimalarial drug discovery—Approaches and progress towards new medicines. Nat. Rev. Microbiol. 11, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fidock D. A.; Rosenthal P. J.; Croft S. L.; Brun R.; Nwaka S. (2004) Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discovery 3, 509–520. [DOI] [PubMed] [Google Scholar]
- Mather M. W.; Darrouzet E.; Valkova-Valchanova M.; Cooley J. W.; McIntosh M. T.; Daldal F.; Vaidya A. B. (2005) Uncovering the molecular mode of action of the antimalarial drug atovaquone using a bacterial system. J. Biol. Chem. 280, 27458–27465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagola S.; Stephens P. W.; Bohle D. S.; Kosar A. D.; Madsen S. K. (2000) The structure of malaria pigment β-haematin. Nature 404, 307–310. [DOI] [PubMed] [Google Scholar]
- Patel V.; Booker M.; Kramer M.; Ross L.; Celatka C. A.; Kennedy L. M.; Dvorin J. D.; Duraisingh M. T.; Sliz P.; Wirth D. F.; Clardy J. (2008) Identification and characterization of small molecule inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J. Biol. Chem. 283, 35078–35085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuthavong Y.; Tarnchompoo B.; Vilaivan T.; Chitnumsub P.; Kamchonwongpaisan S.; Charman S. A.; McLennan D. N.; White K. L.; Vivas L.; Bongard E.; Thongphanchang C.; Taweechai S.; Vanichtanankul J.; Rattanajak R.; Arwon U.; Fantauzzi P.; Yuvaniyama J.; Charman W. N.; Matthews D. (2012) Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. U.S.A. 109, 16823–16828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmgren M. G.; Nissen P. (2011) P-type ATPases. Annu. Rev. Biophys 40, 243–266. [DOI] [PubMed] [Google Scholar]
- Yatime L.; Buch-Pedersen M. J.; Musgaard M.; Morth J. P.; Lund Winther A. M.; Pedersen B. P.; Olesen C.; Andersen J. P.; Vilsen B.; Schiott B.; Palmgren M. G.; M?ller J. V.; Nissen P.; Fedosova N. (2009) P-type ATPases as drug targets: Tools for medicine and science. Biochim. Biophys. Acta 1787, 207–220. [DOI] [PubMed] [Google Scholar]
- Kuhlbrandt W. (2004) Biology, structure, and mechanism of P-type ATPases. Nat. Rev. Mol. Cell Biol. 5, 282–295. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Navarro A.; Benito B. (2010) Sodium or potassium efflux ATPase a fungal, bryophyte, and protozoal ATPase. Biochim. Biophys. Acta 1798, 1841–1853. [DOI] [PubMed] [Google Scholar]
- Spillman N. J.; Allen R. J.; McNamara C. W.; Yeung B. K.; Winzeler E. A.; Diagana T. T.; Kirk K. (2013) Na(+) regulation in the malaria parasite Plasmodium falciparum involves the cation ATPase PfATP4 and is a target of the spiroindolone antimalarials. Cell Host Microbe 13, 227–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeung B. K.; Zou B.; Rottmann M.; Lakshminarayana S. B.; Ang S. H.; Leong S. Y.; Tan J.; Wong J.; Keller-Maerki S.; Fischli C.; Goh A.; Schmitt E. K.; Krastel P.; Francotte E.; Kuhen K.; Plouffe D.; Henson K.; Wagner T.; Winzeler E. A.; Petersen F.; Brun R.; Dartois V.; Diagana T. T.; Keller T. H. (2010) Spirotetrahydro β-carbolines (spiroindolones): A new class of potent and orally efficacious compounds for the treatment of malaria. J. Med. Chem. 53, 5155–5164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse M. T. (2010) Antimalarial drugs: Speeding to a new lead. Nat. Rev. Drug Discovery 9, 842. [DOI] [PubMed] [Google Scholar]
- White N. J.; Pukrittayakamee S.; Phyo A. P.; Rueangweerayut R.; Nosten F.; Jittamala P.; Jeeyapant A.; Jain J. P.; Lefevre G.; Li R.; Magnusson B.; Diagana T. T.; Leong F. J. (2014) Spiroindolone KAE609 for falciparum and vivax malaria. N. Engl. J. Med. 371, 403–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flannery E. L.; Fidock D. A.; Winzeler E. A. (2013) Using genetic methods to define the targets of compounds with antimalarial activity. J. Med. Chem. 56, 7761–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoepfner D.; McNamara C. W.; Lim C. S.; Studer C.; Riedl R.; Aust T.; McCormack S. L.; Plouffe D. M.; Meister S.; Schuierer S.; Plikat U.; Hartmann N.; Staedtler F.; Cotesta S.; Schmitt E. K.; Petersen F.; Supek F.; Glynne R. J.; Tallarico J. A.; Porter J. A.; Fishman M. C.; Bodenreider C.; Diagana T. T.; Movva N. R.; Winzeler E. A. (2012) Selective and specific inhibition of the Plasmodium falciparum lysyl-tRNA synthetase by the fungal secondary metabolite cladosporin. Cell Host Microbe 11, 654–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam T. G.; McNamara C. W.; Bopp S.; Dharia N. V.; Meister S.; Bonamy G. M.; Plouffe D. M.; Kato N.; McCormack S.; Bursulaya B.; Ke H.; Vaidya A. B.; Schultz P. G.; Winzeler E. A. (2011) A chemical genomic analysis of decoquinate, a Plasmodium falciparum cytochrome b inhibitor. ACS Chem. Biol. 6, 1214–1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bopp S. E. R.; Manary M. J.; Bright A. T.; Johnston G. L.; Dharia N. V.; Luna F. L.; McCormack S.; Plouffe D.; McNamara C. W.; Walker J. R.; Fidock D. A.; Denchi E. L.; Winzeler E. A. (2013) Mitotic evolution of Plasmodium falciparum shows a stable core genome but recombination in antigen families. PLoS Genet. 9, e1003293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson P. J.; Lardy H. A. (1970) Bongkrekic acid. An inhibitor of the adenine nucleotide translocase of mitochondria. J. Biol. Chem. 245, 1319–1326. [PubMed] [Google Scholar]
- Nkrumah L. J.; Muhle R. A.; Moura P. A.; Ghosh P.; Hatfull G. F.; Jacobs W. R. Jr.; Fidock D. A. (2006) Efficient site-specific integration in Plasmodium falciparum chromosomes mediated by mycobacteriophage Bxb1 integrase. Nat. Methods 3, 615–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg T.; Burrows J. N.; Kowalczyk P.; McDonald S.; Wells T. N.; Willis P. (2013) The open access malaria box: A drug discovery catalyst for neglected diseases. PLoS One 8, e62906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Pelt-Koops J.; Pett H.; Graumans W.; van der Vegte-Bolmer M.; van Gemert G.; Rottmann M.; Yeung B.; Diagana T.; Sauerwein R. (2012) The spiroindolone drug candidate NITD609 potently inhibits gametocytogenesis and blocks Plasmodium falciparum transmission to Anopheles mosquito vector. Antimicrob. Agents Chemother. 56, 3544–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blagborough A. M.; Delves M. J.; Ramakrishnan C.; Lal K.; Butcher G.; Sinden R. E. (2013) Assessing transmission blockade in Plasmodium spp. Methods Mol. Biol. 923, 577–600. [DOI] [PubMed] [Google Scholar]
- Kirk K.; Lehane A. M. (2014) Membrane transport in the malaria parasite and its host erythrocyte. Biochem. J. 457, 1–18. [DOI] [PubMed] [Google Scholar]
- Kirk K. (2001) Membrane transport in the malaria-infected erythrocyte. Physiol Rev. 81, 495–537. [DOI] [PubMed] [Google Scholar]
- Nagle A.; Wu T.; Kuhen K.; Gagaring K.; Borboa R.; Francek C.; Chen Z.; Plouffe D.; Lin X.; Caldwell C.; Ek J.; Skolnik S.; Liu F.; Wang J.; Chang J.; Li C.; Liu B.; Hollenbeck T.; Tuntland T.; Isbell J.; Chuan T.; Alper P. B.; Fischli C.; Brun R.; Lakshminarayana S. B.; Rottmann M.; Diagana T. T.; Winzeler E. A.; Glynne R.; Tully D. C.; Chatterjee A. K. (2012) Imidazolopiperazines: Lead optimization of the second-generation antimalarial agents. J. Med. Chem. 55, 4244–4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guiguemde W. A.; Shelat A. A.; Garcia-Bustos J. F.; Diagana T. T.; Gamo F. J.; Guy R. K. (2012) Global phenotypic screening for antimalarials. Chem. Biol. 19, 116–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry M.; Pudney M. (1992) Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4′-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80). Biochem. Pharmacol. 43, 1545–1553. [DOI] [PubMed] [Google Scholar]
- Srivastava I. K.; Morrisey J. M.; Darrouzet E.; Daldal F.; Vaidya A. B. (1999) Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol. Microbiol. 33, 704–711. [DOI] [PubMed] [Google Scholar]
- Bueno J. M.; Herreros E.; Angulo-Barturen I.; Ferrer S.; Fiandor J. M.; Gamo F. J.; Gargallo-Viola D.; Derimanov G. (2012) Exploration of 4(1H)-pyridones as a novel family of potent antimalarial inhibitors of the plasmodial cytochrome bc1. Future Med. Chem. 4, 2311–2323. [DOI] [PubMed] [Google Scholar]
- Nilsen A.; LaCrue A. N.; White K. L.; Forquer I. P.; Cross R. M.; Marfurt J.; Mather M. W.; Delves M. J.; Shackleford D. M.; Saenz F. E.; Morrisey J. M.; Steuten J.; Mutka T.; Li Y.; Wirjanata G.; Ryan E.; Duffy S.; Kelly J. X.; Sebayang B. F.; Zeeman A.-M.; Noviyanti R.; Sinden R. E.; Kocken C. H. M.; Price R. N.; Avery V. M.; Angulo-Barturen I.; Jiménez-Díaz M. B.; Ferrer S.; Herreros E.; Sanz L. M.; Gamo F.-J.; Bathurst I.; Burrows J. N.; Siegl P.; Guy R. K.; Winter R. W.; Vaidya A. B.; Charman S. A.; Kyle D. E.; Manetsch R.; Riscoe M. K. (2013) Quinolone-3-diarylethers: A new class of antimalarial drug. Sci. Transl. Med. 5, 177ra137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radloff P. D.; Philipps J.; Nkeyi M.; Hutchinson D.; Kremsner P. G. (1996) Atovaquone and proguanil for Plasmodium falciparum malaria. Lancet 347, 1511–1514. [DOI] [PubMed] [Google Scholar]
- Perlin D. S.; Seto-Young D.; Monk B. C. (1997) The plasma membrane H(+)-ATPase of fungi. A candidate drug target?. Ann. N.Y. Acad. Sci. 834, 609–617. [DOI] [PubMed] [Google Scholar]
- Kortagere S.; Welsh W. J.; Morrisey J. M.; Daly T.; Ejigiri I.; Sinnis P.; Vaidya A. B.; Bergman L. W. (2010) Structure-based design of novel small-molecule inhibitors of Plasmodium falciparum. J. Chem. Inf. Model. 50, 840–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douse C. H.; Green J. L.; Salgado P. S.; Simpson P. J.; Thomas J. C.; Langsley G.; Holder A. A.; Tate E. W.; Cota E. (2012) Regulation of the Plasmodium motor complex: Phosphorylation of myosin A tail-interacting protein (MTIP) loosens its grip on MyoA. J. Biol. Chem. 287, 36968–36977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green J. L.; Martin S. R.; Fielden J.; Ksagoni A.; Grainger M.; Yim Lim B. Y.; Molloy J. E.; Holder A. A. (2006) The MTIP-myosin A complex in blood stage malaria parasites. J. Mol. Biol. 355, 933–941. [DOI] [PubMed] [Google Scholar]
- Goodyer I. D.; Taraschi T. F. (1997) Plasmodium falciparum: A simple, rapid method for detecting parasite clones in microtiter plates. Exp. Parasitol. 86, 158–160. [DOI] [PubMed] [Google Scholar]
- Manary M. J.; Singhakul S. S.; Flannery E. L.; Bopp S. E.; Corey V. C.; Bright A. T.; McNamara C. W.; Walker J. R.; Winzeler E. A. (2014) Identification of pathogen genomic variants through an integrated pipeline. BMC Bioinf. 15, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory J. A.; Li F.; Tomosada L. M.; Cox C. J.; Topol A. B.; Vinetz J. M.; Mayfield S. (2012) Algae-produced Pfs25 elicits antibodies that inhibit malaria transmission. PLoS One 7, e37179. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.