

Abstract
A new metabolomics database and query algorithm for the analysis of 13C–1H HSQC spectra is introduced, which unifies NMR spectroscopic information on 555 metabolites from both the Biological Magnetic Resonance Data Bank (BMRB) and Human Metabolome Database (HMDB). The new database, termed Complex Mixture Analysis by NMR (COLMAR) 13C–1H HSQC database, can be queried via an interactive, easy to use web interface at http://spin.ccic.ohio-state.edu/index.php/hsqc/index. Our new HSQC database separately treats slowly exchanging isomers that belong to the same metabolite, which permits improved query in cases where lowly populated isomers are below the HSQC detection limit. The performance of our new database and query web server compares favorably with the one of existing web servers, especially for spectra of samples of high complexity, including metabolite mixtures from the model organisms Drosophila melanogaster and Escherichia coli. For such samples, our web server has on average a 37% higher accuracy (true positive rate) and a 82% lower false positive rate, which makes it a useful tool for the rapid and accurate identification of metabolites from 13C–1H HSQC spectra at natural abundance. This information can be combined and validated with NMR data from 2D TOCSY-type spectra that provide connectivity information not present in HSQC spectra.
Introduction
The rapid and reliable identification of tens to hundreds of different metabolites in a single biological sample is a principal task of any metabolomics investigation.1 Nuclear Magnetic Resonance (NMR) spectroscopy has become one of the standard tools to study metabolic complex mixtures without requiring extensive extraction, purification, and physical separation procedures.2 The high-resolution information provided by NMR is key for the identification and quantification of metabolites.3 One-dimensional (1D) 1H NMR spectroscopy is the most commonly used NMR technique in metabolomics studies, which can involve the analysis of hundreds of samples in high-throughput mode requiring only a few minutes per sample, for example, with the help of automatic sample changers. However, 1D 1H NMR spectra of complex metabolic mixtures often display strong peak overlaps that can severely hamper unambiguous metabolite identification. These issues can be addressed via the use of two-dimensional (2D) NMR techniques by spreading out cross-peaks of resonances along the indirect dimension that overlap in a 1D NMR spectrum, which considerably reduces the likelihood of peak overlap. A popular 2D NMR experiment for this task is the 13C–1H HSQC experiment4 as it provides excellent spectral resolution along the indirect 13C dimension allowing separation of many of the peaks that overlap in the 1D 1H NMR spectrum. In the recent past, several different metabolite identification5−9 and quantification10−13 strategies have been proposed for the analysis of HSQC spectra.
Metabolomics studies based on 2D 13C–1H HSQC spectra generally follow these steps. First, a manual or automated peak picking is performed, which provides a list of all cross-peaks of all detectable compounds in the sample. The peak list is then queried against HSQC databases, typically in batch mode, which returns a list of potential mixture components.14,15 The advantage of this strategy is that it is fast and has the potential for high-throughput, since it does not require the identification of sets of NMR signals that belong to the same metabolite in the mixture prior to querying. However, the approach is prone to false positive identifications particularly for metabolites with chemical shift values that lie mostly in the crowded regions of the HSQC spectrum, which are typically around 3.2–4.5 ppm in the 1H dimension and 62–82 ppm in the 13C dimension, or for the metabolites having very similar structures and chemical shifts such as saccharides and mono-, di-, and triphosphorylated nucleotides (e.g., AMP, ADP, ATP). Identification of these compounds can be achieved by combining HSQC data with TOCSY-based16 connectivity information, which allows the identification of the subsets of HSQC peaks that belong to each metabolite. The HSQC peaks of each metabolite can be directly queried against a HSQC database. Moreover, the combination of HSQC and TOCSY spectra yields information about chemical bonds and the possibility for de novo elucidation of the backbone topology and eventually the structure of metabolites, which is particularly valuable for unknown metabolites that are not catalogued in any of the existing NMR databases.17
Identification of metabolites from 2D 13C–1H HSQC is a very active area of research.5−9 The performance of 2D 13C–1H HSQC databases is still far from optimum with true positive identification rates of the best performing databases around 45–65% as compared to manual identification and false discovery rates at 0–18%,6 which suggests significant room for improvement. The major 2D 13C–1H HSQC metabolomics databases, each with its own query algorithm, are the BMRB (Biological Magnetic Resonance Data Bank),5 HMDB (Human Metabolome Database),8 MMCD (Madison Metabolomics Consortium Database),6 PRIMe (Platform for RIKEN Metabolomics) database,9 and the Metabominer database.7 All of these databases were compiled by recording the 2D 13C–1H HSQC spectra of solutions of isolated (pure) compounds and they all perform cross-peak by cross-peak matching of the database spectra to the experimental 2D 13C–1H HSQC spectrum to identify metabolites. These databases differ from each other in terms of metabolite content and the underlying querying algorithm. A common feature of all these databases is that the HSQC spectrum of each isolated compound has been measured at high concentration and all HSQC cross-peaks of a metabolite are treated together. For example, cross-peaks stemming from different, slowly interconverting isomers are not assigned to individual isomers.
In our own efforts to construct increasingly accurate NMR metabolomics databases,18,19 we found that higher and more accurate metabolite identification rates from HSQC spectra is possible by improving both the data structure of the database and the querying algorithm. On the data structure side, we observed that ∼10% of all metabolites in these databases consist of more than one isomeric state. For the majority of these metabolites, the populations of the different isomers are quite different. For instance, the metabolite pyruvate, which is important in energy metabolism, has two isomeric states with 84% and 16% relative abundance, glucose exists in two isomeric states with 63% and 37% relative abundance, coenzyme A exists in two isomeric states, and ribose exists in four different isomeric states. These metabolites along with six other metabolites and their relative isomer populations are shown in Figure 1. The chemical shifts of these isomers can be found in Supporting Information Table S-1. Since in real-world metabolic samples, metabolite concentrations are often much lower, in a 2D 13C–1H HSQC spectrum one often only detects the isomer(s) with the highest population. As a consequence, this creates a mismatch when HSQC peak lists are queried against conventional HSQC databases: if an HSQC database has two isomers of a metabolite stored as a single entry and if only one of the isomers is experimentally detected, while the other isomer is below the detection limit, only 50% of the expected cross-peaks are detected, which creates a 50% mismatch. Since most of the HSQC query programs use mismatch as a key criterion for identification, a 50% mismatch might result in no identification or misidentification of the target metabolite. This problem can be addressed by sorting the molecules and their cross-peaks into their slowly exchanging isomers for separate queries. For this purpose, we present a HSQC database where we systematically assign each HSQC peak to its specific isomeric state. This allows the accurate identification of metabolites regardless whether all or only some of the isomers can be observed in the HSQC spectrum of the mixture. In this way, the database allows improved identification of metabolites existing in multiple isomeric states by providing an optimal match for each compound. On these principles, we constructed a unified database from entries of two of the largest public databases, namely the BMRB and the HMDB. Combining these with our improved query algorithm, we reach a higher correct identification rate (37% increase in true positives) with fewer false identifications (false positives) than the best performing existing HSQC databases. We name this new database and query tool COLMAR (Complex Mixture Analysis by NMR) 13C–1H HSQC database.
Figure 1.

Relative isomer populations of ten representative metabolites. The isomer populations are calculated by integrating 1D NMR spectra of the metabolites taken from the BMRB and HMDB databases. The 1D NMR spectra were recorded in H2O/D2O at pH 7.0–7.4 at 298 K.
Results and Discussion
1. Generation of the COLMAR 13C–1H HSQC Database and Query
The new HSQC database contains (presently) 555 compounds derived primarily from the BMRB5 and HMDB8 metabolomics databases. A complete list of database compounds with their number of isomeric states is provided on our web server. With 52 of these compounds existing in multiple isomeric states, an estimated 10% of all metabolites cannot be accurately matched by conventional HSQC databases when one or more of their isomeric states falls below the detection limit.
In the new HSQC database, the assignment of the 13C–1H HSQC peaks of all metabolites is performed by using the NMR spectra of isolated compounds in the BMRB, the HMDB, and the literature. Next, the cross-peaks of the HSQC spectrum are sorted into the different isomeric states allowing separate querying of each isomeric state. Only NMR data of compounds dissolved in H2O/D2O at pH 7.0 or 7.4 were included in the new database.
The querying algorithm used for the matching of compounds was developed by testing the query criteria used by each different existing HSQC database. For each database compound, or isomer, the average of 1H and 13C chemical shift differences (1st and 2nd output parameters) are computed to the closest cross-peaks of the mixture. If the database cross-peak is within a given frequency cutoff, it is considered a “matched peak”. The “matching ratio” is then defined as the ratio of the matched peaks to the total number of peaks (3rd output parameter). For example, if a certain metabolite has 5 cross-peaks in the database and 4 of them have corresponding “matched” peaks in the HSQC spectrum of the mixture, the matching ratio for this metabolite is 0.8. While these 3 parameters have been already part of many HSQC databases, they are often not sufficiently selective to reliably discriminate between true and false identifications (see false positive rates of various databases in the results section). In addition, some HSQC databases use a “database uniqueness value”, which reflects how unique a matched peak is in the database.7,9 This parameter is not as selective as the first 3 parameters and the combination of the 4 parameters still produces many false positives (see also Metabominer false positive rates in the results section).
To address these short-comings, we introduce an additional parameter, which we term the ‘assignment uniqueness value’. It gives the number of cross-peaks in the HSQC spectrum of the mixture that are uniquely assigned to metabolite A. For instance, a uniqueness value of 3 means that 3 of the HSQC peaks of the mixture are only assigned to metabolite A. Three unique matches out of 4 total matches for metabolite A is considered a good hit. We find that the combination of this novel parameter with the other parameters significantly reduces the number of false positives.
We set the default cutoff parameters for the average 1H and 13C chemical shift differences at 0.03 and 0.3 ppm, respectively. For a matching ratio of 1.0, the cutoff for the assignment uniqueness value is set to 1 (i.e., at least one of the cross-peaks must be unique). For a matching ratio below 1.0 but higher than 0.6, the cutoff for the assignment uniqueness value is set to 3. An exception is made for metabolites with a total number of 3 or 4 cross-peaks for which the cutoff for the assignment uniqueness is set to 2. If none of the database entries satisfies the above criteria, the query returns “no match”.
2. Application to Model Mixture and Model Organisms
The difficulty to detect all isomeric states of a metabolite exists both in model mixtures and biological cell extracts. In the 13C–1H HSQC spectrum (Supporting Information Figure S-1) of a fully 13C-labeled sugar mixture consisting of glucose, galactose, ribose, fructose, and galactono-1,4-lactone, we observed two isomers each of glucose and galactose, four isomers of ribose, three isomers of fructose, and one isomer of galactono-1,4-lactone. Although the HSQC spectrum of isolated galactono-1,4-lactone shows two isomeric states (see HSQC taken from BMRB in Supporting Information Figure S-2A), one of the isomers was below the detection limit in the mixture spectrum as shown in Figure 2A, where the cross-peak locations of the highly populated and lowly populated isomeric states of galactono-1,4-lactone are indicated by yellow and cyan boxes, respectively. Other problems with differentially populated isomeric states have been observed in HSQC spectra of fruit fly (Drosophila melanogaster). In Figure 2B, in a metabolite extract of wild-type fruit fly, one can see two isomers of fructose, which are the two highest populated isomers, but not the third isomer (Figure 2B). The cross-peaks of all three fructose isomers are indicated by colored boxes in Figure 2B, where the blue boxes belong to the missing isomer and the red and green boxes belong to the detectable isomers with higher populations. All three isomers of fructose can be seen in the HSQC spectrum of isolated fructose taken from the BMRB database (Supporting Information Figure S-2B). When only one or two isomers of fructose are detected, the query of these peaks against a database peaks containing fructose without discriminating between the different isomers will lead to an ambiguity because of cross-peak mismatch. Another example from Drosophila is shown in Figure 2C, D where HSQC spectra of two different metabolic extracts show different pyruvate concentrations (Figure 2C, D). The second isomer of pyruvate is seen only in the second sample, but not in the first one, whereas both isomers of pyruvate can be seen in the HSQC spectrum of pyruvate taken from the BMRB database (Supporting Information Figure S-2C).
Figure 2.

Illustration of the challenge to detect all isomeric states of a metabolite in 2D 13C–1H HSQC spectra. (A) In the sugar mixture, a highly populated isomer of galactono-1,4-lactone is detected (peaks inside of yellow boxes), whereas the lowly populated isomer is below the limit of detection and hence cannot be observed (cyan boxes). (B) In Drosophila melanogaster metabolite extract, two highly populated fructose isomers are observed (peaks inside of red and green boxes), whereas the lowest populated isomer is not observed (blue boxes). (C) For two different Drosophila melanogaster metabolite extracts, in the first sample only a highly populated isomer of pyruvate (inside of orange box) is observed, (D) whereas in the second sample in addition to the highly populated isomer, a lowly populated isomer of pyruvate (purple box) is also detected.
We manually picked HSQC cross-peaks of the sugar model mixture (Supporting Information Figure S-1), the Drosophila melanogaster extract (Supporting Information Figure S-3), and an Escherichia coli cell extract (Supporting Information Figure S-4), which resulted in 88, 165, and 567 HSQC cross-peaks, respectively. We queried these three peak lists one by one against various HSQC databases. The results provide some insights into how current HSQC databases perform for metabolic mixtures of different levels of complexity. The query results for our new database, COLMAR 13C–1H HSQC, are shown for the sugar mixture in Supporting Information Table S-2, for the Drosophila extract in Supporting Information Table S-3, and for E. coli cell extract in Supporting Information Table S-4. Our query program identified 5, 28, and 56 metabolites in these samples, respectively (whereby compounds with multiple isomers are counted only once). For reference, we also overlaid the 13C–1H spectra of isolated standards over the experimental spectrum and performed manual identification. Manual identifications identified 5, 28, and 58 metabolites in the sugar mixture, Drosophila, and E. coli, respectively. Therefore, our database identified more than 95% of what can be identified manually. It should be noted that even manual identification cannot fully assign all peaks in these spectra. We identified 88% of all HSQC peaks in Drosophila and 58% of all HSQC peaks in E. coli, with the remaining peaks belonging to unknown compounds, that is, compounds not contained in the databases.
We compared our results with the other HSQC databases each with its own querying algorithm. HSQC query of HMDB is excluded from our comparison, because it only allows querying of HSQC peaks of individual metabolites. Since in HSQC spectra of a metabolic mixture, peak assignment of individual compounds is not known in advance, this query is not suitable for the analysis of mixtures. All the other HSQC databases, including ours, permit querying of both individual compounds and of mixtures (batch mode). When we queried our three sets of HSQC peaks (see above), the PRIMe and BMRB databases returned an unusually large number of false positives. For instance, when we queried the 165 peaks of Drosophila against the PRIMe and BMRB databases with chemical shift tolerances of 0.02 ppm for 1H and 0.2 ppm for 13C, they returned 99 and 123 metabolites, respectively. By contrast, based on manual analysis we were able to assign 88% of the 165 peaks to only 28 metabolites (and DSS). Therefore, only ∼33 metabolites are expected to be detectable in this spectrum.
The false positive identification rates of Metabominer and MMCD database are much lower. When we queried the 88 peaks of the sugar mixture using Metabominer and MMCD, they still returned 3 and 4 true positives, respectively (Supporting Information Table S-5). The fact that even for a 5-compound sugar mixture the accuracy is less than 100% demonstrates the intrinsic challenge for the querying of metabolites that exist in multiple isomers.
Querying of the 165 HSQC peaks of Drosophila in Metabominer and MMCD (Supporting Information Table S-6) identified 14 and 21 metabolites, respectively. COLMAR correctly identified 28 metabolites, which is a 33% (or higher) improvement. Querying of the 567 HSQC peaks of the E. coli mixture with Metabominer and MMCD (Supporting Information Table S-7) identified 22 and 40 metabolites, respectively. COLMAR identified 56 metabolites corresponding to a 40% (or higher) improvement. Therefore, for the real-world metabolic extracts, our new database allowed an increase of true positive identifications by 37%.
Besides the highest true positive rate achieved by our new database, a low false positive rate is similarly important. As mentioned in the introduction, HSQC spectra are particularly prone to false positives; therefore, the task is to minimize the false positives and at the same time maximize the true positives. Highest true positive rates were achieved by COLMAR, which is followed by MMCD. However, MMCD also provided alarmingly high false positives rates, 5 and 14 false positives in Drosophila and E. coli, respectively. False positive rates in COLMAR were much lower, 0 and 5 false positives in Drosophila and E. coli, respectively. Therefore, COLMAR reduced the false positive rates by 82% as compared to MMCD. A summary of the overall true positive and false positive rates of all databases tested is compiled in Figure 3. In addition, we use a new category termed “ambiguous identifications” (Figure 3 and Supporting Information Table S-8), which comprises identifications whose verification requires additional NMR experiments such as TOCSY, HSQC-TOCSY, or HMBC, because the cross-peaks appear mostly or exclusively in the crowded regions of the HSQC spectrum or their chemical shifts and structures are very similar to a handful of other compounds (such as saccharides and nucleotide mono-, di-, and triphosphates e.g., AMP, ADP, ATP). Therefore, additional connectivity information can differentiate between these molecules and resolve ambiguities.
Figure 3.
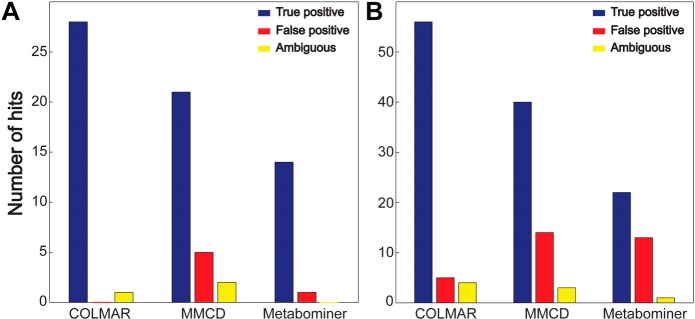
Comparison of the performance of the COLMAR, MMCD and Metabominer 13C–1H HSQC databases for the query of (A) Drosophila and (B) E. coli metabolic extracts.
Although, in this paper, we only used HSQC cross-peak lists that were manually picked, the COLMAR 13C–1H HSQC server also accepts automatically generated cross-peak lists. It is recommended, however, that users visually inspect their peak lists and curate them before uploading for query to avoid problems with peaks originating from t1 noise and other artifacts.
A specifically designed web portal at http://spin.ccic.ohio-state.edu/index.php/hsqc/index allows querying of the 2D 13C–1H HSQC spectra of metabolic mixtures in batch mode as well as querying of the 2D 13C–1H HSQC spectra against individual metabolites in the COLMAR 13C–1H HSQC database. As an example, the 165 peaks of Drosophila were queried against the COLMAR in batch mode on the web server (Figure 4A). The query successfully returned the list of compounds in the sample (Figure 4B). The interactive user interface based on a JavaScript allowed overlaying of 13C–1H HSQC peaks of individual compounds in the database with the experimental peaks upon clicking the “Show Me” button (Figure 4B) of the matched compound, which allows direct visual inspection of the presence of the matched compound in the experimental 2D spectrum. To our knowledge, this is the first 13C–1H HSQC metabolomics database allowing such quick visual checks online, which is very useful to maximize confidence of identifications. Matched compounds are always shown in ‘Number_Metabolite Name’ format, where the integer in front of the metabolite name is used to denote different isomeric states of the metabolite. Metabolites with only one isomeric state are always shown as ‘1_Metabolite-name’, such as ‘1_Alanine’, whereas metabolites with more than one isomeric state are shown as ‘n_Metabolite-name’, where n = 1, 2, 3, ... are different isomers of the metabolite (e.g., 1_Maltose and 2_Maltose).
Figure 4.

Screenshots taken from the interactive COLMAR 13C–1H HSQC web server. (A) The HSQC peak list with 165 cross-peaks of Drosophila melanogaster metabolite extract (upper left) is queried against the database. The lower left shows the cross-peak positions (blue circles and black crosses) in a 2D plane corresponding to the 2D HSQC spectrum. The thin blue circles correspond to HSQC cross-peaks that match a single compound in the database, the thick blue circles correspond cross-peaks that match multiple compounds, and the black crosses correspond to cross-peaks that do not match any compound in the database. (B) List of matching compounds returned by the query. The results served as input for Supporting Information Table S-3.
Reliable identification of metabolites is one of the most critical steps in metabolomics. Here, we introduced the first HSQC metabolomics database, which allows querying of individual states of metabolites. This allows identification of metabolites regardless of the contribution of each isomeric state to the acquired HSQC spectrum. Combining this more accurate and more specific database with a more selective querying approach provided the highest true positive and the lowest false positive identification rate among all the databases and their querying algorithms tested. The new database serves as an alternative to conventional HSQC databases, which rely on the simultaneous query of all cross-peaks of all isomers.
Ideally, the most accurate HSQC query should take into account absolute concentrations of the compounds in the sample with their relative isomeric populations. Such information can be combined with the detection limit of HSQC peaks so that the query can assess how many of the isomeric states of each compound are expected in the experimental HSQC spectra before actually performing the query. Such information can be used to adaptively combine detectable isomeric states of a compound as a single entry for querying to boost the matching accuracy. However, this requires additional information that may not be readily available. For these cases, our approach, which queries individual isomeric states of compounds independently, will provide the highest accuracy.
Currently, many researchers use multiple HSQC databases for the querying of their data in order to maximize the number of identified metabolites in their samples, because the metabolites of different databases only partially overlap requiring an additional effort by the user to go back and forth between databases. Here, we combined database information from the BMRB and the HMDB into a unified database, which can be queried in an isomer-specific manner in a single step. This facilitates the analysis of complex mixtures, while at the same time increasing the number of correct hits. We expect this resource to be of wide usefulness to metabolomics researchers.
Methods
Sample Preparation
The uniformly 13C-labeled carbohydrate mixture was prepared by dissolving ribose, galactose, glucose and fructose in D2O each with a 10 mM final concentration. The final mixture was transferred to a 3 mm NMR tube. Galactono-1,4-lactone appeared as a degradation product or impurity in the spectrum with a relatively lower concentration as compared to the other four carbohydrates in the mixture.
Two male Drosophila samples were prepared, one from 50 wild type (w1118) flies and one from 100 wild type flies. Four to six-day old flies were reared at 25 °C on standard cornmeal/yeast/molasses food for 5 days with 12 h light (6:00 AM to 6:00 PM) and 12 h dark cycles and then harvested at 10:00 AM on the fifth day for metabolite extraction. For each sample, the flies were collected and snap-frozen in liquid nitrogen. The flies were then placed in 400 μL ice-cold 50% acetonitrile and subjected to homogenization with Bullet Blender 24 Gold (Next Advance) for metabolite extraction. The resulting mixture was centrifuged at 10 000g for 5 min. The supernatant was then filtered by centrifugation at 14 000g at 4 °C for 30 min with Amicon Ultra-0.5 mL 10 K (EMD Millipore). The resulting filtrate was lyophilized and resuspended in 50 mM phosphate buffer at pH 7.4 in D2O for NMR measurements.
E. coli DH5α cells were cultured at 37 °C, at 250 rpm in M9 minimum medium with glucose (natural abundance, 5g/L) added as sole carbon source. One liter of culture at OD ∼3 was centrifuged at 5000g for 20 min at 4 °C, and the cell pellet was resuspended in 50 mL of 50 mM phosphate buffer at pH 7.0. The cell suspension was then subjected to centrifugation for cell pellet collection. The cell pellet was resuspended in 10 mL of ice cold water and exposed to freeze–thaw procedure 3 times. The sample was centrifuged at 20 000g at 4 °C for 15 min to remove the cell debris. Prechilled methanol and chloroform were sequentially added to the supernatant under vigorous vortex at H2O:methanol:chloroform ratios of 1:1:1 (v/v/v). The mixture was then left at −20 °C overnight for phase separation. Next, it was centrifuged at 4000g for 20 min at 4 °C, and the clear top hydrophilic phase was collected and subjected to rotary evaporator processing to have the methanol content reduced. Finally, the liquid was lyophilized. The NMR sample was prepared by dissolving the lyophilized material in D2O and transferred to a 5 mm NMR tube.
NMR Experiments and Processing
The 2D 13C–1H HSQC spectrum of the carbohydrate mixture was collected with N1 = 512 and N2 = 1024 complex points by using a cryogenically cooled probe at 800 MHz proton frequency. The spectral widths along the indirect and the direct dimensions were 22135.197 and 9615.385 Hz, respectively. The number of scans per t1 increment was set to 4. The transmitter frequency offsets were 55 ppm in the 13C dimension and 4.7 ppm in the 1H dimension. The total measurement time was 3 h.
2D 13C–1H HSQC spectra of Drosophila extracts were collected with N1 = 512 and N2 = 1024 complex points. The spectral widths along the indirect and the direct dimensions were 34209.9 and 8802.8 Hz, respectively. The number of scans per t1 increment was set to 16. The transmitter frequency offsets were 85 ppm in the 13C dimension and 4.7 ppm in the 1H dimension. The total measurement time for each sample was 10 h. The NMR spectrum was collected using a cryogenically cooled probe at 800 MHz proton frequency.
The 2D 13C–1H HSQC spectrum of E. coli extract was collected with N1 = 512 and N2 = 1024 complex points. The spectral widths along the indirect and the direct dimensions were 29934.5 and 7692.3 Hz, respectively. The number of scans per t1 increment was 64. The transmitter frequency offsets were 85 ppm in the 13C dimension and 4.7 ppm in the 1H dimension. The total measurement time was 36 h. The NMR spectrum was collected using a cryogenically cooled probe at 700 MHz proton frequency. All NMR spectra were collected at 298 K, and the data were zero-filled, Fourier transformed, and phase and baseline corrected using NMRPipe.20
Acknowledgments
We thank the authors of the BMRB and HMDB databases for publicly sharing their data. This work was supported by the National Institutes of Health (grant R01 GM 066041 and SECIM (Southeast Center for Integrated Metabolomics) grant U24 DK097209-01A1).
Supporting Information Available
13C–1H HSQC spectra of carbohydrates and their mixture; entire 13C–1H HSQC spectra of Drosophila melanogaster samples and of E. coli cell lysate; four tables with 13C–1H HSQC cross-peak list and COLMAR HSQC query results of carbohydrate model mixture, drosophila sample, and E. coli cell lysate; four tables with comparison of COLMAR query results with results obtained using other public query web servers. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Nicholson J. K.; Holmes E.; Kinross J. M.; Darzi A. W.; Takats Z.; Lindon J. C. (2012) Metabolic phenotyping in clinical and surgical environments. Nature 491, 384–392. [DOI] [PubMed] [Google Scholar]
- Lenz E. M.; Wilson I. D. (2007) Analytical strategies in metabonomics. J. Proteome Res. 6, 443–458. [DOI] [PubMed] [Google Scholar]
- Bingol K.; Brüschweiler R. (2014) Multidimensional approaches to NMR-based metabolomics. Anal. Chem. 86, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenhausen G.; Ruben D. J. (1980) Natural abundance nitrogen-15 NMR by enhanced heteronuclear spectroscopy. Chem. Phys. Lett. 69, 185–189. [Google Scholar]
- Ulrich E. L.; Akutsu H.; Doreleijers J. F.; Harano Y.; Ioannidis Y. E.; Lin J.; Livny M.; Mading S.; Maziuk D.; Miller Z.; Nakatani E.; Schulte C. F.; Tolmie D. E.; Wenger R. K.; Yao H. Y.; Markley J. L. (2008) BioMagResBank. Nucleic Acids Res. 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q.; Lewis I. A.; Hegeman A. D.; Anderson M. E.; Li J.; Schulte C. F.; Westler W. M.; Eghbalnia H. R.; Sussman M. R.; Markley J. L. (2008) Metabolite identification via the Madison Metabolomics Consortium Database. Nat. Biotechnol. 26, 162–164. [DOI] [PubMed] [Google Scholar]
- Xia J.; Bjorndahl T. C.; Tang P.; Wishart D. S. (2008) MetaboMiner- semi-automated identification of metabolites from 2D NMR spectra of complex biofluids. BMC Bioinf. 9, 507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart D. S.; Knox C.; Guo A. C.; Eisner R.; Young N.; Gautam B.; Hau D. D.; Psychogios N.; Dong E.; Bouatra S.; Mandal R.; Sinelnikov I.; Xia J. G.; Jia L.; Cruz J. A.; Lim E.; Sobsey C. A.; Shrivastava S.; Huang P.; Liu P.; Fang L.; Peng J.; Fradette R.; Cheng D.; Tzur D.; Clements M.; Lewis A.; De Souza A.; Zuniga A.; Dawe M.; Xiong Y. P.; Clive D.; Greiner R.; Nazyrova A.; Shaykhutdinov R.; Li L.; Vogel H. J.; Forsythe I. (2009) HMDB: A knowledgebase for the human metabolome. Nucleic Acids Res. 37, D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikayama E.; Sekiyama Y.; Okamoto M.; Nakanishi Y.; Tsuboi Y.; Akiyama K.; Saito K.; Shinozaki K.; Kikuchi J. (2010) Statistical indices for simultaneous large-scale metabolite detections for a single NMR spectrum. Anal. Chem. 82, 1653–1658. [DOI] [PubMed] [Google Scholar]
- Lewis I. A.; Schommer S. C.; Hodis B.; Robb K. A.; Tonelli M.; Westler W. M.; Sussman M. R.; Markley J. L. (2007) Method for determining molar concentrations of metabolites in complex solutions from two-dimensional 1H-13C NMR spectra. Anal. Chem. 79, 9385–9390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronwald W.; Klein M. S.; Kaspar H.; Fagerer S. R.; Nurnberger N.; Dettmer K.; Bertsch T.; Oefner P. J. (2008) Urinary metabolite quantification employing 2D NMR spectroscopy. Anal. Chem. 80, 9288–9297. [DOI] [PubMed] [Google Scholar]
- Rai R. K.; Tripathi P.; Sinha N. (2009) Quantification of metabolites from two-dimensional nuclear magnetic resonance spectroscopy: Application to human urine samples. Anal. Chem. 81, 10232–10238. [DOI] [PubMed] [Google Scholar]
- Hu K.; Westler W. M.; Markley J. L. (2011) Simultaneous quantification and identification of individual chemicals in metabolite mixtures by two-dimensional extrapolated time-zero 1H-13C HSQC (HSQC0). J. Am. Chem. Soc. 133, 1662–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halouska S.; Fenton R. J.; Zinniel D. K.; Marshall D. D.; Barletta R. G.; Powers R. (2014) Metabolomics analysis identifies d-alanine–d-alanine ligase as the primary lethal target of d-cycloserine in mycobacteria. J. Proteome Res. 13, 1065–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei S.; Zavala-Flores L.; Garcia-Garcia A.; Nandakumar R.; Huang Y.; Madayiputhiya N.; Stanton R. C.; Dodds E. D.; Powers R.; Franco R. (2014) Alterations in energy/redox metabolism induced by mitochondrial and environmental toxins: A specific role for glucose-6-phosphate-dehydrogenase and the pentose phosphate pathway in paraquat toxicity. ACS Chem. Biol. 9, 2032–2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweiler L.; Ernst R. R. (1983) Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 53, 521–528. [Google Scholar]
- Bingol K.; Zhang F.; Bruschweiler-Li L.; Brüschweiler R. (2012) Carbon backbone topology of the metabolome of a cell. J. Am. Chem. Soc. 134, 9006–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol K.; Zhang F.; Bruschweiler-Li L.; Brüschweiler R. (2012) TOCCATA: A customized carbon total correlation spectroscopy NMR metabolomics database. Anal. Chem. 84, 9395–9401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol K.; Bruschweiler-Li L.; Li D. W.; Brüschweiler R. (2014) Customized metabolomics database for the analysis of NMR 1H-1H TOCSY and 13C-1H HSQC-TOCSY spectra of complex mixtures. Anal. Chem. 86, 5494–5501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaglio F.; Grzesiek S.; Vuister G. W.; Zhu G.; Pfeifer J.; Bax A. (1995) NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.