

Abstract
The cardiac glycosides ouabain and digitoxin, established Na+/K+ ATPase inhibitors, were found to inhibit MDA-MB-231 breast cancer cell migration through an unbiased chemical genetics screen for cell motility. The Na+/K+ ATPase acts both as an ion-transporter and as a receptor for cardiac glycosides. To delineate which function is related to breast cancer cell migration, structure–activity relationship (SAR) profiles of cardiac glycosides were established at the cellular (cell migration inhibition), molecular (Na+/K+ ATPase inhibition), and atomic (computational docking) levels. The SAR of cardiac glycosides and their analogs revealed a similar profile, a decrease in potency when the parent cardiac glycoside structure was modified, for each activity investigated. Since assays were done at the cellular, molecular, and atomic levels, correlation of SAR profiles across these multiple assays established links between cellular activity and specific protein–small molecule interactions. The observed antimigratory effects in breast cancer cells are directly related to the inhibition of Na+/K+ transport. Specifically, the orientation of cardiac glycosides at the putative cation permeation path formed by transmembrane helices αM1–M6 correlates with the Na+ pump activity and cell migration. Other Na+/K+ ATPase inhibitors that are structurally distinct from cardiac glycosides also exhibit antimigratory activity, corroborating the conclusion that the antiport function of Na+/K+ ATPase and not the receptor function is important for supporting the motility of MDA-MB-231 breast cancer cells. Correlative SAR can establish new relationships between specific biochemical functions and higher-level cellular processes, particularly for proteins with multiple functions and small molecules with unknown or various modes of action.
Correlative SAR is a strategy to expand the use of existing biologically active compounds to address some of the major challenges in the postgenomic era: annotating protein functions and linking those functions to an observed phenotype. Traditional SAR studies guide improvements of small molecule potency and selectivity for a target. Correlative SAR, on the other hand, takes advantage of a distinct pattern of structural requirements for activity by a group of related molecules (SAR profile) across a series of assays. The SAR profiles are then used to establish relationships between a protein or cellular system and bona fide biological processes, that is, linking specific protein function to a cellular phenotype. Direct correlation of SAR profiles for protein function and a cellular phenotype then implicates a role for the protein in the phenotype. This is especially useful for multifunctional proteins and moonlighting proteins.1 Correlative SAR employs small molecule probes and thus shares the complementarity and advantages of small molecules over genetics and RNA interference techniques for annotating protein functions. In this study, we illustrate the correlative SAR approach by exploring the involvement of Na+/K+ ATPase ion transport function in breast cancer cell migration.
Cell migration is a fundamental biological process of physiological and pathological phenomena such as wound healing, tissue remodeling, angiogenesis, and cancer metastasis. In response to soluble signaling factors and mechanical cues, the cytoskeleton generates protrusive forces and adhesion receptor-linked traction against the extracellular matrix.2 Because of the highly complex nature of the mechanisms of cell migration, no complete model that incorporates all of its components currently exists. One of the major problems in determining the precise roles of each of the proteins involved in cell migration is the lack of potent and selective inhibitors for the functions of these proteins.3 Besides their therapeutic potential, small-molecule inhibitors of cell migration serve as valuable tools to probe the molecular basis of cell motility.4−6
In the course of screening libraries of ca. 12000 total compounds for cell migration inhibitors using a wound-closure assay, cardiac glycosides were found to inhibit the migration of the human triple-negative breast cancer cell line MDA-MB-231 at submicromolar concentrations. The MDA-MB-231 line is a cellular model for triple-negative breast cancer, which is refractory to current chemotherapeutic options.7 Cardiac glycosides are a group of tripartite natural products consisting of a steroid core, an α,β-unsaturated lactone ring, and a sugar moiety. They are used in the treatment of congestive heart failure,8 acting through inhibition of the Na+ and K+ antiport pumping activity of the Na+/K+ ATPase.9 Over the last 20 years, cardiac glycosides have also garnered increasing interest as potential anticancer agents.10 The minimum pharmacophore (Scheme 1) for inhibition of the Na+/K+ ATPase consists of the steroid core, which possesses a distinct cis-configuration of the junctions between the AB and CD rings. The lactone is not essential for activity but increases the affinity between the steroid core and the protein.11 The sugar moiety affects the potency as well as the pharmacokinetic and pharmacodynamic properties of cardiac glycosides.12−14
Scheme 1. Semisynthesis of Ouabain Analogs.

Upon binding of cardiac glycosides at the well-conserved binding site in the α1 subunit of the Na+/K+ ATPase,15 various effects are observed depending on the concentration of cardiac glycosides and the cell type used.16 At concentrations >100 nM, cardiac glycosides cause “classical” inhibition of the ion transport function of the Na+/K+ ATPase, leading to an increase in intracellular Na+ ion concentration; this activates the Na+/Ca2+ exchanger resulting in an increased intracellular Ca2+ concentration, explaining the positive inotropic effect of cardiac glycosides.17 Recent studies have shown that the Na+/K+ ATPase also acts as a receptor for endogenous cardiac glycosides as natural ligands.18 At concentrations too low to affect the ion transport function of the pump (i.e., 10–100 nM), cardiac glycosides have a distinctly different effect: they activate signaling cascades, one of which involves Src tyrosine kinase in a complex with the Na+/K+ ATPase that affects diverse cellular functions such as cell proliferation.19 A pool of nonpumping Na+/K+ ATPase exists in the caveolae and forms a complex with caveolin-1 and Src. Binding of cardiac glycosides to this pool of the Na+/K+ ATPase activates Src-dependent signaling pathways, leading to activation of the mitogen-activated protein kinase cascade.20
Other cardiac glycosides inhibit the migration and proliferation of various cell types without affecting the ion-transport function of the Na+/K+ ATPase, thus supporting the nonclassical mode of action. UNBS-1450, for example, a novel cardenolide (structure in Supplementary Scheme 1, Supporting Information), inhibits the proliferation and migration of several types of cancer cells21−23 at concentrations that do not affect the ion transport activity of the Na+/K+ ATPase. Inhibition of the α1 subunit of the Na+/K+ ATPase by UNBS-1450 leads to a sharp decrease in intracellular ATP concentration, causing disorganization of the actin cytoskeleton and induction of proautophagic cytotoxic effects.22
After discovering that cardiac glycosides inhibit migration of MDA-MB-231 cells, we endeavored to define the specific role of the Na+/K+ ATPase (classical versus nonclassical) in cell migration using correlative SAR. Analogs of ouabain and digitoxin were synthesized and SAR was established with respect to inhibition of cell migration (cellular), inhibition of Na+/K+ ATPase activity in vitro (molecular), and orientation in the cardiac glycoside binding site of Na+/K+ ATPase (atomic). We evaluated whether inhibition of cell migration was separable from inhibition of the Na+/K+ antiport pump by correlating the SAR profiles of ouabain in each of these assays. In addition, to determine whether the inhibition of cell migration resulted from general inhibition of the Na+/K+ ATPase or is confined to an effect of cardiac glycosides, other structurally unique Na+/K+ ATPase inhibitors were tested for their antimigratory activity. A small-molecule inhibitor that does not activate the Src signaling pathway but inhibits the ion pump24 was also tested in the cell migration assay. To our knowledge, no SAR study on the antimigratory activity of cardiac glycosides in MDA-MB-231 breast cancer cells has been reported prior to this work. Furthermore, the current study shows the utility of correlative SAR to establish links between specific protein functions and cellular phenotypes.
Results and Discussion
Ouabain and Analogs: Correlative SAR Between Inhibition of MDA-MB-231 Breast Cancer Cell Migration and the Na+/K+ ATPase
Three analogs of ouabain 1 were synthesized (2–4, Scheme 1) by methods that had been reported previously in the literature.25,26 The derivatives were prepared, starting from ouabain itself, by removal of the sugar moiety (2), reduction of the lactone ring (3) and modification of the steroid core (4). Hydrolysis of the glycosidic linkage between the l-rhamnose and steroid core provided ouabagenin 2. Reduction of the butenolide olefin by hydrogenation provided analog 3. Compound 4 contains an acetonide group that links the C1 and C19 hydroxyl groups. The analogs were chosen because they were synthetically accessible and because they consisted of variations of the key structural segments of the parent compound in a way that would allow for a reasonably comprehensive SAR profile. Ouabain 1 and analogs 2–4 were assayed for cell migration inhibition using a quantitative scratch-wound assay.27
The MDA-MB-231 breast cancer cell line was chosen for the scratch-wound cell migration assay. MDA-MB-231, a cellular model for triple negative breast cancer, has an invasive phenotype and is metastatic in animal models. The assay involved mechanical wounding of a confluent monolayer of cells with a pipet tip after treatment with different concentrations of compounds or DMSO (vehicle) alone. The progress of wound closure was monitored by analysis of digital microscope images of the wounds at different times postwounding. The area of the wound at each time point was calculated with the public-domain ImageJ program, and statistical significance was tested with a two-tailed Student’s t-test.
Based on the IC50 values for inhibition of wound closure (Figure 1 and Table 1), none of the synthesized analogs were more potent than the parent compound, ouabain 1 (with an IC50 of 173 nM). However, the potency of the analogs varied with respect to the type of modification made to the structure of 1. Ouabagenin 2, which lacks the sugar moiety, had an IC50 value of 790 nM. The olefin of the butenolide was reduced to prepare compound 3; its IC50 of 4.3 μM demonstrates that a simple modification to the lactone segment results in a drop in potency by a factor of 25. It further suggests that the olefin is relatively important to the ability of cardiac glycosides to inhibit cell migration. A major decrease in potency (178-fold) was observed for ouabain analog 4, which contains a C1, C19 acetonide group. Modification to the steroid core and specifically the introduction of a bulky group on the concave face of the cup-shaped steroid greatly reduced the antimigratory activity of ouabain. The SAR profile (pattern of structural requirement for activity modulation) for antimigratory activity is: modification on steroid core > modification on lactone ring > modification on sugar in decreasing impact on potency (Figure 2a).
Figure 1.
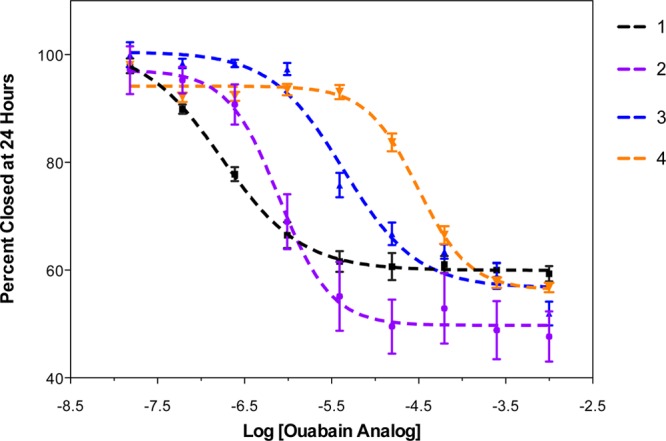
IC50 curves for cell migration inhibition by 1–4.
Table 1. IC50 Values (μM) for the Inhibitory Activity of Ouabain and Analogs in the Na+/K+ ATPase Assay and Cell Migration Assaya.
| Compound | Na+/K+ ATPase | Cell migration | ||
|---|---|---|---|---|
| IC50 | 95% CI | IC50 | 95% CI | |
| 1 | 0.105 | 0.031-0.352 | 0.173 | 0.100-0.300 |
| 2 | 0.669 | 0.526-0.850 | 0.790 | 0.637-0.979 |
| 3 | 2.6 | 0.823-8.46 | 4.3 | 3.02-6.21 |
| 4 | 12.4 | 8.98-17.2 | 30.9 | 25.2-38.0 |
In the trypan blue assay, compounds 1–4 were not toxic to MDA-MB-231 breast cancer cells during the time frame of the wound-closure assay. However, for compound 1, some cells lifted off of the monolayer at concentrations above 60 μM.
Figure 2.

(A) Decrease in potency of ouabain analogs in the cell migration assay and the Na+/K+ ATPase assay (SAR profile). (B) Pearson correlation plot for IC50 values of ouabain and analogs in the antimigratory assay and the Na+/K+ ATPase assay (correlative SAR). (C) Pearson correlation plot for ouabain’s inhibition of Na+/K+ ATPase and cell migration at different concentrations. (D) Pearson correlation plot for inhibition of Na+/K+ ATPase and cell migration by ouabain and analogs at different concentrations.
Ouabain is a potent and specific inhibitor of the Na+/K+ ATPase.28 The Na+/K+ ATPase is a transmembrane antiport ion transporter that pumps three Na+ ions out of the cell for every two K+ ions pumped into the cell against their concentration gradients for each molecule of ATP hydrolyzed. Besides its ion-transport function, this protein complex also functions as a receptor for ouabain and has been implicated in processes and key cellular elements that affect cell motility such as cell tight junctions,29 cell contacts,30 and actin dynamics.31 We investigated whether the antimigratory effect of ouabain was linked to the classical inhibition of Na+/K+ ATPase17 or stimulation of its receptor function20 using correlative SAR. Specifically, we looked at the SAR profile of ouabain in an Na+/K+ ATPase activity assay and correlated it to the SAR profile for antimigratory activity.
The SAR profile in Na+/K+ ATPase inhibition is similar to inhibition of cell migration (Table 1 and Figure 2A). Compound 2, with the cleaved sugar moiety, had a potency that is relatively similar to the parent compound ouabain 1. This implies that for inhibition of Na+/K+ ATPase activity, as for inhibition of cell migration, the sugar moiety is less important than other parts of the molecule. Compound 3, with the saturated lactone ring, displays the next lowest decrease in potency, while 4, with the bulky acetonide group on the steroid core, shows the greatest decrease in potency. The IC50 values of ouabain and its analogs for inhibition of Na+/K+ ATPase activity and the values for inhibition of cell migration fall within the same range. The distinct pattern of modulation in Na+/K+ ATPase activity by ouabain and the analogs, as measured by both IC50 (Figure 2B) and dose–response for ouabain alone (Figure 2C) and for combined ouabain and analogs (Figure 2D) correlates well with the pattern observed for their effect on cell migration. The correlation suggests that the inhibition of cell migration is directly related to the inhibition of Na+/K+ ATPase activity. This result was corroborated by the same strong correlation (r = 0.9998, p = 0.0002, 95% CI = 0.99–1.00) observed for correlative SAR of digitoxin, a hydrophobic digitalis cardiac glycoside (see Supporting Information). The direct relationship between inhibition of Na+/K+ ATPase ion transport function and inhibition of cell migration seemed to be applicable to other members of the cardiac glycoside family of natural products.
Ouabain and Analogs: Ouabain–Na+/K+ ATPase Interaction at Atomic Level Resolution via Molecular Modeling
The ouabain binding site on the extracellular side of the α-subunit of the Na+/K+ ATPase is located in a cleft between the transmembrane segments αM1 and αM6.32−35 Two orientations of ouabain to this site were originally proposed, one with the lactone ring facing toward the cytoplasmic side, as revealed by X-ray crystal structure studies32−34 (Figure 3A,B) and the other one with the lactone ring facing toward the extracellular side, as suggested by a lanthanide-based resonance energy transfer (LRET) experiment.35 Regardless of the two orientations of ouabain, all structural and functional studies agreed that the location of the cardiotonic steroid binding site is in the deep vestibule formed by the tansmembrane helices αM1–M6. Most of these transmembrane helices constitute the cation permeation path (mapped in Figure 3A as residues in cyan) that traverses the Na+/K+ pump from one side of the membrane to the other and passes through the ion-binding site II36 (Figure 3A, red circle), suggesting that ouabain acts by blocking the ion path and directly preventing cations from accessing their binding sites. Another possibility is that ouabain binding may lock the cation transport region into a configuration that prevents the pump from undergoing the conformational change required for ion transport.37
Figure 3.

(A) Crystal structure (4HYT)32 of Na+/K+ ATPase with bound ouabain (orange) showing residues involved in the cation permeation path36 (cyan) and the K+ binding site II. Below is the cup-shaped ouabain structure showing the hydrophobic α-surface and the polar β-surface. Orientation of ouabain (B) and docked analog 2 (C), docked analog 3 (D), and docked analog 4 (E) to cardiac glycoside binding site in the Na+/K+ ATPase.
To our knowledge, no experimental evidence linking ouabain’s orientation to the cation permeation path and inhibition of Na+/K+ ATPase currently exists. We investigated the interactions of ouabain and its analogs to Na+/K+ ATPase at the atomic level (Figure 3B–E) by molecular docking experiments in an effort to rationalize the SAR results with respect to inhibition of Na+/K+ ATPase ion-transport function and cell migration. We explored how each modification on the ouabain structure affects binding energy (evaluated in the form of a docking score) and orientation relative to ouabain’s structure in the cocrystal (evaluated in terms of a root-mean-square-deviation, RMSD) (Table 2). We then correlated the SAR profile for calculated RMSD (Figure 4) and binding energy (Supplementary Figure S5, Supporting Information) to the SAR profile for inhibition of Na+/K+ ATPase activity and inhibition of cell migration.
Table 2. Calculated Binding Energies and RMSD of Ouabain and Analogs from Molecular Docking Experiments.
| docked ligand | binding energy (Amber score), kcal/mol | RMSD (non-hydrogen atoms) | Na+/K+ ATPase | cell migration |
|---|---|---|---|---|
| 1 | –5420.51 | 0.0000 | 0.105 | 0.173 |
| 2 | –5399.66 | 0.5807 | 0.669 | 0.790 |
| 3 | –5412.08 | 0.9367 | 2.6 | 4.3 |
| 4 | –5584.89 | 6.5933 | 12.4 | 30.9 |
Figure 4.

(A) Pearson correlation plot for IC50 values of ouabain and analogs in the Na+/K+ ATPase assay and the RMSD. (B) Pearson correlation plot for IC50 values of ouabain and analogs in the cell migration assay and the RMSD.
The observations of the structural interactions of ouabain and its analogs with the Na+/K+ ATPase are consistent with the observed experimental SAR profile for their inhibition of Na+/K+ ATPase ion-transport function and breast cancer cell migration. The docked analogs 2 and 3 (Figure 3C,D), which are the two most potent analogs (5× and 25× less potent than ouabain, respectively) adapted a similar orientation to the crystal structure of ouabain bound to Na+/K+ ATPase (Figure 3B). The concave nonpolar α-surface of ouabain32 and analogs 2 and 3 is stabilized by hydrophobic residues such as I315, F316, F783, and F786 (Figure 3B–D). The hydroxyl groups of the polar β-surface, on the other hand, are stabilized by H-bonding interactions (between C19-OH and the highly conserved C14-OH of cardiac glycosides and N111 and T797 of αM1 and αM6, respectively). The C14-hydroxyl–T797 interaction for analog 3, however, was slightly shifted, and thus the H-bonding angle was less than ideal (Supplementary Figure S3, Supporting Information), possibly due to reduction of lactone ring olefin. This could explain the 25× decrease in potency observed for both the pump and cell migration inhibition. On the other hand, the least potent analog 4 (Figure 3E) adapted a different pose in the ion permeation path altogether (see Supplementary Figure 2, Supporting Information, for overlaid crystal structure of ouabain and docked ouabain and analogs). Analog 4 took up a shallow position and was oriented in the opposite direction with its polar β-surface facing the helices M4 and M5, while the apolar α-surface faced the helices M1, M2, and M6. This orientation does not allow for H-bonding interactions with Q111 and T797, residues identified by mutagenesis studies38,39 to be critical for ouabain’s inhibition of Na+/K+ ATPase. However, the orientation of the docked analog 4 is possibly stabilized by hydrophobic interactions of I315, F316, and F783 with the steroid rings C and D, and H-bonding interaction of C19-hydroxyl with E117 instead of Q111 (Supplementary Figure S4, Supporting Information).
The binding energy and RMSD were calculated for the docked compounds to quantify the effect of each structural modification on the ouabain-Na+/K+ ATPase interaction (Table 2). These parameters were then correlated to the experimental results from in vitro Na+/K+ ATPase inhibition and breast cancer cell migration assays. Considering that the docking score is only an approximate measure of true free energies of binding, both the IC50 values for inhibition of Na+/K+ ATPase and inhibition of cell migration by ouabain and analogs correlate reasonably well with the calculated binding energy (Supplementary Figure S5, Supporting Information) for analogs 2 and 3. That is, analogs 2 and 3 have less negative energy of binding compared with 1, although with similar orders of magnitude, which is consistent with lower potency. A proper orientation relative to the cardiac glycoside binding site seemed to be highly critical to Na+-pump inhibition. The more negative binding energy calculated for the least potent analog 4 probably results from a stable analog 4–Na+/K+ ATPase complex (possibly resulting from some hydrophobic and H-bonding interactions discussed earlier), but the orientation of analog 4 to the ion permeation path might not be as effective in blocking ion permeation as that of ouabain. In other words, despite analog 4 being more tightly bound, it is noticeably positioned in a region of the binding site much closer to the surface, and therefore less able to stop the permeation of ions deep inside the pocket (Figure 4E). On the other hand, both the IC50 values for inhibition of Na+/K+ ATPase and inhibition of cell migration by ouabain analogs correlate very well with the RMSD (Figure 4), implying that deviation from ouabain’s orientation in the cation permeation of Na+/K+ ATPase is directly proportional to the decrease in its inhibitory activity.
Inhibition of MDA-MB-231 Breast Cancer Cell Migration and Na+/K+ ATPase Activity by Other Na+/K+ ATPase Inhibitors and Potential Involvement of Na+/K+ ATPase in MDA-MB-231 Breast Cancer Cell Migration
To explore whether the inhibition of cell migration results from general inhibition of the Na+/K+ ATPase or is specific for cardiac glycosides, the antimigratory effect of other Na+/K+ ATPase inhibitors that are structurally distinct from cardiac glycosides was determined. All of the non-cardiac-glycoside Na+/K+ ATPase inhibitors tested also inhibited cell migration with varying potencies (Table 3). Compound 9, a nonspecific inhibitor of Na+/K+ ATPase, is the most potent; however, it was also toxic and its IC50 is very close to its minimum lethal concentration, which is likely due to its effect on other cellular targets. The next most potent is compound 11. This Na+/K+ ATPase inhibitor is particularly interesting because it was reported that, unlike ouabain, it does not activate the Na+/K+ ATPase/Src complex nor does it stimulate the protein kinase cascades.24 Compound 11 is a useful small molecule probe since it can inhibit only one function (Na+/K+ ATPase enzymatic activity) of the pump without activating its receptor function. This allows exploration and differentiation of the functional consequences of the enzymatic versus receptor function of the Na+/K+ ATPase. Since 11 also inhibits cell migration, the antimigratory activity of cardiac glycosides does not primarily entail activation of the receptor function of Na+/K+ ATPase. Furthermore, any of the compounds tested that inhibit Na+/K+ ATPase also inhibit MDA-MB-231 breast cancer cell migration. Therefore, the cardiac glycosides do not have a unique antimigratory effect among inhibitors of the Na+/K+ ATPase.
Table 3. Inhibitory Effect of Na+/K+ ATPase Inhibitors That Are Structurally Different from Cardiac Glycoside in the Na+/K+ ATPase Assay and the Cell Migration Assay.

The increase in [Ca2+]i, through the activation of the reverse Na+/Ca2+ exchanger resulting from the “classical” inhibition of the ion transport function of the Na+/K+ ATPase, might explain the observed inhibitory activity of Na+/K+ ATPase inhibitors in MDA-MB-231 breast cancer cell migration. Ca2+ signaling is a crucial coordinator of cell migration through various mechanisms. Some of these mechanisms include activation of myosin light chain kinase (MLCK), modulation of nascent focal adhesions, and retraction–adhesion of lamellipodia through local calcium pulses near the leading edge of migrating cells.42,43 Consequently, inhibition of key regulators of calcium homeostasis affects cell migration. In the wound-closure assay, thapsigargin, a specific and irreversible inhibitor of sarco/endoplasmic reticulum Ca2+ ATPase (SERCA), inhibited the migration of bovine aortic endothelial cells (BAEC). In this migrating BAEC, thapsigargin treatment elicited a rapid increase in [Ca2+]i due to depletion of Ca2+ store sites.44 Interestingly, through chemical genetic screening using the same wound-closure assay, we found that thapsigargin and an analog inhibited MDA-MB-231 breast cancer cell migration below their cytotoxic concentration (Figure S6, Supporting Information). Besides small molecule inhibition, it was reported that siRNA knockdown of SERCA and other key Ca2+ regulators such as plasma membrane calcium ATPase (PMCA) and components of store-operated Ca2+ (SOC) influx pathway also altered the basal [Ca2+]i. An inverse correlation between the effect of small molecule and siRNA knockdown to [Ca2+]i and cell migration was observed, where an increase in [Ca2+]i reduced the speed of human umbilical vein endothelial cells (HUVECs) and vice versa.45 The aim of the present study is to illustrate the utility of correlative SAR as a strategy to link a specific protein function of a multifunctional protein to an observed cellular phenotype. Although the present study did not explore the effect of Na+/K+ ATPase inhibitors in Ca2+ signaling, we showed that the antimigratory effect of Na+/K+ ATPase inhibitors in MDA-MB-231 breast cancer cell migration is directly related to inhibition of the ion-transport function of the Na+ pump. It is established that this classical inhibition of Na+/K+ ATPase leads to an increase in intracellular [Ca2+]i,17 and thus, it is possible that the observed antimigratory effect of Na+/K+ ATPase inhibitors in MDA-MB-231 breast cancer cells might be due to the increase in [Ca2+]i, probably in a similar manner that thapsigargin inhibited the migration of this cell line.
Conclusion
In this study, correlative SAR of cardiac glycosides and analogs were used to link the ion transport function of Na+/K+ ATPase to MDA-MB-231 breast cancer cell migration. Analogs of representative cardiac glycosides ouabian and digitoxin that contain modifications in each part of the tripartite cardiac glycoside were synthesized. The effect of each structural modification on cardiac glycosides to their activities in both inhibition of Na+/K+ ATPase (molecular) and cell migration (cellular) provided a distinct pattern (SAR profile). Our results showed that modification on steroid core > modification on lactone ring > modification on sugar in decreasing impact on potency. These SAR profiles correlated in both experimental assays, showing a direct relationship between inhibition of MDA-MB-231 breast cancer cell migration and inhibition of Na+/K+ ATPase ion transport function. The interactions of ouabain and analogs to Na+/K+ ATPase were also investigated at the atomic level using molecular docking experiments. Their activities in both inhibition of Na+/K+ ATPase and cell migration correlated quite well with the degree of similarity in orientation with respect to the known ouabian orientation and reasonably well with binding energy calculations, provided that the analog completely blocks the permeation path. A cardiac glycoside analog may have a higher binding energy to Na+/K+ ATPase, but if its orientation does not allow for effective blocking of the ion permeation path, binding of that analog may not result in efficient inhibition.
Based on these results, we conclude that it is the ion-transport function of Na+/K+ ATPase that is involved in MDA-MB-231 breast cancer cell migration and not the receptor function. This conclusion is further corroborated by the observed inhibition of breast cancer cell migration by other Na+/K+ ATPase inhibitors that are structurally distinct from cardiac glycosides. In particular, a Na+/K+ ATPase inhibitor that does not activate Na+/K+ ATPase/Src complexes nor the protein kinase cascades still inhibited cell migration, implying that the antimigratory effect of Na+/K+ ATPase inhibitors does not involve activation of its receptor function. We speculate that a mechanism in which perturbation of intracellular Na+ and K+ and therefore Ca+ ion concentrations leads to inhibition of breast cancer cell migration. Further studies on how exactly perturbations of ion homeostasis affect the migration of MDA-MB-231 cells could help contribute to understanding the overall mechanism of triple negative breast cancer cell migration. This report also provides an example of how correlative SAR can help establish new relationships between specific biochemical functions and higher-level cellular processes by providing a quantitative way to link the effect of small molecules at the cellular, molecular, and atomic level.
Methodology
Quantitative Cell Migration Assay
Quantitative wound closure assay27 was performed on compounds that were active at sub-micromolar concentrations during the primary chemical genetic screening assay to evaluate their concentration–response profiles. Confluent monolayers of MDA-MB-231 breast cancer cells were wounded 30 min after treatment with different concentrations of these compounds or DMSO alone. The progress of the wound closure was followed quantitatively by taking digital images of the wounds at 3, 6, 12, and 24 h postwounding. Using the NIH ImageJ software (http://rsbweb.nih.gov/ij/), the remaining open area of the wound at each time point was calculated by tracing the wound margin in each image. Microsoft Excel and GraphPad Prism software were used to tabulate and analyze the corresponding data. GraphPad Prism software was used to determine the IC50 for inhibition of wound closure from dose–response data, and two-tailed Student’s t-test (p > 0.05) was used to statistically determine significant differences. Among the antimigratory compound hits from screening of MDA-MB-231 breast cancer cell migration inhibitors, cardiac glycosides were chosen for further characterization of their mechanism of action in relation to inhibition of MDA-MB-231 breast cancer cell migration. Analogs of reperesentative cardiac glycosides, ouabain and digitoxin, were synthesized and analyzed for their effect on MDA-MB-231 breast cancer cell migration using the same quantitative cell migration assay. Dose–response (using nine concentrations from 1 mM to 15 nM) for inhibition of cell migration by cardiac glycosides and analogs was obtained for each test compound for three trials with three replicates each trial.
In Vitro Na+/K+ ATPase Assay
The enzymatic activity of Na+/K+ ATPase (purchased from Sigma as lyophilized powder from porcine cerebral cortex) was measured by colorimetric quantification of Pi released during ATP hydrolysis. A previously published procedure46 was adapted with some modifications. The reaction was started by incubating 10 μL of Na+/K+ ATPase (600 units/mL) with 2.5 μL of KCl/NaCl solution (45 mM KCl and 2 M NaCl) at 37 °C for 30 min with either 5 μL of DMSO (control) or 5 μL of test compounds in 67.5 μL of buffer (24 mM Tris HCl buffer with 0.68 mM ethylenediaminetetraacetic acid and 6.0 mM magnesium chloride, pH 7.8). ATP (5 μL of 80 mM solution) was then added, and the reaction mixture was incubated again at 37 °C for 15 min. Tricholoroacetic acid (30 μL of 100% w/v) was then added to the reaction mixture followed by centrifugation for 3 min. Supernatant (50 μL aliquot) was transferred to a 96-well plate containing 100 μL of Taussky–Shorr reagent. The absorbance at 660 nm was read after incubation at RT for 5 min. The concentration of phosphate released in the enzymatic reaction was measured from the absorbance at 660 nm using a standard curve. The effect of test compounds on enzymatic activity (percent inhibition) was calculated by taking the ratio of the concentration of liberated phosphate after compound treatment against that of DMSO control. Dose–response (using nine concentrations from 1 mM to 15 nM) for Na+/K+ ATPase inhibition was obtained for each test compound for three trials with three replicates each trial. GraphPad Prism software was used to determine the IC50 for Na+/K+ ATPase inhibition from dose–response data and two-tailed Student’s t-test (p > 0.05) was used to statistically determine significant differences. GraphPad Prism software was also used to calculate and generate correlation plots for the inhibitory effect of test compounds on the Na+/K+ ATPase enzymatic activity and MDA-MB-231 breast cancer cell migration using Pearson correlation.
Molecular Docking
A model of the complex between oubain bound sodium–potassium transporting ATPase was obtained from the available crystallographic data at the RSCB Web site (4HYT) and refined using Chimera GUI. Duplicate chains (C, D, and E) and chain G were deleted, because they were not needed in the ligand docking. All residues were protonated based on neutral pH after the assignment of bond orders using the Dock prep tool in Chimera. Histidine tautomers were determined based on the ability to form hydrogen bonds with the neighboring residues. Ions, solvent molecules, extraneous molecules, and substitution codes for the side chains were also deleted.
The protein was assigned Amber force field parameters, while antechamber was used to assign the ligand and nonstandard residue parameters. The charge set used for the ligand was AM1-BCC charges; the structure was then minimized and duplicated; one part had the ligand deleted (the receptor), while the other had the protein part deleted (the ligand). The ligand was duplicated again, and one part was modified and minimized, while the other served as a control.
Docking followed the standard docking procedure in the Dock6 manual. Molecular surface was constructed using the dms program inbuilt in UCSF’s Chimera using a probe radius of 1.4 Å. The spheres were then generated using sphgen_cc implemented in Dock6 suite and the X-ray structure (reference) ligand was used to select the hotspot up to 10 Å from the position of the heavy atoms of the reference ligand. A site box and the generated spheres were then prepared using the showbox and showsphere modules. The grid box enclosing the selected spheres had an extra 5 Å added in every dimension. Grids were computed using a grid spacing of 0.3 Å and distance dielectric of 4 with a combination of contact and energy scores using the van der Waals parameters of Amber99 force field.
Docking was performed through grid scoring and automated ligand orientation and matching first with a rigid ligand and then choosing 10 best poses and redocking while keeping the ligand flexible. To account for receptor flexibility, an ensemble docking was attempted with Amber force field parameters and running a molecular dynamics simulation; this instead did not give an improvement in the docking poses but produced fluctuations in the structure. The final rescoring was done on the topmost poses from the grid score, using amber score (binding energy).
Acknowledgments
This work was supported by National Institutes of Health Grant GM077622 (to G. Fenteany). J. Johnson was a UConn Honors Scholar and Summer Undergraduate Research Fellowship (SURF) awardee. Narenjan Perera is acknowledged for assisting in the generation of graphics associated with docking experiments.
Supporting Information Available
Correlative SAR data for digitoxin and additional details on the cardiac glycoside interactions with the Na+/K+ ATPase. The material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Jeffrey C. J. (2003) Multifunctional proteins: Examples of gene sharing. Ann. Med. 35, 28–35. [DOI] [PubMed] [Google Scholar]
- Petrie R. J.; Doyle A. D.; Yamada K. M. (2009) Random versus directionally persistent cell migration. Nat. Rev. Mol. Cell Biol. 10, 538–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenteany G.; Zhu S. (2003) Small-molecule inhibitors of actin dynamics and cell motility. Curr. Top. Med. Chem. 3, 593–616. [DOI] [PubMed] [Google Scholar]
- Zhu S.; Mc Henry K. T.; Lane W. S.; Fenteany G. (2005) A chemical inhibitor reveals the role of Raf kinase inhibitor protein in cell migration. Chem. Biol. 12, 981–991. [DOI] [PubMed] [Google Scholar]
- Kahsai A. W.; Zhu S.; Wardrop D. J.; Lane W. S.; Fenteany G. (2006) Quinocarmycin analog DX-52–1 inhibits cell migration and targets radixin, disrupting interactions of radixin with actin and CD44. Chem. Biol. 13, 973–983. [DOI] [PubMed] [Google Scholar]
- Kahsai A. W.; Cui J.; Kaniskan H. U.; Garner P. P.; Fenteany G. (2008) Analogs of tetrahydroisoquinoline natural products that inhibit cell migration and target galectin-3 outside of its carbohydrate-binding site. J. Biol. Chem. 283, 24534–24545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez K. J.; Garimella S. V.; Lipkowitz S. (2010) Triple negative breast cancer cell lines: One tool in the search for better treatment of triple negative breast cancer. Breast Dis. 32, 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahimtoola S. H.; Tak T. (1996) The use of digitalis in heart failure. Curr. Probl. Cardiol. 21, 781–853. [DOI] [PubMed] [Google Scholar]
- Schatzmann H. J.; Rass B. (1965) Inhibition of the active Na+/K+-transport and Na+/K+-activated membrane ATPase of erythrocyte stroma by ouabain. Helv. Physiol. Pharmacol. Acta 65, C47–49. [PubMed] [Google Scholar]
- Mijatovic T.; Van Quaquebeke E.; Delest B.; Debeir O.; Darro F.; Kiss R. (2007) Cardiotonic steroids on the road to anti-cancer therapy. Biochim. Biophys. Acta 1776, 32–57. [DOI] [PubMed] [Google Scholar]
- Schonfeld W.; Weiland J.; Lindig C.; Masnyk M.; Kabat M. M.; Kurek A.; Wicha J.; Repke K. R. (1985) The lead structure in cardiac glycosides is 5 beta, 14 beta-androstane-3 beta 14-diol. Naunyn Schmiedeberg’s Arch. Pharmacol. 329, 414–426. [DOI] [PubMed] [Google Scholar]
- Elbaz H.; Stueckle T. A.; Wang H.-Y. L.; O’Doherty G. A.; Lowry D. T.; Sargent L. M.; Wang L.; Dinu C. Z.; Rojanasakul Y. (2012) Digitoxin and a synthetic monosaccharide analog inhibit cell viability in lung cancer cells. Toxicol. Appl. Pharmacol. 258, 51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.-Y. L.; Rojanasakul Y.; O’Doherty G. A. (2011) Synthesis and evaluation of the α-d-/α-l-rhamnosyl and amicetosyl digitoxigenin oligomers as anti-tumor agents. ACS Med. Chem. Lett. 2, 264–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer A. K.; Zhou M.; Azad N.; Elbaz H.; Wang L.; Rogalsky D. K.; Rojanasakul Y.; O’Doherty G. A.; Langenhan J. M. (2010) A direct comparison of the anticancer activities of digitoxin MeON-neoglycosides and O-glycosides: Oligosaccharide chain length-dependent induction of caspase-9-mediated apoptosis. ACS Med. Chem. Lett. 1, 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Velotta J. B.; McDonough A. A.; Farley R. A. (2001) All human Na+/K+ ATPase α-subunit isoforms have a similar affinity for cardiac glycosides. Am. J. Physiol.: Cell Physiol. 281, C1336–C1343. [DOI] [PubMed] [Google Scholar]
- Schoner W.; Scheiner-Bobis G. (2007) Endogenous and exogenous cardiac glycosides: Their roles in hypertension, salt metabolism, and cell growth. Am. J. Physiol.: Cell Physiol. 293, C509–C536. [DOI] [PubMed] [Google Scholar]
- Dostanic I.; Schultz J. J.; Lorenz J. N.; Lingrel J. B. (2004) The alpha 1 isoform of Na,K-ATPase regulates cardiac contractility and functionally interacts and co-localizes with the Na/Ca exchanger in heart. J. Biol. Chem. 279, 54053–54061. [DOI] [PubMed] [Google Scholar]
- Pierre S. V.; Xie Z. (2006) The Na,K-ATPase receptor complex: its organization and membership. Cell Biochem. Biophys. 46, 303–316. [DOI] [PubMed] [Google Scholar]
- Aydemir-Koksoy A.; Abramowitz J.; Allen J. C. (2001) Ouabain induced signaling and vascular smooth muscle cell proliferation. J. Biol. Chem. 276, 46605–46611. [DOI] [PubMed] [Google Scholar]
- Haas M.; Wang H.; Tian J.; Xie Z. (2002) Src-mediated inter-receptor cross-talk between the Na+/K+-ATPase and the epidermal growth factor receptor relays the signal from ouabain to mitogen-activated protein kinases. J. Biol. Chem. 277, 18694–18702. [DOI] [PubMed] [Google Scholar]
- Mijatovic T.; Roland I.; Van Quaquebeke E.; Nilsson B.; Mathieu A.; Van Vynckt F.; Darro F.; Blanco G.; Facchini V.; Kiss R. (2007) The alpha1 subunit of the sodium pump could represent a novel target to combat non-small cell lung cancers. J. Pathol. 212, 170–179. [DOI] [PubMed] [Google Scholar]
- Lefranc F.; Mijatovic T.; Kondo Y.; Sauvage S.; Roland I.; Debeir O.; Kristic D.; Vasic V.; Gailly P.; Kondo S.; Blanco G.; Kiss R. (2008) Targeting the alpha 1 subunit of the sodium pump to combat glioblastoma cells. Neurosurgery 62, 211–221. [DOI] [PubMed] [Google Scholar]
- Mathieu V.; Pirker C.; de Lassalle E. M.; Vernier M.; Mijatovic T.; DeNeve N.; Gaussin J.-F.; Dehoux M.; Lefranc F.; Berger W.; Kiss R. (2009) The sodium pump α1 sub-unit: A disease progression-related target for metastatic melanoma treatment. J. Cell. Mol. Med. 13, 3960–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Li Z.; Tian J.; Jiang W.; Wang Y.; Zhang X.; Li Z.; You Q.; Shapiro J. I.; Si S.; Xie Z. (2010) Identification of hydroxyxanthones as Na/K-ATPase ligands. Mol. Pharmacol. 77, 961–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong B.-C.; Kim S.; Kim T.-S.; Corey E. J. (2006) Synthesis and properties of several isomers of the cardioactive steroid ouabain. Tetrahedron Lett. 47, 2711–2715. [Google Scholar]
- Middleton D. A.; Rankin S.; Esmann M.; Watts A. (2000) Structural insights into the binding of cardiac glycosides to the digitalis receptor revealed by solid-state NMR. Proc. Natl. Acad. Sci. U.S.A. 97, 13602–13607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mc Henry K. T.; Ankala S. V.; Ghosh A. K.; Fenteany G. (2002) A Non-Antibacterial Oxazolidinone Derivative that Inhibits Epithelial Cell Sheet Migration. ChemBioChem 3, 1105–1111. [DOI] [PubMed] [Google Scholar]
- Lingrel J. B.; Arguello J. M.; Van Huysse J.; Kuntzweiler T. A. (1997) Cation and cardiac glycoside binding sites of the Na,K-ATPase. Ann. N.Y. Acad. Sci. 834, 194–206. [DOI] [PubMed] [Google Scholar]
- Larre I.; Lazaro A.; Contreras R. G.; Balda M. S.; Matter K.; Flores-Maldonado C.; Ponce A.; Flores-Benitez D.; Rincon-Heredia R.; Padilla-Benavides T.; Castillo A.; Shoshani L.; Cereijido M. (2010) Ouabain modulates epithelial cell tight junction. Proc. Natl. Acad. Sci. U.S.A. 107, 11387–11392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cereijido M.; Contreras R. G.; Shoshani L.; Larre I. (2012) The Na+-K+-ATPase as self-adhesion molecule and hormone receptor. Am. J. Physiol.: Cell Physiol. 302, C473–C481. [DOI] [PubMed] [Google Scholar]
- Barwe S. P.; Anilkumar G.; Moon S. Y.; Zheng Y.; Whitelegge J. P.; Rajasekaran S. A.; Rajasekaran A. K. (2005) Novel role for Na,K-ATPase in phosphatidylinositol 3-kinase signaling and suppression of cell motility. Mol. Biol. Cell 16, 1082–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laursen M.; Yatime L.; Nissen P.; Fedosova N. U. (2013) Crystal structure of the high-affinity Na+,K+-ATPase–ouabain complex with Mg2+ bound in the cation binding site. Proc. Natl. Acad. Sci. U.S.A. 110, 10958–10963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H.; Shinoda T.; Cornelius F.; Toyoshima C. (2009) Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. U.S.A. 106, 13742–13747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatime L.; Laursen M.; Morth J. P.; Esmann M.; Nissen P.; Fedosova N. U. (2011) Structural insights into the high affinity binding of cardiotonic steroids to the Na+/K+ ATPase. J. Struct. Biol. 174, 296–306. [DOI] [PubMed] [Google Scholar]
- Sandtner W.; Egwolf B.; Khalili-Araghi F.; Saanchez-Rodriguez J. E.; Roux B.; Bezanilla F.; Holmgren M. (2011) Ouabain binding site in a functioning Na+/K+ ATPase. J. Biol. Chem. 286, 38177–38183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A.; Reyes N.; Artigas P.; Gadsby D. (2008) The ion pathway through the opened Na/K ATPase pump. Nature 456, 413–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingrel J. B.; Croyle M. L.; Woo A. L.; Argüello J. M. (1998) Ligand binding sites of Na+/K+ ATPase. Acta Physiol. Scand. Suppl. 643, 69–77. [PubMed] [Google Scholar]
- Feng J.; Lingrel J. B. (1994) Analysis of amino acid residues in the H5-H6 transmembrane and extracellular domains of Na,K-ATPase alpha subunit identifies threonine 797 as a determinant of ouabain sensitivity. Biochemistry 33, 4218–4224. [DOI] [PubMed] [Google Scholar]
- Qiu L. Y.; Koenderink J. B.; Swarts H. G. P.; Willems P.; De Pont J. (2003) Phe783, Thr797, and Asp804 in transmembrane hairpin M5-M6 of Na+/K+ ATPase play a key role in ouabain binding. J. Biol. Chem. 278, 47240–47244. [DOI] [PubMed] [Google Scholar]
- Cantley L. C. Jr.; Josephson L.; Warner R.; Yanagisawa M.; Lechene C.; Guidotti G. (1977) Vanadate is a potent Na+/K+ ATPase inhibitor found in ATP derived from muscle. J. Biol. Chem. 252, 7421–7423. [PubMed] [Google Scholar]
- Ogan J. T.; Reifenberger M. S.; Milanick M. A.; Gatto C. (2007) Kinetic characterization of Na+/K+ ATPase inhibition by eosin. Blood Cells, Mol., Dis. 38, 229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannone G.; Dubin-Thaler B. J.; Rossier O.; Cai Y.; Chaga O.; Jiang G.; Beaver W.; Dobereiner H.-G.; Freund Y.; Borisy G.; Sheetz M. P. (2007) Lamellipodial actin mechanically links myosin activity with adhesion-site formation. Cell 128, 561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai F.-C.; Meyer T. (2012) Ca2+ pulses control local cycles of lamellipodia retraction and adhesion along the front of migrating cells. Curr. Biol. 22, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura C.; Oike M.; Koyama T.; Ito Y. (2001) Alterations of Ca2+ mobilizing properties in migrating endothelial cells. Am. J. Physiol.: Heart Circ. Physiol. 281, H745–H754. [DOI] [PubMed] [Google Scholar]
- Tsai F.-C.; Seki A.; Yang H. W.; Hayer A.; Carrasco S.; Malmersjö S.; Meyer T. (2014) A polarized Ca2+, diacylglycerol and STIM1 signalling system regulates directed cell migration. Nat. Cell Biol. 16, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taussky H. H.; Shorr E. (1953) A microcolorimetric method for the determination of inorganic phosphorus. J. Biol. Chem. 202, 675–682. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.