

Background: Microtubule dynamics is involved in podocyte morphogenesis.
Results: The microtubule-associated proteins Tau and CRMP2 are regulated by GSK3β in podocytes. Blockade of GSK3β ameliorates podocyte injury and proteinuria.
Conclusion: The GSK3β-dictated microtubule remodeling is crucial for the compensatory glomerular adaptation to podocyte loss.
Significance: Inhibition of GSK3β might represent a novel therapeutic strategy to treat podocytopathy.
Keywords: Cytoskeleton, Microtubule, Microtubule-associated Protein (MAP), Podocyte, Tau Protein (Tau), Glycogen Synthase Kinase 3β, Proteinuria
Abstract
Reminiscent of neural repair, following podocyte depletion, remnant-surviving podocytes exhibit a considerable adaptive capacity to expand and cover the denuded renal glomerular basement membrane. Microtubules, one of the principal cytoskeletal components of podocyte major processes, play a crucial role in podocyte morphogenesis and podocyte process outgrowth, branching, and elongation. Here, we demonstrated that the microtubule-associated proteins Tau and collapsin response mediator protein (CRMP) 2, key regulators of microtubule dynamics, were abundantly expressed by glomerular podocytes in vivo and in vitro, interacted with glycogen synthase kinase (GSK)3β, and served as its putative substrates. GSK3β overactivity induced by adriamycin injury or by a constitutively active mutant of GSK3β augmented phosphorylation of Tau and CRMP2, concomitant with microtubule depolymerization, cell body shrinkage, and shortening of podocyte processes. Conversely, inhibition of GSK3β by a dominant negative mutant or by lithium, a Food and Drug Administration-approved neuroprotective mood stabilizer, diminished Tau and CRMP2 phosphorylation, resulting in microtubule polymerization, podocyte expansion, and lengthening of podocyte processes. In a mouse model of adriamycin-induced podocyte depletion and nephropathy, delayed administration of a single low dose of lithium attenuated proteinuria and ameliorated progressive glomerulosclerosis despite no correction of podocytopenia. Mechanistically, lithium therapy obliterated GSK3β overactivity, mitigated phosphorylation of Tau and CRMP2, and enhanced microtubule polymerization and stabilization in glomeruli in adriamycin-injured kidneys, associated with elongation of podocyte major processes. Collectively, our findings suggest that the GSK3β-dictated podocyte microtubule dynamics might serve as a novel therapeutic target to reinforce the compensatory glomerular adaptation to podocyte loss.
Introduction
Reduced podocyte density, due to either absolute podocyte depletion or relative podocyte inadequacy following glomerular hypertrophy, is a fundamental pathogenic mechanism that drives the development and progression of proteinuria and progressive glomerulosclerosis (1–4). Recent evidence indicates that depletion of a small number of podocytes, however, only leads to transient proteinuria with subsequent spontaneous resolution despite the absence of podocyte replenishment, suggesting a mechanism to compensate for podocyte loss (5). Similarly, in experimental glomerular hypertrophy secondary to aging and body weight gain, overt proteinuria and glomerulosclerosis did not develop until the decompensated stage (6). These findings are in agreement with clinical data demonstrating that even glomerulopathies with severe podocyte injury could remit and regress (7).
Several reparative mechanisms might be involved in the compensation for podocyte depletion. Proliferation of the surviving podocytes is, however, less likely responsible, because podocytes are terminally differentiated cells and thus have a very limited capacity to divide or proliferate. Even when being forced into cell cycle, podocytes will undergo cell death via mitotic catastrophe (8, 9). Alternatively, a hierarchical population of progenitor cells expressing certain podocyte markers has been identified to either line the Bowman's capsule or reside in the juxtaglomerular apparatus and might possibly serve as a stem cell reservoir to replenish lost podocytes (10–12). However, it remains uncertain whether these cells are capable of differentiating into mature podocytes equipped with foot processes and slit diaphragms. So far, compensatory adaptation of surviving podocytes, characterized by increased cell size and elongated cellular processes, has been regarded as one of the most important mechanisms for glomerular repair following podocyte loss (13).
Podocytes are highly specialized and polarized epithelial cells resembling neurons in the large cell body and expression of many neuronal proteins as well as neurite-like extensions, which are sustained by an intricate and dynamic cytoskeleton machinery (14, 15). Similar to neurons, upon podocyte loss, remnant-intact podocytes exhibit a considerable adaptive capacity to extend cellular processes or develop collateral branches to cover the denuded glomerular basement membrane. Central to outgrowth, branching, and elongation of the cellular processes in arborized cells, like neurons and podocytes, is the remodeling of microtubules (15), which are the principal cytoskeletal components in podocyte cell body and major processes (16). The dynamic equilibrium of microtubules between polymerization and depolymerization or assembly and disassembly is under a close and orchestral control by microtubule-associated proteins (MAPs)2 (17), including Tau, which stabilizes microtubules against depolymerization (18), and collapsin response mediator protein (CRMP)2, which promotes microtubule polymerization (19). The activity of MAPs is regulated by a myriad of signaling pathways. In many of these pathways, glycogen synthase kinase (GSK) 3β has emerged as the integration point and plays a crucial role in microtubule remodeling (20, 21).
GSK3β is a well conserved, ubiquitously expressed serine/threonine protein kinase originally characterized as one that regulates glucose metabolism (22). Interest in GSK3β expanded greatly with the realization that it is a key regulator of multiple pathophysiological processes extending well beyond glycogen metabolism to cell morphogenesis, neuroplasticity, neurodegeneration, and repair (23). Indeed, the GSK3β-induced phosphorylation of Tau and other MAPs impairs their microtubule stabilizing activity and causes tauopathy and microtubule depolymerization, accounting for neurodegenerative injuries (20, 24–26). Conversely, selective inhibition of GSK3β reinforces the activity of MAPs, favors microtubule assembly in neurons, and potently promotes neurite growth and sprouting (27, 28). Consistently, lithium, an inhibitor of GSK3β and Food and Drug Administration-approved mood stabilizer, has been well known to have a potent promotional effect on neural repair in both neurodegenerative diseases and acute neuronal injuries (28, 29). More recently, a plethora of evidence suggests that GSK3β plays a pivotal role in kidney injury, repair, and regeneration (30–32). Nevertheless, its role in podocytopathy and glomerular repair remains unknown and was thus examined in this study by using a murine model of adriamycin-induced podocyte depletion and nephropathy. The potential effect of delayed administration of a low dose of lithium on podocyte adaptation following adriamycin injury was delineated.
EXPERIMENTAL PROCEDURES
Cell Culture and Transient Transfection
Conditionally immortalized mouse podocytes in culture were provided by Dr. Stuart Shankland (33). The cells between passages 21 and 25 were used. Podocytes were cultured in RPMI 1640 medium (Invitrogen) supplemented with 10% FBS in a humidified incubator with 5% CO2. The cells were cultured at 33 °C with 50 units/ml recombinant mouse interferon-γ (Millipore, Billerica, MA) on collagen-coated plastic Petri dishes and were transferred to a 37 °C incubator without interferon-γ to induce differentiation for 14 days. The expression vectors encoding the constitutively active (S9A) GSK3β mutant (S9A-GSK3β-HA/pcDNA3), kinase-dead (KD) GSK3β mutant (KD-GSK3β-HA/pcDNA3) and wild type (WT) GSK3β (WT-GSK3β-HA/pcDNA3) were provided by Dr. Johnson (Birmingham, AL) (34). The plasmids encoding the green fluorescent protein (GFP) and the pIRESneo-PA-GFP-α-tubulin were a gift from Dr. Patricia Wadsworth (35). Transient transfection was carried out by using Lipofectamine 2000 (Invitrogen) as described previously (36).
Time-lapse Microscopy
The cell cultures were placed in a heated chamber (37 °C) on the stage of an inverted phase contrast microscope (Axiovert, Zeiss, Cologne, Germany). Images were taken at 10-min intervals. Cell sizes were measured by the ImageJ analysis program (National Institutes of Health, Bethesda, MD).
Measurement of Cellular Volume and Height in Live Podocytes
Cellular volume and cellular height were measured as described before (37). In brief, podocytes were transfected to express GFP and treated with vehicle or adriamycin for 8 h followed by lithium chloride or sodium chloride treatment. After adriamycin injury for 14 h, cells were examined by laser scanning confocal fluorescence microscopy. To measure cellular volume and height, z-stacks were acquired from the base of the cells (at the coverslip) to beyond the top of the cells and processed for three-dimensional reconstruction by ImageJ software.
Immunofluorescent Staining
Podocytes or kidney cryosections were fixed with 4% paraformaldehyde, permeabilized, and stained with primary antibodies against detyrosinated tubulin (Millipore), GSK3β (Santa Cruz Biotechnology, Dallas), Tau (Santa Cruz Biotechnology), CRMP2 (Abcam, Cambridge, MA), phosphorylated Tau (Ser-396) (Sigma), phosphorylated CRMP2 (Abcam), HA (Santa Cruz Biotechnology), synaptopodin (Santa Cruz Biotechnology), and WT-1 (Santa Cruz Biotechnology), and then with the Alexa Fluorophore-conjugated secondary antibody (Invitrogen). Finally, cells were counterstained with DAPI, mounted with Vectashield mounting medium (Vector Laboratories, Burlingame, CA), and visualized using a fluorescence microscope. As a negative control, the primary antibody was replaced by nonimmune serum from the same species; no staining occurred.
Microtubule Dynamic Analysis
Podocytes were transfected to express both GFP-tubulin and WT, KD, or S9A mutant of GSK3β. Changes in the GFP-labeled microtubules in live podocytes were examined and recorded by time-lapse fluorescence microscopy. The microtubule length, assembly, and disassembly rate was measured by ImageJ software using the kymographs constructed from each microtubule as described previously (38). Microtubule catastrophe and rescue frequency were measured for individual dynamic microtubules by dividing the number of events by the duration of microtubule assembly and disassembly.
Western Immunoblot
Cultured cells were lysed and animal tissues homogenized in RIPA buffer supplemented with protease inhibitors. Samples were processed for immunoblot as described previously (39). The antibodies against Tau, GSK3β, phosphorylated GSK3β (Ser-9), synaptopodin, and GAPDH were purchased from Santa Cruz Biotechnology, and those against phosphorylated Tau (Ser-396) and tyrosinated tubulin were acquired from Sigma. Antibodies against CRMP2, phosphorylated CRMP2, and detyrosinated tubulin were acquired from Cell Signaling, Abcam, and Millipore, respectively.
GSK3β Activity Assay
GSK3β kinase activity was measured using the Tau (Ser(P)-396) phospho-ELISA kit commercially available from Invitrogen according to the manufacturer's instructions as described previously (40).
Tetrazolium Assay for Cellular Viability
One hour before termination of experiments, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was added (final concentration, 0.5 mg/ml) to each cell culture. Tetrazolium was released by dimethyl sulfoxide, and the optical density was determined with a spectrophotometer at 570 nm reader as before (41).
Animal Experimental Design
Experiments were performed in male BALB/c mice at age 8 weeks. Animal studies were approved by the Rhode Island Hospital Animal Care and Use Committee, and they conformed to the United States Department of Agriculture regulations and the National Institutes of Health guidelines for the humane care and use of laboratory animals. Mice were randomly assigned to four groups. Adriamycin (10 mg/kg; Sigma) or an equal volume of saline (controls) was given as a tail vein injection on day 0. A single dose of lithium chloride (40 mg/kg) or an equal molar amount (1 mEq/kg) of sodium chloride as saline was given via intraperitoneal injection on day 4. Spot urine was collected on days 0, 1, 3, 5, 7, 10, 14, and 28. Urine albumin was measured using a mouse albumin ELISA kit (Bethyl Laboratories, Inc., Montgomery, TX). Urine creatinine was measured by a creatinine assay kit (BioAssay Systems, Hayward, CA). Blood pressure was measured using the tail cuff method as described previously (41). Mice were sacrificed on day 3, 7, 14, or 28. Six mice were randomly assigned to each of the above groups for each observed time point.
Morphological Studies
Formalin-fixed kidneys were embedded in paraffin and prepared in 3-μm-thick sections. For general histology, sections were processed for Masson's trichrome staining. Morphology of all sections was assessed by a single observer in a blinded manner. A semi-quantitative score was used to evaluate the degree of glomerular sclerosis. The severity of sclerosis for each glomerulus was graded from 0 to 4 as follows: 0 represents no lesion; 1 represents sclerosis of <25% of the glomerulus; and 2, 3, and 4 represent sclerosis of 25 to 50%, 50 to 75%, and >75% of the glomerulus, respectively. A whole kidney average sclerosis score was obtained by averaging scores from all glomeruli on one section.
Glomerular Isolation
Glomerular isolation was carried out according to the method described previously (42). In brief, mice were anesthetized and perfused by infusing the abdominal artery with 5 ml of PBS containing 8 × 107 Dynabeads M-450 (Dynal Biotech ASA, Oslo, Norway). After perfusion, the kidneys were removed and cut into 1-mm3 pieces and digested in collagenase A (1 mg/ml) at 37 °C for 30 min with gentle shaking. The tissue was pressed gently through a 100-mm cell strainer (Falcon). The glomeruli containing Dynabeads were then gathered using a magnetic particle concentrator. The isolated glomeruli were washed three times with cold PBS and used for subsequent studies.
Scanning and Transmission Electron Microscopy
For electron microscopy, the kidney cortical specimens were cut into small pieces (1 mm3), fixed with glutaraldehyde solution, and embedded in Epon 812 (Polysciences, Warrington, PA). Transmission electron micrographs were obtained using a Zeiss EM-10 microscope operated at 60 kV. For scanning electron microscopy, kidney tissues were further fixed with osmium tetroxide following dehydration, critical point drying, mounting on stubs, coating with gold, and observation with a Hitachi 2700 scanning electron microscope (Hitachi High Technologies America, IL). The podocyte foot process density was estimated by dividing the length of glomerular basement membrane by the total number of foot processes present in each transmission electron micrograph.
Stereological Analysis of Podocyte Number and Process Length
Estimation of Total Lengths of Major Process Per Kidney
Kidneys were decapsulated and weighed before analysis. Kidneys were sliced transversely at 1 mm, and kidney volume V(kid) was estimated using the Cavalieri Principle (43).
Every second kidney slice from the above was fixed in 10% formalin and then embedded in paraffin, and one complete 3-μm section was cut from each block and stained with hematoxylin and eosin. Each section was viewed with a stereological test grid with an area per point (a(p)) of 104 μm2. The volume density of cortex in the kidney VV(cortx,kid) was estimated using Equation 1,
where P(cortx) was the total number of grid points overlying cortex, and P(kid) was the total number of grid points overlying kidney.
Each section was then viewed with an orthogonal grid (a(p) of 177.78 μm2), and the volume density of glomerulus VV(glom,cortx) in the cortex was estimated using Equation 2,
where P(glom) was the total number of grid points overlying the glomerulus.
The total volume of glomerulus V(glom,kid) was estimated using Equation 3,
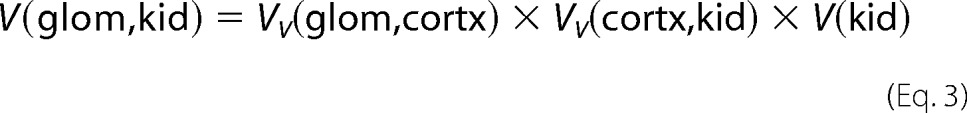 |
The tissue blocks for electron microscopy were obtained from the cortex of every other slide with a 1-mm interval in half of the sliced kidney, which resulted in about three slides for each mouse. Each block was sectioned at 1 μm, and as a new glomerulus came out, thin sections were cut about 35 μm after the previous section. Glomeruli located less than one glomerular diameter from the section edge were excluded from analysis (44). Two glomeruli were sampled from each block of the three blocks in this way, thus resulting in the analysis of about six glomeruli/mouse (44). Glomeruli were selected using the modulus sampling method (45). Every time a podocyte major process was observed that was not sectioned longitudinally and did not touch the exclusion lines, the number of the major processes was counted. From these counts, the mean number of major process profiles per glomerular profile [Q(pp,glom)] was calculated as shown in Equation 4,
where ∑A(ref) was the total area of glomerular profiles analyzed.
The total length of major processes per kidney L(pp,kid) was estimated using Equation 5 based on a cascade experimental design,
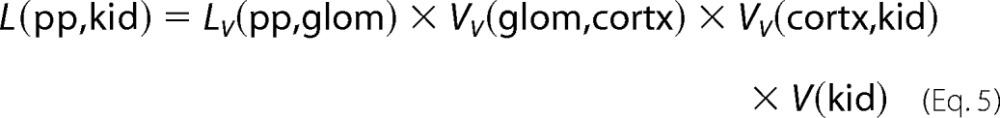 |
Estimation of Total Podocyte Number per Glomerulus
The method used to estimate the number of podocytes per glomerulus was previously described (46). The section pairs were collected every 20 μm from throughout the glomerulus with a 1-μm interval between sample section and consecutive section, and one-twentieth of the glomeruli were sampled in this way. The number of podocytes in a glomerulus was calculated using Equation 6,
where N is the number of podocytes in the glomerulus; 20 was the reciprocal of the fraction of the glomeruli sampled, and ∑Q(podo) was the sum over all pairs of profiles from nuclei seen in the sampling section but not present in the look-up section.
Estimation of Total Glomerular Numbers per Kidney
N(glom,kid) was estimated using another half of kidney by the physical dissector/fractionator combination method described in detail elsewhere (47).
Estimation of Average Major Process Length per Podocyte
The average major process length was calculated using Equation 7,
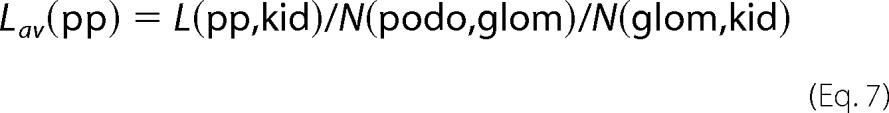 |
Statistical Analysis
For immunoblot analysis, bands were scanned, and the integrated pixel density was determined using a densitometer and the image analysis program (from National Institutes of Health). All data are expressed as means ± S.D. Statistical analysis of the data from multiple groups was performed by analysis of variance followed by Student-Newman-Keuls tests. Data from two groups were compared by t test. p < 0.05 was considered significant.
RESULTS
Delayed Lithium Treatment Overrides GSK3β Overactivity and Restores Cellular Shape and Microtubule Integrity in Adriamycin-injured Podocytes
Adriamycin (0.25 μg/ml) injury in conditionally immortalized mouse podocytes elicited cellular shrinkage (Fig. 1A) and progressively reduced cell size (Fig. 1B). A delayed lithium treatment (10 mm) 8 h following adriamycin injury abrogated this effect and largely restored the shape and size of podocytes (Fig. 1, A and B). Morphometric analysis of cellular height and cellular volume based on laser scanning confocal fluorescence microscopy of GFP-expressing podocytes revealed that adriamycin injury markedly reduced the cell volume, and this effect was significantly attenuated by the delayed treatment with lithium. No statistical significance in cellular height was noted among the differently treated podocytes. (Fig. 1, C and D). Podocyte shape and process formation are mechanically dependent on microtubule cytoskeleton structure (15, 48). Consistently, microtubules were observed to radiate outward from the centrosome, distribute throughout the whole cytoplasm, and extend to the cell edge in podocytes as probed by detyrosinated tubulin (Fig. 2A), which is a marker of stable microtubules and microtubule assembly (49, 50). Lithium treatment alone induced the level of microtubules rich in detyrosinated tubulin, denoting elongated microtubule fibers and facilitated microtubule assembly. In contrast, adriamycin injury disrupted microtubule integrity and drastically shortened microtubule fibers, resulting in the change of microtubule localization mainly to perinuclear area. This effect was prominently attenuated by the rescue treatment with lithium (Fig. 2A). To quantitate the changes in microtubule cytoskeleton, parameters of microtubule organization and dynamics in live podocytes expressing the GFP-tubulin were examined next. The parameters include microtubule average length, microtubule assembly rate, disassembly rate, catastrophe frequency, and rescue frequency. Shown in Fig. 2, B–E, the average length of microtubules was remarkably reduced following adriamycin injury, and lithium protected podocytes against microtubule shortening (Fig. 2B). The microtubule dynamic assay revealed that adriamycin promoted microtubule disassembly rate and catastrophe frequency, but it reduced microtubule assembly rate and rescue frequency, whereas lithium chloride attenuated the alterations of microtubule dynamics elicited by adriamycin (Fig. 2, C–E). To validate the morphological observations, microtubules rich in detyrosinated tubulin, namely stable microtubules, were quantified by immunoblot analysis and normalized to tyrosinated tubulin (Fig. 2, F and I), a marker of bulk unstable microtubules and free tubulin subunits. In noninjured podocytes, lithium dose-dependently induced inhibitory phosphorylation of GSK3β and increased the level of stable microtubules that were rich in detyrosinated tubulin (Fig. 2, F and G). Adriamycin injury mitigated the inhibitory phosphorylation of GSK3β, denoting an augmented GSK3β activity, and diminished the level of detyrosinated tubulin, suggestive of microtubule disassembly. These effects were abrogated by rescue treatment with lithium in a dose-dependent fashion (Fig. 2, F and G). However, at a high dose of 30 mm, lithium impaired podocyte viability, as evidenced by the MTT assay for cellular viability (Fig. 2H). Thus, a median dose of lithium at 10 mm was chosen for subsequent experiments in this study. Consistent with the morphological findings of microtubule organization and dynamics, a time course experiment (Fig. 2, I and J) indicated that adriamycin injury resulted in a diminished level of detyrosinated tubulin, denoting that the equilibrium of microtubule dynamics shifted to disassembly. This effect was associated with GSK3β overactivity as reflected by a decrease in the inhibitory phosphorylation of GSK3β (Fig. 2, I and J) and confirmed by the GSK3β activity assay (Fig. 2K). Rescue treatment with lithium counteracted GSK3β overactivity (Fig. 2, I–K) and markedly obliterated the adriamycin-reduced level of detyrosinated tubulin.
FIGURE 1.
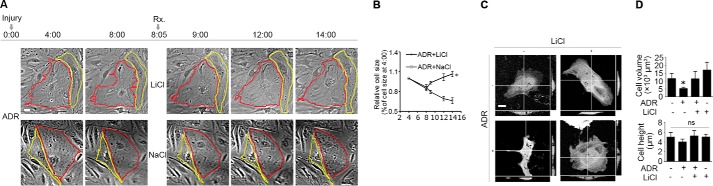
Rescue treatment with lithium chloride restores cellular shape in adriamycin-injured podocytes. A, time-lapse microscopy of differentiated and conditionally immortalized mouse podocytes following injury with adriamycin (ADR, 0.25 μg/ml) or vehicle. Lithium chloride (LiCl, 10 mm) or sodium chloride (NaCl, 10 mm) was added 8 h after ADR injury when cell shrinkage had occurred. Bar, 10 μm. B, morphometric quantification of cell sizes as relative to the original cell sizes at the 4th h (n = 30 cells per group). *, p < 0.05 versus sodium treatment at the same time point. C, podocytes were transfected to express GFP and then treated as stated in A. Representative micrographs of laser scanning confocal microscopy demonstrated the orthogonal views of podocytes at the 14th h following injury with adriamycin or vehicle. Bar, 10 μm. D, quantification of podocyte volume and height at the 14th h. *, p < 0.05 versus all other groups. ns, not significant.
FIGURE 2.

Delayed treatment with lithium chloride improves microtubule dynamics and reinstates microtubule cytoskeletal integrity in adriamycin-injured podocytes. A, phase contrast fluorescent microscopy illustrated the distribution pattern of microtubule in podocytes treated with lithium chloride (LiCl, 10 mm) or sodium chloride (NaCl, 10 mm) 8 h after injury with vehicle or ADR (0.25 μg/ml). Micrographs were taken 14 h after ADR injury. Bar, 10 μm. B–E, podocytes were transfected to express GFP-tubulin and then treated as stated in A. Time-lapse fluorescence microscopy was performed 14 h following injury with adriamycin or vehicle to examine changes in the organization and dynamics of the GFP-labeled microtubules in live podocytes. B, quantification of microtubule (MT) length. *, p < 0.05 versus all other groups. C, kymographs of assembly and disassembly of single dynamic microtubules. D, quantification of microtubule assembly and disassembly rates. *, p < 0.05 versus all other groups; #, p < 0.05 versus control group. E, quantification of microtubule rescue and catastrophe frequency. *, p < 0.05 versus all other groups; #, p < 0.05 versus control group. F, differentiated podocytes were treated with different concentrations of LiCl 8 h after injury with ADR (0.25 μg/ml) or vehicle. Cells were collected 14 h after injury with ADR or vehicle, and cell lysates were prepared for immunoblot analysis for phosphorylated GSK3β (serine 9), total GSK3β, detyrosinated tubulin (D-tubulin), tyrosinated tubulin (T-tubulin), and GAPDH. G, arbitrary units of p-GSK3β/GSK3β ratios or detyrosinated tubulin/tyrosinated tubulin ratios expressed as immunoblot densitometric ratios as fold induction over the control group. *, p < 0.05 versus other groups; #, p < 0.05 versus ADR treatment only (n = 3 representative experiments). H, tetrazolium (MTT) assay of podocytes treated with different concentrations of LiCl. *, p < 0.05 versus the control group. I, after ADR (0.25 μg/ml) injury for 8 h, conditionally immortalized mouse podocytes were treated with LiCl (10 mm) or NaCl (10 mm) for 1, 6, and 16 h corresponding to 9, 14, and 24 h post-ADR injury. Cells were harvested at the indicated time, and cell lysates were subjected to immunoblot analysis for the indicated molecules. J, arbitrary units of p-GSK3β/GSK3β ratios or detyrosinated tubulin/tyrosinated tubulin ratios expressed as immunoblot densitometric ratios of the molecules as folds of the control group. *, p < 0.05 versus control group; #, p < 0.05 versus group ADR + LiCl (n = 3 representative experiments). K, GSK3β activity was assayed using the Tau (Ser(P)-396) phospho-ELISA kit, which measures the GSK3β-catalyzed phosphorylation of recombinant human Tau. The results were expressed as fold induction over the control group. *, p < 0.05 versus the control group (n = 4 representative experiments).
Inhibition of GSK3β by Lithium Obliterates Tau and CRMP2 Phosphorylation and Reinstates Their Association with Microtubules in Adriamycin-injured Podocytes
Dynamic equilibrium of microtubules is tightly controlled by MAPs (17). To understand the mechanism by which GSK3β might be involved in adriamycin- and lithium-regulated microtubule integrity, MAPs in podocytes were next examined. First, expression of various MAPs, including MAP2c, MAP1b, MAP4, Tau, and CRMP2, were screened, and Tau and CRMP2 were identified as major MAPs in podocytes expressed in high abundance. Shown in Fig. 3A, immunoblot analysis, where mouse brain homogenates served as a positive control, demonstrated that Tau and CRMP2 were evidently expressed in mouse kidney and in glomeruli isolated from mouse kidneys. The Tau proteins detected in whole kidney homogenates were mainly short isoforms (<56 kDa) of small Tau, consistent with a previous observation (51). In contrast, in homogenates of isolated glomeruli, an ∼70-kDa Tau component predominated, corresponding to the long isoforms of small Tau. The Tau and CRMP2 detected in glomerular homogenates were likely to be attributable to glomerular podocytes, because both the long isoforms of small Tau and CRMP2 were also abundantly probed in differentiated podocytes in culture. To corroborate the immunoblot data, peroxidase immunohistochemistry of mouse kidney specimens was performed and revealed that both Tau and CRMP2 were evidently detected in glomeruli and distributed in a pattern of podocyte-specific expression (Fig. 3B). Furthermore, in cultured podocytes, fluorescent immunocytochemistry staining of Tau and CRMP2 demonstrated a distribution pattern similar to microtubules (Fig. 3, C and D). Thus, Tau was closely associated with stable microtubules longitudinally along the whole length as illustrated by staining of detyrosinated tubulin, whereas CRMP2 clustered at the leading edges of podocyte extensions, in localization consistent with the plus end of microtubules as described previously in neurons (21). In addition, perinuclear distributions of both Tau and CRMP2 were also noted and closely associated with microtubules in centrosome (Fig. 3, C and D).
FIGURE 3.

Microtubule-associated proteins Tau and CRMP2 are evidently expressed in podocytes in glomeruli, associated with microtubules, and regulated by adriamycin or lithium treatment. A, homogenates of mouse brain, kidney, isolated mouse glomeruli, and lysates of the differentiated mouse podocytes in culture were subjected to immunoblot analysis for Tau and CRMP2. Note that the long forms of Tau were probed as the major forms in isolated glomeruli and in cultured podocytes, whereas the short forms of Tau predominated in whole kidney homogenates. B, representative micrographs of peroxidase immunohistochemistry staining of mouse kidney for Tau and CRMP2 in glomeruli. Immunoperoxidase staining of Tau and CRMP2 was consistent with a pattern of podocyte-specific distribution. The primary antibody was replaced by nonimmune serum from the same species as a negative control, and no staining occurred. Bar, 20 μm. C, representative micrographs of dual color fluorescent immunocytochemistry staining of the differentiated mouse podocytes in culture indicated an association of Tau with stable microtubules as probed by detyrosinated tubulin (D-tubulin). Tau was closely associated with microtubule along the whole length. Bar, 10 μm. D, representative micrographs of dual color fluorescent immunocytochemistry staining of the differentiated mouse podocytes in culture indicated an association of CRMP2 with stable microtubules as probed by detyrosinated tubulin (D-tubulin). CRMP2 was associated with microtubules and clustered at the leading edge of podocyte extensions. Bar, 10 μm. E, after ADR (0.25 μg/ml) or vehicle treatment for 8 h, conditionally immortalized mouse podocytes were treated with or without lithium chloride (10 mm) or sodium chloride (10 mm) for 1, 6, and 16 h corresponding to 9, 14, and 24 h post-ADR injury. Cells were harvested at the indicated time points, and cell lysates were subjected to immunoblot analysis for the indicated proteins. F, arbitrary units of p-CRMP2/CRMP2 ratios or p-Tau/Tau ratios expressed as immunoblot densitometric ratios of the molecules as folds of the control group. *, p < 0.05 versus control group; #, p < 0.05 versus ADR + LiCl group, (n = 3 representative experiments). G, representative micrographs of fluorescent immunocytochemistry staining of phosphorylated Tau and phosphorylated CRMP2 in podocytes 14 h following different treatments as stated in Fig. 2A. Bar, 5 μm. H, morphometric analysis of relative fluorescence intensity of p-Tau and p-CRMP2 in podocytes. *, p < 0.05 versus control group; #, p < 0.05 versus ADR + LiCl group; (n = 6).
Under basal conditions, phosphorylated Tau and CRMP2, which cannot interact with and stabilize microtubules, were constitutively expressed in podocytes as shown by both immunoblot analysis and immunocytochemistry staining (Fig. 3, E and G). Adriamycin injury significantly augmented Tau and CRMP2 phosphorylation (Fig. 3, E and F), concomitant with GSK3β overactivity (Fig. 2, I–K). Moreover, the adriamycin-elicited phosphorylation of Tau and CRMP2 was accompanied with a diminished association of Tau (Fig. 4A) or CRMP2 (Fig. 4B) with microtubules in podocyte extensions, which was further confirmed by morphometric analysis of the fluorescence intensity of microtubule-associated Tau or CRMP2 per unit length of microtubule (Fig. 4, C and D). Delayed lithium treatment induced inhibitory phosphorylation of GSK3β and substantially suppressed Tau and CRMP2 phosphorylation (Fig. 3, E–H). Furthermore, delayed lithium treatment abolished the adriamycin-induced phosphorylation of Tau and CRMP2 in adriamycin-injured podocytes (Fig. 3, E–H), in parallel with a restoration of the association of Tau (Fig. 4, A and C) or CRMP2 (Fig. 4, B and D) with microtubules, suggesting a corrected microtubule stabilizing activity of these MAPs.
FIGURE 4.

Lithium treatment reinstates the association of Tau and CRMP2 with microtubules and improves microtubule integrity in adriamycin-injured podocytes. A, differentiated podocytes were treated as stated in Fig. 2A and prepared at 14 h for immunocytochemistry staining. Representative micrographs of dual color fluorescent immunocytochemistry staining of detyrosinated tubulin and Tau. Tau was associated with microtubules in control and lithium (LiCl)-treated podocytes. ADR injury induced podocyte shrinkage, disrupted microtubule integrity, and lessened the association between Tau and microtubule. The effect of ADR was abrogated by rescue treatment with LiCl. Sodium treatment (NaCl) served as a control for LiCl treatment. Bar, 10 μm. B, representative micrographs of dual color fluorescent immunocytochemistry staining of detyrosinated tubulin (D-tubulin) and CRMP2 in podocytes. There were some clusters of CRMP2 at the leading edges in control and LiCl-treated cells. Adriamycin injury reduced podocyte sizes, disrupted microtubule networks, and diminished CRMP2 clusters at the leading edges of cell projections. Rescue treatment with lithium chloride restored cell shape and reinstated CRMP2 clustering at the leading edges of podocytes. Bar, 10 μm. C, quantitative morphometric analysis of the fluorescence intensity of microtubule-associated Tau per unit length of microtubules. *, p < 0.05 versus all other groups (n = 3 representative experiments). D, quantitative morphometric analysis of the fluorescence intensity of microtubule-associated CRMP2 per unit length of microtubule. *, p < 0.05 versus all other groups (n = 3 representative experiments).
GSK3β Overactivity Is Necessary and Sufficient for Microtubule Disorganization and Phosphorylation of Tau and CRMP2
To explore a possible causal relationship between GSK3β and the regulation of Tau and CRMP2 as well as microtubule dynamics, the activity of GSK3β was selectively manipulated by forced expression of vectors encoding the hemagglutinin (HA)-conjugated WT GSK3β or a KD or S9A mutant of GSK3β (Fig. 5, A–C). Immunofluorescent staining for HA revealed a satisfactory transfection efficiency (>70%) (Fig. 5C). Ectopic expression of S9A prominently induced phosphorylation of Tau and CRMP2, associated with a reduced level of stable microtubules rich in detyrosinated tubulin (Fig. 5, A and B). In parallel, in podocytes overexpressing S9A, stable microtubules were limited only to the perinuclear area; microtubule integrity in cellular extensions was drastically disrupted, and microtubule length was decreased, concomitant with a reduced cell size and shortened cellular extensions (Fig. 5C). In contrast, forced expression of KD mutant of GSK3β decreased phosphorylation of Tau and CRMP2 and promoted microtubule polymerization as reflected by increased levels of detyrosinated tubulin and increased length of microtubules (Fig. 5, A and B), reminiscent of the effect of lithium. Morphologically, cells expressing the KD mutant of GSK3β exhibited more elongated and more intense stable microtubules in the extended cellular processes (Fig. 5C). To further quantitate the regulatory effect of GSK3β on microtubule dynamics, parameters of microtubule organization and dynamics, including microtubule average length, microtubule assembly rate, disassembly rate, catastrophe frequency, and rescue frequency, were further examined in live podocytes ectopically expressing both GFP-tubulin and WT, KD, or S9A mutants of GSK3β. Shown in Fig. 5, D–G, microtubule length, assembly rate, and rescue frequency were increased, although microtubule disassembly rate and catastrophe frequency were diminished in podocytes expressing the KD mutant of GSK3β. Conversely, forced expression of the S9A mutant of GSK3β exerted an opposing effect on microtubule length and microtubule dynamics in podocytes.
FIGURE 5.

GSK3β controls microtubule dynamics by regulating Tau and CRMP2. A, differentiated podocytes were transfected with vectors encoding the hemagglutinin (HA)-conjugated wild type GSK3β (WT), kinase-dead mutant of GSK3β (KD), or constitutively active mutant of GSK3 β (S9A). Cells were collected 48 h after transfection and prepared for immunoblot analysis or fluorescent immunocytochemistry staining. B, densitometric analysis of Western immunoblot in A. *, p < 0.05 versus all other groups (n = 3). C, representative micrographs of dual color fluorescent immunocytochemistry staining of detyrosinated tubulin (D-tubulin) and HA. Bar, 10 μm. D–G, differentiated podocytes were transfected to express both GFP-tubulin and WT, KD, or S9A mutant of GSK3β. Time-lapse fluorescence microscopy of live podocytes was performed 48 h after transfection, and micrographs were recorded and analyzed. D, quantification of microtubule (MT) length. *, p < 0.05 versus all other groups (n = 3). E, kymographs of assembly and disassembly of single dynamic microtubules. F, quantification of microtubule assembly and disassembly rate. *, p < 0.05 versus all other groups (n = 3). G, quantification of microtubule rescue and catastrophe frequency. *, p < 0.05 versus all other groups; (n = 3).
Tau and CRMP2 Colocalize and Physically Interact with GSK3β as Its Putative Substrates in Podocytes
To understand the mechanistic essence of the GSK3β-regulated Tau and CRMP2 phosphorylation, we next examined the intracellular physical relationship between GSK3β and Tau or CRMP2. Laser scanning confocal fluorescence microscopy of dual color fluorescent immunocytochemistry staining of GSK3β and Tau or CRMP2 indicated that GSK3β distributed mainly in cytoplasm and, to a much lesser extent, in nucleus of podocytes. A discrete pool of GSK3β staining was noted to colocalize with both Tau and CRMP2 staining (Fig. 6A). To validate this morphological finding, immunoprecipitation was carried out and demonstrated that GSK3β evidently coprecipitated with both Tau and CRMP2, suggesting that GSK3β physically interacts with Tau and CRMP2 (Fig. 6B). Adriamycin injury enhanced the association of GSK3β with both Tau and CRMP2, and this effect was substantially abrogated by rescue treatment with lithium. The Tau and CRMP2 detected in the immunoprecipitates were less likely to originate from those bound to cytoskeletons that were nonspecifically coprecipitated, because immunoblot analysis of the immunoprecipitates indicated no detection of actin or tubulin, suggesting no contamination by the actin or microtubule cytoskeletons. To further define the possible mechanism by which GSK3β regulates Tau and CRMP2 phosphorylation, the amino acid sequences of the longest isoform of small Tau (NCBI Reference Sequence, NP_005901) and CRMP2 (NCBI Reference Sequence, NP_034085.2) were subjected to the computational active site analysis for putative consensus phosphorylation motifs for GSK3β (Fig. 6C). In silico analysis indicated that residues Thr-181, Ser-195, Ser-208, Thr-231, and Ser-396 of Tau and residues Ser-304, Thr-514, and Ser-518 of CRMP2 reside in the consensus motifs for phosphorylation by GSK3β with statistically significant prediction scores, denoting that Tau and CRMP2 are putative cognate substrates for GSK3β (Fig. 6D).
FIGURE 6.

Tau and CRMP2 colocalize and physically interact with GSK3β as its putative substrates in podocytes. A, representative micrographs of laser scanning confocal microscopy of dual color fluorescent immunocytochemistry staining of GSK3β and Tau or CRMP2 in differentiated conditionally immortalized mouse podocytes. Evident colocalization of Tau or CRMP2 with GSK3β was noted in the cytoplasm of differentiated podocytes. Bar, 5 μm. B, podocytes were treated as described in Fig. 2A, and cell lysates were prepared at 14 h for immunoprecipitation followed by immunoblot analysis for indicated molecules. LC, IgG light chain. An aliquot of cell lysates from each group served as input control. C, in silico analysis indicated that amino acid residues Thr-181, Ser-195, Ser-208, Thr-231, and Ser-396 of the longest isoform of small Tau and amino acid residues Ser-304, Thr-514, and Ser-518 of CRMP2 reside in the consensus motifs for phosphorylation by GSK3β. D, characteristics of consensus GSK3β phosphorylation motifs in Tau and CRMP2, including the predicted phosphorylation sites, prediction confidence scores, and sequences, as estimated by in silico analysis.
Rescue Treatment with Lithium Attenuates Proteinuria and Ameliorates Glomerulosclerosis in Experimental Adriamycin Nephropathy
To further validate the role of GSK3β in podocytopathy in vivo and also to assess the possible effect of therapeutic targeting of GSK3β by lithium, we employed the mouse model of adriamycin-induced podocyte depletion and nephropathy, which recapitulates key features of podocytopathy and glomerulosclerosis in humans, including podocyte injury and depletion, podocyte cytoskeleton disarrangement, massive proteinuria, and progressive glomerulosclerosis (52, 53). Mice injured with an intravenous injection of adriamycin (10 mg/kg) presented massive proteinuria that peaked between days 5 and 7. Lithium chloride treatment was given as an intraperitoneal injection 4 days after adriamycin injury. Thus, this delayed treatment was unlikely to have impacted the degree to which podocytes were injured by the adriamycin. Lithium chloride rescue treatment at a low dose of 40 mg/kg as compared with an equal molar amount of sodium chloride (∼0.15 ml normal saline) prominently reduced albuminuria by more than 80% on day 5 and by more than 50% on day 7 (Fig. 7, A and B) without affecting blood pressure. Consistently, renal lesions of the adriamycin-induced glomerulopathy, including glomerulosclerosis and protein casts as revealed by Masson's trichrome staining, were significantly ameliorated following lithium rescue treatment on days 7, 14, and 28 (Fig. 7C). These morphological findings were corroborated by the semi-quantitative morphometric scoring of glomerulosclerosis (Fig. 7D).
FIGURE 7.

Delayed administration of a single low dose of lithium induces proteinuria remission and ameliorates glomerulosclerosis in experimental adriamycin nephropathy. Mice were subjected to ADR injury (10 mg/kg, tail vein injection) on day 0 and received a single intraperitoneal injection of lithium chloride (40 mg/kg) or an equal molar amount (1 meq/kg) of sodium chloride (NaCl) as saline on day 4. A, urine was collected at the indicated time points and subjected to SDS-PAGE followed by Coomassie Brilliant Blue staining. Bovine serum albumin (BSA) of 5, 10, 20, and 40 μg served as standard controls. Urine (1.5 μl) from each group was loaded. B, quantification of urine albumin levels adjusted for urine creatinine concentrations. *, p < 0.05 versus control group; #, p < 0.05 versus group ADR + LiCl. (n = 6). C, representative micrographs demonstrating Masson's trichrome staining of mouse kidneys. Bars, 20 μm. D, morphometric analysis of glomerulosclerosis score. *, p < 0.05 versus day 0; #, p < 0.05 versus group ADR + LiCl (n = 6).
Delayed Lithium Therapy Counteracts GSK3β Overactivity, Diminishes Tau and CRMP2 Phosphorylation, and Reinstates Microtubule Integrity in Adriamycin-injured Glomeruli
Glomeruli were isolated from kidneys by the magnetic bead-based approach and homogenized for immunoblot analysis. Shown in Fig. 8, A and B, adriamycin injury significantly diminished the inhibitory phosphorylation of GSK3β on days 3, 7, and 14, denoting GSK3β overactivity. Consistent with in vitro findings, this GSK3β overactivity was associated with increased phosphorylation of Tau and CRMP2 and the ensuing microtubule disassembly, marked by a reduced level of detyrosinated microtubules. Lithium treatment prominently overrode the adriamycin-elicited GSK3β overactivity, mitigated Tau and CRMP2 phosphorylation (Fig. 8, A and B), and resulted in an improvement in microtubule integrity, as reflected by a reinstated level of detyrosinated microtubules in isolated glomeruli.
FIGURE 8.

Lithium counteracts GSK3β overactivity, diminishes Tau and CRMP2 phosphorylation, and reinstates microtubule integrity in adriamycin-injured glomeruli. A, immunoblot analysis of homogenates of isolated glomeruli for indicated molecules. B, arbitrary units of p-GSK3β/GSK3β ratios, p-Tau/Tau ratios, p-CRMP2/CRMP2 ratios, and detyrosinated tubulin (d-tubulin)/tyrosinated tubulin (t-tubulin) ratios expressed as immunoblot densitometric ratios of the molecules as folds of the control groups. *, p < 0.05 versus day 0; #, p < 0.05 versus group ADR + LiCl (n = 6).
Delayed Inhibition of GSK3β Promotes Podocyte Process Elongation to Compensate for Podocyte Depletion in Adriamycin Nephropathy
Next, podocyte injury in vivo was evaluated by evaluating podocyte phenotypes (Fig. 9) and by electron microscopy (Fig. 10). Adriamycin injury substantially mitigated glomerular expression of synaptopodin (Fig. 9, A and C), a marker of podocyte foot processes, consistent with massive foot process effacement on electron microscopy (Fig. 10). Podocyte depletion as indicated by the reduced expression of WT-1, a nuclear marker of podocyte, was also evidently noted in adriamycin-injured kidneys (Fig. 9, A and B). Rescue treatment with lithium minimally attenuated podocytopenia and barely induced podocyte repopulation, based on the absolute counting of the number of WT-1 positive cells in each glomerulus (Fig. 9B). Surprisingly, the adriamycin-diminished synaptopodin expression was prominently abrogated following lithium treatment as demonstrated by both immunohistochemistry staining (Fig. 9A) and immunoblot analysis of homogenates of isolated glomeruli (Fig. 9C), in agreement with a marked amelioration of foot process effacement as quantified by measuring the number of foot processes per unit length of glomerular basement membrane on electron microscopy (Fig. 10, B and C). This finding implies that despite no evidence of podocyte replenishment, the processes of the surviving podocytes might outgrow and elongate after lithium therapy. Indeed, as shown by scanning electron microscopy (Fig. 10A), lithium therapy as compared with saline treatment remarkably elongated podocyte major processes and significantly improved podocyte morphology in adriamycin-injured kidneys, despite residual lesions of foot process effacement and minimal microvillous transformation. To quantify the findings of scanning electron microscopy, the lengths of podocyte major processes were measured by stereological approaches based on transmission electron microscopy (Fig. 10B). Shown in Table 1, the average length of podocyte major processes in adriamycin-injured kidneys increased significantly following lithium treatment as compared with that in the saline-treated group, even though the number of podocytes in each glomerulus was not statistically different between lithium- and saline-treated groups.
FIGURE 9.

Delayed lithium therapy potentiates glomerular adaptation to podocyte depletion in adriamycin nephropathy. A, fluorescence immunohistochemistry staining of kidney specimens procured on day 14 for synaptopodin and WT-1. Lithium chloride therapy abrogated the ADR-diminished synaptopodin expression in the glomerulus but barely improved the adriamycin-mitigated WT-1 expression. Bar, 30 μm. B, absolute count of WT-1-positive podocytes per glomerulus on kidney specimens procured from mice on days 0, 3, 7, and 14. *, p < 0.05 versus other time points, NS, not significant versus group ADR + LiCl (n = 6). C, Western immunoblot analysis of homogenates of isolated glomeruli for synaptopodin. D, densitometric analysis of the immunoblots of synaptopodin. *, p < 0.05 versus ADR-treated groups; #, p < 0.05 versus group ADR + LiCl (n = 6).
FIGURE 10.

Rescue treatment with a single low dose of lithium, an inhibitor of GSK3β and Food and Drug Administration-approved mood stabilizer, in experimental adriamycin nephropathy promotes major process elongation in remnant surviving podocytes to compensate for the loss of podocytes and to cover the denuded glomerular basement membrane. Kidney cortical tissues were procured on day 7 from ADR-injured mice receiving rescue treatment of lithium chloride (40 mg/kg) or an equal molar amount of sodium chloride as saline. Kidney specimens were processed from scanning or transmission electron microscopy. A, scanning electron microscopy of glomeruli. White arrows indicate podocyte foot process effacement; white arrowheads indicate podocyte microvillous transformation. B, transmission electron microscopy of glomeruli. Major processes were outlined by red lines for the purpose of stereological analysis to estimate the lengths of podocyte major processes. Bar, 10 μm. C, absolute count of the number of foot processes per unit length of glomerular basement membrane as evaluated by transmission electron microscopy. *, p < 0.05 versus all other groups.
TABLE 1.
Stereological analysis of glomeruli in mouse kidneys
Lv(pp,glom), length density of podocyte major processes; V(kid), kidney volume; L(pp,kid), total length of podocyte major processes in a kidney; L(pp,podo)(μm), average length of podocyte major processes per podocyte; NS, not significant. Data are presented as means ± S.E.
| Control (n = 6) | Group ADR + NaCl (n = 6) | Group ADR + LiCl (n = 6) | p valuea | |
|---|---|---|---|---|
| Lv(pp,glom)(× 10−2 μm−2) | 3.5 ± 0.115 | 1.5 ± 0.098 | 3.7 ± 0.074 b | <0.01 |
| V(kid) (mm3) | 131.1 ± 7.50 | 150.9 ± 10.51 | 138.8 ± 8.35 | NS |
| L(pp,kid) (× 106 μm) | 122.1 ± 7.74 | 61.2 ± 9.05 | 134.5 ± 8.42b | <0.01 |
| Podocyte number/glomerular | 88.50 ± 8.81 | 44.83 ± 4.69 | 53.17 ± 4.67c | <0.01 |
| Glomerular number/kidney (×102) | 111.17 ± 5.10 | 104.00 ± 3.26 | 110.00 ± 4.88 | NS |
| L(pp,podo) (μm) | 130.3 ± 14.02 | 132.2 ± 11.98dd | 236.6 ± 17.92 | <0.01 |
a p value was based on analysis of variancefollowed by Student-Newman-Keuls test for any differences among the threegroups.
b There was no significant difference betweencontrol and group ADR + LiCl.
c There was no significant difference between groupADR + NaCl and group ADR + LiCl.
d There was no significant difference betweencontrol and group ADR + NaCl.
DISCUSSION
Mismatch between podocytes and glomerular tufts due to absolute or relative podocytopenia is a culprit of progressive glomerulosclerosis and proteinuria and serves as a therapeutic target to treat glomerulopathies and retard the progression of glomerulosclerosis (2, 54, 55). Accordingly, correction of podocytopenia and alleviation of glomerular hypertrophy are general principles of treatment of podocytopathy. As highly specialized and terminally differentiated cells, podocytes have minimal capacity of proliferation. Moreover, podocyte repopulation via differentiation of progenitor cells in the glomeruli warrants further investigation. To date, the therapeutic approaches available for clinical practice are limited to the following: 1) angiotensin blockades, which relieve the glomerular hypertension and thus slow or even regress the hypertrophy of glomerular tufts (56); 2) removal of the original etiological factors that cause podocyte injuries, including nephrotoxins, hyperglycemia, circulating permeability factors, and more, so that further podocyte injury and depletion could be prevented (2). To the best of our knowledge, this study is the first to demonstrate that enhancement of surviving podocyte adaptation by promoting microtubule assembly through lithium-mediated inhibition of GSK3β potentiates the compensatory glomerular adaptation to podocyte loss, attenuates proteinuria, and ameliorates glomerulosclerosis.
The exact role of GSK3β in the pathogenesis of podocytopathy remains unclear. We recently found that expression of GSK3β is aberrantly up-regulated in diseased human kidneys in injured tubules and glomeruli (57). Consistently, Waters and Koziell (58) also noted up-regulation of GSK3β in human podocytes in association with specific NPHS1 mutations, suggesting that GSK3β dysregulation might be involved in podocyte dysfunction. In concert with these observations, inhibition of GSK3β by a selective small molecule inhibitor 2′Z,3′E-6-bromoindirubin-3′-oxime (BIO) at a low dose dramatically normalized proteinuria and attenuated histological injury of glomeruli in rat models of diabetic nephropathy even though hyperglycemia was not corrected, denoting a direct anti-proteinuric and renoprotective action (59). In contrast, Matsui et al. (60) found that high dose BIO exacerbated proteinuria and loss of glomerular nephrin in puromycin-injured rats. In addition, another study by Dai et al. (61) reported that transient albuminuria was rapidly induced by an ultrahigh dose of lithium in normal mice but was spontaneously restored to normal levels within 24 h. Both studies suggest that GSK3β is protective and GSK3β inhibition worsens podocytopathy. These data sharply contradict the findings from gene-targeted knock-in mice with mutated un-inhibitable GSK3 which developed spontaneous albuminuria and podocyte injury, suggesting a detrimental role of GSK3 in podocyte injury (62). To reconcile these conflicting findings, one plausible explanation would be the difference in the dose of GSK3β inhibitors used. As mentioned above, Matsui et al. (60) used a high dose BIO, which is about 10 times higher than that used in the earlier study of diabetic nephropathy. Dai et al. (61) used an ultrahigh dose of lithium chloride (16mmol/kg), which is almost two times the median lethal dose of lithium chloride in mice, and surprisingly, no lethality was reported in that study. At high doses both inhibitors could have nonspecific off-target effects on a wide variety of essential kinases and thus could induce cytotoxicity and even lethality (63). Collectively, despite a couple of conflicting findings, evidence suggests that GSK3β is involved in the pathogenesis of proteinuria and podocytopathy and inhibition of GSK3β might be beneficial. In support of this viewpoint, this study indicated that delayed inhibition of GSK3β by a low dose of lithium ameliorated proteinuria and glomerulosclerosis.
Historically, lithium, a first generation GSK3β inhibitor, has been safely used for decades as a mood stabilizer (64). Lithium is well known for its potent pro-reparative activities in the central nervous system (65). It has been known for a long time that lithium has a beneficial effect in both acute neural injury and organic neurological degenerative diseases, including Alzheimer disease (66). The mechanism underlying this neuroprotective effect has been attributed to an enhanced neurite outgrowth and branching following lithium-promoted microtubule assembly in neurons (67, 68). Podocytes resemble neurons in many aspects. Thus, it is not surprising to find that long isoforms of small Tau, which are typically present in neural tissues, predominated in cultured podocytes and in glomeruli. Of note, glomeruli account for only ∼2% of the total kidney volume (69). This might explain why the podocyte-specific long isoforms of small Tau were barely detected in whole kidney homogenates as shown by our work and other studies (51). Consistent with the beneficial effect of lithium in neural tissues, our study showed that inhibition of GSK3β by lithium overrode the adriamycin-elicited GSK3β overactivity, subsequently diminished phosphorylation of Tau and CRMP2, enhanced microtubule polymerization in podocytes, and resulted in process outgrowth and elongation of surviving podocytes and a strengthened glomerular adaptation to podocyte loss in adriamycin nephropathy models. Our findings suggest that similar to neurons, microtubule assembly is also essential for podocyte adaptation and repair. In support of this, taxol, a quintessential microtubule stabilizer, has been shown to reduce proteinuria and attenuate glomerulosclerosis in remnant kidney disease (70), a standard model of post-adaptive focal segmental glomerulosclerosis, and also in the adriamycin-induced podocytopenia and nephropathy (71). Clinical use of lithium has been complicated by some renal side effects. However, epidemiology data suggest that for psychiatric patients who received lithium therapy for a long period of time (usually more than 10 years), kidney diseases, like glomerular or interstitial nephropathy are uncommon (72). Of note, the dose of lithium used in our study (40 mg/kg) is much lower than the standard dose of lithium (120 mg/kg) that has been safely and routinely used for neurobiology research in rodents. Nevertheless, more clinical studies are needed to determine the appropriate dose of lithium for humans to suppress GSK3β activity and promote podocyte adaptation in glomerular diseases.
In summary, blockade of GSK3β by low dose lithium overrides the adriamycin-elicited GSK3β overactivity in podocytes, diminishes phosphorylation of Tau and CRMP2, restores podocyte microtubule cytoskeleton integrity, and thereby promotes podocyte process outgrowth and elongation, reinforces the compensatory adaptation to podocyte loss, and attenuates proteinuria and glomerulosclerosis in experimental adriamycin podocytopathy. Our data suggest that the GSK3β-dictated microtubule dynamics might serve as a novel and feasible therapeutic target to treat podocytopathy and slow the progression of glomerulosclerosis.
This work was supported, in whole or in part, by National Institutes of Health Grant R01DK092485. This work was also supported by China 973 Program 2012CB517600 and the International Society of Nephrology Sister Renal Center Trio Program.
- MAP
- microtubule-associated protein
- GSK3β
- glycogen synthase kinase 3β
- CRMP2
- collapsin response mediator protein
- ADR
- adriamycin
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- BIO
- 2′Z,3′E-6-bromoindirubin-3′-oxime.
REFERENCES
- 1. Fukuda A., Chowdhury M. A., Venkatareddy M. P., Wang S. Q., Nishizono R., Suzuki T., Wickman L. T., Wiggins J. E., Muchayi T., Fingar D., Shedden K. A., Inoki K., Wiggins R. C. (2012) Growth-dependent podocyte failure causes glomerulosclerosis. J. Am. Soc. Nephrol. 23, 1351–1363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kriz W. (2012) Glomerular diseases: podocyte hypertrophy mismatch and glomerular disease. Nat. Rev. Nephrol. 8, 618–619 [DOI] [PubMed] [Google Scholar]
- 3. D'Agati V. D., Kaskel F. J., Falk R. J. (2011) Focal segmental glomerulosclerosis. N. Engl. J. Med. 365, 2398–2411 [DOI] [PubMed] [Google Scholar]
- 4. D'Agati V. D. (2012) Pathobiology of focal segmental glomerulosclerosis: new developments. Curr. Opin. Nephrol. Hypertens. 21, 243–250 [DOI] [PubMed] [Google Scholar]
- 5. Wharram B. L., Goyal M., Wiggins J. E., Sanden S. K., Hussain S., Filipiak W. E., Saunders T. L., Dysko R. C., Kohno K., Holzman L. B., Wiggins R. C. (2005) Podocyte depletion causes glomerulosclerosis: diphtheria toxin-induced podocyte depletion in rats expressing human diphtheria toxin receptor transgene. J. Am. Soc. Nephrol. 16, 2941–2952 [DOI] [PubMed] [Google Scholar]
- 6. Wiggins J. E., Goyal M., Sanden S. K., Wharram B. L., Shedden K. A., Misek D. E., Kuick R. D., Wiggins R. C. (2005) Podocyte hypertrophy, “adaptation,” and “decompensation” associated with glomerular enlargement and glomerulosclerosis in the aging rat: prevention by calorie restriction. J. Am. Soc. Nephrol. 16, 2953–2966 [DOI] [PubMed] [Google Scholar]
- 7. Macconi D., Sangalli F., Bonomelli M., Conti S., Condorelli L., Gagliardini E., Remuzzi G., Remuzzi A. (2009) Podocyte repopulation contributes to regression of glomerular injury induced by ACE inhibition. Am. J. Pathol. 174, 797–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lasagni L., Lazzeri E., Shankland S. J., Anders H. J., Romagnani P. (2013) Podocyte mitosis–a catastrophe. Curr. Mol. Med. 13, 13–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Liapis H., Romagnani P., Anders H. J. (2013) New insights into the pathology of podocyte loss: mitotic catastrophe. Am. J. Pathol. 183, 1364–1374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ronconi E., Sagrinati C., Angelotti M. L., Lazzeri E., Mazzinghi B., Ballerini L., Parente E., Becherucci F., Gacci M., Carini M., Maggi E., Serio M., Vannelli G. B., Lasagni L., Romagnani S., Romagnani P. (2009) Regeneration of glomerular podocytes by human renal progenitors. J. Am. Soc. Nephrol. 20, 322–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shankland S. J., Anders H. J., Romagnani P. (2013) Glomerular parietal epithelial cells in kidney physiology, pathology, and repair. Curr. Opin. Nephrol. Hypertens. 22, 302–309 [DOI] [PubMed] [Google Scholar]
- 12. Pippin J. W., Sparks M. A., Glenn S. T., Buitrago S., Coffman T. M., Duffield J. S., Gross K. W., Shankland S. J. (2013) Cells of renin lineage are progenitors of podocytes and parietal epithelial cells in experimental glomerular disease. Am. J. Pathol. 183, 542–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Guzman J., Jauregui A. N., Merscher-Gomez S., Maiguel D., Muresan C., Mitrofanova A., Diez-Sampedro A., Szust J., Yoo T. H., Villarreal R., Pedigo C., Molano R. D., Johnson K., Kahn B., Hartleben B., Huber T. B., Saha J., Burke G. W., 3rd, Abel E. D., Brosius F. C., Fornoni A. (2014) Podocyte-specific GLUT4-deficient mice have fewer and larger podocytes and are protected from diabetic nephropathy. Diabetes 63, 701–714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kobayashi N. (2002) Mechanism of the process formation; podocytes versus neurons. Microsc. Res. Tech. 57, 217–223 [DOI] [PubMed] [Google Scholar]
- 15. Kobayashi N., Mundel P. (1998) A role of microtubules during the formation of cell processes in neuronal and non-neuronal cells. Cell Tissue Res. 291, 163–174 [DOI] [PubMed] [Google Scholar]
- 16. Drenckhahn D., Franke R. P. (1988) Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab. Invest. 59, 673–682 [PubMed] [Google Scholar]
- 17. Maccioni R. B., Cambiazo V. (1995) Role of microtubule-associated proteins in the control of microtubule assembly. Physiol. Rev. 75, 835–864 [DOI] [PubMed] [Google Scholar]
- 18. Bunker J. M., Wilson L., Jordan M. A., Feinstein S. C. (2004) Modulation of microtubule dynamics by Tau in living cells: implications for development and neurodegeneration. Mol. Biol. Cell 15, 2720–2728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fukata Y., Itoh T. J., Kimura T., Ménager C., Nishimura T., Shiromizu T., Watanabe H., Inagaki N., Iwamatsu A., Hotani H., Kaibuchi K. (2002) CRMP-2 binds to tubulin heterodimers to promote microtubule assembly. Nat. Cell Biol. 4, 583–591 [DOI] [PubMed] [Google Scholar]
- 20. Mandelkow E. M., Drewes G., Biernat J., Gustke N., Van Lint J., Vandenheede J. R., Mandelkow E. (1992) Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein Tau. FEBS Lett. 314, 315–321 [DOI] [PubMed] [Google Scholar]
- 21. Yoshimura T., Kawano Y., Arimura N., Kawabata S., Kikuchi A., Kaibuchi K. (2005) GSK-3β regulates phosphorylation of CRMP-2 and neuronal polarity. Cell 120, 137–149 [DOI] [PubMed] [Google Scholar]
- 22. Embi N., Rylatt D. B., Cohen P. (1980) Glycogen synthase kinase-3 from rabbit skeletal muscle. Separation from cyclic-AMP-dependent protein kinase and phosphorylase kinase. Eur. J. Biochem. 107, 519–527 [PubMed] [Google Scholar]
- 23. Hur E. M., Zhou F. Q. (2010) GSK3 signalling in neural development. Nat. Rev. Neurosci. 11, 539–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tan M., Ma S., Huang Q., Hu K., Song B., Li M. (2013) GSK-3α/β-mediated phosphorylation of CRMP-2 regulates activity-dependent dendritic growth. J. Neurochem. 125, 685–697 [DOI] [PubMed] [Google Scholar]
- 25. Sánchez C., Pérez M., Avila J. (2000) GSK3β-mediated phosphorylation of the microtubule-associated protein 2C (MAP2C) prevents microtubule bundling. Eur. J. Cell Biol. 79, 252–260 [DOI] [PubMed] [Google Scholar]
- 26. Goold R. G., Gordon-Weeks P. R. (2001) Microtubule-associated protein 1B phosphorylation by glycogen synthase kinase 3β is induced during PC12 cell differentiation. J. Cell Sci. 114, 4273–4284 [DOI] [PubMed] [Google Scholar]
- 27. Namekata K., Harada C., Guo X., Kimura A., Kittaka D., Watanabe H., Harada T. (2012) Dock3 stimulates axonal outgrowth via GSK-3β-mediated microtubule assembly. J. Neurosci. 32, 264–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hong M., Chen D. C., Klein P. S., Lee V. M. (1997) Lithium reduces Tau phosphorylation by inhibition of glycogen synthase kinase-3. J. Biol. Chem. 272, 25326–25332 [DOI] [PubMed] [Google Scholar]
- 29. Caberlotto L., Carboni L., Zanderigo F., Andreetta F., Andreoli M., Gentile G., Razzoli M. (2013) Differential effects of glycogen synthase kinase 3 (GSK3) inhibition by lithium or selective inhibitors in the central nervous system. Naunyn Schmiedebergs Arch. Pharmacol. 386, 893–903 [DOI] [PubMed] [Google Scholar]
- 30. Wang Z., Havasi A., Gall J., Bonegio R., Li Z., Mao H., Schwartz J. H., Borkan S. C. (2010) GSK3β promotes apoptosis after renal ischemic injury. J. Am. Soc. Nephrol. 21, 284–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Obligado S. H., Ibraghimov-Beskrovnaya O., Zuk A., Meijer L., Nelson P. J. (2008) CDK/GSK-3 inhibitors as therapeutic agents for parenchymal renal diseases. Kidney Int. 73, 684–690 [DOI] [PubMed] [Google Scholar]
- 32. Wang Z., Ge Y., Bao H., Dworkin L., Peng A., Gong R. (2013) Redox-sensitive glycogen synthase kinase 3β-directed control of mitochondrial permeability transition: rheostatic regulation of acute kidney injury. Free Radic. Biol. Med. 65, 849–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Shankland S. J., Pippin J. W., Reiser J., Mundel P. (2007) Podocytes in culture: past, present, and future. Kidney Int. 72, 26–36 [DOI] [PubMed] [Google Scholar]
- 34. Cho J. H., Johnson G. V. (2004) Primed phosphorylation of Tau at Thr231 by glycogen synthase kinase 3β (GSK3β) plays a critical role in regulating Tau's ability to bind and stabilize microtubules. J. Neurochem. 88, 349–358 [DOI] [PubMed] [Google Scholar]
- 35. Tulu U. S., Rusan N. M., Wadsworth P. (2003) Peripheral, non-centrosome-associated microtubules contribute to spindle formation in centrosome-containing cells. Curr. Biol. 13, 1894–1899 [DOI] [PubMed] [Google Scholar]
- 36. Gong R., Rifai A., Ge Y., Chen S., Dworkin L. D. (2008) Hepatocyte growth factor suppresses proinflammatory NFκB activation through GSK3β inactivation in renal tubular epithelial cells. J. Biol. Chem. 283, 7401–7410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hevia D., Marta Alonso-Gervós A., Quirós-González I., Cimadevilla H. M., Gómez-Cordovés C., Sainz R. M., Mayo J. C. (2011) Cell volume and geometric parameters determination in living cells using confocal microscopy and 3D reconstruction. Protocol Exchange 10.1038/protex.2011.272 [DOI] [Google Scholar]
- 38. Al-Bassam J., Kim H., Brouhard G., van Oijen A., Harrison S. C., Chang F. (2010) CLASP promotes microtubule rescue by recruiting tubulin dimers to the microtubule. Dev. Cell 19, 245–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gong R., Rifai A., Tolbert E. M., Centracchio J. N., Dworkin L. D. (2003) Hepatocyte growth factor modulates matrix metalloproteinases and plasminogen activator/plasmin proteolytic pathways in progressive renal interstitial fibrosis. J. Am. Soc. Nephrol. 14, 3047–3060 [DOI] [PubMed] [Google Scholar]
- 40. Khanfar M. A., Asal B. A., Mudit M., Kaddoumi A., El Sayed K. A. (2009) The marine natural-derived inhibitors of glycogen synthase kinase-3β phenylmethylene hydantoins: in vitro and in vivo activities and pharmacophore modeling. Bioorg. Med. Chem. 17, 6032–6039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Si J., Ge Y., Zhuang S., Wang L. J., Chen S., Gong R. (2013) Adrenocorticotropic hormone ameliorates acute kidney injury by steroidogenic-dependent and -independent mechanisms. Kidney Int. 83, 635–646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Takemoto M., Asker N., Gerhardt H., Lundkvist A., Johansson B. R., Saito Y., Betsholtz C. (2002) A new method for large scale isolation of kidney glomeruli from mice. Am. J. Pathol. 161, 799–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gundersen H. J., Bendtsen T. F., Korbo L., Marcussen N., Møller A., Nielsen K., Nyengaard J. R., Pakkenberg B., Sørensen F. B., Vesterby A. (1988) Some new, simple and efficient stereological methods and their use in pathological research and diagnosis. APMIS 96, 379–394 [DOI] [PubMed] [Google Scholar]
- 44. Kett M. M., Bergström G., Alcorn D., Bertram J. F., Anderson W. P. (2001) Renal vascular resistance properties and glomerular protection in early established SHR hypertension. J. Hypertens. 19, 1505–1512 [DOI] [PubMed] [Google Scholar]
- 45. Nyengaard J. R. (1993) The quantitative development of glomerular capillaries in rats with special reference to unbiased stereological estimates of their number and sizes. Microvasc. Res. 45, 243–261 [DOI] [PubMed] [Google Scholar]
- 46. Bai X. Y., Basgen J. M. (2011) Podocyte number in the maturing rat kidney. Am. J. Nephrol. 33, 91–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cullen-McEwen L. A., Douglas-Denton R. N., Bertram J. F. (2012) Estimating total nephron number in the adult kidney using the physical dissector/fractionator combination. Methods Mol. Biol. 886, 333–350 [DOI] [PubMed] [Google Scholar]
- 48. Mundel P., Reiser J., Zúñiga Mejía Borja A., Pavenstädt H., Davidson G. R., Kriz W., Zeller R. (1997) Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Exp. Cell Res. 236, 248–258 [DOI] [PubMed] [Google Scholar]
- 49. Kreis T. E. (1987) Microtubules containing detyrosinated tubulin are less dynamic. EMBO J. 6, 2597–2606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Gundersen G. G., Kalnoski M. H., Bulinski J. C. (1984) Distinct populations of microtubules: tyrosinated and nontyrosinated α tubulin are distributed differently in vivo. Cell 38, 779–789 [DOI] [PubMed] [Google Scholar]
- 51. Gu Y., Oyama F., Ihara Y. (1996) Tau is widely expressed in rat tissues. J. Neurochem. 67, 1235–1244 [DOI] [PubMed] [Google Scholar]
- 52. Lee V. W., Harris D. C. (2011) Adriamycin nephropathy: a model of focal segmental glomerulosclerosis. Nephrology 16, 30–38 [DOI] [PubMed] [Google Scholar]
- 53. Wang Y., Wang Y. P., Tay Y. C., Harris D. C. (2000) Progressive adriamycin nephropathy in mice: sequence of histologic and immunohistochemical events. Kidney Int. 58, 1797–1804 [DOI] [PubMed] [Google Scholar]
- 54. Barisoni L., Schnaper H. W., Kopp J. B. (2007) A proposed taxonomy for the podocytopathies: a reassessment of the primary nephrotic diseases. Clin. J. Am. Soc. Nephrol. 2, 529–542 [DOI] [PubMed] [Google Scholar]
- 55. Kretzler M. (2005) Role of podocytes in focal sclerosis: defining the point of no return. J. Am. Soc. Nephrol. 16, 2830–2832 [DOI] [PubMed] [Google Scholar]
- 56. Zoja C., Corna D., Camozzi D., Cattaneo D., Rottoli D., Batani C., Zanchi C., Abbate M., Remuzzi G. (2002) How to fully protect the kidney in a severe model of progressive nephropathy: a multidrug approach. J. Am. Soc. Nephrol. 13, 2898–2908 [DOI] [PubMed] [Google Scholar]
- 57. Gong R., Ge Y., Chen S., Liang E., Esparza A., Sabo E., Yango A., Gohh R., Rifai A., Dworkin L. D. (2008) Glycogen synthase kinase 3β: a novel marker and modulator of inflammatory injury in chronic renal allograft disease. Am. J. Transplant. 8, 1852–1863 [DOI] [PubMed] [Google Scholar]
- 58. Waters A., Koziell A. (2009) Activation of canonical Wnt signaling meets with podocytopathy. J. Am. Soc. Nephrol. 20, 1864–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Lin C. L., Wang J. Y., Huang Y. T., Kuo Y. H., Surendran K., Wang F. S. (2006) Wnt/β-catenin signaling modulates survival of high glucose-stressed mesangial cells. J. Am. Soc. Nephrol. 17, 2812–2820 [DOI] [PubMed] [Google Scholar]
- 60. Matsui I., Ito T., Kurihara H., Imai E., Ogihara T., Hori M. (2007) Snail, a transcriptional regulator, represses nephrin expression in glomerular epithelial cells of nephrotic rats. Lab. Invest. 87, 273–283 [DOI] [PubMed] [Google Scholar]
- 61. Dai C., Stolz D. B., Kiss L. P., Monga S. P., Holzman L. B., Liu Y. (2009) Wnt/β-catenin signaling promotes podocyte dysfunction and albuminuria. J. Am. Soc. Nephrol. 20, 1997–2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Boini K. M., Amann K., Kempe D., Alessi D. R., Lang F. (2009) Proteinuria in mice expressing PKB/SGK-resistant GSK3. Am. J. Physiol. Renal Physiol. 296, F153–F159 [DOI] [PubMed] [Google Scholar]
- 63. Meijer L., Flajolet M., Greengard P. (2004) Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol. Sci. 25, 471–480 [DOI] [PubMed] [Google Scholar]
- 64. Marmol F. (2008) Lithium: bipolar disorder and neurodegenerative diseases. Possible cellular mechanisms of the therapeutic effects of lithium. Prog. Neuropsychopharmacol. Biol. Psychiatry 32, 1761–1771 [DOI] [PubMed] [Google Scholar]
- 65. Yang E. S., Wang H., Jiang G., Nowsheen S., Fu A., Hallahan D. E., Xia F. (2009) Lithium-mediated protection of hippocampal cells involves enhancement of DNA-PK-dependent repair in mice. J. Clin. Invest. 119, 1124–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Forlenza O. V., de Paula V. J., Machado-Vieira R., Diniz B. S., Gattaz W. F. (2012) Does lithium prevent Alzheimer's disease? Drugs Aging 29, 335–342 [DOI] [PubMed] [Google Scholar]
- 67. Li B., Chohan M. O., Grundke-Iqbal I., Iqbal K. (2007) Disruption of microtubule network by Alzheimer abnormally hyperphosphorylated Tau. Acta Neuropathol. 113, 501–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Muñoz-Montaño J. R., Lim F., Moreno F. J., Avila J., Díaz-Nido J. (1999) Glycogen synthase kinase-3 modulates neurite outgrowth in cultured neurons: possible implications for neurite pathology in Alzheimer's disease. J. Alzheimers Dis. 1, 361–378 [DOI] [PubMed] [Google Scholar]
- 69. Luippold G., Beilharz M., Mühlbauer B. (2004) Chronic renal denervation prevents glomerular hyperfiltration in diabetic rats. Nephrol. Dial. Transplant. 19, 342–347 [DOI] [PubMed] [Google Scholar]
- 70. Sun L., Zhang D., Liu F., Xiang X., Ling G., Xiao L., Liu Y., Zhu X., Zhan M., Yang Y., Kondeti V. K., Kanwar Y. S. (2011) Low-dose paclitaxel ameliorates fibrosis in the remnant kidney model by down-regulating miR-192. J. Pathol. 225, 364–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Xu W., Ge Y., Liu Z., Gong R. (2014) Microtubule stabilization by taxol promotes podocyte process elongation and enhances compensatory glomerular adaptation to podocyte depletion. J. Am. Soc. Nephrol. 25, 92A24029422 [Google Scholar]
- 72. Bendz H., Schön S., Attman P. O., Aurell M. (2010) Renal failure occurs in chronic lithium treatment but is uncommon. Kidney Int. 77, 219–224 [DOI] [PubMed] [Google Scholar]