

Background: Oligomers of pathogenic proteins are implicated in the pathomechanisms of neurodegenerative diseases.
Results: Depletion of p62 delays the degradation of polyglutamine protein oligomers via autophagy and exacerbates neurodegeneration in polyglutamine disease model flies.
Conclusion: p62 plays a protective role via autophagic degradation of polyglutamine protein oligomers.
Significance: p62 should be a therapeutic target for the polyglutamine diseases.
Keywords: Autophagy, Drosophila, Neurodegenerative Disease, Polyglutamine Disease, Protein Aggregation, Oligomer, p62
Abstract
Oligomer formation and accumulation of pathogenic proteins are key events in the pathomechanisms of many neurodegenerative diseases, such as Alzheimer disease, ALS, and the polyglutamine (polyQ) diseases. The autophagy-lysosome degradation system may have therapeutic potential against these diseases because it can degrade even large oligomers. Although p62/sequestosome 1 plays a physiological role in selective autophagy of ubiquitinated proteins, whether p62 recognizes and degrades pathogenic proteins in neurodegenerative diseases has remained unclear. In this study, to elucidate the role of p62 in such pathogenic conditions in vivo, we used Drosophila models of neurodegenerative diseases. We found that p62 predominantly co-localizes with cytoplasmic polyQ protein aggregates in the MJDtr-Q78 polyQ disease model flies. Loss of p62 function resulted in significant exacerbation of eye degeneration in these flies. Immunohistochemical analyses revealed enhanced accumulation of cytoplasmic aggregates by p62 knockdown in the MJDtr-Q78 flies, similarly to knockdown of autophagy-related genes (Atgs). Knockdown of both p62 and Atgs did not show any additive effects in the MJDtr-Q78 flies, implying that p62 function is mediated by autophagy. Biochemical analyses showed that loss of p62 function delays the degradation of the MJDtr-Q78 protein, especially its oligomeric species. We also found that loss of p62 function exacerbates eye degeneration in another polyQ disease fly model as well as in ALS model flies. We therefore conclude that p62 plays a protective role against polyQ-induced neurodegeneration, by the autophagic degradation of polyQ protein oligomers in vivo, indicating its therapeutic potential for the polyQ diseases and possibly for other neurodegenerative diseases.
Introduction
The polyglutamine (polyQ)2 diseases are inherited intractable neurodegenerative diseases, including Huntington disease, several spinocerebellar ataxias (SCA1, -2, -6, -7, and -17 and SCA3/MJD), dentatorubral-pallidoluysian atrophy, and spinobulbar muscular atrophy, which are caused by the expansion of a CAG repeat encoding for polyQ stretch within specific genes (1). PolyQ proteins are prone to misfold, oligomerize, and form aggregates and eventually accumulate as inclusion bodies in affected neurons (2, 3). Whereas the formation of polyQ protein inclusion bodies is believed to be protective, by sequestering the toxic polyQ proteins (4), the intermediate structures formed during the aggregation process, such as monomers or oligomers, are reported to be more toxic for the cells, leading to neuronal dysfunction or neuronal cell death (5, 6). The polyQ diseases are thus considered as one of the protein-folding diseases, together with Alzheimer disease, Parkinson disease, and ALS. Because there are currently no effective therapies for the polyQ diseases, establishment of a novel therapy based on the disease pathomechanism is a challenging theme. Considering the pathomechanism of the polyQ diseases, the clearance of toxic forms of the polyQ proteins should be a promising therapeutic strategy.
Although the precise mechanisms of how polyQ proteins are degraded in the cell are not clearly understood, the two major cellular degradation systems (i.e. the autophagy-lysosome system and the ubiquitin-proteasome system (UPS)) are both thought to be involved in polyQ protein degradation (7). However, the UPS may be inadequate for degrading polyQ protein oligomers or aggregates, because substrate proteins of the UPS need to be unfolded when entering the narrow proteasomal pore (8). Furthermore, the mammalian UPS might not have a protease activity to efficiently degrade the polyQ stretch (9, 10). Alternatively, autophagy can degrade even large aggregates by sequestering and delivering them to the lysosome (11). Although autophagy was considered a non-selective degradation system in the past, emerging evidence suggests that it can specifically degrade some ubiquitinated proteins, organelles, and intracellular pathogens; this is now known as “selective autophagy” (12). The specific autophagic degradation of polyQ proteins, including large sized aggregates, would be a preferable therapeutic strategy, because nonspecific degradation of cytosolic proteins may cause adverse effects due to the loss of normal protein functions.
The p62/sequestosome 1 protein (hereafter called p62) was initially identified as an adaptor molecule for the selective autophagic degradation of ubiquitinated proteins, because p62 has domains that bind both ubiquitinated proteins and autophagosomes, giving selectivity to autophagy (13, 14). Neuropathological studies revealed that p62 co-localizes with ubiquitin-positive inclusions consisting of disease-causative proteins within neurons and glia of patients with various neurodegenerative diseases (15, 16). This evidence suggests that p62 is associated with various abnormal proteins, including the polyQ protein. However, whether p62 recognizes these pathogenic proteins as substrates for p62-associated selective autophagy has remained unclear.
In this study, we explored the role of p62 in the polyQ diseases, using Drosophila polyQ disease models. We demonstrated that p62 plays an important role in the autophagic degradation of polyQ protein oligomers, resulting in protection against polyQ protein toxicity in vivo. Furthermore, we demonstrated the protective role of p62 in various neurodegenerative disease models, indicating that p62 could be a therapeutic target for various neurodegenerative diseases.
EXPERIMENTAL PROCEDURES
Fly Stocks
Flies were raised and maintained on standard cornmeal-agar-yeast-based food at 25 °C. The transgenic fly lines bearing the gmr-GAL4 (17) or UAS-human TDP-433 transgene have been described previously. The transgenic fly lines bearing the gmr-GeneSwitch or gmr-grim transgene and the mutant fly line bearing the Atg600096 mutation were obtained from the Bloomington Drosophila Stock Center. The transgenic fly lines bearing the UAS-MJDtr-Q78 and UAS-MJDtr-Q27 transgene were gifts from Drs. N. M. Bonini (18), and the UAS-Httex1p97QP (19), UAS-Aβ arc2 (20), and UAS-R406W tau (21) transgene were gifts from J. L. Marsh, D. C. Crowther, and M. B. Feany, respectively. The mutant fly lines bearing a ref(2)P mutation, namely ref(2)Pod2 or ref(2)Pod3, were described previously (22). The RNAi fly lines bearing the UAS-ref(2)P-IR, UAS-Atg12-IR, UAS-alfy-IR, or UAS-Prosβ2-IR transgene were obtained from the Vienna Drosophila Resource Center.
Fly Eye Imaging
Light microscopic images were taken using a stereoscopic microscope model SZX10 (Olympus, Tokyo, Japan) with a CCD camera (PD21, Olympus, Tokyo, Japan). Scanning electron microscopic (SEM) images were taken using an electron microscope (model TM1000, Hitachi, Tokyo, Japan).
Calculation of Eye Pigmentation Score
The eye images of adult flies were obtained. To quantitatively evaluate the degree of eye degeneration in the MJDtr-Q78 flies, the area of remaining normal pigment in their eyes were measured using the National Institutes of Health ImageJ software as follows: 1) extraction of the green color to produce grayscale images and determination of the area of compound eye as the region of interest (Fig. 2, Q and R); 2) smoothing of the images by Gaussian blur (Fig. 2S), production of binary images, and adjustment of the threshold of the binary images to determine the area of remaining normal pigment in the eyes (Fig. 2T); and 3) calculation of the mean area of remaining normal pigment within the region of interest. For the TDP-43 flies, the region of interest was determined in the anterior half of the eye to avoid the necrotic tissues appeared in the posterior half because the necrotic tissues could be misjudged as normal pigment. More than four eyes were analyzed in each experiment.
FIGURE 2.

Loss of p62 function causes exacerbation of eye degeneration in MJDtr-Q78 flies. A and B, Western blot analysis of the monomeric MJDtr-Q78 protein in larval eye disc lysates of the MJDtr-Q78S flies (MJDs) and the MJDtr-Q78W (MJDw) flies expressing the MJDtr-Q78 protein under the gmr-GAL4 driver, using an anti-HA antibody to detect the MJDtr-Q78 protein. The expression level of actin was used as a protein-loading control. The graph shows the relative ratio of the MJDtr-Q78 protein to actin. The relative amount of each protein was measured by densitometric analysis of the bands in A. Data are presented as the mean ± S.E. (**, p < 0.01, versus the MJDw flies) (n = 3). Fly genotypes were gmr-GAL4/+;;UAS-MJDtr-Q78W/+ and gmr-GAL4/+;UAS-MJDtr-Q78S/+. C and D, knockdown efficiency of RNAi-mediated knockdown of ref(2)P, the Drosophila ortholog of the p62 gene. The p62 protein in lysates prepared from adult fly heads expressing an IR targeted to ref(2)P/p62 under the tub-GAL4 driver was detected with the anti-Ref(2)P/p62 antibody by Western blot analysis. The expression level of actin was used as a protein-loading control. The graph shows the relative ratio of p62 protein to actin. The relative amount of each protein was measured by densitometric analyses of the bands in C. Data are presented as the mean ± S.E. (**, p < 0.01, versus the control flies expressing the GAL4 protein alone) (n = 3). Fly genotypes were tub-GAL4/+ and UAS-p62-IR/+;tub-GAL4/+. E–P, light microscopic images of the external compound eyes of 1-day-old adult flies of two different MJDtr-Q78 fly lines, MJDtr-Q78S flies (E, MJDs) and MJDtr-Q78W flies (I, MJDw), expressing p62-IR (F and J) or bearing p62 mutations, namely, ref(2)Pod2 (G and K) and ref(2)Pod3 (H and L), and the control MJDtr-Q27 flies (M–P). Note that the p62 mutant flies also possess a wild-type p62 allelle in trans to the mutant allele. Fly genotypes were as follows: gmr-GAL4/+;UAS-MJDtr-Q78S/+ (E); gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-p62-IR (F); gmr-GAL4/+;UAS-MJDtr-Q78S/ref(2)Pod2 (G); gmr-GAL4/+;UAS-MJDtr-Q78S/ref(2)Pod3 (H); gmr-GAL4/+;;UAS-MJDtr-Q78W/+ (I); gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q78W/+ (J); gmr-GAL4/+;ref(2)Pod2/+;UAS-MJDtr-Q78W/+ (K); gmr-GAL4/+;ref(2)Pod3/+;UAS-MJDtr-Q78W/+ (L); gmr-GAL4/+;;UAS-MJDtr-Q27/+ (M); gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q27/+ (N); gmr-GAL4/+;ref(2)Pod2/+;UAS-MJDtr-Q27/+ (O); and gmr-GAL4/+;ref(2)Pod3/+;UAS-MJDtr-Q27/+ (P). Q–T, calculation of eye pigmentation score. Shown are light microscopic images of the external compound eye of an MJDtr-Q78W fly (Q; MJDw), the grayscale images extracted from Q (R), the smoothed image of R (S), and the binary image of R (T). Red outlines show the region of interest defined to evaluate the eye pigmentation. Note that the images shown here are representative images to explain this procedure clearly. The fly genotype used was gmr-GAL4/+;;UAS-MJDtr-Q78W/+. U and V, quantitative imaging analyses of eye pigmentation in the 1-day-old adult MJDtr-Q78W flies (U) or the control 1-day-old adult MJDtr-Q27 flies (V) expressing p62-IR alone or bearing p62 mutations, namely ref(2)Pod2 and ref(2)Pod3. More than four eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (**, p < 0.01; ***, p < 0.001; n.s., not significant, versus the MJDtr-Q78W flies in U and the MJDtr-Q27 flies in V, respectively). W and X, light microscopic images of the external compound eyes of 1-day-old adult flies expressing the Grim protein with or without p62 knockdown. Fly genotypes were as follows: gmr-GAL4/+;gmr-grim/+ (W) and gmr-GAL4/+;gmr-grim/UAS-p62-IR (X). Y, quantitative imaging analyses of eye pigmentation in the 1-day-old adult grim flies with or without expressing p62-IR. More than five eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars) (n.s., not significant, versus the grim flies).
Calculation of the Number of Interommatidial Bristles
The SEM images of adult fly eyes were obtained. The number of interommatidial bristles within a 150-μm2 area in the eye was counted (23). Seven eyes were analyzed in each genotype.
Calculation of Eye Size
The light microscopic images of adult fly eyes were obtained. The eye size was measured using ImageJ software (24). Ten eyes were analyzed in each genotype.
Immunohistochemistry
Eye discs were dissected from third instar larvae, fixed with 4% paraformaldehyde, and then immunostained with a rat monoclonal anti-HA antibody (clone 3F10, Roche Applied Science), a rabbit polyclonal anti-Ref(2)P/p62 protein antibody (22), or a mouse monoclonal anti-elav antibody (clone 9F8A9, Developmental Studies Hybridoma Bank, Iowa City, IA) at 1:200 dilution as the primary antibody. As the secondary antibody, an Alexa 568-conjugated anti-rat antibody, an Alexa 488-conjugated anti-rabbit antibody, or an Alexa 488-conjugated anti-mouse antibody was used at 1:1000 dilution. Nuclei were stained using DAPI (Bio-Rad) after the secondary antibody staining. An Alexa 647-conjugated wheat germ agglutinin (Molecular Probes, Inc., Eugene, OR) staining to define the nuclear membrane was performed after DAPI staining at 1:500 dilution. Images were then taken by confocal laser-scanning microscopy (FV1000, Olympus, Tokyo, Japan). The number of MJDtr-Q78 protein aggregates in the eye discs was quantitatively measured using the FV10-ASW 2.0 Viewer software (Olympus, Tokyo, Japan), as follows: 1) selection of photoreceptor neurons within the 13 developing ommatidia in row 2 and row 3 at the posterior tip of the eye discs, by anti-elav staining (Fig. 4, E and F), because these ommatidia are in approximately the same developing stage and can be easily identified; 2) counting of the number of MJDtr-Q78 protein aggregates localized in either the cytoplasm or the nucleus, judged by whether they merge with the nuclei stained with DAPI or not (Fig. 4, G and H). More than five eye discs were analyzed in each experiment.
FIGURE 4.

Loss of p62 function results in an increase in cytoplasmic MJDtr-Q78 protein aggregates. A–D, confocal microscopic images of the larval eye discs of flies expressing the MJDtr-Q78 protein alone (A and B, MJDs) or co-expressing p62-IR (C and D), stained with an anti-HA antibody to detect the MJDtr-Q78 protein (red) and DAPI for nuclear staining (blue). B and D, high magnification images of the indicated areas of A and C, respectively. The arrows and arrowheads indicate cytoplasmic and nuclear MJDtr-Q78 protein aggregates, respectively. Bars, 10 μm. Fly genotypes were as follows: gmr-GAL4/+;UAS-MJDtr-Q78S/+ (A and B) and gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-p62-IR (C and D). E–H, quantitative analyses of MJDtr-Q78 protein aggregates in the eye discs. Confocal microscopic images of an MJDtr-Q78S fly (MJDs) larval eye disc stained with an anti-HA antibody to detect the MJDtr-Q78 protein (red) and with an anti-elav antibody to detect the photoreceptor neurons (E and F, green). Nuclei were stained with DAPI (G and H, blue). Thirteen ommatidia in row 2 and row 3 at the posterior tip of the eye discs were selected (yellow outlines). F and H, high magnification images of the indicated areas of E and G, respectively. The MJDtr-Q78 protein aggregates were counted as either cytoplasmic aggregates (arrow) or nuclear aggregates (arrowhead), depending on whether they merged with the nucleus or not. The fly genotype used was gmr-GAL4/+;UAS-MJDtr-Q78S/+. Bars, 10 μm. I–K, the number of MJDtr-Q78 protein aggregates in the eye discs of MJDtr-Q78S flies with or without expression of p62-IR. The total numbers of MJDtr-Q78 protein aggregates (I), cytoplasmic aggregates (J), and nuclear aggregates (K) are presented. More than five eye discs were analyzed for both genotypes. Data are presented as the mean ± S.E. (error bars) (**, p < 0.01; ***, p < 0.001, versus the MJDtr-Q78S flies). L, the -fold change in the number of cytoplasmic or nuclear MJDtr-Q78 protein aggregates by p62 knockdown.
Western Blot Analysis
Five heads of adult flies or 20 eye discs of larvae were lysed in 100 μl of SDS sample buffer using a pestle, sonicated, boiled for 5 min, and centrifuged at 10,000 × g for 3 min at 25 °C. The supernatants were run on a 5–20% gradient polyacrylamide gel (Wako, Osaka, Japan) and then transferred onto an Immun-Blot PVDF membrane (Bio-Rad). The membrane was blocked with 5% skim milk in PBS containing 0.1% Tween 20 for 30 min at room temperature and then incubated overnight with a rat monoclonal anti-HA antibody (clone 3F10, Roche Applied Science), a rabbit polyclonal anti-Ref(2)P/p62 antibody (22), or a mouse monoclonal anti-actin antibody (clone AC-40, Sigma-Aldrich) at 1:1000 dilution as primary antibody. After overnight incubation, the membranes were incubated with HRP-conjugated secondary antibodies. Membranes were then treated with SuperSignal West Dura chemiluminescent substrate (Thermo Fisher Scientific), and images were taken by the LAS-4000 imaging system (Fujifilm, Tokyo, Japan). Quantification of each signal was performed using the MultiGauge software (Fujifilm).
SDS-Agarose Gel Electrophoresis (SDS-AGE)
SDS-AGE was performed according to the previous reports (25–27). Briefly, adult fly head lysates were run on a 1.5% agarose, 0.1% SDS gel and then transferred onto a nitrocellulose membrane (Schleicher & Schuell BioScience). Blocking with skim milk, reaction with the primary or secondary antibody, and detection were done with the same protocols as Western blot analysis.
Gene Switch Protocol
RU486 (mifepristone, Sigma-Aldrich) was dissolved in 100% ethanol, further diluted in water, and then mixed with Instant Drosophila medium at a final concentration of 10 μg/ml (Carolina Biological Supply Company, Burlington, NC). For RU486 treatment, flies were in RU486-containing medium from the larval stage until adulthood.
Statistical Analyses
For comparisons between two groups, statistical differences were analyzed by Student's t test. Data are presented as the mean ± S.E. A p value of <0.05 was considered to indicate a statistically significant difference between groups.
RESULTS
p62 Co-localizes with Cytoplasmic PolyQ Protein Aggregates
To evaluate whether p62 affects the polyQ protein in vivo, we used the MJDtr-Q78S transgenic fly line, which expresses a truncated form of the mutant MJD protein with an expanded Gln-78 repeat (MJDtr-Q78). As a control, we used the MJDtr-Q27 transgenic fly line, which expresses a truncated form of the MJD protein with a normal-length Gln-27 repeat (MJDtr-Q27). The MJDtr-Q78S flies showed severe compound eye degeneration when the MJDtr-Q78 protein was selectively expressed in the eye by the gmr-GAL4 driver, as revealed by light microscopic analyses (18) (Fig. 1B). On the contrary, the MJDtr-Q27 flies did not show any eye degenerative phenotypes (Fig. 1A). In these MJDtr-Q78S flies, the MJDtr-Q78 protein was found to accumulate as aggregates in both the cytoplasm and nucleus, although nuclear aggregates were more abundant than cytoplasmic aggregates (Fig. 1, I and J). To evaluate the relationship between p62 and the MJDtr-Q78 protein, we performed immunohistochemical analyses of larval eye discs. In the MJDtr-Q27 flies, p62 was predominantly present in the cytoplasm (Fig. 1, C–H), and punctate dotlike structures were also observed, which are known as “p62 bodies” (28) (Fig. 1F, open arrows). On the other hand, in the MJDtr-Q78S flies, p62 co-localized with cytoplasmic MJDtr-Q78 protein aggregates but not with nuclear aggregates (Fig. 1, I–N), although most MJDtr-Q78 protein aggregates were present in the nucleus. Wheat germ agglutinin staining to define the nuclear membrane revealed that p62-positive MJDtr-Q78 protein aggregates are present in the cytoplasm, and p62-negative MJDtr-Q78 protein aggregates are present in the nucleus (Fig. 1, O–Q). The size of p62-positive MJDtr-Q78 protein aggregates was much larger than that of the p62 bodies seen in the control flies, indicating that these p62-positive MJDtr-Q78 protein aggregates are different from the p62 bodies (Fig. 1, F and L). These results suggest that p62 is associated with the MJDtr-Q78 protein, especially with its cytoplasmic aggregates.
FIGURE 1.

p62 co-localizes with cytoplasmic aggregates of the MJDtr-Q78 protein. A and B, light microscopic images of the external compound eyes of the control 1-day-old adult MJDtr-Q27 flies (A) and MJDtr-Q78S flies (MJDs) (B). C–N, confocal microscopic images of the larval eye discs of the control MJDtr-Q27 flies (C–H) and MJDtr-Q78S flies (I–Q), stained with an anti-HA antibody to detect the MJDtr-Q27 or MJDtr-Q78 protein (red), an anti-Ref(2)P/p62 antibody (white in E, F, K, and L and green in G, H, M, N, and O), DAPI for nuclear staining (blue), and wheat germ agglutinin to define the nuclear membrane (white in P and green in Q). D, F, H, J, L, and N, high magnification images of the indicated areas of C, E, G, I, K, and M, respectively. The white arrows and arrowheads indicate cytoplasmic and nuclear MJDtr-Q78 protein aggregates, respectively, and the open arrows indicate punctate dotlike structures of p62. Bars, 10 μm. Fly genotypes were as follows: gmr-GAL4/+;;UAS-MJDtr-Q27/+ (A and C–H) and gmr-GAL4/+;UAS-MJDtr-Q78S/+ (B and I–Q).
Loss of p62 Function Causes Exacerbation of Eye Degeneration in PolyQ Disease Model Flies
To evaluate the role of p62 in the pathomechanisms of the polyQ disease model flies, we examined the effect of the loss of p62 function on eye degeneration in two different MJDtr-Q78 fly lines: MJDtr-Q78S and MJDtr-Q78W flies. The latter flies express a lower expression level of the MJDtr-Q78 protein and show milder eye degeneration than the MJDtr-Q78S flies (Fig. 2, A, B, E, and I). We used a transgenic RNAi fly line that expresses an inverted repeat RNA (IR) of ref(2)P, the Drosophila ortholog of the p62 gene, and two different mutant fly lines of p62: ref(2)Pod2, bearing a deletion of the Phox and Bem1p (PB1) domain, and ref(2)Pod3, bearing a deletion of the ubiquitin-associated domain (22). We confirmed the efficient knockdown of p62 protein expression when p62-IR is expressed throughout the whole fly body by the tub-GAL4 driver (Fig. 2, C and D). When we crossed the MJDtr-Q78S flies with the p62-IR flies or p62 mutant flies, we found that p62 knockdown or p62 mutations significantly aggravate the compound eye degeneration of the MJDtr-Q78S flies, resulting in more severe depigmentation and the appearance of necrotic tissue (Fig. 2, E–H). Knockdown of p62 caused more severe eye degeneration than p62 mutations, probably because p62 mutant flies also possess a wild-type p62 allele in trans to the mutant allele. Next, we examined the effect of loss of p62 function in the MJDtr-Q78W fly line. Knockdown of p62 or p62 mutations caused the exacerbation of eye depigmentation (Fig. 2, I–L), suggesting a protective role of p62 against polyQ-induced eye degeneration. Upon quantification of the eye pigmentation by imaging analyses (Fig. 2, Q–T), the exacerbation of eye depigmentation by p62 knockdown or p62 mutations was statistically significant (Fig. 2U). The exacerbation by loss of p62 function became even more evident upon the age-related progression of eye degeneration (Fig. 3). We confirmed that either p62 knockdown or p62 mutation does not cause any deleterious effects on the eyes of the flies expressing the MJDtr-Q27 protein (Fig. 2, M–P and V) or expressing the GAL4 protein alone (data not shown), excluding the possibility that the exacerbation of eye degeneration could be a simple additive effect. Furthermore, to exclude the possibility that p62 generally affects any degeneration regardless of the cause, we examined the effect of p62 knockdown on the flies expressing Grim, which causes eye degeneration by apoptosis (Fig. 2W). We found that eye degeneration in the grim flies is not affected by p62 knockdown (Fig. 2, W–Y), suggesting that the protective role of p62 is specific to polyQ protein toxicity.
FIGURE 3.
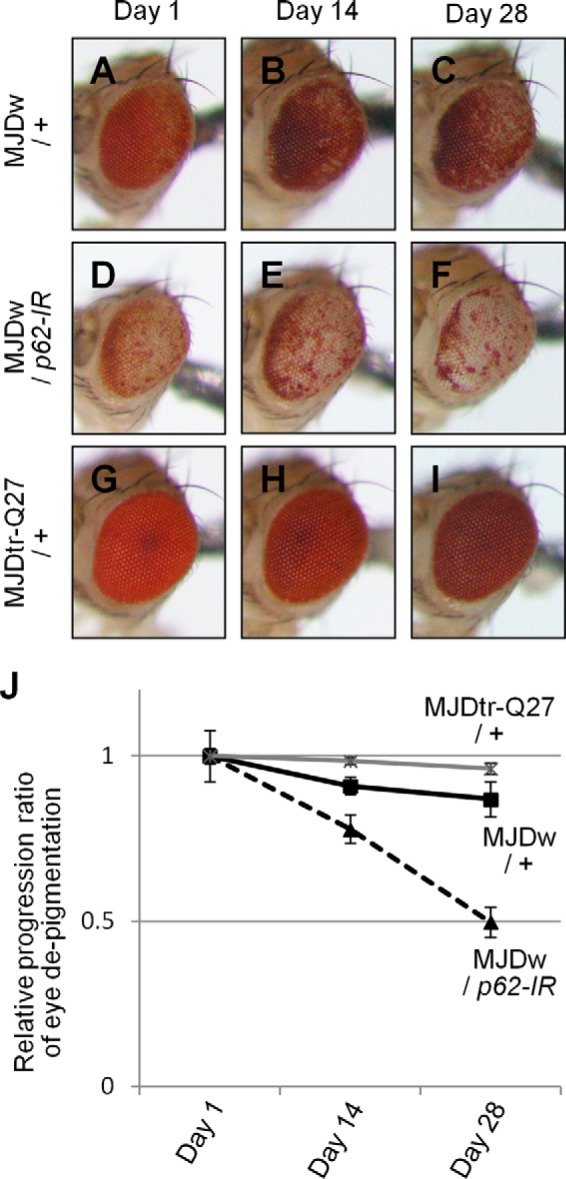
The effect of p62 knockdown on age-related progression of eye degeneration in the MJDtr-Q78W flies. A–F, light microscopic images of the external compound eyes of 1-, 14-, and 28-day-old adult flies expressing the MJDtr-Q78 protein alone (A–C, MJDw/+) or co-expressing p62-IR (D–F, MJDw/p62-IR) and the control MJDtr-Q27 flies (G–I, MJDtr-Q27/+). J, relative progression ratio of eye depigmentation by performing quantitative imaging analyses of eye pigmentation in the MJDw flies with or without expressing p62-IR and the MJDtr-Q27 flies, respectively. More than five eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars). Fly genotypes were as follows: gmr-GAL4/+;;UAS-MJDtr-Q78W/+ (A–C); gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q78W/+ (D–F); and gmr-GAL4/+;;UAS-MJDtr-Q27/+ (G–I).
Loss of p62 Function Results in an Increase in Cytoplasmic PolyQ Protein Aggregates
To clarify the mechanisms underlying the protective role of p62 against polyQ protein toxicity, we evaluated the effect of p62 knockdown on MJDtr-Q78 protein aggregates in the MJDtr-Q78S flies. Immunohistochemical analyses revealed that p62 knockdown results in a robust increase in MJDtr-Q78 protein aggregates, especially those in the cytoplasm, compared with the control MJDtr-Q78S flies (Fig. 4, A–D). Upon quantification of the number of MJDtr-Q78 protein aggregates by imaging analyses (Fig. 4, E–H), we found that the increase in the number of MJDtr-Q78 protein aggregates by p62 knockdown is statistically significant (Fig. 4, I–K), but the -fold change in the number of cytoplasmic aggregates was much higher than the -fold change in the number of nuclear aggregates (Fig. 4L). These results are consistent with the observation that p62 is predominantly localized in the cytoplasm and co-localizes with cytoplasmic MJDtr-Q78 protein aggregates (Fig. 1). These results suggest that the exacerbation of eye degeneration by p62 knockdown in the MJDtr-Q78S flies is accompanied by the enhanced accumulation of cytoplasmic MJDtr-Q78 protein aggregates.
Loss of Autophagic Function Causes the Exacerbation of Eye Degeneration Accompanied by the Enhanced Accumulation of Cytoplasmic PolyQ Protein Aggregates
To evaluate whether the intrinsic protein degradation systems are involved in the toxicity of the polyQ protein, we examined the effect of both autophagic deficiency and UPS deficiency in the MJDtr-Q78 flies. For this purpose, we used the RNAi fly lines that express IRs of Atg12 (autophagy-related gene 12), alfy (29), and Prosβ2 (proteasome β2 subunit) and a mutant fly line of Atg6 (Atg600096). We found that knockdown of the autophagy-related genes Atg12 and alfy and the Atg6 mutation result in significant exacerbation of eye degeneration in both the MJDtr-Q78S and MJDtr-Q78W flies (Fig. 5, A–D, F–I, and P). We also found that Prosβ2 knockdown results in more severe exacerbation of the phenotypes of the MJDtr-Q78 flies, showing a lethal phenotype in the MJDtr-Q78S flies and more severe depigmentation in the MJDtr-Q78W flies (Fig. 5, E, J, and P). We confirmed that knockdown of Atg12, alfy, or Prosβ2 or the Atg6 mutation does not cause any deleterious effects on the eyes of the flies expressing the MJDtr-Q27 protein (Fig. 5, K–O and Q) or expressing the GAL4 protein alone (data not shown), excluding the possibility that the exacerbation of eye degeneration could be a simple additive effect.
FIGURE 5.
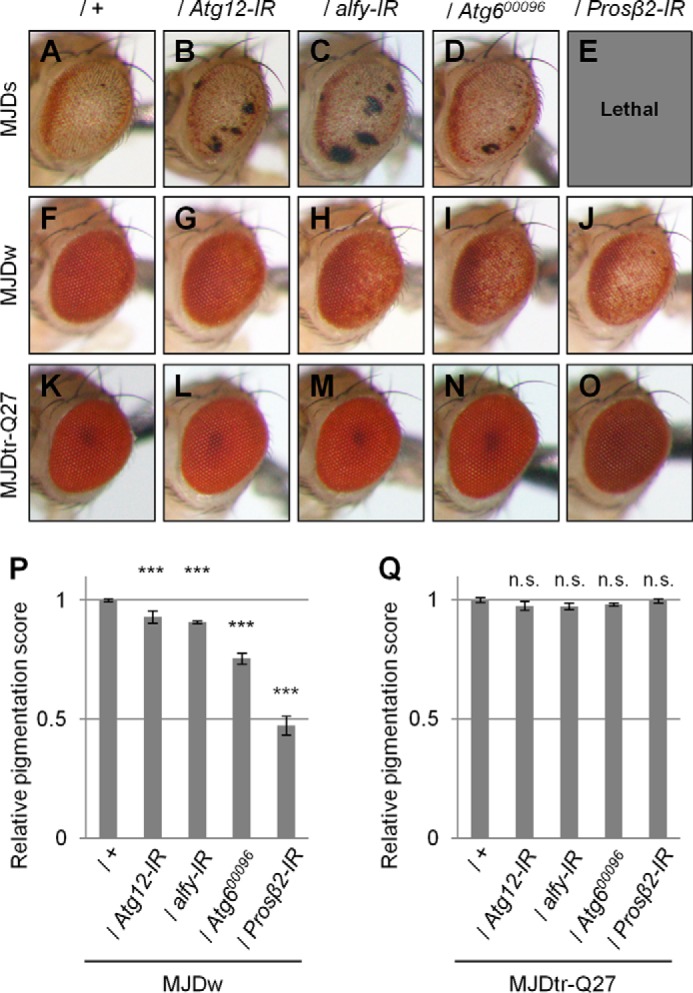
Loss of autophagic or proteasomal function causes exacerbation of eye degeneration in MJDtr-Q78 flies. A–O, light microscopic images of the external compound eyes of 1-day-old adult flies of two different MJDtr-Q78 fly lines, MJDtr-Q78S flies (A, MJDs) and MJDtr-Q78W flies (F, MJDw); MJDtr-Q78 flies co-expressing Atg12-IR (B and G), alfy-IR (C and H), or the proteasome β2 subunit (Prosβ2)-IR (E and J) or bearing an Atg6 mutation (Atg600096) (D and I); and the control 1-day-old adult MJDtr-Q27 flies (K–O). Note that Prosβ2 knockdown showed a lethal phenotype in the MJDtr-Q78S flies. Fly genotypes were as follows: gmr-GAL4/+;UAS-MJDtr-Q78S/+ (A); gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-Atg12-IR (B); gmr-GAL4/+;UAS-MJDtr-Q78S/alfy-IR (C); gmr-GAL4/+;UAS-MJDtr-Q78S/+;Atg600096/+ (D); gmr-GAL4/+;UAS-MJDtr-Q78S/+;Prosβ2-IR/+ (E); gmr-GAL4/+;;UAS-MJDtr-Q78W/+ (F); gmr-GAL4/+;UAS-Atg12-IR/+;UAS-MJDtr-Q78W/+ (G); gmr-GAL4/+;UAS-alfy-IR/+;UAS-MJDtr-Q78W/+ (H); gmr-GAL4/+;;UAS-MJDtr-Q78W/Atg600096 (I); gmr-GAL4/+;;UAS-MJDtr-Q78W/Prosβ2-IR (J); gmr-GAL4/+;;UAS-MJDtr-Q27/+ (K); gmr-GAL4/+;UAS-Atg12-IR/+;UAS-MJDtr-Q27/+ (L); gmr-GAL4/+;alfy-IR/+;UAS-MJDtr-Q27/+ (M); gmr-GAL4/+;;UAS-MJDtr-Q27/Atg600096 (N); and gmr-GAL4/+;;UAS-MJDtr-Q27/Prosβ2-IR (O). P and Q, quantitative imaging analyses of eye pigmentation in the 1-day-old adult MJDtr-Q78W flies (P) or the control 1-day-old adult MJDtr-Q27 flies (Q) expressing Atg12-IR, alfy-IR, or Prosβ2-IR or bearing an Atg6 mutation. More than four eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars) (***, p < 0.001; n.s., not significant, versus the MJDtr-Q78W flies in P and versus the MJDtr-Q27 flies in Q, respectively).
We next evaluated the effects of autophagic or UPS function on MJDtr-Q78 protein aggregates. We found that Atg12 knockdown significantly increases the number of MJDtr-Q78 protein aggregates, especially those in the cytoplasm, similarly to p62 knockdown (Fig. 6, A–D and G–J). In contrast, Prosβ2 knockdown resulted in a significant increase in MJDtr-Q78 protein aggregates only in the nucleus (Fig. 6, E–J). These results are consistent with the fact that protein degradation by autophagy takes place in the cytoplasm. These results imply that p62 plays a role similar to autophagy in the suppression of cytoplasmic polyQ protein aggregate formation.
FIGURE 6.
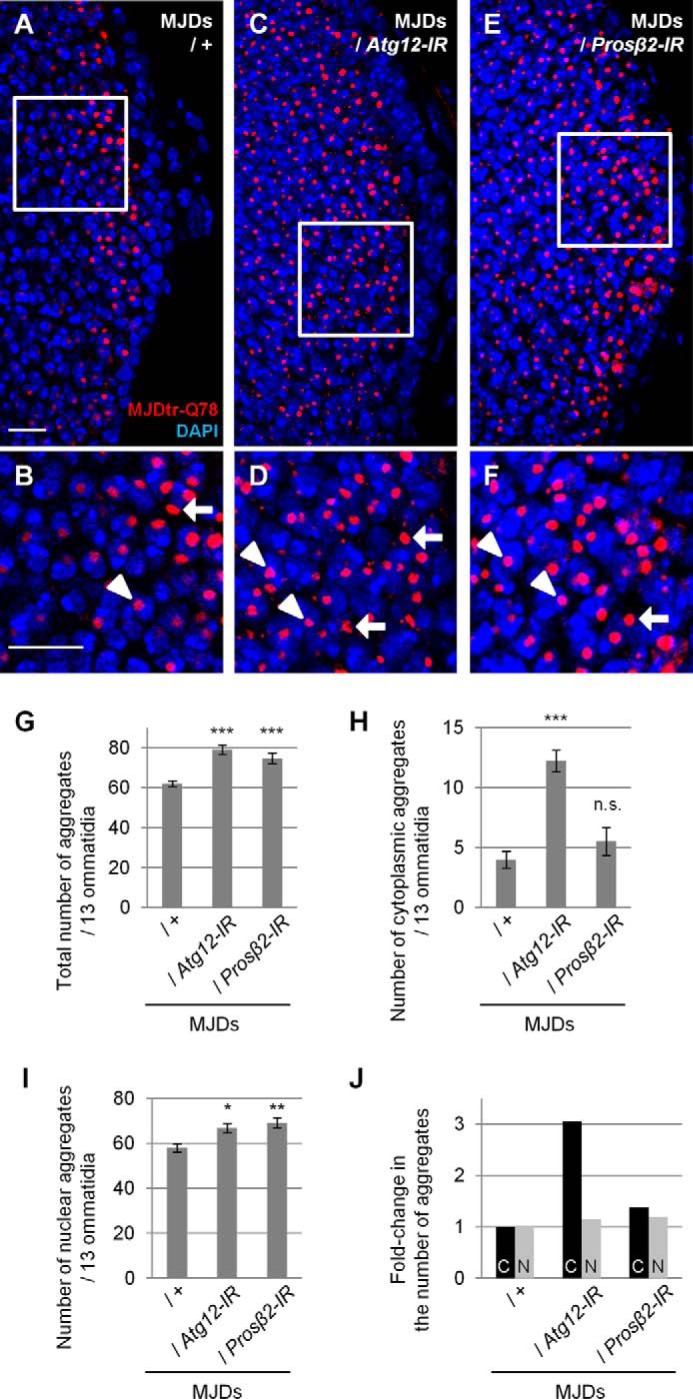
Loss of autophagic function results in an increase in cytoplasmic MJDtr-Q78 protein aggregates. A–F, confocal microscopic images of larval eye discs of flies expressing the MJDtr-Q78S protein alone (A and B, MJDs) or co-expressing either Atg12-IR (C and D) or the proteasome β2 subunit (Prosβ2)-IR (E and F), stained with an anti-HA antibody to detect the MJDtr-Q78 protein (red) and DAPI for nuclear staining (blue). B, D, and F, high magnification images of the indicated areas of A, C, and E, respectively. The arrows and arrowheads indicate cytoplasmic and nuclear MJDtr-Q78 protein aggregates, respectively. Bars, 10 μm. Fly genotypes used were as follows: gmr-GAL4/+;UAS-MJDtr-Q78S/+ (A and B); gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-Atg12-IR (C and D); and gmr-GAL4/+;UAS-MJDtr-Q78S/+;Prosβ2-IR/+ (E and F). G–I, number of MJDtr-Q78 protein aggregates in the eye discs of MJDtr-Q78S flies with or without expression of Atg12-IR or Prosβ2-IR. The total numbers of MJDtr-Q78 protein aggregates (G), cytoplasmic aggregates (H), and nuclear aggregates (I) are presented. More than five eye discs were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars) (*, p < 0.05; **, < 0.01; ***, p < 0.001; n.s., not significant, versus the MJDtr-Q78S flies). J, -fold change in the number of cytoplasmic (C) or nuclear (N) MJDtr-Q78 protein aggregates by knockdown of Atg12 or Prosβ2.
Protective Role of p62 against PolyQ Protein Toxicity Is Dependent on Autophagy
To clarify whether the effects of p62 on polyQ protein toxicity are dependent on autophagy or the UPS, we performed genetic analyses to examine the interaction between p62 and genes involved in these two degradation systems. We crossed MJDtr-Q78W flies co-expressing p62-IR with flies expressing IRs targeted to either autophagy- or UPS-related genes. We found that additional knockdown of either Atg12 or alfy in the MJDtr-Q78W flies together with p62 knockdown does not show any additive effects on eye degeneration compared with that in the MJDtr-Q78W flies with p62 knockdown alone (Fig. 7, A–C and I). On the contrary, additional knockdown of Prosβ2 resulted in further exacerbation of eye degeneration in the MJDtr-Q78W flies with p62 knockdown, as confirmed by quantitative imaging analyses (Fig. 7, D and I). We confirmed that knockdown of Atg12, alfy, or Prosβ2 together with p62 knockdown does not cause any deleterious effects on the eyes of the flies expressing the MJDtr-Q27 protein (Fig. 7, E–H and J) or expressing the GAL4 protein alone (data not shown), excluding the possibility that the exacerbation of eye degeneration could be a simple additive effect. These results suggest that p62 plays a protective role against the toxicity of the MJDtr-Q78 protein via autophagy.
FIGURE 7.
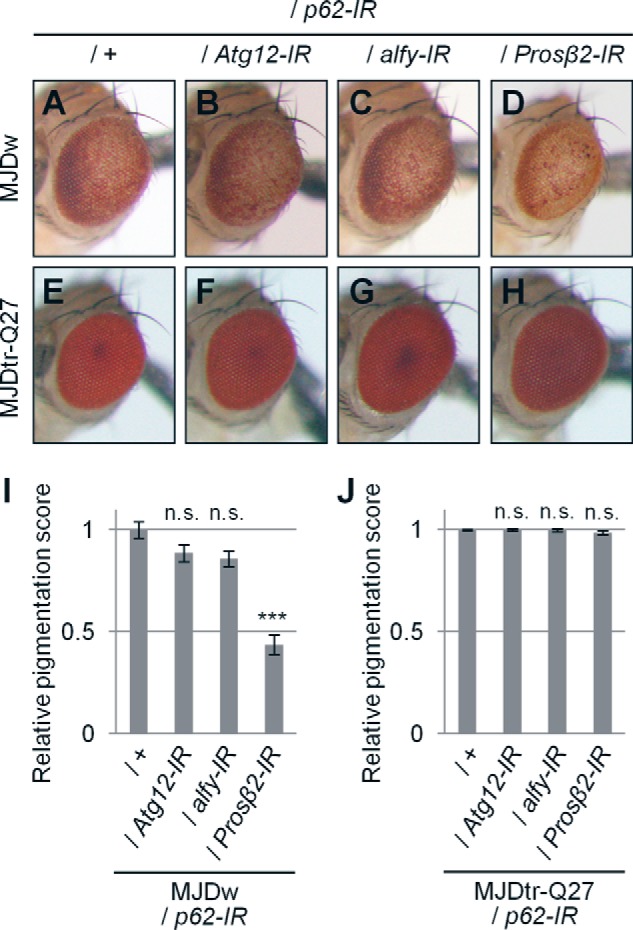
Knockdown of autophagy-related genes in addition to p62 knockdown does not cause additive exacerbation of eye degeneration in MJDtr-Q78W flies. A–H, light microscopic images of the external compound eyes of 1-day-old adult MJDtr-Q78W (MJDw) flies with p62 knockdown alone (A) or together with Atg12 knockdown (B), alfy knockdown (C), or knockdown of Prosβ2 (D) and the control 1-day-old adult MJDtr-Q27 flies (E–H). Fly genotypes used were as follows: gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q78W/+ (A); gmr-GAL4/+;UAS-p62-IR/UAS-Atg12-IR/+;UAS-MJDtr-Q78W/+ (B) gmr-GAL4/+;UAS-p62-IR/UAS-alfy-IR;UAS-MJDtr-Q78W/+ (C); gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q78W/Prosβ2-IR (D); gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q27/+ (E); gmr-GAL4/+;UAS-p62-IR/UAS-Atg12-IR;UAS-MJDtr-Q27/+ (F); gmr-GAL4/+;UAS-p62-IR/alfy-IR;UAS-MJDtr-Q27/+ (G); and gmr-GAL4/+;UAS-p62-IR/+;UAS-MJDtr-Q27/Prosβ2-IR (H). I and J, quantitative imaging analyses of eye pigmentation in the 1-day-old adult MJDtr-Q78W flies (I) or the control 1-day-old adult MJDtr-Q27 flies (J) with p62 knockdown alone or together with Atg12 knockdown, alfy knockdown, or Prosβ2 knockdown. More than five eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars) (***, p < 0.001; n.s., not significant, versus the MJDtr-Q78W flies with p62 knockdown in I and versus the MJDtr-Q27 flies in J, respectively).
Loss of p62 Function Delays the Degradation of the PolyQ Protein in Vivo
To determine the role of p62 in degradation of the polyQ protein, we analyzed the MJDtr-Q78 protein expression level in the MJDtr-Q78S flies with or without p62 knockdown. Western blot analyses of larval eye disc lysates of the MJDtr-Q78S flies constitutively expressing the MJDtr-Q78 protein by the gmr-GAL4 driver revealed that knockdown of neither p62 nor Atg12 affects the MJDtr-Q78 protein expression level (Fig. 8, A and B). Because the MJDtr-Q78 protein expression level is dependent on the equilibrium between its expression and degradation, constitutive expression of the MJDtr-Q78 protein by the gmr-GAL4 driver may make it difficult to detect alterations in MJDtr-Q78 protein turnover. Therefore, to evaluate MJDtr-Q78 protein turnover, we next used the inducible MJDtr-Q78S fly line, namely “ind-MJDtr-Q78S,” which expresses the MJDtr-Q78 protein only in the presence of RU486 (mifepristone) under the control of the gmr-GeneSwitch (gmr-GS) driver (30). To validate this drug-inducible system in vivo, we evaluated the decay of the MJDtr-Q78 protein by Western blot analysis. RU486 was administered orally to ind-MJDtr-Q78 flies during the larval stage to induce MJDtr-Q78 protein expression. After emergence, RU486 was withdrawn to abolish MJDtr-Q78 protein expression. Western blot analyses of lysates prepared from these adult fly heads demonstrated the gradual reduction of MJDtr-Q78 protein levels in the 7-day-old flies compared with the 1-day-old flies (Fig. 8C, +). These results suggest that both the monomeric MJDtr-Q78 protein and its high molecular weight complexes are efficiently degraded during the 6 days after emergence. The ind-MJDtr-Q78S flies without RU486 treatment showed slight leak expression of the MJDtr-Q78 protein. In this condition, we next analyzed the effects of p62 or autophagic deficiency on MJDtr-Q78 protein turnover. We crossed the ind-MJDtr-Q78S flies with p62 or Atg6 mutant flies, which are deficient in their functions independent of RU486 treatment. Western blot analyses showed that loss of p62 function clearly delays the degradation of both monomeric and high molecular weight complexes of the MJDtr-Q78 protein, similarly to loss of Atg6 function (Fig. 8C, top and middle panels). Surprisingly, the amounts of high molecular weight complexes of the MJDtr-Q78 protein were rather increased by the loss of p62 or Atg6 function in the 7-day-old flies, implying that the monomeric MJDtr-Q78 protein that escaped from degradation assembles into oligomers during the 6 days. To further characterize these high molecular weight complexes of the MJDtr-Q78 protein, we performed the SDS-AGE blotting, allowing for the detection of SDS-soluble oligomeric species (25–27). The amounts of the slowly migrating SDS-soluble oligomeric species of the MJDtr-Q78 protein were clearly increased by the loss of p62 or Atg6 function in the 7-day-old flies, consistent with the results of Western blot analyses (Fig. 8C, bottom panel). These results suggest that loss of p62 function impairs autophagic degradation of the MJDtr-Q78 protein, especially its oligomeric species.
FIGURE 8.
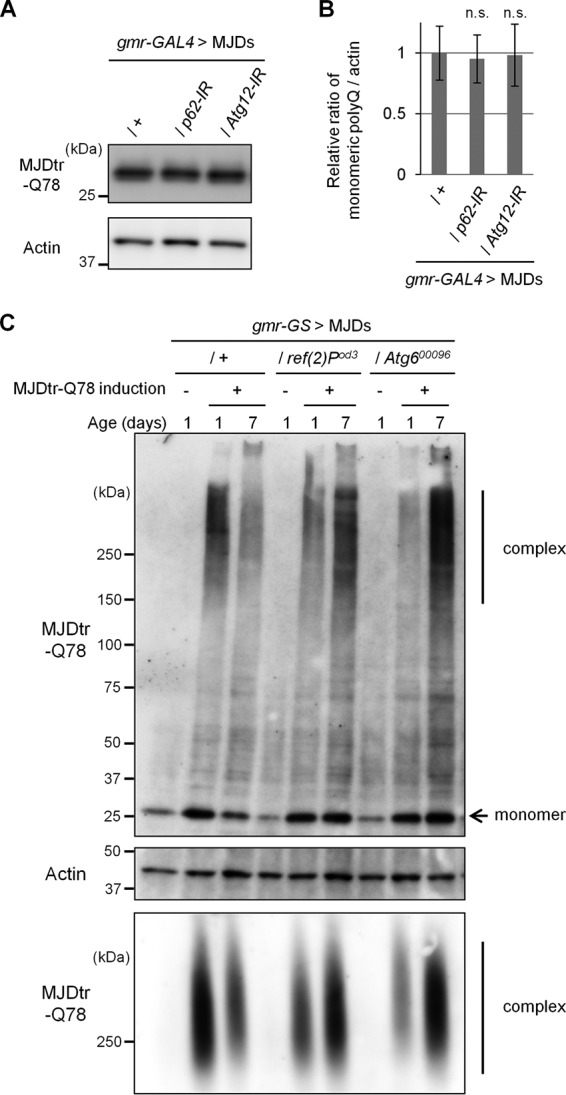
Loss of p62 function delays the degradation of the MJDtr-Q78 protein in vivo. A and B, Western blot analysis of the monomeric MJDtr-Q78 protein in larval eye disc lysates of the MJDtr-Q78S flies (MJDs) expressing the MJDtr-Q78 protein alone (+) or co-expressing either p62-IR or Atg12-IR under the gmr-GAL4 driver, using an anti-HA antibody to detect the MJDtr-Q78 protein. The expression level of actin was used as a protein-loading control. The graph shows the ratio of the MJDtr-Q78 protein to actin. The relative amount of each protein was measured by densitometric analysis of the bands in A. Data are presented as the mean ± S.E. (error bars) (n.s., not significant, versus the MJDtr-Q78S flies) (n = 3–4). Fly genotypes used were gmr-GAL4/+;UAS-MJDtr-Q78S/+, gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-p62-IR, and gmr-GAL4/+;UAS-MJDtr-Q78S/UAS-Atg12-IR. C, Western blot analysis (top and middle panels) and SDS-AGE (bottom panel) of the MJDtr-Q78 protein in lysates prepared from 1-day-old and 7-day-old adult fly heads, expressing the MJDtr-Q78 protein alone (+) or bearing either the p62 mutation (ref(2)Pod3) or Atg6 mutation (Atg600096), under the gmr-GeneSwitch (gmr-GS) driver. MJDtr-Q78 protein expression was induced only during the larval stage by RU486 treatment (10 μg/ml) for the evaluation of MJDtr-Q78 protein turnover. In Western blot analysis, the monomeric MJDtr-Q78 protein (monomer) and its high molecular weight complexes (complex) were detected with an anti-HA antibody. The expression level of actin was used as a protein-loading control. The groups without MJDtr-Q78 protein induction (−) were evaluated for leak MJDtr-Q78 protein expression. In SDS-AGE, the high molecular weight complexes of the MJDtr-Q78 protein were detected with an anti-HA antibody. Fly genotypes used were gmr-GS/+;UAS-MJDtr-Q78S/+, gmr-GS/+;UAS-MJDtr-Q78S/ref(2)Pod3, and gmr-GS/+;UAS-MJDtr-Q78S/+;Atg600096/+.
p62 Plays a Protective Role in Various Neurodegenerative Disease Model Flies
To evaluate whether the protective role of p62 is specific to MJDtr-Q78 flies, we examined the effects of p62 knockdown in various neurodegenerative disease model flies. We used the Huntington disease model flies expressing a mutant huntingtin protein with an expanded Gln-97 repeat (19), the Alzheimer disease model flies expressing the mutant amyloid-β (Aβ) protein (20), the ALS model flies expressing the human TDP-43 protein, and the tauopathy model flies expressing the mutant Tau protein (21). In the huntingtin flies, p62 knockdown resulted in significant exacerbation of eye degeneration, similarly to the MJDtr-Q78 flies (Fig. 9, A–C). Upon quantification of the eye pigmentation by imaging analyses in this model flies, the exacerbation of eye depigmentation by p62 knockdown was statistically significant (Fig. 9D). In the TDP-43 flies, p62 knockdown also resulted in significant exacerbation of eye degeneration (Fig. 9, E–G). Upon quantification of the eye pigmentation by imaging analyses in flies of this model, the exacerbation of eye depigmentation by p62 knockdown was also statistically significant (Fig. 9H). On the contrary, the Aβ flies and the Tau flies did not show exacerbation of eye degeneration by p62 knockdown, as revealed by both light microscopic and SEM analyses, respectively (Fig. 9, I–N and P–U). Upon quantification of the number of interommatidial bristles in the Aβ flies (Fig. 9, L–N) or the eye size in the Tau flies (Fig. 9, P–R), the exacerbation of eye degeneration by p62 knockdown in both flies was not statistically significant (Fig. 9, O and V). These results suggest that p62 exerts protective effects not only against the polyQ protein but also against other proteins associated with neurodegenerative diseases.
FIGURE 9.
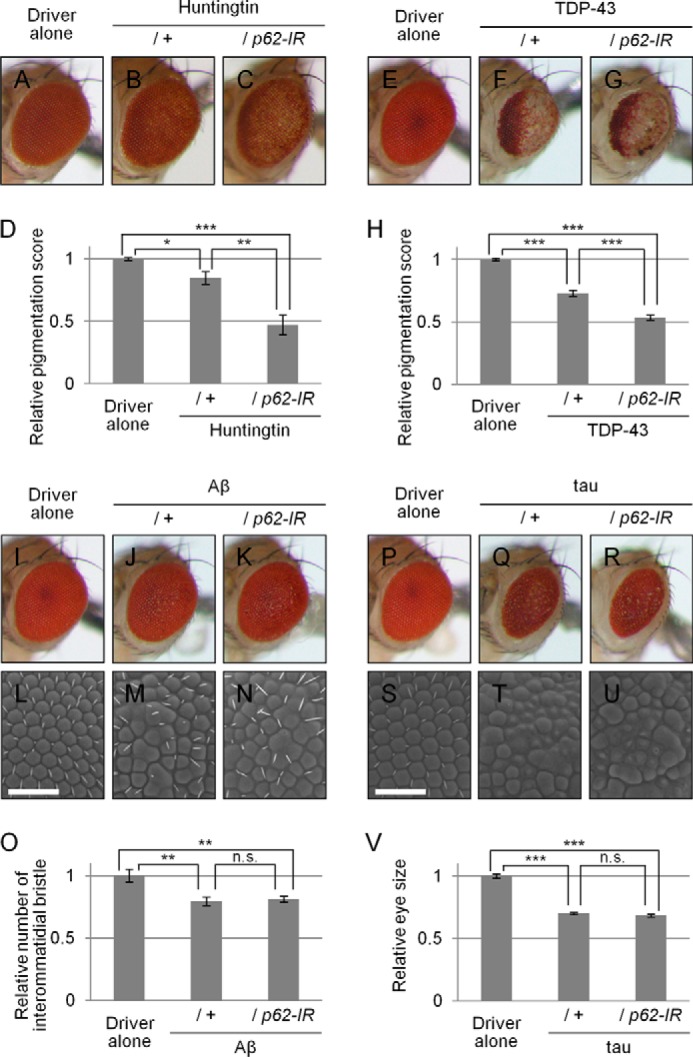
Effects of loss of p62 function in various neurodegenerative disease model flies. A–C, light microscopic images of the external compound eyes of 1-day-old adult flies expressing the mutant huntingtin protein with an expanded Gln-97 repeat with or without p62 knockdown (B and C) and the control flies expressing the GAL4 protein alone (A). Fly genotypes used were as follows: gmr-GAL4/+ (A); gmr-GAL4/+;;UAS-Httex1p97QP/+ (B); gmr-GAL4/+;UAS-p62-IR/+;UAS-Httex1p97QP/+ (C). D, quantitative imaging analyses of eye pigmentation in A–C. More than five eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (error bars) (*, p < 0.05; **, p < 0.01; ***, p < 0.001, versus the control flies expressing the GAL4 protein alone). E–G, light microscopic images of the external compound eyes of 7-day-old adult flies expressing the human TDP-43 protein with or without p62 knockdown (F and G), and the control flies expressing the GAL4 protein alone (E). Fly genotypes used were as follows: gmr-GAL4/+ (E); gmr-GAL4/+;UAS-human TDP-43/+ (F); and gmr-GAL4/+;UAS-human TDP-43/UAS-p62-IR (G). H, quantitative imaging analyses of eye pigmentation in E–G. More than five eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (***, p < 0.001, versus the control flies expressing the GAL4 protein alone). I–N, light microscopic images (I–K) and SEM images (L–N) of the external compound eyes of 1-day-old adult flies expressing the mutant Aβ protein with or without p62 knockdown (J, K, M, and N) and the control flies expressing the GAL4 protein alone (I and L). Bars, 50 μm. Fly genotypes used were as follows: gmr-GAL4/+ (I and L); gmr-GAL4/+;;UAS-Aβ arc2 (J and M); and gmr-GAL4/+;UAS-p62-IR/+;UAS-Aβ arc2/+ (K and N). O, quantitative imaging analyses of the number of the interommatidial bristles in L–N. More than seven eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (**, p < 0.01; n.s., not significant, versus the control flies expressing the GAL4 protein alone). P–U, light microscopic images (P–R) and SEM images (S–U) of the external compound eyes of 1-day-old adult flies expressing the mutant Tau protein with or without p62 knockdown (Q, R, T, and U) and the control flies expressing the GAL4 protein alone (P and S). Bars, 50 μm. Fly genotypes used were as follows: gmr-GAL4/+ (P and S); gmr-GAL4/+;;UAS-R406W tau/+ (Q and T); and gmr-GAL4/+;UAS-p62-IR/+;UAS-R406W tau/+ (R and U). V, quantitative imaging analyses of the eye size in P–R. More than 10 eye images were analyzed for each genotype. Data are presented as the mean ± S.E. (***, p < 0.001; n.s., not significant, versus the control flies expressing the GAL4 protein alone).
DISCUSSION
In the present study, we provide evidence that p62 plays a protective role in the polyQ diseases in vivo. We demonstrated that the loss of p62 function causes a delay in the autophagic degradation of the polyQ protein, resulting in the enhanced accumulation of polyQ protein oligomers and cytoplasmic aggregates, and eventually leads to the exacerbation of the eye degeneration of polyQ disease model flies. We also genetically showed that these functions of p62 are dependent on autophagy. These results suggest that degradation of polyQ protein oligomers is one of the important roles of p62 in protecting against polyQ protein toxicity.
Because p62 is delivered to the autophagosome by interaction with autophagosome membrane light chain 3 via the light chain 3-interacting region of p62 and recognizes ubiquitinated proteins via the ubiquitin-associated domain, p62 was suggested to be engaged in the selective autophagic degradation of ubiquitinated proteins, including the polyQ protein (13, 14). However, there have been substantial controversies regarding the role of p62 in polyQ cell culture models; one study reported that p62 depletion accelerates polyQ-induced cytotoxicity and it is rescued by p62 overexpression (28), whereas another study reported that p62 overexpression accelerates polyQ-induced cytotoxicity (31). In addition, some studies reported that p62 depletion does not affect the formation of polyQ protein aggregates (31–33). In this study, using polyQ disease model flies, we demonstrated that p62 depletion delays the autophagic degradation of the polyQ protein, including its oligomeric species, not merely its monomer. Our results are consistent with a recent report that depletion of p62 induces the accumulation of polyQ proteins in spinobulbar muscular atrophy model mice (34). These results suggest that p62-mediated autophagic degradation of the polyQ protein contributes to the protective effects of p62 against polyQ protein toxicity.
Our study clearly indicates the tight correlation between the amount of polyQ protein oligomers and the degree of neurodegeneration upon the loss of p62 function. Although several studies reported that the amount of polyQ protein inclusions does not correlate with neurodegeneration or that these inclusions are even protective, oligomeric species of misfolded polyQ proteins have been suggested as the principal culprit of neurodegeneration (6, 35). In other protein-misfolding neurodegenerative diseases, soluble oligomers composed of Aβ or α-synuclein have also been shown to exert cytotoxicity, suggesting the intrinsic toxicity of oligomeric structures, regardless of their primary amino acid sequences (36).
Although autophagy is known to act in the cytoplasm, our immunohistochemical analyses showed that p62 depletion also results in a modest increase in nuclear polyQ protein aggregates (Figs. 4 and 6). These results may be a secondary consequence of inefficient autophagic degradation of the polyQ protein, because Atg12 depletion also resulted in a similar consequence. A possible explanation for these results is that inefficient degradation of the polyQ protein in the cytoplasm may also result in an increase of polyQ proteins in the nucleus and their accumulation as nuclear aggregates.
In this study, we showed the protective role of p62 not only in the MJDtr-Q78 flies but also in other neurodegenerative disease flies, such as Huntington disease and TDP-43 proteinopathies (Fig. 9). These results are consistent with the previous reports that huntingtin and TDP-43 are degraded by autophagy (37–40). However, we could not detect the protective effect of p62 in the Aβ flies and the Tau flies. It is known that these Tau-expressing flies do not develop tangles consisting of misfolded Tau proteins (21). Moreover, the Tau protein is reported to be mainly degraded by the UPS rather than autophagy (41–43). In addition, the Aβ peptides are not ubiquitinated in the Alzheimer disease brain, and they are known to be degraded by endopeptidases, such as neprilysin and insulin-degrading enzyme (44, 45). Therefore, p62 could be involved in neurodegeneration in the polyQ and TDP-43 flies but not in the Aβ and Tau flies, although further studies are required to elucidate the mechanisms of the selectivity of p62.
Our study provides new evidence that p62 is involved in the autophagic degradation of the polyQ protein, including its oligomers, in vivo, indicating its therapeutic potential for the polyQ diseases. Considering the selectivity of p62 for ubiquitinated proteins, the next step toward developing a p62-based therapy for the polyQ diseases should be establishing a method for the efficient recognition and degradation of polyQ proteins and possibly other misfolded proteins. A recent study revealed that phosphorylation of p62 at serine 403 renders a higher affinity to the polyubiquitin chain, resulting in the efficient degradation of ubiquitinated proteins (33). Selective and efficient degradation of misfolded proteins, especially oligomers, by p62 is expected to be developed as a general therapeutic strategy against protein-misfolding neurodegenerative diseases in the future.
Acknowledgments
We thank Drs. E. N. Minakawa (National Center of Neurology and Psychiatry), T. Takeuchi (Kyoto University), M. Komatsu (Niigata University), and T. Yoshimori (Osaka University) for helpful discussions; Dr. D. Contamine (deceased; Université Versailles-St Quentin-en-Yvelines) for kindly providing the mutant fly lines ref(2)Pod2 and ref(2)Pod3 and the rabbit polyclonal anti-Ref(2)P/p62 antibody; and the Bloomington Drosophila Stock Center and the Vienna Drosophila Resource Center for the other fly lines. We also thank T. Okada for technical assistance.
This work was supported in part by Grants-in-Aid for Scientific Research (B) (to Y. N.) from the Japan Society for the Promotion of Science (JSPS), Japan; by Grants-in-Aid for Scientific Research on Priority Areas (Proteolysis) (to Y. N.) and on Innovative Areas (Synapse and Neurocircuit Pathology) (to Y. N.) from the Ministry of Education, Culture, Sports, Science, and Technology, Japan; by Health Labor Sciences Research Grants for Research on Development of New Drugs and the Research Committee for Ataxic Diseases (to Y. N.) from the Ministry of Health, Labor, and Welfare, Japan; and by a grant from Core Research for Evolutional Science and Technology (CREST) of the Japan Science and Technology Agency (to Y. N.).
N. Fujikake, N. Kimura, Y. Saitoh, S. Nagano, Y. Hatanaka, T. Ishiguro, T. Takeuchi, M. Suzuki, H. A. Popiel, E. N. Minakawa, M. Ueyama, G. Matsumoto, A. Yokoseki, N. Nukina, T. Araki, O. Onodera, K. Wada, and Y. Nagai, submitted for publication.
- polyQ
- polyglutamine
- UPS
- ubiquitin-proteasome system
- MJDtr
- truncated form of mutant MJD
- IR
- inverted repeat RNA
- Aβ
- amyloid-β
- AGE
- agarose gel electrophoresis
- SEM
- scanning electron microscopic.
REFERENCES
- 1. Gatchel J. R., Zoghbi H. Y. (2005) Diseases of unstable repeat expansion: mechanisms and common principles. Nat. Rev. Genet. 6, 743–755 [DOI] [PubMed] [Google Scholar]
- 2. Davies S. W., Turmaine M., Cozens B. A., DiFiglia M., Sharp A. H., Ross C. A., Scherzinger E., Wanker E. E., Mangiarini L., Bates G. P. (1997) Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell 90, 537–548 [DOI] [PubMed] [Google Scholar]
- 3. Scherzinger E., Lurz R., Turmaine M., Mangiarini L., Hollenbach B., Hasenbank R., Bates G. P., Davies S. W., Lehrach H., Wanker E. E. (1997) Huntingtin-encoded polyglutamine expansions form amyloid-like protein aggregates in vitro and in vivo. Cell 90, 549–558 [DOI] [PubMed] [Google Scholar]
- 4. Arrasate M., Mitra S., Schweitzer E. S., Segal M. R., Finkbeiner S. (2004) Inclusion body formation reduces levels of mutant huntingtin and the risk of neuronal death. Nature 431, 805–810 [DOI] [PubMed] [Google Scholar]
- 5. Nagai Y., Inui T., Popiel H. A., Fujikake N., Hasegawa K., Urade Y., Goto Y., Naiki H., Toda T. (2007) A toxic monomeric conformer of the polyglutamine protein. Nat. Struct. Mol. Biol. 14, 332–340 [DOI] [PubMed] [Google Scholar]
- 6. Takahashi T., Kikuchi S., Katada S., Nagai Y., Nishizawa M., Onodera O. (2008) Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum. Mol. Genet. 17, 345–356 [DOI] [PubMed] [Google Scholar]
- 7. Rubinsztein D. C. (2006) The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 443, 780–786 [DOI] [PubMed] [Google Scholar]
- 8. Verhoef L. G., Lindsten K., Masucci M. G., Dantuma N. P. (2002) Aggregate formation inhibits proteasomal degradation of polyglutamine proteins. Hum. Mol. Genet. 11, 2689–2700 [DOI] [PubMed] [Google Scholar]
- 9. Holmberg C. I., Staniszewski K. E., Mensah K. N., Matouschek A., Morimoto R. I. (2004) Inefficient degradation of truncated polyglutamine proteins by the proteasome. EMBO J. 23, 4307–4318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Venkatraman P., Wetzel R., Tanaka M., Nukina N., Goldberg A. L. (2004) Eukaryotic proteasomes cannot digest polyglutamine sequences and release them during degradation of polyglutamine-containing proteins. Mol. Cell 14, 95–104 [DOI] [PubMed] [Google Scholar]
- 11. Ravikumar B., Duden R., Rubinsztein D. C. (2002) Aggregate-prone proteins with polyglutamine and polyalanine expansions are degraded by autophagy. Hum. Mol. Genet. 11, 1107–1117 [DOI] [PubMed] [Google Scholar]
- 12. Johansen T., Lamark T. (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 14. Ichimura Y., Kumanomidou T., Sou Y. S., Mizushima T., Ezaki J., Ueno T., Kominami E., Yamane T., Tanaka K., Komatsu M. (2008) Structural basis for sorting mechanism of p62 in selective autophagy. J. Biol. Chem. 283, 22847–22857 [DOI] [PubMed] [Google Scholar]
- 15. Kuusisto E., Salminen A., Alafuzoff I. (2001) Ubiquitin-binding protein p62 is present in neuronal and glial inclusions in human tauopathies and synucleinopathies. Neuroreport 12, 2085–2090 [DOI] [PubMed] [Google Scholar]
- 16. Kuusisto E., Kauppinen T., Alafuzoff I. (2008) Use of p62/SQSTM1 antibodies for neuropathological diagnosis. Neuropathol. Appl. Neurobiol. 34, 169–180 [DOI] [PubMed] [Google Scholar]
- 17. Yamaguchi M., Hirose F., Inoue Y. H., Shiraki M., Hayashi Y., Nishi Y., Matsukage A. (1999) Ectopic expression of human p53 inhibits entry into S phase and induces apoptosis in the Drosophila eye imaginal disc. Oncogene 18, 6767–6775 [DOI] [PubMed] [Google Scholar]
- 18. Warrick J. M., Paulson H. L., Gray-Board G. L., Bui Q. T., Fischbeck K. H., Pittman R. N., Bonini N. M. (1998) Expanded polyglutamine protein forms nuclear inclusions and causes neural degeneration in Drosophila. Cell 93, 939–949 [DOI] [PubMed] [Google Scholar]
- 19. Steffan J. S., Agrawal N., Pallos J., Rockabrand E., Trotman L. C., Slepko N., Illes K., Lukacsovich T., Zhu Y. Z., Cattaneo E., Pandolfi P. P., Thompson L. M., Marsh J. L. (2004) SUMO modification of Huntingtin and Huntington's disease pathology. Science 304, 100–104 [DOI] [PubMed] [Google Scholar]
- 20. Crowther D. C., Kinghorn K. J., Miranda E., Page R., Curry J. A., Duthie F. A., Gubb D. C., Lomas D. A. (2005) Intraneuronal Aβ, non-amyloid aggregates and neurodegeneration in a Drosophila model of Alzheimer's disease. Neuroscience 132, 123–135 [DOI] [PubMed] [Google Scholar]
- 21. Wittmann C. W., Wszolek M. F., Shulman J. M., Salvaterra P. M., Lewis J., Hutton M., Feany M. B. (2001) Tauopathy in Drosophila: neurodegeneration without neurofibrillary tangles. Science 293, 711–714 [DOI] [PubMed] [Google Scholar]
- 22. Wyers F., Petitjean A. M., Dru P., Gay P., Contamine D. (1995) Localization of domains within the Drosophila Ref(2)P protein involved in the intracellular control of σ rhabdovirus multiplication. J. Virol. 69, 4463–4470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ueyama M., Takemae H., Ohmae Y., Yoshida H., Toyoda H., Ueda R., Nishihara S. (2008) Functional analysis of proteoglycan galactosyltransferase II RNA interference mutant flies. J. Biol. Chem. 283, 6076–6084 [DOI] [PubMed] [Google Scholar]
- 24. Menzies F. M., Garcia-Arencibia M., Imarisio S., O'Sullivan N. C., Ricketts T., Kent B. A., Rao M. V., Lam W., Green-Thompson Z. W., Nixon R. A., Saksida L. M., Bussey T. J., O'Kane C. J., Rubinsztein D. C. (2014) Calpain inhibition mediates autophagy-dependent protection against polyglutamine toxicity. Cell Death Differ. 10.1038/cdd.2014.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Weiss A., Klein C., Woodman B., Sathasivam K., Bibel M., Régulier E., Bates G. P., Paganetti P. (2008) Sensitive biochemical aggregate detection reveals aggregation onset before symptom development in cellular and murine models of Huntington's disease. J. Neurochem. 104, 846–858 [DOI] [PubMed] [Google Scholar]
- 26. Legleiter J., Mitchell E., Lotz G. P., Sapp E., Ng C., DiFiglia M., Thompson L. M., Muchowski P. J. (2010) Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J. Biol. Chem. 285, 14777–14790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sontag E. M., Lotz G. P., Yang G., Sontag C. J., Cummings B. J., Glabe C. G., Muchowski P. J., Thompson L. M. (2012) Detection of mutant huntingtin aggregation conformers and modulation of SDS-soluble fibrillar oligomers by small molecules. J. Huntingtons Dis. 1, 127–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bjørkøy G., Lamark T., Brech A., Outzen H., Perander M., Overvatn A., Stenmark H., Johansen T. (2005) p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 171, 603–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Filimonenko M., Isakson P., Finley K. D., Anderson M., Jeong H., Melia T. J., Bartlett B. J., Myers K. M., Birkeland H. C., Lamark T., Krainc D., Brech A., Stenmark H., Simonsen A., Yamamoto A. (2010) The selective macroautophagic degradation of aggregated proteins requires the PI3P-binding protein Alfy. Mol. Cell 38, 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Roman G., Davis R. L. (2002) Conditional expression of UAS-transgenes in the adult eye with a new gene-switch vector system. Genesis 34, 127–131 [DOI] [PubMed] [Google Scholar]
- 31. Korolchuk V. I., Mansilla A., Menzies F. M., Rubinsztein D. C. (2009) Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol. Cell 33, 517–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nagaoka U., Kim K., Jana N. R., Doi H., Maruyama M., Mitsui K., Oyama F., Nukina N. (2004) Increased expression of p62 in expanded polyglutamine-expressing cells and its association with polyglutamine inclusions. J. Neurochem. 91, 57–68 [DOI] [PubMed] [Google Scholar]
- 33. Matsumoto G., Wada K., Okuno M., Kurosawa M., Nukina N. (2011) Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol. Cell 44, 279–289 [DOI] [PubMed] [Google Scholar]
- 34. Doi H., Adachi H., Katsuno M., Minamiyama M., Matsumoto S., Kondo N., Miyazaki Y., Iida M., Tohnai G., Qiang Q., Tanaka F., Yanagawa T., Warabi E., Ishii T., Sobue G. (2013) p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J. Neurosci. 33, 7710–7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schaffar G., Breuer P., Boteva R., Behrends C., Tzvetkov N., Strippel N., Sakahira H., Siegers K., Hayer-Hartl M., Hartl F. U. (2004) Cellular toxicity of polyglutamine expansion proteins: mechanism of transcription factor deactivation. Mol. Cell 15, 95–105 [DOI] [PubMed] [Google Scholar]
- 36. Kayed R., Head E., Thompson J. L., McIntire T. M., Milton S. C., Cotman C. W., Glabe C. G. (2003) Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 300, 486–489 [DOI] [PubMed] [Google Scholar]
- 37. Ravikumar B., Vacher C., Berger Z., Davies J. E., Luo S., Oroz L. G., Scaravilli F., Easton D. F., Duden R., O'Kane C. J., Rubinsztein D. C. (2004) Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat. Genet. 36, 585–595 [DOI] [PubMed] [Google Scholar]
- 38. Martin D. D., Ladha S., Ehrnhoefer D. E., Hayden M. R. (2014) Autophagy in Huntington disease and huntingtin in autophagy. Trends Neurosci. 10.1016/j.tins.2014.09.003 [DOI] [PubMed] [Google Scholar]
- 39. Brady O. A., Meng P., Zheng Y., Mao Y., Hu F. (2011) Regulation of TDP-43 aggregation by phosphorylation and p62/SQSTM1. J. Neurochem. 116, 248–259 [DOI] [PubMed] [Google Scholar]
- 40. Scotter E. L., Vance C., Nishimura A. L., Lee Y. B., Chen H. J., Urwin H., Sardone V., Mitchell J. C., Rogelj B., Rubinsztein D. C., Shaw C. E. (2014) Differential roles of the ubiquitin proteasome system and autophagy in the clearance of soluble and aggregated TDP-43 species. J. Cell Sci. 127, 1263–1278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Petrucelli L., Dickson D., Kehoe K., Taylor J., Snyder H., Grover A., De Lucia M., McGowan E., Lewis J., Prihar G., Kim J., Dillmann W. H., Browne S. E., Hall A., Voellmy R., Tsuboi Y., Dawson T. M., Wolozin B., Hardy J., Hutton M. (2004) CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 13, 703–714 [DOI] [PubMed] [Google Scholar]
- 42. Shimura H., Schwartz D., Gygi S. P., Kosik K. S. (2004) CHIP-Hsc70 complex ubiquitinates phosphorylated Tau and enhances cell survival. J. Biol. Chem. 279, 4869–4876 [DOI] [PubMed] [Google Scholar]
- 43. Lee M. J., Lee J. H., Rubinsztein D. C. (2013) Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog. Neurobiol. 105, 49–59 [DOI] [PubMed] [Google Scholar]
- 44. Iwata N., Tsubuki S., Takaki Y., Watanabe K., Sekiguchi M., Hosoki E., Kawashima-Morishima M., Lee H. J., Hama E., Sekine-Aizawa Y., Saido T. C. (2000) Identification of the major Aβ1–42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nat. Med. 6, 143–150 [DOI] [PubMed] [Google Scholar]
- 45. Qiu W. Q., Walsh D. M., Ye Z., Vekrellis K., Zhang J., Podlisny M. B., Rosner M. R., Safavi A., Hersh L. B., Selkoe D. J. (1998) Insulin-degrading enzyme regulates extracellular levels of amyloid β-protein by degradation. J. Biol. Chem. 273, 32730–32738 [DOI] [PubMed] [Google Scholar]