

Background: ISG15 is a Ub-like protein that conjugates cellular and pathogenic proteins during the innate immune response.
Results: ISG15 associates with the selective autophagic factors HDAC6 and p62, leading to degradation of ISG15 conjugates.
Conclusion: The IFN response leads to ISG15 expression allowing for its association with HDAC and p62. This may mark proteins for autophagy.
Significance: This finding provides evidence of an interferon-stimulated pathway linked to autophagy.
Keywords: Autophagy, Histone Deacetylase 6 (HDAC6), Lysosome, Post-translational Modification (PTM), Ubiquitin, Virus, ISG15, SQSTM1, Infection, p62
Abstract
The ubiquitin-like interferon (IFN)-stimulated gene 15 (ISG15) and its specific E1, E2, and E3 enzymes are transcriptionally induced by type I IFNs. ISG15 conjugates newly synthesized proteins. ISG15 linkage to proteins appears to be an important downstream IFN signaling event that discriminates cellular and pathogenic proteins synthesized during IFN stimulation from existing proteins. This eliminates potentially pathogenic proteins as the cell attempts to return to normal homeostasis after IFN “stressed” conditions. However, the molecular events that occur in this process are not well known. Here, we show that the C-terminal LRLRGG of ISG15 interacts with the binder of ubiquitin zinc finger (BUZ) domain of histone deacetylase 6 (HDAC6). Because HDAC6 is involved in the autophagic clearance of ubiquitinated aggregates during which SQSTM1/p62 plays a major role as a cargo adapter, we also were able to confirm that p62 binds to ISG15 protein and its conjugated proteins upon forced expression. Both HDAC6 and p62 co-localized with ISG15 in an insoluble fraction of the cytosol, and this co-localization was magnified by the proteasome inhibitor MG132. In addition, ISG15 was degraded via the lysosome. Overexpression of ISG15, which leads to an increased conjugation level of the cellular proteome, enhanced autophagic degradation independently of IFN signaling transduction. These results thus indicate that ISG15 conjugation marks proteins for interaction with HDAC6 and p62 upon forced stressful conditions likely as a step toward autophagic clearance.
Introduction
The type I interferon (IFN) signaling pathway is the quintessential intracellular innate defense mechanism against invading pathogens. It sets up a robust antiviral state, rendering cells non-permissive for infection and replication. Upon binding of IFNs to their receptors (type I interferon α/β receptors), plasma membrane-resident STATs are activated through phosphorylation by JAK1/TYK2 kinases. This leads to subsequent translocation of STATs into nuclei where they bind to a cis-acting DNA element upstream of IFN-stimulated genes (ISGs),2 leading to ISG transcriptional activation. The ISG family of proteins activates IFN signaling, virus recognition factors or pattern recognition receptors, protein kinase R, ribonuclease L, Mx1, and ISG15 (1). ISG15 is a diubiquitin-like protein able to conjugate to lysine residues of its substrate via isopeptide bonds, utilizing the same enzymatic cascade as that of ubiquitin (2). ISG15 conjugation (referred to as “ISGylation”) onto de novo synthesizing polypeptides on polysomes proceeds through a series of enzymatic reactions mediated by the E1-activating UbE1L, E2-conjugating UbcH8, and HECT (homologous to E6-AP carboxyl terminus)-type E3 ligase Herc5, which are also all IFN-inducible. There is also an inverse reaction where the IFN-inducible UBP43/USP18 protease deconjugates ISG15 from conjugated substrates (3). ISG15 broadly conjugates IFN-induced ISGs and progeny viral proteins (2). It has been shown that ISG15 knock-out mice are broadly susceptible to RNA and DNA viruses (4). Several studies have found that free ISG15 and ISGylated host and viral proteins interfere in the processes of virion egress and release from the cell during the viral life cycle (4). The fate(s) of ISG15 and ISG15-conjugated proteins, particularly those that belong to pathogenic organelles and particles, is assumed to be elimination, but the pathway of this degradation is not well understood. The 26 S proteasome, which is responsible for degrading ubiquitinated proteins as part of the canonical ubiquitin-proteasome system (UPS), is not thought to play a role in degradation of ISG15 conjugates (5, 6).
Although in general the UPS is involved primarily in the degradation of single molecules, the lysosome in the process of (macro-) autophagy is more involved in eliminating large multiple molecules, organelles, and microbes/viruses. Type I interferons have also been reported to induce autophagy, which plays a role in cellular homeostasis through sequestration of intracellular materials including pathogens within double membrane vesicles (autophagosomes) that fuse with lysosomes, leading to degradation of delivered cellular contents (7). Although starvation-induced, non-selective autophagy recycles cytosolic contents and organelles during limited nutrient supply, nutrient-independent, basal, or selective autophagy eliminates misfolded proteins and damaged organelles before the accumulation of excessive amounts of cytosolic aggregates or inclusion bodies at the microtubule-organizing center (8–10). It has been reported that aggregate-prone proteins become marked by the lysine 63 (Lys63)-linked polyubiquitin chain that functionally distinguishes them from UPS-targeted lysine 48 (Lys48)-linked polyubiquitin chains (11, 12). In this context, the cargo adaptor protein SQSTM1/p62 plays an important role by preferentially binding Lys63-linked polyubiquitinated proteins via a ubiquitin-associated domain at its C terminus. To facilitate ubiquitin (Ub)-protein aggregates and autophagic degradation, p62 undergoes self- and/or hetero-oligomerization at its N-terminal PB1 domain and recruits the LC3 membrane protein that is anchored in phagophore membranes for autophagosome maturation (13–15). During selective autophagy, the cytosolic deacetylase HDAC6 is involved in clearance of ubiquitin-prone aggregates (10, 16). Its C-terminal binder of ubiquitin zinc finger (BUZ) domain can bind free mono- and poly-Ub chains (17, 18). Both the catalytic activity and the BUZ domain are required for HDAC6 to facilitate clearance of ubiquitin-prone aggregates (also referred to as “aggrephagy”), and HDAC6 promotes autophagosome-lysosome fusion through the cortactin-dependent actin remodeling machinery in mouse embryonic fibroblast cells (19).
Here, we report that forced expression of HDAC6 leads to interaction with p62 and ISG15 that structurally resembles Lys63-di-Ub and its ISGylated substrates, rendering them targets for lysosomal degradation. ISG15 localized to cytosolic inclusion bodies with HDAC6 and p62. Pharmacological induction of aggrephagy became more prominent if ISG15 was overexpressed. Therefore, our results suggest that ISG15 augments p62-mediated aggresome formation and their autophagic degradation under conditions of cellular stress such as forced expression of genes, implying an important role during intrinsic cellular defense.
EXPERIMENTAL PROCEDURES
Materials
Reagents
Human IFNβ 1a (11410) was obtained from PBL Assay Science. Proteasome inhibitor MG132 (40 mm stock in DMSO) was from EMD Millipore. Lysosomal protease inhibitors pepstatin A (20 mm in DMSO) and E64d (10 mg/ml in DMSO) were from Enzo Life Science. Tubacin was kindly obtained from Dr. Stuart Schreiber (Broad Institute of Massachusetts Institute of Technology/Harvard, Cambridge, MA) (20). Doxycycline hyclate was purchased from Sigma. Protease inhibitor mixture Complete EDTA-free and phosphatase inhibitor mixture PhosSTOP were from Roche Applied Science. Recombinant GST-tagged HDAC6 protein was from EMD Millipore. GST-tagged p62 was from Enzo Life Science. Human ISG15 protein was from Boston Biochem. Anti-FLAG M1-agarose affinity gel and monoclonal anti-FLAG M1 antibody were from Sigma-Aldrich. Immobilized GST-agarose and GST protein were from Thermo Fisher Scientific.
cDNA and Lentiviral Vectors
HDAC6 Flag (13823), pcDNA-hISG15 (12447), pFlagCMV2-UbcH8 (12442), pCAGGS-HA-hUBE1L (12438), and HA-p62 (28027) were purchased from Addgene. Human HERC5 cDNA (GenBankTM accession number BC140716) was obtained from Mammalian Gene Collection cDNA library. The Tet3G system and lentivirus vectors (pLenti CMV rtTA3G Blast, pLenti CMVTRE3G Neo DEST, and pLenti CMVTRE3G Puro DEST) were purchased from Addgene. pGIPZ vector was purchased from Open Biosystems, Inc.
Antibodies
Antibodies against ISG15 (sc-50366) and HDAC6 (sc-28386) were from Santa Cruz Biotechnology. Antibodies against FLAG M2 (F3165), α-tubulin (T6074), acetylated α-tubulin (T6793), and SQSTM1/p62 (WH0008878M1) were from Sigma. Antibodies against ubiquitin (3936), LC3B (2775), and lamin A/C (2032) were from Cell Signaling Technology. Antibodies against HA (11-867) and GST (ab34589) were from Roche Applied Science and Abcam, respectively, and GFP (598) was from MBL International Corp.
Cell Culture and Transfection
293A (Invitrogen) and U251 cells (ATCC) were cultured in DMEM supplemented with 10% FBS, 100 μg/ml penicillin/streptomycin, and 10 mm HEPES at 37 °C in a humidified incubator with 5% CO2. For transfections, 293A cells were seeded at 2 × 106 cells on 60-mm dishes and transfected with vectors using Lipofectamine 2000 (Invitrogen) following the supplier's protocol.
DNA Constructs
For mutant constructs, HDAC6 point mutation at histidine 611 to alanine (H611A) and deletions from residues 841 to 1195 (ΔCter), from 2 to 837 (ΔCD1/2), from 841 to 1053 (ΔSE14), and from 1133 to 1195 (ΔBUZ) were generated from HDAC6 Flag plasmid DNA by PCR (QuikChange site-directed mutagenesis kit, Agilent Technologies). For lentivirus packaging, some HDAC6 constructs were also cloned in pGIPZ at blunt-ended NotI and XbaI sites by inserting the SpeI-XbaI fragments that contain a CMV intermediate-early promoter and an HDAC6 coding sequence. ISG15 truncated mutants in the C-terminal end peptides LRLRGG and aspartic acid (D) insert (LRLRGGD) were also generated from a pcDNA-hISG15 plasmid by PCR. The sequences of all mutant constructs were verified by DNA sequencing analyses. For the ISG15-cyan fluorescent protein (CFP) construct, three repeated FLAG sequences were added in ISG15 by PCR using the primers 5′-caccctcgagctccatatggactacaaagaccatgacggtgatta-3′ and 5′-ttaggatcccgggcccgcccacccctcaggcgcaga-3′ and pcDNA-hISG15 as template. These were cloned in pENTR/D-TOPO (Invitrogen). The FLAG-ISG15 fragment was inserted in pmTurquoise2-Golgi (Addgene, 36205) at XhoI/BamHI sites. FLAG-tagged mTurquoise2 (referred to CFP in this study) was engineered previously.3 Gene transfer was performed either by Lipofectoamine 2000 (for 293), Lipofectoamine 3000 (for U251), or lentiviral infection. For transduction of genes via lentiviral infection, vectors were packaged in lentiviral vector systems using 293T (ATCC) or 293FT cells (Invitrogen) by co-transfection with packaging vectors psPAX2 and pMD2.G (a gift from Dr. Didier Trono, Switzerland) followed by lentivirus infection. U251 cells expressing FLAG-HDAC6 wild type (WT) and derivative mutants were stably selected by G418 treatment after infection.
Doxycycline (Dox)-inducible ISG15 Conjugation System
Construction of the Dox-inducible ISG15 conjugation system was as follows. 2A peptide linkage constructs between Ube1L and ISG15 and between Herc5 and UbcH8 were generated in pENTR/D-TOPO vectors with two-step PCR using the following primer sets as described previously (21): UBE1L, 5′-caccaagcttgccagcatggatgccctggacgcttcg-3′ and 5′-gtctcctgcttgctttaacagagagaagttcgtggctccggatcccagctcatagtgcagaggtgggaagg-3′; ISG15, 5′-gccacgaacttctctctgttaaagcaagcaggagacgtggaagaaaaccccggtcctatggaattccatatgggctgggacc-3′ and 5′-caatgtatcttatcatgtctggatccccg-3′; Herc5, 5′-caccgtcgacgccagcatggagcggaggtcgcggag-3′ and 5′-gtctcctgcttgctttaacagagagaagttcgtggctccggatccgccaaatcctctgttgttgttgatgg-3′; UbcH8, 5′-gccacgaacttctctctgttaaagcaagcaggagacgtggaagaaaaccccggtcctgcgagcatgcgagtggtgaag-3′ and 5′-gctcgagttaggagggccggtccactcc-3′. Tet-On constructs of these 2A peptide-linked dicistronic vectors were generated in pLenti CMVTRE3G Neo DEST and pLenti CMVTRE3G Puro DEST using the Gateway cloning system (Invitrogen). Lentivirus-mediated transformed cell lines along with rtTA3G were initially selected by G418, puromycin, and blasticidin S. Clonal cells were analyzed by immunoblots using antibody against ISG15, and we used clone.7–8 of U251 (referred to U251.ISG7–8) and clone.8 of 293A (referred to 293.ISG8) in this study.
Protein Soluble and Insoluble Fractions
Cells were lysed in soluble lysis buffer (50 mm Tris-Cl (pH 7.4), 150 mm NaCl, 2.5 mm EDTA, 0.5% Triton X-100, 40 μm MG132, PhosSTOP, and protease inhibitor mixture) and incubated on ice for 5 min. After centrifugation at 13,000 rpm, the supernatant was collected in the tube as the soluble fraction, whereas the pellet was dissolved in insoluble lysis buffer (1% SDS in soluble lysis buffer) followed by sonication and centrifugation to collect the supernatant as the insoluble fraction.
Protein Interaction Assays
For the in vitro GST pulldown assay of p62, each 2 μg of recombinant GST-p62 or GST protein was incubated with immobilized glutathione-agarose in binding buffer (20 mm Tris-Cl (pH 7.4), 50 mm NaCl, 0.1% Nonidet P-40, 25 μg/ml BSA, 1 mm ATP, 20 μm MG132, and protease inhibitor mixture) followed by three washes with binding buffer. The recombinant mature form of human ISG15 protein (2 μg) was incubated with GST protein-bound agarose overnight at 4 °C before analysis by immunoblotting. The GST-HDAC6 pulldown assay was performed using the cell lysate of 293T cells that were transfected with mutant ISG15 constructs. For the co-immunoprecipitation assay, cells were lysed in a buffer containing 50 mm Tris-Cl (pH 7.4), 150 mm NaCl, 1% Triton X-100, 2 mm EGTA, 40 μm MG132, PhosSTOP, and protease inhibitor mixture. The lysates were incubated with anti-FLAG M2 affinity gel overnight at 4 °C. Immunocomplexes were collected by centrifugation and analyzed by immunoblotting.
Immunofluorescence Microscopy Analysis
Cells were cultured on German glass round coverslip (number 1 thickness) and then washed with 37 °C PBS once before fixation with 4% paraformaldehyde in PBS. For prepermeabilized cell preparation, washed cells were incubated in extraction buffer (100 mm PIPES (pH 6.9), 0.5 mm MgCl2, 0.1 mm EDTA, and 0.5% Triton X-100) for ∼20 s before 4% paraformaldehyde fixation. The samples were quenched with 50 mm NH4Cl in PBS and permeabilized with 50 μg/ml digitonin (EMD Millipore) for the non-prepermeabilized condition. The samples were washed with 0.1% gelatin in PBS and incubated with diluted antibodies (1:200, α-tubulin and FLAG M; 1:50, p62 and ISG15) followed by Alexa Fluor antibodies (1:200; Invitrogen). Nuclei were counterstained with DAPI during the wash step, and then coverslips were mounted with VectaShield. Fluorescence images were acquired with a Zeiss LSM510 META or LSM710 confocal microscopy system and processed using NIH ImageJ (1.48 or earlier version). Figure panels were assembled using Adobe Illustrator CS5.
RESULTS
HDAC6 Associates with IFN-induced ISG15-conjugated Proteins
We first attempted to analyze whether an interaction between ISG15-conjugated substrates and HDAC6 occurred upon IFN treatment. We attempted to utilize two commercially available antibodies raised against endogenous HDAC6 to detect co-immunoprecipitates of HDAC6 with ISG15 conjugates in U251 exposed to type I IFN. However, we were unable to visualize co-immunoprecipitation of endogenous HDAC6 with ISGylated substrates (data not shown). Although there could be multiple reasons for this, we reasoned that perhaps we needed to overexpress HDAC6 and/or the HDAC6 antibody was not sufficiently avid to allow co-immunoprecipitations.
We thus performed a second series of experiments whereby the lysates of human U251 glioma cells forced to express FLAG-tagged HDAC6 by lentivirus transduction were immunoprecipitated with an antibody against FLAG-peptides and the co-immunoprecipitates of FLAG-HDAC6 were analyzed by Western blotting using antibodies against ISG15 or ubiquitin. Fig. 1a shows that IFNβ treatment led to ISG15 conjugation. FLAG-HDAC6 co-immunoprecipitated with ISGylated substrates, and this was even more prominent when cells were treated with MG132, a protease inhibitor that induces formation of intracellular aggresomes. In contrast, in comparison with ISGylated substrates, there was a specific effect of MG132 on FLAG-HDAC6 association with ubiquitinated substrates that was independent of IFN treatment (17, 18). These data thus suggested that there was a previously undescribed but visible association of forcibly expressed FLAG-HDAC6 with IFNβ-induced ISGylated substrates even in the absence of MG132 (i.e. when the UPS is functional).
FIGURE 1.
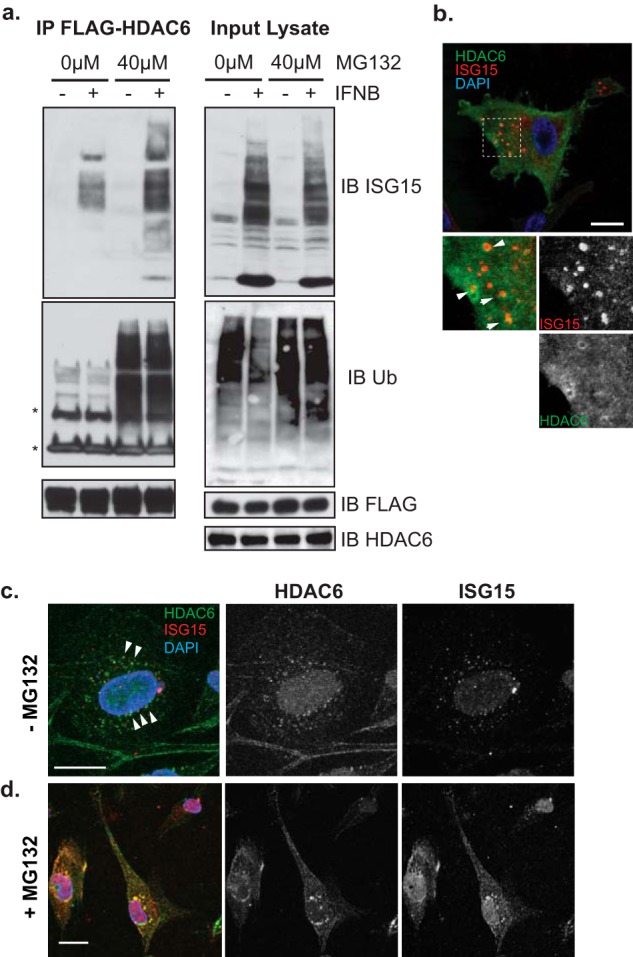
HDAC6 interacts with ISG15-conjugated cellular proteins. a, U251 expressing FLAG-HDAC6 WT (full length) were treated with IFNβ (1,000 units/ml; 16 h) with and without MG132 (40 μm; 4 h). Anti-FLAG-immunoprecipitated (IP) proteins and input cell lysates were immunoblotted (IB) with anti-FLAG, anti-ISG15, and anti-Ub antibodies. b–d, U251 cells expressing FLAG-HDAC6 WT were treated with IFNβ (1,000 units/ml; 16 h) in the absence (b and c) or the presence (d) of MG132 (40 μm; 5 h). Cytoskeletal insoluble fractions were visible in cells prepermeabilized with Triton X-100 (c and d). Fluorescence signals for Alexa Fluor 488 (green) for FLAG, Alexa Fluor 555 (red) for ISG15, and DAPI (blue) were visualized using confocal microscopy. High magnification views highlighted by broken line boxes are shown in the bottom panels of the image (b). Scale bars, 20 μm.
Next, we visualized the subcellular localization of FLAG-HDAC6 and ISG15 in IFNβ-treated cells utilizing immunofluorescence staining with antibodies against FLAG and ISG15 (Fig. 1b). ISG15 displayed small punctate stainings in the cytoplasm with a concentration in the perinuclear region where FLAG-HDAC6 vacuoles also appeared to partially localize, although co-localization was minimal if any. To examine whether this lack of localization reflected insoluble aggregates, live cells were permeabilized with a pulse of Triton X-100 to remove soluble proteins from cytosols prior to fixation (Fig. 1, c and d). In permeabilized cells in the absence of proteasomal inhibition, ISG15 punctate stainings were visualized in the same perinuclear region as that of small HDAC6 subfractions (Fig. 1c). However, in the presence of MG132 proteasome inhibition, there was visually intensified HDAC6 immunofluorescence with a perinuclear aggregation pattern that now also co-localized with ISG15 speckles (Fig. 1d). Taken together, these results suggested that the interaction between HDAC6 and ISG15 was localized to detergent-resistant aggregates in the perinuclear region and that proteasome inhibition led to a prominent co-localization of ISG15 and HDAC6 in the same aggregates.
HDAC6 Binds ISG15 at the C-terminal LRLRGG Motif via a BUZ Domain
To find out which motif of HDAC6 interacts with ISG15, we engineered U251 cells that stably overexpress FLAG-tagged full-length HDAC6 WT or its C-terminal deletion mutant (ΔCter; Δ841–1195) or its catalytic subsite mutant (H611A) (Fig. 2a). Subsequently, we performed co-immunoprecipitation assays with an antibody against FLAG peptides using cell lysates that had been treated with IFNβ. Fig. 2b shows that ISG15 was bound to FLAG-HDAC6 WT and FLAG-HDAC6H611A but not to FLAG-HDAC6ΔCter. To further map the ISG15 binding site in residues 841–1195 of the HDAC6 C-terminal region, we transiently transfected FLAG-tagged full-length HDAC6 and truncation mutant constructs of HDAC6 that lacked the BUZ domain (ΔBUZ), the Ser-Glu-containing tetradecapeptide repeat (SE14) domain (ΔSE14), or both catalytic domains (ΔCD1/2) (see Fig. 2a) together with the HA-tagged mature form of ISG15 in 293 cells. In addition to FLAG-HDAC6 WT, Fig. 2c shows that ISG15 co-immunoprecipitated with ΔCD1/2 and ΔSE14 but not with the ΔBUZ truncation mutant of HDAC6. Therefore, these findings indicated that residues 1133–1195, corresponding to a BUZ domain, were responsible for ISG15 binding.
FIGURE 2.

Mapping the interacting domains of HDAC6 and ISG15. a, schematic of FLAG-tagged HDAC6 and its deletion mutants. The two catalytic domains (CD1 and CD2), SE14 domain, and BUZ domain are shown as closed boxes. An asterisk (*) represents the H611A point mutation in WT. b, U251 cells that express FLAG-HDAC6 WT, mutant without an active site histidine (H611A), an SE14-BUZ-truncated mutant (ΔCter), or empty vector (Vec) were treated with IFNβ (1,000 units/ml; 16 h). Lysates were subjected to co-immunoprecipitation (IP) with antibody against FLAG and analysis using antibodies against FLAG and ISG15. c, 293 cells were transiently co-transfected with expression vectors for FLAG-HDAC6 WT, a C-terminal truncation (ΔCter) mutant, an N-terminal truncation (ΔCD1/2) mutant, a BUZ domain truncation (ΔBUZ) mutant, an SE14 domain truncation (ΔSE14) mutant, or an empty vector (Vec) along with an HA-tagged ISG15 expression vector. Lysates and immunoprecipitates were analyzed with antibodies against FLAG and HA. d, schematic of HA-tagged ISG15 with wild type or mutants. The two Ub-like domains are shown as closed boxes. Peptides were tagged with HA at the N terminus, and amino acids were deleted from the LRLRGG peptide sequences at the C-terminal end (c-end) as indicated by asterisks (*). e, 293A cells were transiently transfected with plasmids carrying HA-tagged ISG15 vectors with mutants. In a pulldown assay, cell lysates were incubated with purified recombinant GST-tagged HDAC6 or control GST, bound to glutathione-Sepharose beads, and subjected to analysis with antibodies against HA and GST. IB, immunoblotting.
Based on this, we then sought to find the motif in ISG15 responsible for binding to HDAC6. It has been reported that the BUZ domain of HDAC6 interacts with a terminal conjugation motif of Ub composed of LRLRGG. This motif appears in Ub aggregates that contain free mono- or polyubiquitin chains (22). The mature form of ISG15 also has a C-terminal LRLRGG motif, and thus we asked whether it also bound to HDAC6 BUZ. We utilized a full-length mature ISG15 (ISG15 WT) and four mutants of ISG15 (ISG15-LRLRG, ISG15-LRLR, ISG15-LRLRGGD, and ISG15-LRGG) with deletions or additions at the C-terminal residues (Fig. 2d). In vitro protein affinity assays revealed that the full-length, wild-type LRLRGG motif was the most efficient at interacting with recombinant GST-tagged HDAC6, whereas all the other mutant forms of ISG15 failed to bind to GST-HDAC6 (Fig. 2e). There was some relatively weak interaction of the LRLRGGD mutant with HDAC6. Overall, these findings thus confirmed that the ISG15 LRLRGG peptide motif is indispensable for binding to the BUZ domain of HDAC6.
ISG15 Associates with p62 Likely to Facilitate Autophagosome/Lysosome Clearance
The Ub binding property of HDAC6 is associated with formation and clearance of organized aggregates (also called aggresomes) as part of the autophagy-lysosome degradation pathway (12, 17, 23). During this process, the SQSTM1/p62 cargo protein forms aggregates of ubiquitinated proteins and recruits LC3 anchored in the phagophore membrane (24). p62 preferentially binds with higher affinity to Lys63-linked di-Ub when compared with binding to monoubiquitin and Lys48-linked di-Ub (25). Our in silico analysis using the flexible structure alignment (available on the Protein Data Bank site) predicted that the structure of ISG15 (Protein Data Bank code 1z2m) resembles that of Lys63-linked di-Ub (Protein Data Bank code 2w9n) rather than that of Lys48-linked di-Ub (Protein Data Bank code 3m3j) (26–29). We thus sought to determine whether ISG15 also bound to p62. In fact, a pulldown assay using recombinant GST-tagged p62 did lead to direct binding of GST-p62 to ISG15 (Fig. 3a). This indicated that ISG15, like Lys63-linked di-Ub, also directly bound to p62.
FIGURE 3.

ISG15 up-regulation of aggregate formation of p62. a, purified recombinant ISG15 proteins were pulled down with glutathione-Sepharose bead-bound purified recombinant GST tag-p62 fusion or control GST and analyzed as indicated. b, schematic inducible co-expression vectors of ISG15 and the core enzyme genes ISG15, E1 UbE1L, E2 UbcH8, and E3 Herc5 using self-cleavage peptide sequences (P2A) under the control of the Tet-On promoter (Ptet-on). The details of the vector constructs are described under “Experimental Procedures.” c, U251.ISG7–8 exhibited overexpression of ISG15 and ISG15 conjugation to cellular proteins in the presence of Dox (100 ng/ml; 24 h) prior to immunoblotting (IB) with anti-ISG15 antibody. d, immunoblots of detergent-soluble (Soluble) and -insoluble (Insoluble) fractions of U251.ISG7–8 cells with and without Dox treatment (100 ng/ml; 24 h) in the presence or absence of PepA/E64d (each 20 μg/ml; 4 h) were probed for the indicated proteins. e, 293.ISG8 exhibited overexpression of ISG15 and conjugation in the presence of Dox (200 ng/ml; 24 h). f, 293-ISG8 cells were transiently transfected with FLAG-tagged p62 or control empty vector and treated with MG132 (40 μm; 6 h). Anti-FLAG-immunoprecipitated (IP) proteins and input cell lysates were immunoblotted (IB) with anti-FLAG and anti-ISG15 antibodies. g, immunoblots of 0.5% Triton X-100 detergent-soluble (Soluble) and -insoluble (Insoluble) lysate fractions of 293.ISG8 cells treated with Dox (200 ng/ml; 24 h) in the presence or absence of MG132 (6 h) at the indicated doses prior to collecting the cells were probed for the indicated proteins.
To test whether p62 was involved in the formation and clearance of aggregates of ISGylated proteins, we engineered U251 cells that expressed a Dox-inducible transcriptionally regulated ISG15 as well as UbE1L, UbcH8, and Herc5, the enzymatic components of the ISG15 conjugation cascade (Fig. 3b). These cells thus would express the ISG15 conjugation system upon Dox treatment independently of IFN. Fig. 3c shows that one of the isolated clones (U251.ISG7–8) showed evidence of significant induction of ISGylation under Dox treatment. We then sought to determine whether ISGylation affected proteins associated with p62 in soluble versus insoluble cellular subfractions. Uninduced or Dox-induced cells were treated with pepstatin A and E64d (PepA/E64d; inhibitors of lysosomal proteases), and then soluble and insoluble fractions of cell lysates were analyzed by Western blotting. Fig. 3d shows that Dox-treatment induced ISGylation of the proteome in both soluble and insoluble fractions and that this was even more evident when cells were treated with PepA/E64d. Interestingly, Dox treatment resulted in perhaps a mild increase in p62 levels in the soluble fraction, whereas LC3-II levels were increased a bit more convincingly. However, there was a clear increase by Dox of p62 and LC3-II in the insoluble fraction that was even more visible in PepA/E64d-treated cells. Similar findings were also obtained in IFNβ-treated U251 cells (see Fig. 6). These results suggested that induction of ISG15 and ISGylation leads to up-regulation of p62 and LC3-II association with IGS15-conjugated proteins ending up in the lysosomes under condition of both Dox-induced ISGylation and IFN stimulation (from Fig. 6). The association with p62 and LC3-II strongly suggests an autophagic mechanism for degradation of ISG15.
FIGURE 6.
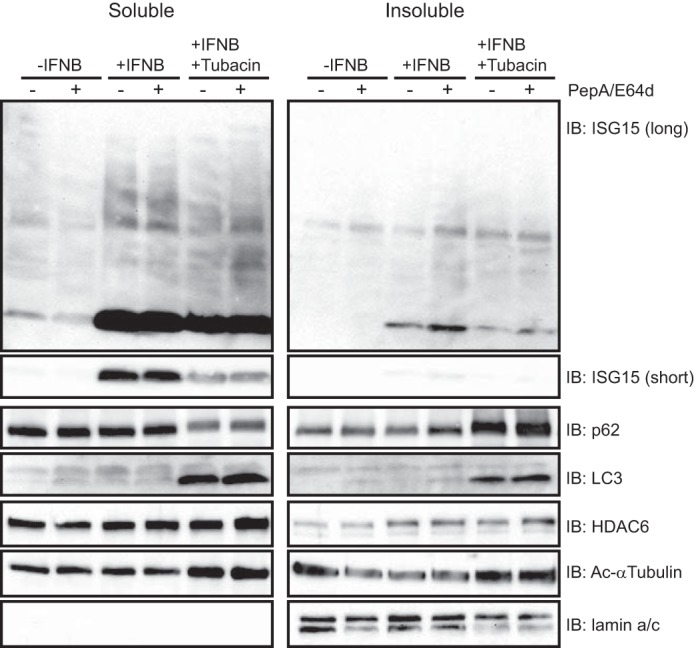
The HDAC6 deacetylase inhibitor, Tubacin, suppresses ISG15 degradation. U251 cells were treated with IFNβ (1,000 units/ml; 12 h) and tubacin (5 μm; 13 h) before treatment with PepA/E64d (20 μg/ml; 6 h). Cell lysates prepared as the soluble and insoluble fractions were immunoblotted (IB) using antibodies against the indicated proteins. An immunoblot against ISG15 shows both a short exposure (short) and a long exposure (long) time to compare the levels in non-conjugated ISG15.
To analyze the in vivo interaction between p62 and ISG15-conjugated proteins, we stably transduced 293 cells to express the ISG15 conjugation system in response to Dox (293.ISG8 cells) as shown in Fig. 3e. These 293.ISG8 cells were then transiently transfected with human p62 containing a C-terminal FLAG epitope tag (FLAG-p62) or empty vector. Fig. 3f shows that FLAG-p62 co-immunoprecipitated with ISG15-conjugated substrates. Interestingly, MG132 led to a visible decrease in FLAG-p62 levels in the input lysates that were extracted from the detergent-containing lysis buffer (Fig. 3f). To further try to understand this, we decided to analyze the detergent-insoluble fraction for p62 levels as a function of MG132 doses with and without Dox treatment. Fig. 3g shows that, in the absence of Dox, p62 was present primarily in the soluble fraction without an effect from proteasome inhibition. With Dox (thus increasing ISGylation activity) and increased doses of MG132, there was a visible shift of p62 from the soluble to insoluble fractions. Because MG132 treatment blocks the autophagic degradation pathway of aggregates, there was also an MG132 dose-dependent increase in LC3, ISGylated proteins, and HDAC6 in the insoluble fraction, although a lot of these were still present in the soluble fraction. These findings thus suggested that ISGylation led to a shift of p62, HDAC6, and LC3 into the insoluble aggregate fraction of cells that was detectable once the proteasome was inhibited.
Therefore, to determine whether p62 interacted and directed ISG15 to accumulate in inclusion bodies in response to IFN, we prepared prepermeabilized IFN-treated U251 cells and immunostained them using antibodies against p62, ISG15, and α-tubulin (Fig. 4). In the absence of MG132, cells contained numerous small punctate areas of staining for p62 bodies in the cytosol, most of which also co-localized with ISG15 (Fig. 4a). The p62-positive puncta became visibly larger in MG132-treated cells, and this was also true for puncta in which ISG15 and p62 co-localized (Fig. 4b). ISG15 puncta were also observed on detergent-resistant stable microtubules regardless of MG132 treatment (Fig. 4, c and d) that accumulated ISG15-positive large aggregates at the microtubule-organizing center (Fig. 4d). These results suggested that p62 contributes to the formation of ISG15-containing aggregates in the microtubule-organizing center.
FIGURE 4.
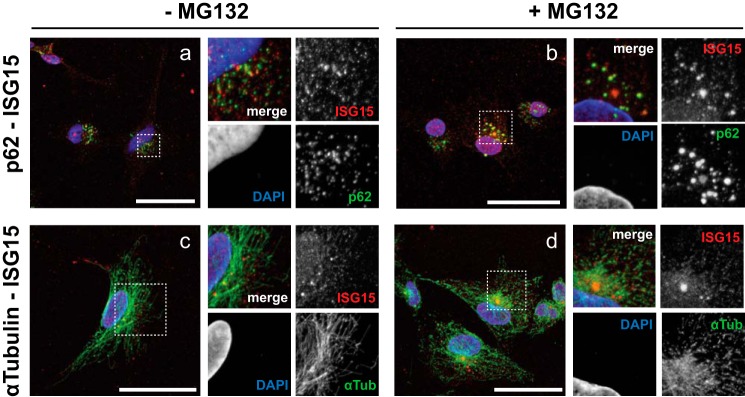
ISG15 and p62 co-localization in insoluble fraction. a–d, U251 cells were treated with IFNβ (1,000 units/ml; 16 h) before treatment with MG132 (40 μm; 5 h) (b and d) or without it (a and c). Prepermeabilized cells were fixed with 4% paraformaldehyde and subjected to fluorescence microscopy analysis using Alexa Fluor 488 (green) against p62 and α-tubulin, Alexa Fluor 555 (red) against ISG15, and DAPI (blue). High magnification rescans of the broken line box regions of interest are shown in the panels on the right of each sample image. Scale bars, 50 μm.
An Engineered ISG15 Protein Is a Target of Lysosomal Degradation
We also attempted to detect ISG15 conjugate degradation using an engineered ISG15 conjugate. For this, we engineered a CFP-fused ISG15 expression vector in which the C-terminal LRLRGG is linked to the N terminus of mTurquoise2 (CFP) protein (Fig. 5a). As shown in Fig. 5b, we could not detect the fusion protein upon DNA transfection in the absence of interferon but instead observed separate immunopositive bands for ISG15 and mTurquoise2 proteins, suggesting fairly prompt cleavage of the expressed fusion protein after transfection into cells. This observation appears to agree with that describing precursor processing of ISG15 to expose LRLRGG-terminal peptides by proteases (30). We then took advantage of this observed cleavage of ISG15-CFP protein to evaluate its lysosomal degradation (Fig. 5c). Similar to the results of Fig. 3, cleaved ISG15 protein was observed in the detergent-insoluble fraction, and PepA/E64d treatment caused accumulation of the ISG15 protein. These findings thus suggested that ISG15 protein can be a target of lysosomal degradation.
FIGURE 5.

Lysosomal degradation of cleaved ISG15. a, schematic of ISG15-CFP and CFP-expressing cDNA vector and its product protein (in amino acids (a.a.)). The LRLRGG C-terminal end of FLAG-tagged human ISG15 is linked to the N terminus of mTurquoise2 (referred to as CFP). Control CFP is tagged with FLAG at the C-terminal end. b, cell lysates were prepared in radioimmune precipitation assay buffer 3 days after transient transfection of the vectors as indicated and immunoblotted using antibodies against GFP, ISG15, HDAC6, and p62. c, immunoblots (IB) of detergent-soluble (Soluble) and -insoluble (Insoluble) fractions of U251 with 24-h transient transfection of the indicated vectors in the presence or absence of PepA/E64d (each 20 μg/ml; 6 h) were probed for the indicated proteins. Volumes of insoluble fractions are 5-fold (×5) higher concentration than those of the soluble fractions. Numbers shown on the left indicate protein sizes (kDa). Arrows indicate the bands of cleaved ISG15 or CFP protein.
The Deacetylase Activity of HDAC6 Contributes to ISG15 Degradation
We showed in Fig. 2 that the BUZ domain of HDAC6 was essential for binding to ISG15 protein, and it has also been shown that the deacetylase activity of HDAC6 is involved in the degradation of the aggresome, which contains ubiquitinated proteins, via autophagy-lysosome degradation (10). In a similar fashion, we thus sought to determine whether HDAC6 activity was involved in ISG15 degradation. Fig. 6 shows that type I IFN as expected visibly increased ISG15 levels and also mildly increased ISG15 conjugates in both soluble and insoluble fractions. When PepA/E64D lysosomal protease inhibitors were added, there was a visible increase in ISG15 levels in the insoluble fraction. This suggests that ISG15 can be degraded by lysosomal proteases.
We then utilized tubacin (20), a relatively selective HDAC6 inhibitor, and observed a visible decrease of ISG15 levels in both soluble and insoluble fractions without much of a change in ISG15 conjugate levels. However, when PepA/E64D lysosomal protease inhibitors were also added, there was no change in ISG15 levels. This suggested that HDAC6 was needed for the degradation of ISG15 by lysosomal proteases. Tubacin also led to an increase of p62 and LC3 levels in the insoluble fraction. Therefore, inhibition of HDAC6 activity led to the accumulation of p62 and LC3-II in the insoluble fraction, suggesting inhibition of lysosome-autophagosome fusion. It also reduced total ISG15 levels in cells in response to type I IFN, but this did not lead to an increase in ISG15 when lysosomal proteases were inhibited. Those results suggested that the deacetylase activity of HDAC6 is important for degradation of ISG15 via the lysosome.
DISCUSSION
The ubiquitin-like post-translational modifier ISG15 is up-regulated during type I IFN stimulation of cells, and it becomes conjugated to newly synthesized proteins of the host cell and invading pathogens. Although this conjugation can be reversed by a specific protease, UBP43/USP18 (3), it is not known whether other cellular pathways may also be operative. In this report, for the first time, we show that 1) ISG15 interacts via its LRLRGG C-terminal motif with a BUZ domain of cytosolic Ub-binding HDAC6, implicating HDAC6 for a relevant role in the function of ISG15. That this interaction is the same as that known to occur for (poly-) ubiquitin chains binding to HDAC6 also suggests that the ISG15 pathway may represent the IFN-stimulated counterpart to the normal homeostatic ubiquitin clearance pathway. 2) ISG15 also interacts with the cargo adapter protein SQSTM1/p62. The knowledge that both p62 and HDAC6 facilitate aggregate formation (aggresomes) and clearance of ubiquitinated cargo through the LC3-II-marked autophagosome and lysosome also suggested that a clearance process (similar to that of Ubiquitination) may be occurring with ISGylation of proteins during the innate immune response. Therefore, our data strongly suggest that ISG15 conjugation marks target substrates for clearance via autophagy-lysosome pathway as proposed in the model shown in Fig. 7.
FIGURE 7.

Schematic model of ISG15-mediated degradation. a, IFNs are known to stimulate the JAK-STAT pathway to induce a number of interferon-stimulated genes including ISG15, UbE1L, UbcH8, and Herc5 that lead to the ISGylation process and UBP43 that deconjugates ISGylated proteins. b, the Lys63-linked di-Ub-like protein ISG15 conjugates to lysine (K) residues of newly synthesized proteins at the LRLRGG-terminal end (2). Some ISG15 conjugates are also deconjugated to remove ISG15. c, our findings indicate that HDAC6 and SQSTM1/p62 can independently bind ISG15 to recognize ISG15 and ISG15 conjugates directly or indirectly. ISG15 aggregates or aggresomes would be formed through oligomerization and recruitment of autophagosome-bound LC3 by p62. The proteasome inhibitor MG132 induces and enhances this aggregate formation and accumulation in the insoluble fraction. d, the deacetylase activity of HDAC6 can mediate lysosomal fusion of ISG15-containing aggregates for clearance, and this is inhibited by the specific HDAC6 inhibitor tubacin. PepA/E64d inhibit lysosomal digestion of peptide contents.
We should note that the reported data were mostly obtained under experimental conditions of forced overexpression, and we have not been able to detect this under normal, homeostatic physiological conditions. This implies that the observed results may occur primarily when the cell is stressed (forced overexpression of cDNAs, interferon stimulation, and inhibition of the proteasome and lysosomal proteases).
The structural conformation of ISG15 resembles that of Lys63-linked di-Ub rather than that of Lys48-linked di-Ub, which is known to be associated with the proteasomal pathway. Lys63 linkage appears to function in homeostatic cellular processes including signal transduction (31). In addition, when molecular chaperones and proteasome degradation systems are unable to unfold and thread misfolded/aggregated proteins through the narrow interior proteolytic chamber of the 26 S proteasome, these proteins get tagged with Lys63, designating them for the autophagy-lysosome pathway (11, 13). Molecularly, this happens through p62, which preferentially binds to polyubiquitinated proteins with Lys63-linked chains instead of those with Lys48-linked chains, resulting in the formation of oligomeric aggregates of ubiquitinated proteins, organelles, and intracellular pathogens that lead to autophagic rather than proteasomal degradation (15, 25, 32, 33). This is also in agreement with evidence indicating that Lys63-linked polyubiquitin substrates are enriched in intracellular inclusion bodies, which are also sequestered into p62-LC3-marked autophagosomes (34). Results in the present study show that p62 also binds to non-covalent ISG15 by pulldown assay and to ISG15-conjugated proteins by co-immunoprecipitation assay. In addition, using a ISG15 fusion protein with CFP via linkage of LRLRGG, we clearly showed that ISG15 itself is a target of lysosomal degradation (Fig. 5c). Therefore, this suggests that p62 also directly targets ISG15 and ISGylated peptides to form aggregates and autophagosomes in a manner that is similar to p62 interaction with Lys63-linked polyubiquitinated proteins being targeted to autophagosomes.
ISG15 and its conjugates have been shown to possess activity against several viruses using cultured cells or animal models (4). The antiviral mechanisms of ISG15 include inhibition of viral release and budding as well as direct ISG15 conjugation of cellular proteins including antiviral factors (e.g. IRF3, retinoic acid induced gene-I (RIG-I), and protein kinase R) and other ISG proteins. Also, ISG15 can be directly conjugated with capsid proteins of human papillomavirus and Sindbis virus (2, 35). However, it is unknown how ISG15 modification of viral proteins acts mechanistically. Our findings show that ISG15 not only co-localizes with p62 and HDAC6 in the same intracellular punctate areas (puncta) but also associates with them. p62 and other sequestrome-1-like receptors function as cargo adaptor proteins that target substrates to autophagosomes via the LC3-interacting region motif. In xenophagy, ubiquitination of invading bacteria is required for antimicrobial autophagy through p62 cargo recognition and degradation (36). In a similar manner to antiviral autophagy (virophagy), capsids of Sindbis virus and Chikungunya virus are targets of autophagosomes through binding of p62, and Chikungunya virus capsids are also modified by ubiquitin (37, 38). Therefore, future studies should elucidate whether ISG15 enhances degradation or sequestration of viral proteins via p62- and HDAC6-mediated selective autophagy.
HDAC6 functions to deacetylate cytoplasmic proteins (e.g. α-tubulin), and catalytic inhibition leads to hyperacetylation of α-tubulin, which stimulates canonical (bulk) autophagy by kinesin-1 recruitment to microtubules for autophagosome transport toward the microtubule-organizing center (39, 40). In non-canonical (selective) autophagy, HDAC6 regulates the fusion of autophagosome and lysosome through both catalytic deacetylase and ubiquitin binding activities (10). We also observed that this deacetylase activity is associated with ISG15 degradation via the lysosome-autophagosome (Fig. 6). HDAC6 preferentially interacts with Lys63-linked polyubiquitinated protein aggregates in vivo (16). However, there is no biochemical evidence that HDAC6 directly binds to Ub-conjugated proteins in vitro (12). We also identify for the first time in this report that the BUZ domain of HDAC6 interacts with the C-terminal LRLRGG peptide of free ISG15. Coupled with the co-immunoprecipitation evidence of FLAG-HDAC6 interaction with ISG15 conjugates, we speculate that HDAC6 may recognize and associate with unanchored free ISG15 in the p62 cargo, which also sequesters the substrates of ISG15 conjugation.
The multifaceted adapter p62 plays critical roles in the regulation of a broad range of signaling pathways that respond to inflammatory and oxidative stresses through its binding to Lys63-linked Ub proteins and related binding partners such as tumor necrosis factor receptor-associated factor 6 and Kelch-like ECH-associated protein 1 (31, 41). ISG15 is also involved in the innate immune signaling pathway. In fact, IFN-induced ISG proteins including IRF3 are known substrates of ISG15 conjugation, and their function is modulated by ISGylation (42). Our study also shows for the first time that p62 is involved in ISG15-mediated signaling pathways in a manner similar to that of p62 involvement in Lys63-linked Ub-mediated signaling in published reports (15, 25). However, it is not known how p62 distinguishes ISG15-conjugated proteins that remain active to perform a function from those that are targets for a degradation tag. One possible explanation is that, for ISGylated proteins to remain active, p62 scaffolding with ISG15 conjugates forms without free ISG15 and/or free Lys63-linked Ub chains. Conversely, for proteins destined for degradation, sequestration into disorganized aggresomes occurs (and this could be induced by blocking the UPS with MG132) in combination with HDAC6 and LC3 recruitment (12). In fact, we did find accumulation of free ISG15 in the detergent-insoluble fraction of cells with p62 and lipid-bound LC3-II as a function of MG132 dose. Furthermore, the catalytic inhibition of HDAC6 with tubacin also suppressed lysosomal ISG15 degradation. Therefore, it is likely that type I IFN signaling induces ISG15, which in turn strengthens the basal activity of selective autophagy, leading to efficient removal of ubiquitin and ISG15-tagged unwelcome proteins and pathogens through p62 and HDAC6.
In summary, our studies provide novel evidence for ISG15 association with HDAC6 and p62, implying that ISGylation can lead to autophagic degradation in pathways that are similar to those utilized by the Ub system. ISG15-mediated protein conjugation may thus be the IFN-based system that, together with or in lieu of Ub-based selective autophagy, allows the cell to use autophagy as an intrinsic antipathogen defense.
This work was supported, in whole or in part, by National Institutes of Health Grants P01 CA069246 and P01 CA163205 from the NCI. This work was also supported by a sundry fund from the Brigham and Women's Hospital (to E. A. C.).
H. Nakashima, T. Nguyen, W. F. Goins, and E. A. Chiocca, unpublished data.
- ISG
- interferon-stimulated gene
- HDAC
- histone deacetylase
- BUZ
- binder of ubiquitin zinc finger
- Ub
- ubiquitin
- ISGylation
- ISG15 conjugation
- UPS
- ubiquitin-proteasome system
- CD
- catalytic domain
- SE14
- serine-glutamate-containing tetradecapeptide
- PepA
- pepstatin A
- Dox
- doxycycline.
REFERENCES
- 1. Sadler A. J., Williams B. R. (2008) Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 8, 559–568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Durfee L. A., Lyon N., Seo K., Huibregtse J. M. (2010) The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell 38, 722–732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Malakhov M. P., Malakhova O. A., Kim K. I., Ritchie K. J., Zhang D. E. (2002) UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 277, 9976–9981 [DOI] [PubMed] [Google Scholar]
- 4. Morales D. J., Lenschow D. J. (2013) The antiviral activities of ISG15. J. Mol. Biol. 425, 4995–5008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Romijn R. A., Westein E., Bouma B., Schiphorst M. E., Sixma J. J., Lenting P. J., Huizinga E. G. (2003) Mapping the collagen-binding site in the von Willebrand factor-A3 domain. J. Biol. Chem. 278, 15035–15039 [DOI] [PubMed] [Google Scholar]
- 6. Liu M., Li X. L., Hassel B. A. (2003) Proteasomes modulate conjugation to the ubiquitin-like protein, ISG15. J. Biol. Chem. 278, 1594–1602 [DOI] [PubMed] [Google Scholar]
- 7. Schmeisser H., Bekisz J., Zoon K. C. (2014) New function of type I IFN: induction of autophagy. J. Interferon Cytokine Res. 34, 71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johansen T., Lamark T. (2011) Selective autophagy mediated by autophagic adapter proteins. Autophagy 7, 279–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Shaid S., Brandts C. H., Serve H., Dikic I. (2013) Ubiquitination and selective autophagy. Cell Death Differ. 20, 21–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee J. Y., Koga H., Kawaguchi Y., Tang W., Wong E., Gao Y. S., Pandey U. B., Kaushik S., Tresse E., Lu J., Taylor J. P., Cuervo A. M., Yao T. P. (2010) HDAC6 controls autophagosome maturation essential for ubiquitin-selective quality-control autophagy. EMBO J. 29, 969–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Tan J. M., Wong E. S., Kirkpatrick D. S., Pletnikova O., Ko H. S., Tay S. P., Ho M. W., Troncoso J., Gygi S. P., Lee M. K., Dawson V. L., Dawson T. M., Lim K. L. (2008) Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum. Mol. Genet. 17, 431–439 [DOI] [PubMed] [Google Scholar]
- 12. Hao R., Nanduri P., Rao Y., Panichelli R. S., Ito A., Yoshida M., Yao T. P. (2013) Proteasomes activate aggresome disassembly and clearance by producing unanchored ubiquitin chains. Mol. Cell 51, 819–828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pankiv S., Clausen T. H., Lamark T., Brech A., Bruun J. A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282, 24131–24145 [DOI] [PubMed] [Google Scholar]
- 14. Babu J. R., Geetha T., Wooten M. W. (2005) Sequestosome 1/p62 shuttles polyubiquitinated τ for proteasomal degradation. J. Neurochem. 94, 192–203 [DOI] [PubMed] [Google Scholar]
- 15. Long J., Gallagher T. R., Cavey J. R., Sheppard P. W., Ralston S. H., Layfield R., Searle M. S. (2008) Ubiquitin recognition by the ubiquitin-associated domain of p62 involves a novel conformational switch. J. Biol. Chem. 283, 5427–5440 [DOI] [PubMed] [Google Scholar]
- 16. Olzmann J. A., Li L., Chudaev M. V., Chen J., Perez F. A., Palmiter R. D., Chin L. S. (2007) Parkin-mediated K63-linked polyubiquitination targets misfolded DJ-1 to aggresomes via binding to HDAC6. J. Cell Biol. 178, 1025–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kawaguchi Y., Kovacs J. J., McLaurin A., Vance J. M., Ito A., Yao T. P. (2003) The deacetylase HDAC6 regulates aggresome formation and cell viability in response to misfolded protein stress. Cell 115, 727–738 [DOI] [PubMed] [Google Scholar]
- 18. Pai M. T., Tzeng S. R., Kovacs J. J., Keaton M. A., Li S. S., Yao T. P., Zhou P. (2007) Solution structure of the Ubp-M BUZ domain, a highly specific protein module that recognizes the C-terminal tail of free ubiquitin. J. Mol. Biol. 370, 290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zuin A., Carmona M., Morales-Ivorra I., Gabrielli N., Vivancos A. P., Ayté J., Hidalgo E. (2010) Lifespan extension by calorie restriction relies on the Sty1 MAP kinase stress pathway. EMBO J. 29, 981–991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Haggarty S. J., Koeller K. M., Wong J. C., Grozinger C. M., Schreiber S. L. (2003) Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U.S.A. 100, 4389–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Szymczak A. L., Workman C. J., Wang Y., Vignali K. M., Dilioglou S., Vanin E. F., Vignali D. A. (2004) Correction of multi-gene deficiency in vivo using a single 'self-cleaving' 2A peptide-based retroviral vector. Nat. Biotechnol. 22, 589–594 [DOI] [PubMed] [Google Scholar]
- 22. Reyes-Turcu F. E., Horton J. R., Mullally J. E., Heroux A., Cheng X., Wilkinson K. D. (2006) The ubiquitin binding domain ZnF UBP recognizes the C-terminal diglycine motif of unanchored ubiquitin. Cell 124, 1197–1208 [DOI] [PubMed] [Google Scholar]
- 23. Ouyang H., Ali Y. O., Ravichandran M., Dong A., Qiu W., MacKenzie F., Dhe-Paganon S., Arrowsmith C. H., Zhai R. G. (2012) Protein aggregates are recruited to aggresome by histone deacetylase 6 via unanchored ubiquitin C termini. J. Biol. Chem. 287, 2317–2327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kirkin V., McEwan D. G., Novak I., Dikic I. (2009) A role for ubiquitin in selective autophagy. Mol. Cell 34, 259–269 [DOI] [PubMed] [Google Scholar]
- 25. Seibenhener M. L., Babu J. R., Geetha T., Wong H. C., Krishna N. R., Wooten M. W. (2004) Sequestosome 1/p62 is a polyubiquitin chain binding protein involved in ubiquitin proteasome degradation. Mol. Cell. Biol. 24, 8055–8068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ye Y., Godzik A. (2003) Flexible structure alignment by chaining aligned fragment pairs allowing twists. Bioinformatics 19, Suppl. 2, ii246–ii255 [DOI] [PubMed] [Google Scholar]
- 27. Narasimhan J., Wang M., Fu Z., Klein J. M., Haas A. L., Kim J. J. (2005) Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J. Biol. Chem. 280, 27356–27365 [DOI] [PubMed] [Google Scholar]
- 28. Komander D., Reyes-Turcu F., Licchesi J. D., Odenwaelder P., Wilkinson K. D., Barford D. (2009) Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep. 10, 466–473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Trempe J. F., Brown N. R., Noble M. E., Endicott J. A. (2010) A new crystal form of Lys48-linked diubiquitin. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 66, 994–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Potter J. L., Narasimhan J., Mende-Mueller L., Haas A. L. (1999) Precursor processing of pro-ISG15/UCRP, an interferon-β-induced ubiquitin-like protein. J. Biol. Chem. 274, 25061–25068 [DOI] [PubMed] [Google Scholar]
- 31. Komatsu M., Kurokawa H., Waguri S., Taguchi K., Kobayashi A., Ichimura Y., Sou Y. S., Ueno I., Sakamoto A., Tong K. I., Kim M., Nishito Y., Iemura S., Natsume T., Ueno T., Kominami E., Motohashi H., Tanaka K., Yamamoto M. (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 12, 213–223 [DOI] [PubMed] [Google Scholar]
- 32. Itakura E., Mizushima N. (2011) p62 Targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J. Cell Biol. 192, 17–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Searle M. S., Garner T. P., Strachan J., Long J., Adlington J., Cavey J. R., Shaw B., Layfield R. (2012) Structural insights into specificity and diversity in mechanisms of ubiquitin recognition by ubiquitin-binding domains. Biochem. Soc. Trans. 40, 404–408 [DOI] [PubMed] [Google Scholar]
- 34. Wooten M. W., Geetha T., Babu J. R., Seibenhener M. L., Peng J., Cox N., Diaz-Meco M. T., Moscat J. (2008) Essential role of sequestosome 1/p62 in regulating accumulation of Lys63-ubiquitinated proteins. J. Biol. Chem. 283, 6783–6789 [DOI] [PubMed] [Google Scholar]
- 35. Lenschow D. J., Giannakopoulos N. V., Gunn L. J., Johnston C., O'Guin A. K., Schmidt R. E., Levine B., Virgin H. W., 4th (2005) Identification of interferon-stimulated gene 15 as an antiviral molecule during Sindbis virus infection in vivo. J. Virol. 79, 13974–13983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Boyle K. B., Randow F. (2013) The role of 'eat-me' signals and autophagy cargo receptors in innate immunity. Curr. Opin. Microbiol. 16, 339–348 [DOI] [PubMed] [Google Scholar]
- 37. Orvedahl A., MacPherson S., Sumpter R., Jr., Tallóczy Z., Zou Z., Levine B. (2010) Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7, 115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Judith D., Mostowy S., Bourai M., Gangneux N., Lelek M., Lucas-Hourani M., Cayet N., Jacob Y., Prévost M. C., Pierre P., Tangy F., Zimmer C., Vidalain P. O., Couderc T., Lecuit M. (2013) Species-specific impact of the autophagy machinery on Chikungunya virus infection. EMBO Rep. 14, 534–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Geeraert C., Ratier A., Pfisterer S. G., Perdiz D., Cantaloube I., Rouault A., Pattingre S., Proikas-Cezanne T., Codogno P., Poüs C. (2010) Starvation-induced hyperacetylation of tubulin is required for the stimulation of autophagy by nutrient deprivation. J. Biol. Chem. 285, 24184–24194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Reed N. A., Cai D., Blasius T. L., Jih G. T., Meyhofer E., Gaertig J., Verhey K. J. (2006) Microtubule acetylation promotes kinesin-1 binding and transport. Curr. Biol. 16, 2166–2172 [DOI] [PubMed] [Google Scholar]
- 41. Sanz L., Diaz-Meco M. T., Nakano H., Moscat J. (2000) The atypical PKC-interacting protein p62 channels NF-κB activation by the IL-1-TRAF6 pathway. EMBO J. 19, 1576–1586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shi H. X., Yang K., Liu X., Liu X. Y., Wei B., Shan Y. F., Zhu L. H., Wang C. (2010) Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell. Biol. 30, 2424–2436 [DOI] [PMC free article] [PubMed] [Google Scholar]