

Background: Viruses exploit a variety of cell surface receptors to infect their hosts.
Results: An integrin recognition motif in AAV9 was found to play a multifunctional role in systemic transport and cellular uptake.
Conclusion: Recombinant AAV vectors might exploit integrins during cellular uptake in different host tissues.
Significance: Host-receptor interactions influence the biodistribution of recombinant AAV vectors.
Keywords: Cell Surface Receptor, Gene Therapy, Integrin, Viral Protein, Virus Entry, Adeno-Associated Virus
Abstract
Adeno-associated viruses (AAVs) display a highly conserved NGR motif on the capsid surface. Earlier studies have established this tripeptide motif as being essential for integrin-mediated uptake of recombinant AAV serotype 2 (AAV2) in cultured cells. However, functional attributes of this putative integrin recognition motif in other recombinant AAV serotypes displaying systemic transduction in vivo remain unknown. In this study, we dissect the biology of an integrin domain capsid mutant derived from the human isolate AAV9 in mice. The AAV9/NGA mutant shows decreased systemic transduction in mice. This defective phenotype was accompanied by rapid clearance of mutant virions from the blood circulation and nonspecific sequestration by the spleen. Transient vascular hyperpermeability, induced by histamine coinjection, exacerbated AAV9/NGA uptake by the spleen but not the liver. However, such treatment did not affect AAV9 virions, suggesting a potential entry/post-entry defect for the mutant in different tissues. Further characterization revealed modestly decreased cell surface binding but a more pronounced defect in the cellular entry of mutant virions. These findings were corroborated by the observation that blocking multiple integrins adversely affected recombinant AAV9 transduction in different cell types, albeit with variable efficiencies. From a structural perspective, we observed that the integrin recognition motif is located in close proximity to the galactose binding footprint on AAV9 capsids and postulate that this feature could influence cell surface attachment, cellular uptake at the tissue level, and systemic clearance by the reticuloendothelial system.
Introduction
Adeno-associated viruses (AAVs)3 belong to the Dependovirus genus of the Parvoviridae subfamily (1, 2). These small, non-enveloped, single-strand DNA viruses are classified into six distinct clades, A through F, isolated from several different animal sources (3). The subject of this study, AAV serotype 9/AAV isolate Hu.14, belongs to clade F. Of the numerous recombinant AAV strains currently being developed into gene transfer vectors, AAV9 is one of few isolates that has displayed a propensity to traverse the vasculature with high efficiency following systemic administration. Consequently, widespread and robust transduction of multiple tissues, including the heart, liver, skeletal muscle, lung, and, notably, the brain, by AAV9 vectors has been reported (4). Efforts to understand the structural attributes and molecular mechanisms that facilitate the systemic transduction profile of recombinant AAV9 are ongoing. Key findings to date include cryo-EM and x-ray crystallographic determination of the three-dimensional structure of the AAV9 capsid (5), the discovery of galactosylated glycans as the primary cell surface attachment factor for AAV9 (6, 7), elucidation of the residues that form the galactose-binding footprint (8), and functional annotation of several other key residues on the AAV9 capsid (9).
The first critical step in recombinant AAV transduction, much like other non-enveloped viruses, involves recognition of glycans for cell surface attachment (10). Subsequent to binding, cellular uptake of different AAV serotypes appears to involve specific coreceptors on the cell surface. For instance, the FGF receptor is exploited by AAV2 (11), whereas the hepatocyte growth factor receptor/C-Met appears to be utilized by both AAV2 and AAV3 (12, 13). Further, platelet-derived growth factor and epidermal growth factor receptors have been implicated in the cellular uptake of AAV5 and AAV6, respectively (14, 15). In addition to these coreceptors that presumably contribute to the differential transduction profiles of recombinant AAV serotypes, earlier reports have implied an essential role for integrins in the cellular uptake of AAV2 capsids (16, 17). Importantly, the identification of a highly conserved integrin binding motif (NGR) in the major capsid protein (VP3) subunit of a vast majority of AAV serotypes might suggest a nonspecific role for integrins in recombinant AAV transduction (18).
Previous studies by our laboratory demonstrated that glycan binding avidity plays a critical role in determining the systemic fate of different AAV serotypes following intravenous administration (19). In this report, we expand our mechanistic understanding of AAV systemic transport by interrogating the role of integrins in viral uptake in vitro and systemic transport in vivo. Specifically, we validate the functional role of the integrin binding NGR motif within the context of another recombinant AAV serotype, i.e. AAV9, through both in vitro and in vivo experiments. Further, we present data that confirm the essential role played by different integrins in mediating AAV cell entry. Our results clearly demonstrate that the inability to engage integrins in vivo can adversely affect cellular entry of mutant AAV9 within different tissues. This defect is accompanied by rapid clearance of mutant vectors from the systemic circulation because of nonspecific uptake by the reticuloendothelial system.
MATERIALS AND METHODS
Plasmids and Mutants
The R514A mutation resulting in the disruption of the putative integrin recognition motif 512NGR514 to non-functional 512NGA514 was introduced into the AAV9 Cap gene in the plasmid pXR9 using the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). The mutagenesis primer 5′-GCTTCTTCTTGGGCTCTCAATGGAGCCAATAGCTTGATGAATCCTGG-3′ (IDT, Coralville, IA) was utilized to generate individual clones that were sequenced and verified by the UNC Genome Analysis facility. Sequence analysis and alignments were carried out using VectorNTI® software (Invitrogen).
Virus Production and Titers
Recombinant AAV9 and the AAV9/NGA mutant packaging reporter transgenes were generated using an updated triple plasmid transfection and purification method described earlier (20). The plasmids utilized were the AAV9 helper pXR9 or pXR9/NGA, pXX6–80 expressing adenoviral helper genes, and pTR-CBA-Luc containing the chicken β-actin (CBA) promoter-driven firefly luciferase transgene flanked by AAV inverted terminal repeats derived from the AAV2 genome. Viral titers were obtained by quantitative PCR using a Roche Lightcycler® 480 (Roche Applied Sciences). Quantitative PCR primers were designed to specifically recognize the luciferase transgene (forward, 5′-CTT CTT GGG CTC TCA ATG GAG CTA ATA GCT TGA TGA ATC CTG-3′; reverse, 5′-CAG GAT TCA TCA AGC TAT TAG CTC CAT TGA GAG CCC AAG AAG-3′).
Cell Lines, Anti-integrin Antibodies, and Lentiviral shRNA Constructs
CHO Lec2 cells were maintained in α modified Eagle's medium, whereas HEK293 and HeLa cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% FBS, 100 units/ml of penicillin, 100 μg/ml of streptomycin, and 2.5 μg/ml of amphotericin B (Sigma-Aldrich, St. Louis, MO). Cells were maintained in 5% CO2 at 37 °C unless indicated otherwise. Primary human umbilical vein endothelial cells (HUVECs) were cultured in endothelial growth medium 2 with added supplements (Lonza, Allendale, NJ). Unless indicated otherwise, different cell lines were desialylated prior to AAV9 infection by treatment with recombinant sialidase (neuraminidase, Sigma) as described earlier (7) to expose the cell attachment receptor, i.e. terminal galactose.
For competitive inhibition studies, anti-integrin antibodies directed against different α and β subunit combinations were obtained from Abcam (catalog no. ab24694, mouse monoclonal (P1F6) to integrin α V + β 5; catalog no. ab78289, mouse monoclonal (27.1 (VNR-1)) to integrin α V + β 3; catalog no. ab30388, mouse monoclonal (JB1B) to integrin β 1; catalog no. ab16821, mouse monoclonal (272-17E6) to integrin α V; catalog no. ab75472, mouse monoclonal (P1D6) to integrin α 5 + β 1; and catalog no. ab75872, rabbit monoclonal (EPR2417Y) to integrin β 3). This specific subset of antibodies was selected for evaluation on the basis of prior studies implicating different integrins in AAV2 infection (16–18). Desialylated HEK293 cells, HeLa cells, or HUVECs with exposed surface galactose were then pretreated with anti-integrin antibodies (20 μg/ml) for 60 min at 37 °C to facilitate receptor blockade prior to infection with AAV9 (1000 vg/cell). Transduction efficiency was then determined by quantitation of luciferase transgene expression in cell lysates with D-luciferin (Promega) as substrate on a Victor2® luminometer (PerkinElmer Life Sciences).
For stable knockdown of integrins, HeLa cells were plated at a density of 5 × 104 cells/well in a 24-well plate and allowed to attach overnight. Cells were infected with shRNA-expressing lentiviral vectors obtained from the University of North Carolina lenti-shRNA core facility via spin infection in the presence of 4 μm Polybrene. On the basis of results from antibody blocking studies described earlier, only the integrin β 3 subunit was chosen as a target for knockdown (three clones targeted against ITGB3, NM_000212.2, oligo ID no. TRCN0000003236-3238). Detailed information pertaining to these constructs are available on the GenomeRNAi database (21–23). Lentiviral vectors expressing shRNA against the GFP transgene were utilized as a control. The virus was removed 24 h post-infection, and selection with 1 μm puromycin was initiated 48 h post-infection. Selection was continued for 48 h, and then cells were synchronously infected with ssAAV9-CBA-fLuc at a multiplicity of infection of 10,000 μg/cell. Luciferase activity was assayed 24 h post-infection as described above.
Cellular Binding, Uptake, and Transduction Assays
CHO Lec2 cells were seeded in 24-well plates at a density of 1 × 105 cells/well 18 h prior to the experiment. The cells were first prechilled at 4 °C for 30 min, and then AAV9 or AAV9/NGA packaging the CBA-luc transgene was incubated on Lec2 cells at 4 °C for 1 h at 1000 vg/cell, followed by three washes with ice-cold 1× PBS to remove unbound virions. To quantitate the number of cell surface-bound virions, 200 μl of deionized H2O was added to each well, and cells were subjected to three freeze-thaw cycles prior to extraction of total genomic DNA using a DNeasy® kit (Qiagen, Germantown, MD). Viral genome titers were then determined by qPCR as described earlier.
For cellular uptake studies following removal of unbound virions, Lec2 cells in freshly added α modified Eagle's medium + 10% FBS were moved to a 37 °C incubator to synchronize virus internalization. After either 5 or 60 min of incubation, cells were treated with 150 μl/well of 0.05% trypsin at 37 °C for 5 min to release cell surface-associated virions. Trypsin was then quenched by adding 150 μl/well of DMEM with 10% FBS into each well, followed by three washes of the cell pellets with ice-cold PBS. Total genomic DNA was then extracted as described earlier and vg copy numbers were determined by qPCR.
For transduction assays, Lec2 cells in freshly added α modified Eagle's medium + 10% FBS were infected with AAV9 or AAV9/NGA (different multiplicities of infection) and maintained at 37 °C as outlined above. Cells were then lysed at 18 h post-transduction to quantitate luciferase transgene expression using a Victor2® luminometer (PerkinElmer Life Sciences).
Animal Studies
All animal experiments were carried out using 6- to 8-week-old female BALB/c mice purchased from Jackson ImmunoResearch Laboratories. Mice were maintained and treated in accordance with NIH guidelines and as approved by the University of North Carolina Institutional Animal Care and Use Committee. BALB/c mice were injected intravenously with AAV9 or AAV9/NGA vectors (1 × 1011 vg/animal) through the tail vein. To image luciferase transgene expression patterns in mice 3 weeks after vector administration, animals were anesthetized with 2% isoflurane prior to intraperitoneal injection of D-luciferin (120 mg/kg, Nanolight, Pinetop, AZ) and imaged using an Xenogen IVIS Lumina® system (PerkinElmer Life Sciences/Caliper Life Sciences). To quantify luciferase expression, mice subjected to live animal imaging were sacrificed, and different tissues were harvested for analysis. Tissue lysates were generated by adding 150 μl of 2× passive lysis buffer (Promega, Madison, WI) prior to mechanical lysis using a Tissue Lyser II 352 system (Qiagen). Tissue lysates were then pelleted to remove debris by centrifugation, and 50 μl of the supernatant from each lysate was subjected to luminometric analysis using a Victor2® luminometer (PerkinElmer Life Sciences). In addition, 2 μl of supernatant was used for a Bradford assay (Bio-Rad) to determine protein content in each sample.
For viral biodistribution analysis, different organs were harvested from BALB/c mice in the same treatment group that were sacrificed 3 days or 3 weeks post-administration. Approximately 50 mg of tissue from each systemic organ was pretreated with proteinase K prior to being homogenized using a tissue lyser as described earlier. Total genomic DNA was then extracted from tissue lysates using a DNeasy® kit (Qiagen) and viral genome copy numbers determined by qPCR of the luciferase transgene using the same primers listed earlier. The number of cells in each sample was quantified using qPCR with primers specific to the mouse LaminB housekeeping gene (forward, 5′-GGA CCC AAG GAC TAC CTC AAG GG-3′; reverse, 5′-AGG GCA CCT CCA TCT CGG AAA C-3′).
For pharmacokinetic analysis, BALB/c mice were injected intravenously with 1 × 1011 vg copies of AAV9 or AAV9/NGA vectors through the tail vein. 10-μl aliquots of whole blood were then collected by nicking the tail vein at the following time points post injection: 5 min, 15 min, 30 min, 1 h, 3 h, 6 h, 24 h, 48 h, and 72 h. Total DNA was then extracted from each blood sample using a Dneasy® kit, and viral genome copy numbers were measured by qPCR.
For histamine-induced vascular hyperpermeability studies, 5 mg of histamine (Sigma-Aldrich) was dissolved in 200 μl of PBS containing AAV9-CBA-Luc or AAV9/NGA-CBA-Luc, and vector doses ranging from 1–5 × 1011 vg were injected into mice via the tail vein. Control animals received AAV9 or mutant virions in PBS without histamine. Mice were sacrificed 30 min post-administration and perfused transcardially with PBS followed by 4% paraformaldehyde in PBS. The spleen was harvested, fixed overnight with 4% paraformaldehyde at 4 °C, and then transferred to PBS. Tissue sections of 50 μm were processed using a Leica vibrating blade microtome, and immunostaining for AAV9 capsids was carried out as described earlier (19). Briefly, a 10-fold dilution of hybridoma medium containing monoclonal anti-AAV9 antibodies (ADK9) followed by Alexa Fluor 647 goat-anti-mouse secondary antibody was utilized to carry out these studies. Immunostained tissue sections were mounted with ProLong Gold antifade reagent with DAPI (Invitrogen) and imaged with a Zeiss 710 confocal microscope with a ×63 oil objective.
Structural Modeling
Coordinates for the AAV2 and AAV9 viral protein (VP) crystal structures were obtained from the RCSB Protein Data Bank (PDB codes 1LP3 and 3UX1, respectively) (5, 24). Three-dimensional models of the AAV2 and AAV9 VP3 subunit trimers were created using the Oligomer Generator utility in VIPERdb-Virus Particle ExploreR2 (25). Structural models were then visualized using PyMOL, with heparan sulfate binding residues on AAV2 (VP1 numbering Arg484, Arg487, Lys532, Arg585, and Arg588) (26–29) and the galactose binding residues on AAV9 (VP1 numbering Asp271, Asn272, Tyr446, Asn470, and Trp503) (8). The putative integrin recognition motif is highlighted by residues 511NGR513 on AAV2 and 512NGR514 (VP1 numbering) on AAV9.
Statistical Analysis
All data are expressed as mean ± S.E. Unless specified otherwise, a two-tailed unpaired Student's t test was used for all statistical analyses. p < 0.05 was considered significant (*, p < 0.05; **, p < 0.01; ***, p < 0.005).
RESULTS AND DISCUSSION
Mutation of the AAV9 integrin recognition motif (NGR) abrogates transduction in vivo. Intravenously administered recombinant AAV9 virions likely interact first with the vascular endothelium before uptake by other cell types in major systemic organs such as the heart, lung, and liver (19, 30). To address the impact of the NGA mutation in vivo, we studied the transduction profile and pharmacokinetics of recombinant AAV9 and AAV9/NGA vectors in mice. As shown in Fig. 1A, live animal bioluminescent images obtained 3 weeks after vector administration display a significant decrease in luciferase reporter transgene expression in the case of the AAV9/NGA mutant. These data were further corroborated by quantitative assessment of luciferase expression in tissue lysates obtained from major systemic organs, including the brain, heart, liver, lung, and spleen (Fig. 1B). Specifically, the AAV9/NGA mutant displays a more than 100-fold reduction in transduction in the murine heart and liver.
FIGURE 1.
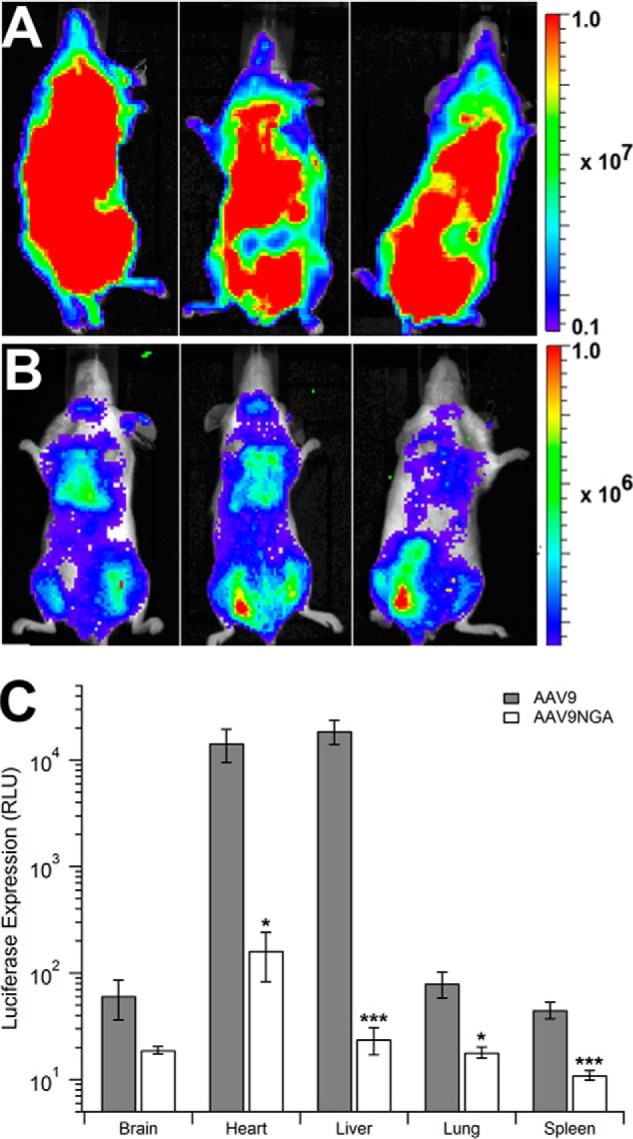
In vivo transduction profile of the AAV9NGA mutant in mice. BALB/c mice (n = 3) were injected intravenously with AAV9 (A) or AAV9/NGA (B) vectors (1 × 1011 vg/mouse) through the tail vein. Luciferase transgene expression patterns in mice were imaged using a Xenogen IVIS Lumina® system 3 weeks post-injection. Note that the rainbow scale indicates total flux (photons/second/square centimeter/steradian) and is displayed at an order of magnitude lower in B compared with A. To quantify luciferase expression (C), mice subjected to live animal imaging were sacrificed at 3 weeks, and different tissues were harvested for analysis. Tissue lysates from AAV9-treated (gray columns) or AAV9NGA-treated (white columns) animals were subjected to luminometric analysis using a Victor2® luminometer and normalized to the mass of each tissue sample. Error bars represent mean ± S.E. *, p < 0.05; ***, p < 0.005. RLU, relative light unit(s).
The AAV9/NGA mutant is cleared rapidly from the blood circulation and sequestered by the spleen. To understand the mechanism(s) resulting in decreased transduction efficiency of AAV9/NGA particles, we first determined viral genome copy numbers in different organs at two different time intervals. As seen in Fig. 2, A and B, we observed a similar biodistribution profile on days 3 or 21 after administration. Although vector genome copy numbers were reduced 2- to 4-fold in major systemic organs, these changes were not statistically significant and could not fully explain the large defect in transduction of the AAV9/NGA mutant. The lower vg copy numbers observed at the later time interval are expected because single-stranded AAV genomes are known to be rapidly degraded following capsid uncoating in different tissues (31–33). Interestingly, despite the reduced transgene expression mediated by AAV9/NGA vectors in the spleen, we observed a statistically significant (> a log order) increase in vector genomes within this organ compared with recombinant AAV9 vectors at both time intervals. This observation suggests that AAV9/NGA particles are potentially sequestered in large numbers by nonspecific mechanisms such as phagocytic uptake by the reticuloendothelial system at early time intervals. Nonspecific uptake of mutant AAV9/NGA virions by endothelial cells lining different tissues could potentially explain the modest decrease in vg copy numbers within each tissue. Alternatively, it is possible that the mutant virions are capable of successful trans-endothelial transport but suffer from a cellular entry defect after entering the tissue interstitium.
FIGURE 2.

Biodistribution and blood clearance of the AAV9NGA mutant in mice. BALB/c mice (n = 4) treated with AAV9 (gray bars) or AAV9NGA (white columns) were sacrificed 3 days (A) or 3 weeks (B) after administration to harvest different organs. Total genomic DNA was then extracted from lysates obtained from homogenized tissue samples. Viral genome copy numbers were normalized to the total number of copies of the mouse lamin gene in each sample as determined by quantitative PCR. For pharmacokinetic analysis (C), BALB/c mice (n = 4) were injected intravenously with 1 × 1011 vg copies of AAV9 (solid circles) or AAV9/NGA (triangles) vectors through the tail vein. Viral genome copy numbers were determined by qPCR from 10-μl aliquots of whole blood collected by nicking the tail vein at different time intervals. Error bars represent mean ± S.E. ***, p < 0.005.
The notion of reticuloendothelial system clearance is further corroborated by the observation that AAV9/NGA virions are rapidly cleared from the blood circulation following a single intravenous bolus injection, as measured by quantitative PCR. As shown in Fig. 2C, over a 72-h period, we observed striking differences in the blood circulation profiles of AAV9 and AAV9/NGA virions. Although tissue distribution profiles overlapped over the initial 6-h period, the AAV9/NGA virions were more rapidly eliminated at later time intervals (24, 48, and 72 h). These results suggest that mutant virions are likely defective in binding/entry, resulting in rapid clearance from the blood circulation by the spleen and endothelial system.
Increasing vascular permeability in tissues does not rescue the AAV9/NGA mutant from splenic sequestration. We hypothesized that if binding was the major defect, then we should be able to reverse reticuloendothelial system clearance of mutant AAV9/NGA virions, possibly by transiently permeabilizing the vascular wall and allowing entry into different tissues. To test this hypothesis, we transiently permeabilized the vasculature by coadministering histamine with AAV vectors (34, 35). Confocal fluorescence microscopy of spleen and liver tissues immunostained for AAV9 or mutant virions was then carried out to assess the impact of vascular hyperpermeability on vascular transport and systemic uptake. As shown in Fig. 3, A and B, histamine coinjection does not appear to affect the accumulation of AAV9 capsids in the spleen or the liver (Fig. 3, E and F), suggesting that systemic tissue uptake of these virions is fully functional and does not benefit from increased vascular permeability. In contrast, the AAV9/NGA mutant shows markedly increased splenic accumulation rather than reversal of this phenomenon in histamine-treated animals (Fig. 3, C and D). Liver uptake of the mutant virions does not appear to be influenced (Fig. 3, G and H). These results corroborate the notion that AAV9/NGA virions suffer from defective cellular entry after entering the tissue interstitial space. Concomitant with this defect, mutant virions are subject to nonspecific, phagocytic uptake mechanisms, predominantly within the spleen. The latter appears to be exacerbated by the administration of histamine because increased vascular permeability does not alleviate a cellular entry defect.
FIGURE 3.
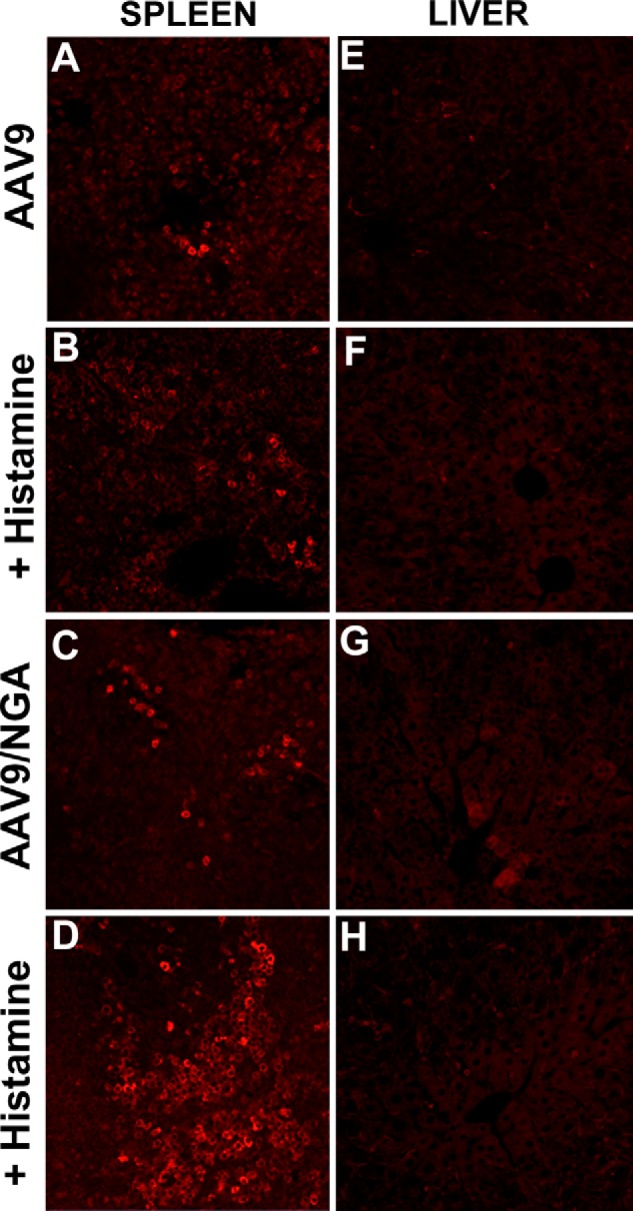
Histamine-induced vascular hyperpermeability affects viral sequestration patterns. BALB/c mice (n = 4) were treated with AAV9 or AAV9/NGA mutant virions alone or coinjected with histamine to concurrently induce vascular hyperpermeability. Spleen (A–D) and liver (E–H) tissue sections were immunostained with the ADK9 mouse monoclonal antibody against AAV9 capsids followed by an Alexa Fluor 647 goat anti-mouse secondary antibody (red). Immunostained tissue sections were mounted with ProLong Gold antifade reagent with×63 oil objective. Representative micrographs obtained from multiple tissue samples are shown.
The AAV9/NGA mutant is defective in cell surface binding as well as uptake. To further examine mechanisms underlying the defective systemic transport displayed by the AAV9/NGA mutant, we carried out transduction, binding, uptake, and confocal microscopy studies in the permissive CHO Lec2 cell line, which overexpresses terminal galactose residues, the primary attachment receptor for AAV9 (7). As shown in Fig. 4A, the AAV9/NGA mutant is transduction-deficient at different multiplicities of infection in Lec2 cells. Assessment of cell surface binding revealed that the AAV9/NGA mutant was defective when compared with parental AAV9 in engaging galactose residues on the Lec2 cell surface (Fig. 4B). Specifically, a comparison of the binding potentials (defined as the ratio of the maximum binding capacity and the relative binding affinity of virions per cell) revealed a value of 4.54 × 10−2 for AAV9 and 2.38 × 10−2 for AAV9/NGA particles. These results suggest that one possible cause for the defective phenotype displayed by the AAV9/NGA mutant is decreased cell surface binding potential.
FIGURE 4.

Transduction, binding, and uptake of the AAV9NGA mutant in CHO Lec2 cells. A, transduction efficiency of AAV9-CBA-Luc (white columns) and AAV9NGA-CBA-Luc (black columns) on CHO Lec2 cells that contain surface-exposed galactose essential for AAV9 infection at increasing multiplicities of infection were determined 24 h post infection (n = 4). B, sialic acid-deficient CHO Lec2 cells were prechilled and incubated with AAV9 or AAV9NGA particles at different multiplicities of infection ranging from 100–500,000 at 4 °C to allow binding without cellular uptake. Quantitative analysis of dose-dependent AAV9 (solid circles) or AAV9NGA (triangles) attachment to cell surfaces was assessed using qPCR to quantify viral genome copy number and binding curves generated using a single-site binding model (n = 5). The inset shows data of capsid binding at earlier time intervals. C, for cellular uptake studies following removal of unbound virions, viral genomes were extracted from Lec2 cells either 5 (gray columns) or 60 min (white columns) after incubation, and copy numbers were determined by qPCR. The percentage of internalized virions is derived from the ratio of total number of internalized virions normalized to the number of bound AAV9, AAV9NGA, or the binding deficient AAV9/W503R virions per cell (n = 5). Error bars represent mean ± S.E. *, p < 0.05; ***, p < 0.005. RLU, relative light unit(s).
To further examine whether an ∼50% decrease in cell surface binding potential was sufficient to explain the defective transport phenotype of AAV9/NGA virions, we subjected different virions to a quantitative PCR-based cellular uptake assay. Briefly, this experiment involves the removal of cell surface-bound virions using trypsin and quantitation of internalized virions alone. As indicated by the recovery of trypsin-resistant virions (Fig. 4C), ∼10% of total AAV9 vector genome copies appear to be internalized 5 min post-incubation, and this amount increases to ∼25% at 60 min. In contrast, <1% of trypsin-resistant AAV9/NGA vector genome copies is recovered at 5 min, indicating a defect in uptake. This effect is exacerbated at 60 min, when no cumulative increase in uptake of the AAV9/NGA mutant is observed. In contrast, although the previously characterized galactose binding-deficient mutant AAV9/W503R (19) displays <0.1% uptake at 5 min, uptake of these virions is doubled at 60 min. Therefore, although AAV9/W503R is a true galactose binding mutant, the AAV9/NGA mutant appears to be marginally deficient in galactose binding and significantly defective in cellular uptake.
Multiple integrins can mediate AAV9 transduction. To examine whether integrins might act as coreceptors for AAV9 transduction, we utilized several anti-integrin antibodies that are known to block integrin function. All cell types described here were treated with sialidase as described earlier to expose cell surface galactose receptors to facilitate efficient AAV9 binding. As shown in Fig. 5, A and B, integrins composed of the α V (but not the α 5 subunit), when combined with β 1, β 3, or β 5 subunits, appear to play a role in AAV9 transduction. Specifically, antibodies targeted against different integrins blocked AAV9 transduction in desialylated HEK293 cells 2- to 5-fold and in desialylated HeLa cells 2- to 10-fold. Inhibition of αVβ3 integrin had the most profound effect on transgene expression, whereas the effect of blockade of αVβ5 or α5β1 integrins in these cell lines was minimal. To define a potential role for integrins in vascular uptake of AAV9, we also assessed the role of integrins in AAV9-mediated transduction of cultured endothelial cells (HUVECs). As seen in Fig. 5C, transgene expression in desialylated HUVECs appears to be modestly reduced when AAV9 entry is blocked by inhibitory antibodies against α5β1, αVβ1, or αVβ3 but not αVβ5 integrins. However, it should be noted that these results are not statistically significant. Because β3 integrin subunits appeared to be involved in both HEK293 and HeLa cells, we further evaluated AAV9 transduction efficiency in stable HeLa cell lines subjected to lenti-shRNA-mediated knockdown of the integrin β3 subunit. The latter target was chosen for knockdown because the αVβ3 integrin appeared to be essential in all cell lines tested so far. As shown in Fig. 5D, the transduction efficiency of AAV9 vectors was decreased 2- to 5-fold in desialylated HeLa integrin β3 knockdown cell lines. Taken together, these results suggest that multiple integrins can influence AAV9 transduction in different cell types, presumably in a redundant fashion and with variable efficiencies.
FIGURE 5.
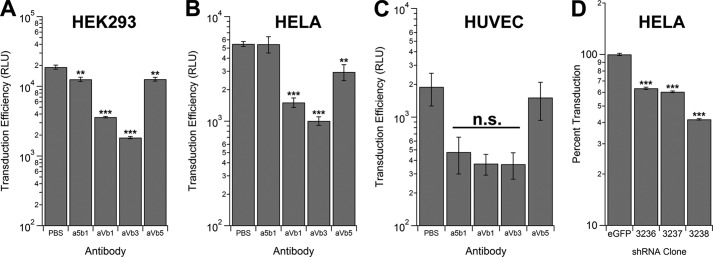
Competitive inhibition and knockdown of integrin subunits affect AAV9 transduction. HEK293 cells (A), HeLa cells (B), and HUVECs (C) were desialylated to expose the galactose receptor and pretreated with anti-integrin monoclonal antibodies directed against different α and β subunits followed by infection with AAV9 vectors packaging a CBA promoter-driven firefly luciferase transgene (1000 vg/cell). Transduction efficiency was then determined by luciferase bioluminescence assays performed 24 h post-infection (n = 4). D, stable knockdown of the integrin β3 subunit was achieved with three different anti-integrin β3 shRNAs (3236, 3237, and 3238) packaged within lentiviral vectors. Lentiviral vectors expressing an shRNA targeted against eGFP were utilized as control. Clonally expanded HeLa integrin β3 knockdown or eGFP control cell lines were infected with AAV9-CBA-Luc (1000 vg/cell), and luciferase assays were carried out 24 h post-infection (n = 4). Error bars represent mean ± S.E. **, p < 0.01; ***, p < 0.005; n.s., not significant. RLU, relative light unit(s).
Structure-function correlates of AAV9-glycan and AAV9-integrin interactions. The structure of the icosahedral AAV9 capsid has recently been determined using x-ray crystallography and cryo-EM (5). Although the AAV9 capsid displays a conserved surface topology similar to that observed in other AAV strains, our understanding of structure-function correlates that are unique to AAV9 continue to evolve. For instance, previous studies have established the galactose receptor binding footprint on the AAV9 capsid through a combination of mutagenesis and structural modeling (8). The amino acid residues that have been directly implicated in galactose recognition include Asn470, Asp271, Asn272, Tyr446, and Trp503, which form a binding pocket at the base of the protrusions around the icosahedral three-fold axes of symmetry. In addition, several other amino acid residues that indirectly affect galactose binding have been mapped through random as well as alanine scanning mutagenesis (9, 36). Mutagenesis of residues that affect glycan recognition, thereby resulting in decreased or increased binding affinity, can have a profound impact on the transduction efficiency, pharmacokinetics, and biodistribution of recombinant AAV9 (9, 19, 36). We recently utilized this information to successfully graft a functional galactose binding footprint onto other AAV capsid templates (20). In addition, several other single amino acid residues have recently been identified as being essential for post-attachment processing and transduction of multiple tissues by recombinant AAV9, although the underlying molecular mechanisms have yet to be determined (9).
In this study, we assign the structure-function correlates of one such subset of amino acid residues, i.e. an apparently pleiotropic role of the integrin-binding NGR motif in binding, cellular uptake and systemic clearance of recombinant AAV9 vectors. A key explanation for this multifunctional attribute hinges on the structural location of the NGR motif adjacent to the galactose binding pocket on the AAV9 capsid (Fig. 6B). Previously, we mapped the location of the NGR motif on the AAV2 capsid to a site distinct from the heparan sulfate binding region (Fig. 6A) at the icosahedral three-fold symmetry axis (18). In contrast to AAV2, where mutation of the NGR motif does not affect heparin binding, it is conceivable that a similar alteration of the NGR motif on AAV9 might indirectly affect glycan binding because of its close proximity to the galactose binding pocket. These observations are corroborated by a closer examination of the different heparan sulfate and galactose binding residues on the capsid surface as well as the NGR residues, which appear to be structurally conserved in AAV2 as well as AAV9 and, most likely, several other AAV serotypes (Fig. 6C). Importantly, we establish, by subsequent analysis, that the NGR motif is more critical for post-attachment processing mediated by integrin receptors and, consequently, cellular entry within different tissues, leading to successful transduction. When these potential receptor interactions are compromised, our results suggest that rapid clearance of recombinant AAV vectors by nonspecific uptake mechanisms, such as by the spleen or endothelial lining, is likely. In addition, although our studies implicate integrins in the vascular uptake of AAV9, it is important to note that these observations are likely distinct from other possible coreceptors and/or mechanisms by which recombinant AAV9 vectors might cross the vascular barrier.
FIGURE 6.
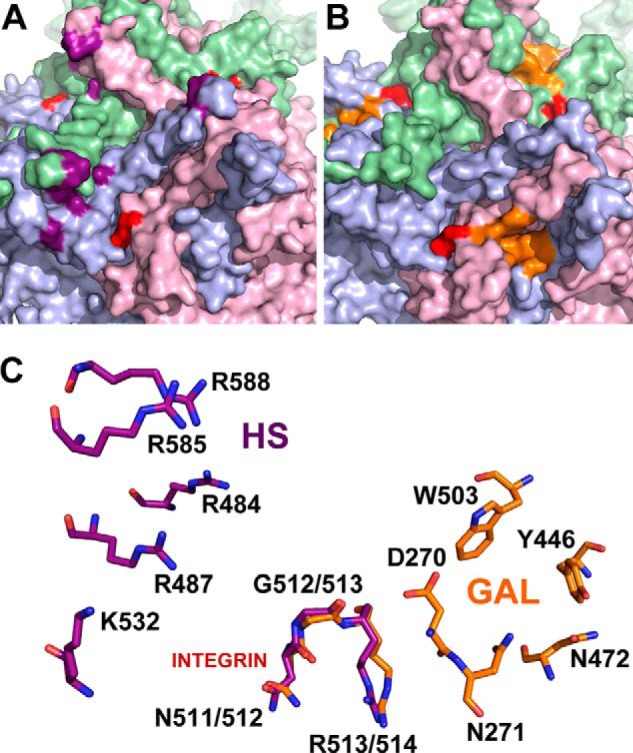
Comparison of glycan and integrin recognition motifs on AAV2 and AAV9 capsid surfaces. Exterior capsid surface representation of VP3 subunit trimers for AAV2 (A) and AAV9 (B) generated using the Oligomer Generator utility in VIPERdb-Virus Particle ExploreR2 and PyMOL. Different VP3 monomers are colored in pale green, light blue, and light pink. The surface location of heparan sulfate binding residues on AAV2 (VP1 numbering Arg484, Arg487, Lys532, Arg585, and Arg588) and the galactose binding residues on AAV9 (VP1 numbering Asp271, Asn272, Tyr446, Asn470, and Trp503) are highlighted in purple and orange, respectively. The integrin recognition motifs, 511NGR513 on AAV2 and 512NGR514 (VP1 numbering) on AAV9 are highlighted in red. C, close-up views of superposed structures of AAV2 and AAV9 trimers with one group of individual residues labeled in each. The overlapping NGR motif is structurally conserved and located in closer proximity to the AAV9 galactose footprint than the AAV2 heparan sulfate binding footprint.
Acknowledgments
We thank Drs. Jürgen Kleinschmidt and Mavis Agbandje-McKenna for the ADK9 monoclonal antibody.
This work was supported by National Institutes of Health Grants R01HL089221 and P01HL112761 (to A. A.) and R01-DK058702-10 (to T. K.).
- AAV
- adeno-associated virus
- CBA
- chicken β-actin
- HUVEC
- human umbilical vein endothelial cell
- qPCR
- quantitative PCR
- VP
- viral protein
- vg
- vector genome copy number.
REFERENCES
- 1. Bowles D. E., Rabinowitz J. E., Samulski R. J. (2006) Parvoviruses, pp. 15–24, Edward Arnold Ltd., New York [Google Scholar]
- 2. Berns K., Parrish C. R. (2007) Fields Virology, 5th Ed., pp. 2437–2477, Lippincott Williams & Wilkins, Philadelphia [Google Scholar]
- 3. Gao G., Vandenberghe L. H., Alvira M. R., Lu Y., Calcedo R., Zhou X., Wilson J. M. (2004) Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78, 6381–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Asokan A., Schaffer D. V., Samulski R. J. (2012) The AAV vector toolkit: poised at the clinical crossroads. Mol. Ther. 20, 699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. DiMattia M. A., Nam H. J., Van Vliet K., Mitchell M., Bennett A., Gurda B. L., McKenna R., Olson N. H., Sinkovits R. S., Potter M., Byrne B. J., Aslanidi G., Zolotukhin S., Muzyczka N., Baker T. S., Agbandje-McKenna M. (2012) Structural insight into the unique properties of adeno-associated virus serotype 9. J. Virol. 86, 6947–6958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bell C. L., Vandenberghe L. H., Bell P., Limberis M. P., Gao G. P., Van Vliet K., Agbandje-McKenna M., Wilson J. M. (2011) The AAV9 receptor and its modification to improve in vivo lung gene transfer in mice. J. Clin. Invest. 121, 2427–2435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Shen S., Bryant K. D., Brown S. M., Randell S. H., Asokan A. (2011) Terminal N-linked galactose is the primary receptor for adeno-associated virus 9. J. Biol. Chem. 286, 13532–13540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bell C. L., Gurda B. L., Van Vliet K., Agbandje-McKenna M., Wilson J. M. (2012) Identification of the galactose binding domain of the AAV9 capsid. J. Virol. 86, 7326–7333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Adachi K., Enoki T., Kawano Y., Veraz M., Nakai H. (2014) Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat. Commun. 5, 3075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Neu U., Bauer J., Stehle T. (2011) Viruses and sialic acids: rules of engagement. Curr. Opin. Struct. Biol. 21, 610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qing K., Mah C., Hansen J., Zhou S., Dwarki V., Srivastava A. (1999) Human fibroblast growth factor receptor 1 is a co-receptor for infection by adeno-associated virus 2. Nat. Med. 5, 71–77 [DOI] [PubMed] [Google Scholar]
- 12. Ling C., Lu Y., Kalsi J. K., Jayandharan G. R., Li B., Ma W., Cheng B., Gee S. W., McGoogan K. E., Govindasamy L., Zhong L., Agbandje-McKenna M., Srivastava A. (2010) Human hepatocyte growth factor receptor is a cellular coreceptor for adeno-associated virus serotype 3. Hum. Gene Ther. 21, 1741–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kashiwakura Y., Tamayose K., Iwabuchi K., Hirai Y., Shimada T., Matsumoto K., Nakamura T., Watanabe M., Oshimi K., Daida H. (2005) Hepatocyte growth factor receptor is a coreceptor for adeno-associated virus type 2 infection. J. Virol. 79, 609–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Weller M. L., Amornphimoltham P., Schmidt M., Wilson P. A., Gutkind J. S., Chiorini J. A. (2010) Epidermal growth factor receptor is a co-receptor for adeno-associated virus serotype 6. Nat. Med. 16, 662–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Di Pasquale G., Davidson B. L., Stein C. S., Martins I., Scudiero D., Monks A., Chiorini J. A. (2003) Identification of PDGFR as a receptor for AAV-5 transduction. Nat. Med. 9, 1306–1312 [DOI] [PubMed] [Google Scholar]
- 16. Kaminsky P. M., Keiser N. W., Yan Z., Lei-Butters D. C., Engelhardt J. F. (2012) Directing integrin-linked endocytosis of recombinant AAV enhances productive FAK-dependent transduction. Mol. Ther. 20, 972–983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Summerford C., Bartlett J. S., Samulski R. J. (1999) αVβ5 integrin: a co-receptor for adeno-associated virus type 2 infection. Nat. Med. 5, 78–82 [DOI] [PubMed] [Google Scholar]
- 18. Asokan A., Hamra J. B., Govindasamy L., Agbandje-McKenna M., Samulski R. J. (2006) Adeno-associated virus type 2 contains an integrin α5β1 binding domain essential for viral cell entry. J. Virol. 80, 8961–8969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shen S., Bryant K. D., Sun J., Brown S. M., Troupes A., Pulicherla N., Asokan A. (2012) Glycan binding avidity determines the systemic fate of adeno-associated virus 9. J. Virol. 86, 10408–10417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shen S., Horowitz E. D., Troupes A. N., Brown S. M., Pulicherla N., Samulski R. J., Agbandje-McKenna M., Asokan A. (2013) Engraftment of a galactose receptor footprint onto adeno-associated viral capsids improves transduction efficiency. J. Biol. Chem. 288, 28814–28823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schmidt E. E., Pelz O., Buhlmann S., Kerr G., Horn T., Boutros M. (2013) GenomeRNAi: a database for cell-based and in vivo RNAi phenotypes, 2013 update. Nucleic Acids Res. 41, D1021–D1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilsdorf M., Horn T., Arziman Z., Pelz O., Kiner E., Boutros M. (2010) GenomeRNAi: a database for cell-based RNAi phenotypes. 2009 update. Nucleic Acids Res. 38, D448–D452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Horn T., Arziman Z., Berger J., Boutros M. (2007) GenomeRNAi: a database for cell-based RNAi phenotypes. Nucleic Acids Res. 35, D492–D497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xie Q., Bu W., Bhatia S., Hare J., Somasundaram T., Azzi A., Chapman M. S. (2002) The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. U.S.A. 99, 10405–10410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carrillo-Tripp M., Shepherd C. M., Borelli I. A., Venkataraman S., Lander G., Natarajan P., Johnson J. E., Brooks C. L., 3rd, Reddy V. S. (2009) VIPERdb2: an enhanced and web API enabled relational database for structural virology. Nucleic Acids Res. 37, D436–D442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kern A., Schmidt K., Leder C., Müller O. J., Wobus C. E., Bettinger K., Von der Lieth C. W., King J. A., Kleinschmidt J. A. (2003) Identification of a heparin-binding motif on adeno-associated virus type 2 capsids. J. Virol. 77, 11072–11081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Opie S. R., Warrington K. H., Jr, Agbandje-McKenna M., Zolotukhin S., Muzyczka N. (2003) Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. J. Virol. 77, 6995–7006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. O'Donnell J., Taylor K. A., Chapman M. S. (2009) Adeno-associated virus-2 and its primary cellular receptor: cryo-EM structure of a heparin complex. Virology 385, 434–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Levy H. C., Bowman V. D., Govindasamy L., McKenna R., Nash K., Warrington K., Chen W., Muzyczka N., Yan X., Baker T. S., Agbandje-McKenna M. (2009) Heparin binding induces conformational changes in adeno-associated virus serotype 2. J. Struct. Biol. 165, 146–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang B., Li S., Wang H., Guo Y., Gessler D. J., Cao C., Su Q., Kramer J., Zhong L., Ahmed S. S., Zhang H., He R., Desrosiers R. C., Brown R., Xu Z., Gao G. (2014) Global CNS Transduction of adult mice by intravenously delivered rAAVrh. 8 and rAAVrh. 10 and nonhuman primates by rAAVrh. 10. Mol. Ther. 22, 1299–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Thomas C. E., Storm T. A., Huang Z., Kay M. A. (2004) Rapid uncoating of vector genomes is the key to efficient liver transduction with pseudotyped adeno-associated virus vectors. J. Virol. 78, 3110–3122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J., Xie J., Lu H., Chen L., Hauck B., Samulski R. J., Xiao W. (2007) Existence of transient functional double-stranded DNA intermediates during recombinant AAV transduction. Proc. Natl. Acad. Sci. U.S.A. 104, 13104–13109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hauck B., Zhao W., High K., Xiao W. (2004) Intracellular viral processing, not single-stranded DNA accumulation, is crucial for recombinant adeno-associated virus transduction. J. Virol. 78, 13678–13686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Petrov M., Malik A., Mead A., Bridges C. R., Stedman H. H. (2011) Gene transfer to muscle from the isolated regional circulation. Methods Mol. Biol. 709, 277–286 [DOI] [PubMed] [Google Scholar]
- 35. Greelish J. P., Su L. T., Lankford E. B., Burkman J. M., Chen H., Konig S. K., Mercier I. M., Desjardins P. R., Mitchell M. A., Zheng X. G., Leferovich J., Gao G. P., Balice-Gordon R. J., Wilson J. M., Stedman H. H. (1999) Stable restoration of the sarcoglycan complex in dystrophic muscle perfused with histamine and a recombinant adeno-associated viral vector. Nat. Med. 5, 439–443 [DOI] [PubMed] [Google Scholar]
- 36. Pulicherla N., Shen S., Yadav S., Debbink K., Govindasamy L., Agbandje-McKenna M., Asokan A. (2011) Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol. Ther. 19, 1070–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]