

Background: mAb Elec410 inhibits acetylcholinesterase by binding at the active site gorge entrance.
Results: Biochemical and structural characterization of Fab410-bound acetylcholinesterase reveals residual catalytic activity, coexisting open/closed states of a back door channel, and a semi-occluded gorge entrance.
Conclusion: Bound antibody restricts substrate access through the gorge and may modulate back door opening.
Significance: This unique molecular template with high frequency back door opening refines our understanding of acetylcholinesterase catalysis.
Keywords: Acetylcholinesterase (AChE), Enzyme Mechanism, Homology Modeling, Monoclonal Antibody, Snake Venom, X-ray Crystallography, Back Door Channel, Peripheral Anionic Site, Regulation of Catalysis
Abstract
The acetylcholinesterase found in the venom of Bungarus fasciatus (BfAChE) is produced as a soluble, non-amphiphilic monomer with a canonical catalytic domain but a distinct C terminus compared with the other vertebrate enzymes. Moreover, the peripheral anionic site of BfAChE, a surface site located at the active site gorge entrance, bears two substitutions altering sensitivity to cationic inhibitors. Antibody Elec410, generated against Electrophorus electricus acetylcholinesterase (EeAChE), inhibits EeAChE and BfAChE by binding to their peripheral sites. However, both complexes retain significant residual catalytic activity, suggesting incomplete gorge occlusion by bound antibody and/or high frequency back door opening. To explore a novel acetylcholinesterase species, ascertain the molecular bases of inhibition by Elec410, and document the determinants and mechanisms for back door opening, we solved a 2.7-Å resolution crystal structure of natural BfAChE in complex with antibody fragment Fab410. Crystalline BfAChE forms the canonical dimer found in all acetylcholinesterase structures. Equally represented open and closed states of a back door channel, associated with alternate positions of a tyrosine phenol ring at the active site base, coexist in each subunit. At the BfAChE molecular surface, Fab410 is seated on the long Ω-loop between two N-glycan chains and partially occludes the gorge entrance, a position that fully reflects the available mutagenesis and biochemical data. Experimentally based flexible molecular docking supports a similar Fab410 binding mode onto the EeAChE antigen. These data document the molecular and dynamic peculiarities of BfAChE with high frequency back door opening, and the mode of action of Elec410 as one of the largest peptidic inhibitors targeting the acetylcholinesterase peripheral site.
Introduction
Acetylcholinesterase (AChE4; EC 3.1.1.7) rapidly terminates cholinergic neurotransmission by catalyzing hydrolysis of the neurotransmitter acetylcholine at neuronal and neuromuscular synapses (1–3). The active site of AChE, which contains the Glu/His/Ser catalytic triad and binds competitive reversible or irreversible inhibitors, is positioned at the center of the subunit at the bottom of a long and narrow gorge (4), a geometry not reflecting the high rate of catalytic activity (turnover number up to 104 s−1) of the enzyme (5). At the enzyme surface and entrance of the active site gorge, the peripheral anionic site (PAS) encompasses overlapping binding loci for a range of positively charged, reversible modulators of catalysis (6–9). Inhibitor binding at the PAS appears to limit the catalytic efficiency by combined steric and electrostatic blockade of ligand trafficking through the gorge and by altering the active site conformation (10–13). The surface topography and conformational flexibility of this site have been characterized in detail; however, the mechanisms involved in its allosteric functioning remain unclear (14–20).
The so-called “back door region,” which is also at the enzyme surface but remote from the gorge entrance, has been another matter of discussion. Indeed, transient enlargement or opening of a back door would permit fractional substrate or product trafficking into or out of the enzyme, thereby contributing to the high catalytic activity of the enzyme (21, 22) and to the residual activity of its complex with the large snake toxin fasciculin 2 (Fas2) bound at the PAS (14, 23). Shutter-like motion of the aromatic side chain of either residue Trp84 or residue Tyr442 (Torpedo californica AChE (TcAChE) numbering), whose aromatic rings make thin walls in the back door region between the active site pocket and the outside solvent, were first visualized by molecular dynamics simulations (21, 22, 24). Subsequent evidence for an open back door channel was found upon crystallographic analysis of Drosophila AChE (DmAChE) where genuine Ile and Asp substitutions to Met83 and Tyr442, respectively, were found to weaken the interaction network in this region (25), and in a combined crystallography and molecular dynamics simulation study of TcAChE in complex with PAS-bound aflatoxin where channel opening was attributed to concerted motions of Tyr442 and Trp84 (26). Complementary crystal structures of mouse AChE (mAChE), an inactive mAChE mutant, and TcAChE bound with a range of substrates, substrate analogues, and reaction products led us and others to picture successive positions and orientations for an incoming substrate, first bound at the PAS and then proceeding within the gorge toward the active site; the conformations of the presumed transition state for acylation and the acyl-enzyme intermediate; the positions and orientations of the dissociating and egressing products (8, 9); and unexpected substrate binding sites at the enzyme surface in the back door region (8). Hence, transient back door opening, likely to be associated with substantial conformational fluctuation in the protein core, is clearly linked to the dynamic properties or breathing motions underlying the catalytic mechanism of AChE.
The venoms of some Elapidae snakes are abundant sources of non-synaptic (non-cholinergic) AChE of an unknown physiological role because it is non-toxic by itself and does not enhance the toxicity of the pharmacologically active venom components (27–29). However, it could be a vestige of the pancreatic origin of the venom gland (30). These snake venom AChEs are inhibited by small, organic PAS ligands such as propidium, albeit at a lower affinity compared with the other species found in neuronal or neuromuscular tissues, but they differ widely in their sensitivity to larger, peptidic PAS ligands such as Fas2 or mAb Elec410 (see below) (27, 31, 32). For example, the venom enzymes from Bungarus fasciatus (BfAChE) and Ophiophagus hannah are inhibited by Fas2 and Elec410, whereas those from Naja haje and Hemachatus haemachatus are not (27). BfAChE is a true AChE (as is its more recently studied Bungarus sindanus ortholog (Ref. 33 and references therein)), and it displays all the structural and catalytic characteristics of AChEs from cholinergic tissues, including the presence of a large permanent dipole moment (Refs. 34–40 and for reviews, see Refs. 41 and 42). However, unlike the AChEs from cholinergic tissues that bear C-terminal tailed (“T”) or hydrophobic (“H”) peptides and can form oligomers (for a review, see Ref. 43), BfAChE is expressed in the venom and in mammalian cell models as a hydrophilic monomer characterized by a short C-terminal soluble (“S”) peptide (38). Compared with Torpedo and mammalian AChEs, BfAChE also presents two non-conservative substitutions at the PAS, corresponding to replacement of Tyr70 (TcAChE numbering) by a Met and of the acidic residue at position 285 by a Lys, on opposite sides of the gorge rim. Comparative analysis of wild-type BfAChE and its “reverse” M70Y and K285D mutants ascertained both the responsibility of these two substitutions for the low sensitivity of BfAChE to various PAS inhibitors, and their absence of effect on its catalytic turnover rate and competitive inhibition by active site ligands (38).
Elec410, one of the three inhibitory mAbs raised against natural Electrophorus electricus AChE (EeAChE), was initially reported to inhibit BfAChE with an apparent Ki or IC50 value “in the nanomolar range” versus the Kd value of ∼0.04 nm reported for the EeAChE antigen (27, 31). This property and availability of the two protein sequences (38, 44) were instrumental in delineating the binding site of Elec410 (and those of its Elec403 and Elec408 congeners) at the EeAChE surface using complementary biochemical and mutagenesis approaches (45). In particular, these studies identified distinct but overlapping loci at the PAS surface as the binding sites for Elec410 and Elec403 and the back door region as the binding site for Elec408. In a preliminary structure-function relationship study of the Fab fragments of these mAbs (46), comparison of BfAChE and EeAChE inhibition by Fab410 pointed to a greater residual activity for the BfAChE complex, suggesting slight variability in the Fab410 position or strength of interaction at the two gorge entrances (as would be expected from the sequence and affinity differences) and/or in the opening width or frequency of the two back doors. More recently, a comprehensive analysis of the molecular determinants dictating the binding specificity and mode of action of these three mAbs led us to document their nucleotidic and peptidic sequences, the biochemical features and functional properties toward EeAChE of their Fab domains, a crystal structure of Fab408 and homology models of Fab403 and Fab410, and a data-driven docking model of an Fab403-EeAChE complex (32). The combining site of Fab410 was pictured as a ∼1000-Å2 surface area mostly populated by cationic side chains with a highly electropositive CDR H2 and a protruding CDR H3 well positioned for interaction with the PAS surface. In turn, comparison of the protein sequences in the two PAS regions pointed to several substitutions, of which some generate or eliminate consensus N-glycosylation sequences, as likely candidates for dictating slightly distinct poses of bound Fab410. Therefore, BfAChE and Fab410 respectively appeared as a new AChE model and a new, non-competitive, peptidic ligand probe suitable for further exploration of the mechanisms for regulation of catalysis triggered by the PAS and the back door region.
In this context, we solved a crystal structure of natural, glycosylated BfAChE in complex with Fab410 and generated an experimentally based homology model of the Fab410-EeAChE complex. This comprehensive study documents the molecular and dynamic peculiarities of BfAChE with high back door opening frequency, along with the mode of action of Elec/Fab410 as one of the largest peptidic inhibitors targeting the PAS of an AChE.
EXPERIMENTAL PROCEDURES
Materials
Dried B. fasciatus venom was a gift from Dr. Cassian Bon (Institut Pasteur, Paris, France). The crude ascitic fluid for Elec410, the sequence of Fab410 prior to publication, and a reference sample of purified BfAChE, respectively, were provided by Dr. Jacques Grassi, Dr. Didier Boquet, and Dr. Christophe Créminon (Commissariat à l'Energie Atomique Saclay, Gif-sur-Yvette, France). The prepacked Superdex-200 HR-10/30 and HL-26/60 columns and protein G- and protein A-Sepharose HiTrap units (1 ml) and the calibration markers for gel filtration were from GE Healthcare. PEG 8000 was from Hampton Research, and sodium citrate from Fluka. The molecular weight standards for SDS-PAGE and all others biochemical reagents were from Sigma-Aldrich.
Protein Purification and Preparation
mAb Elec410 was produced as an IgG1,κ from murine hybridoma (31). Isolation of Elec410 from the ascitic fluid and preparation and purification of Fab410 and its physical characterization by MALDI-TOF MS and various electrophoretic means were described previously (32). Purified Fab410 was dialyzed against 50 mm Tris, pH 7.5, 50 mm NaCl, 0.01% (w/v) NaN3 and concentrated to 5 mg/ml.
BfAChE was isolated from the venom by affinity chromatography on immobilized m-carboxyphenyldimethylethyl using elution by 20 mm decamethonium (38, 47). It was further purified by gel filtration on Superdex-200 in 100 mm sodium phosphate, pH 7.4, 100 mm NaCl, 0.01% (w/v) NaN3. When eluting at ∼0.25 mg/ml from the gel filtration column, BfAChE behaved as a monomer. Injection at very high concentration to explore elution as a dimer and compensate for the ≥10-fold dilution occurring on the column was not attempted. Purified BfAChE was extensively dialyzed against 10 mm Hepes, pH 7.5, 50 mm NaCl, 0.01% (w/v) NaN3 and concentrated to 2.5 mg/ml. Homogeneity was assessed by SDS- and native PAGE (see below and Fig. 1).
FIGURE 1.
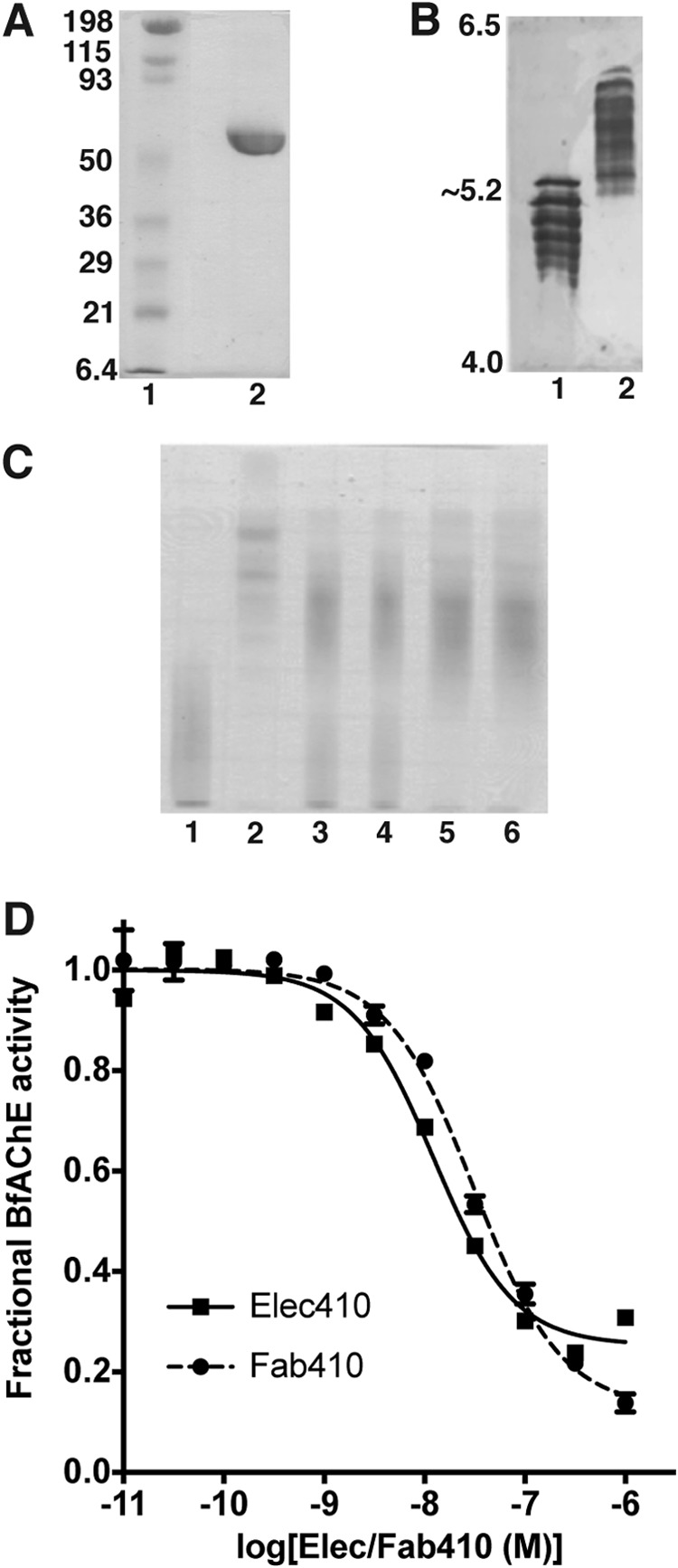
Functional and electrophoretic characterization of BfAChE and its Elec410 and Fab410 complexes. A, SDS-PAGE of BfAChE (non-reducing conditions; 12.5% PhastGel). The molecular weight markers are displayed and labeled. B, comparative isoelectric focusing analysis of mAChE (lane 1) and BfAChE (lane 2) (pI 4.0–6.5 PhastGel). The pI limits of the gel are indicated. BfAChE is as heterogeneous in charge as mAChE, but it displays higher overall pI value. C, native PAGE (7.5% PhastGel) of BfAChE (lane 1), Fab410 (lane 2), and pre-incubated Fab410/BfAChE mixtures in a 0.75:1, 0.9:1, 1:1, and 1.2:1 molar ratio (lanes 3–6) with migration from the cathode (top) toward the anode (bottom). The cationic character of Fab410 is evident. Both BfAChE and Fab410 are homogenous in mass but not in charge, due to heterogeneous N-glycosylation of natural BfAChE (38, 67) and nonspecific C-terminal processing of Fab410 by papain (32); however, all isoforms form complexes as assessed by the inhibition curves (D; below) and further verified by analytical gel filtration (not shown). D, inhibition of BfAChE by Elec410 and Fab410 at equilibrium (individual experiments). Data points correspond to the average ± variation of duplicates. Non-linear fitting used a sigmoidal equation. The slight difference in the IgG (squares) and Fab (circles) affinities for BfAChE may reflect their relative avidity. The significant residual (fractional) activity of BfAChE at near saturating concentrations of Elec410 or Fab410 is evident. Mean IC50 and residual activity values from three to four independent experiments are reported in Table 1 as are those for the inhibition of EeAChE.
The Fab410-BfAChE complex used for crystallogenesis was formed with a slight molar excess of Fab410 over the enzyme and incubated for 2 h at room temperature and then overnight at 4 °C. Unbound Fab410 was removed by gel filtration on Superdex-200 in 10 mm Hepes, pH 7.5, 50 mm NaCl, 0.01% (w/v) NaN3. The complex was concentrated to 10 mg/ml, filtrated, and stored on ice.
Biochemical and Functional Analyses
SDS- and native PAGE used a PhastSystem apparatus (GE Healthcare); homogenous 12.5 and 7.5% PhastGels, respectively; migration toward the anode; and silver or Coomassie Blue staining. SDS-PAGE samples were boiled for 5 min in the presence of 2.5% (w/v) SDS with (reducing conditions) or without (non-reducing conditions) 5% (v/v) β-mercaptoethanol. Isoelectric focusing analysis used the same apparatus and pI 4.0–6.5 PhastGels preloaded with ampholytes. Native PAGE mobility shift assays used complexes formed in solution with a 0.75–1.2 molar excess of Fab410 over BfAChE (3-h incubation at room temperature).
AChE activities were recorded from duplicates for 5 min on a UNICAM 8700 spectrophotometer (Thermo Optek) using 10–25 pm EeAChE or BfAChE, 1.25 mm acetylthiocholine iodide (∼10 × Km), and 0.33 mm dithiobis(2-nitrobenzoic acid) in 100 mm sodium phosphate, pH 8.0, 0.1 mg/ml BSA (λ = 412 nm) (48). The control AChE samples and the IgG/ or Fab/AChE mixtures were incubated overnight under mild agitation at room temperature before recording their residual (fractional) activity. Data analysis used GraphPad Prism 6, a sigmoidal dose-response equation, and one-way analysis of variance. Data are summarized in Table 1.
TABLE 1.
Parameters for inhibition of BfAChE and EeAChE by Elec410 (italics) and Fab410 (plain)
| BfAChEa,b | EeAChE | |
|---|---|---|
| IC50 Elec410 (nm) | 10.9 ± 0.9 | 0.47 ± 0.18a,b |
| (8.1–13.7) | (−1.8–2.76) | |
| IC50 Fab410 (nm) | 33.4 ± 5.0 | 0.42 ± 0.03b,c |
| (11.8–54.9) | (0.33–0.50) | |
| Residual activity Elec410 (%) | 28.1 ± 0.9 | 4.75 ± 0.55b,c |
| (25.2–31.0) | (−2.24–11.74) | |
| Residual activity Fab410 (%) | 13.7 ± 1.9 | 4.50 ± 0.62b,c |
| (5.4–22.1) | (2.52–6.48) |
a This study.
b Means ± S.E. from three (Fab410/BfAChE), four (IgG410/BfAChE and Fab410/EeAChE), and two (Elec410/EeAChE) independent experiments performed with duplicates. The 95% confidence intervals are provided in parentheses (p values <0.05).
c From Ref. 32 with modification.
Crystallization
Crystallization was achieved manually at 20 °C by vapor diffusion using the Fab410-BfAChE complex at 10 mg/ml, 1-μl hanging drops, and a protein-to-well solution ratio of 1:1. Fine plate crystals grew spontaneously within 10 days with 12.5% (v/v) PEG 8000, 200 mm sodium citrate, pH 5.0 as the well solution. Thicker plates were obtained by seeding in sitting drops with 7% (v/v) PEG 8000, 200 mm sodium citrate, pH 5.0. Crystals were flash cooled in a nitrogen gas stream after successive short soaks in the well solution supplemented with 7.5, 15, and 25% (v/v) ethylene glycol, and stored in liquid nitrogen. Diffraction data were collected at 100 K at the European Synchrotron Radiation Facility (Grenoble, France), processed with MOSFLM (49), and scaled and merged with SCALA (50).
Solution and Refinement of the Fab410-BfAChE Complex
Initial phases were obtained by molecular replacement with the AMoRe program package (51) using as search models the structures of apo-mAChE (Protein Data Bank (PDB) code 1J06 (18)) and antibody HFE7A (PDB code 1IQW (52)). Various normal mode-perturbed models of the Fab template harboring a range of elbow angles were generated using the ElNemo server (53). Among them, five models were selected based on their lowest correlation coefficient and R-factor values during molecular replacement searches and subsequently used as a Phaser ensemble (54). This procedure yielded a final solution with a log-likelihood gain score of 2455 in the 40–2.75-Å resolution range. The model was improved by manual adjustment with the graphics program Coot (55) and refined with REFMAC (56) and autoBUSTER (57), including non-crystallographic symmetry restraints and translation/libration/screw refinement with each AChE subunit and Fab410 variable and constant domain defining a translation/libration/screw group. Data collection and refinement statistics are summarized in Table 2.
TABLE 2.
Data collection and refinement statistics
| Fab410-BfAChE complex | |
|---|---|
| Data collectiona | |
| Beamline (ESRFb) | ID29 |
| Space group | P21212 |
| Cell parameters (Å) | a = 156.59, b = 251.34, c = 73.98 |
| Resolution range (Å) | 78–2.7 |
| Total observations | 357,674 |
| Unique reflections | 81,126 |
| Multiplicity | 4.4 (4.2) |
| Completeness (%) | 99.9 (99.4) |
| ((I)/σ(I)) | 10.2 (2.0) |
| Rsymc | 10.5 (59.4) |
| B Wilson (Å2) | 64.1 |
| Refinementd | |
| R-factor /Rfree (%) | 20.2 (22.8)/24.1 (29.2) |
| r.m.s.d.e | |
| Bonds (Å)/angles (°) | 0.01/1.25 |
| Chiral volume (Å3) | 0.087 |
| Mean B-factors (Å) | |
| Main/side chains | 45.9/48.1 |
| Solvent | 46.6 |
| Ramachandran plotf | |
| Residues in favored/outlier regions (%) | 96.9/0.2 |
| PDB accession code | 4QWW |
a Values in parentheses are those for the highest resolution shell.
b European Synchrotron Radiation Facility.
c Rsym = Σhkl(Σi|Ihkl − 〈Ihkl〉|)/Σhkl|〈Ihkl〉|.
d R-factor = Σhkl‖Fo| − |Fc‖/Σhkl|Fo|. Rfree is calculated for 2% of randomly selected reflections excluded from refinement.
e Root mean square deviations from ideal geometry.
f Ramachandran plot statistics have been calculated with the MolProbity server.
The final structure of the Fab410-BfAChE complex comprises 528 residues (Gly4–Thr535; Fig. 2A) for each subunit in the crystalline BfAChE dimer (0.3 Å for 528 Cα atoms); 212 and 225 residues for each of the two Fab410 light (L) and heavy (H) chains, respectively; and 188 water molecules. The BfAChE gorge is free of any component from the purification, crystallization, or flash cooling solutions. The Fab410 molecule shows an elbow angle of 142° between the pseudodyad axes relating the variable (V) and constant (C) domains of the H and L chains. The Fab410 CDR boundaries were defined according to the IMGT standards as described (32). However, to avoid virtual gaps in the structure coordinates and inconsistencies related to definition of β-strands, the consecutive numbering of residues and Greek letter labeling of β-strands and α-helices will be used herein (Fig. 2B). CDRs L1, L2, and H1 clearly belong to the canonical structural class 1, whereas CDR L3 is only similar to class 1; CDR H2, which is the most exposed to solvent, belongs to class 4; and CDR H3, which encompasses 14 residues, is a non-canonical CDR. High temperature factors and weak electron densities are associated with the surface loop region Asp487–Gly490 and segments Gly4–Val57, Cys254–Cys265, Lys315–Gln318, Ala412–Asn415, and Pro482–Ala486 in BfAChE and with the surface loop region Ser140–Asn145 in the H chain of Fab410 (Fig. 2). Stereochemistry of the structure was analyzed with MolProbity (58); only Fab410 L chain residue Ala51 was found in the disallowed regions of the Ramachandran plot.
FIGURE 2.

Sequences of the AChE species mentioned in this study and of Fab410. A, the sequences of BfAChE from snake venom, TcAChE and EeAChE from electric fishes, and mAChE from mouse are displayed (38, 45, 78, 79). The residue numbering and secondary structure elements displayed above the alignment are those of BfAChE. Conserved residues are shown on a black background, and non-conserved residues are shown on a white background. The symbols above the alignment point to BfAChE-specific PAS residues Met70 and Lys285 (squares) and to EeAChE residues whose substitution by rat AChE residues abolished (S75L for BfAChE Ser74) or reduced (L282H for BfAChE Ser280) Elec410 binding (45) (vertical bars). The symbols below the alignment point to the conserved catalytic triad residues (stars), to BfAChE residues buried at the Fab410 complex interface (triangles), and to BfAChE Asn residues within consensus N-glycosylation sequences (38) (closed circles). The arrow at the BfAChE C terminus denotes the end of the mature protein (38). B, sequences of the Fab410 L (top) and H (bottom) chains showing the CDR positions (shaded) and secondary structure elements. Fab410 residues buried at the BfAChE complex interface are indicated by triangles. In the structure, a Trp was found at position 200 of the H chain instead of an Arg as published previously (32). For an alignment of Fab410 with Fab403 and Fab408, see Fig. 1 in Bourne et al. (32).
Structural Analysis and Comparisons
Electrostatic surface potentials of BfAChE and Fab410 were calculated using APBS (59) with the PyMOL APBS tools. The molecular surface area buried to a 1.4-Å-radius probe at the Fab410-BfAChE complex interface was calculated using PISA (60). Tunnels and channels in AChE were computed with CAVER 3.0 (61). Structural superimposition, based on the respective AChE subunits, of the Fab410-BfAChE complex with the Fas2-mAChE complex (PDB code 1KU6 (14); root mean square deviation value, 0.83 Å for 516 Cα atoms), propidium-mAChE complex (PDB code 1N5R (18); 0.82 Å for 520 Cα atoms), acetylcholine/acetylthiocholine-mAChE complexes (PDB codes 2HA4/2HA5 (8); 0.83/0.79 Å for 521/518 Cα atoms), decamethonium-TcAChE complex (PDB code 1ACL (62); 0.86 Å for 525 Cα atoms), BW284C51-TcAChE complex (PDB code 1E3Q (63); 0.89 Å for 527 Cα atoms) and unliganded DmAChE (PDB code 1DX4 (64); 1.47 Å for 490 Cα atoms) and comparison of the bound Fab410 structure with the theoretical model of unbound Fab410 (Ref. 32; 1.8 and 2.6 Å for 208/203 Cα atoms for the L and H chains, respectively) used Coot (55). Figs. 3–6 were generated using PyMOL (65).
FIGURE 3.
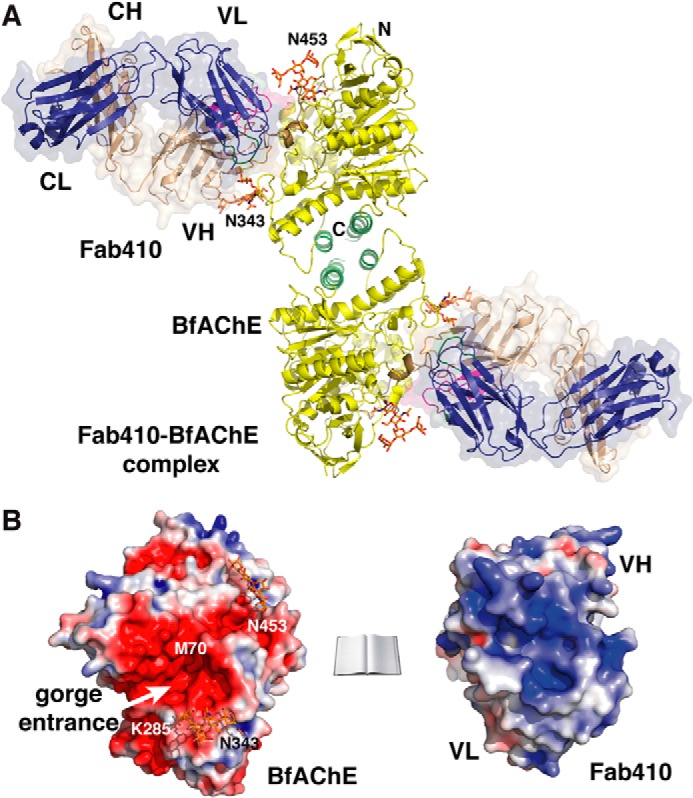
Overall structure of the Fab410-BfAChE complex and electrostatic properties of the binding surfaces. A, two BfAChE subunits related by a 2-fold symmetry axis and linked through a tightly packed four-helix bundle made of their α37,8 and α10 helices assemble as a non-covalent antiparallel dimer. Two Fab410 molecules are bound on opposite faces of the dimer with their CDRs tightly apposed to the PAS regions. The BfAChE subunits are displayed in yellow with labeled N and C termini, a green four-helix bundle at the dimer interface, a brown long Ω loop at the complex interface, red catalytic residues at the center of the subunits, and orange N-glycan moieties linked to Asn343 and Asn453 (labeled). The Fab410 L and H chains and their molecular surfaces are displayed in dark blue and wheat, respectively. CDRs L1, L2, and L3 are displayed in blue, light green, and dark green, and CDRs H1, H2, and H3 are displayed in red, orange, and purple, respectively, with a clearly visible extended CDR H2 (see also Figs. 4A and 5A, right panel). B, distribution of the electrostatic potentials mapped onto the molecular surfaces of BfAChE (left) and Fab410 (right) at −3kT/e (red) to +3kT/e (blue) (molecules not on scale). The charge complementarity of the electronegative binding surface on BfAChE, which is centered on the gorge entrance (white arrow), versus the electropositive combining site in Fab410, which is centered on the H chain CDRs, is evident. The N and C termini of the BfAChE subunit, key residues Met70 and Lys285, and the N-glycosylated Asn453 and Asn343 are labeled.
FIGURE 4.

The back door region in Fab410-bound BfAChE and comparison with other AChEs. A, overall view of the Fab410-BfAChE complex (one subunit only) slightly rotated from Fig. 3A (top subunit) to better show the solvent-accessible paths in BfAChE. The lining walls of the active site gorge and back door channel are shown as transparent green and red surfaces, and their pathways are shown as green and red center lines, respectively (other color codes are as in Fig. 3A). B, omit electron density maps contoured at 3.0σ of the back door region in BfAChE showing the two equally represented alternate conformations of Tyr442 and their respective interactions with neighboring residues stabilized by H-bonds (dashed lines). C, close-up views of the continued path from the PAS at the gorge entrance to the back door region in BfAChE (left), TcAChE (center) (PDB code 2XI4) and DmAChE (right) (PDB code 1DX4) in the same orientation with green and red lining walls for the active site gorge and back door channel (see A). BfAChE and TcAChE Tyr442 in the rotated conformation and DmAChE Asp482 are displayed in pink, whereas BfAChE and TcAChE Tyr442 in the usual conformation are in white. Key side chains from the long Ω loop are in brown, and those lining the channel are in yellow for BfAChE, light orange for TcAChE, and green for DmAChE. In BfAChE and TcAChE, Tyr442 in the rotated conformation points toward a neighboring hydrophobic pocket and leaves the back door channel open, whereas in DmAChE, Asp482 is too small to occlude the channel. D, profiles of the back door channel in BfAChE (red line), TcAChE (orange line), and DmAChE (brown line) and of the gorge path in BfAChE up the constricted region midway in the gorge (green line) as calculated by CAVER. (The dotted line denotes the gorge path between the constricted region and the gorge entrance that was ignored by the program.)
FIGURE 5.

The Fab410-BfAChE complex interface. A, overall views of the buried interfaces at the molecular surfaces of BfAChE (left) (same color codes as in Figs. 3A and 4A) and Fab410 (center) (yellow buried surface overlaid onto the CDRs colored as in Figs. 3A and 4A and in the right panel) in the complex (molecules oriented 90° from each other; not drawn on scale). The positions for Met70 and Lys285, which distinguish the PAS of BfAChE from those of other AChEs, are shown in violet; that for Trp279 (Trp279/280/286 in TcAChE/EeAChE/mAChE) is in light blue; those for Ser74 and Ser280, corresponding to EeAChE Ser75 and Leu282 whose substitution by rat AChE residues alters Fab410 binding, are in green; and that for Asn343, corresponding to EeAChE Asn345 whose deglycosylation enhances Fab410 binding, is in pink. Right, the Fab410 combining surface with only the colored CDRs. B, close-up views of the Fab410-BfAChE complex interface with (left) and without (right) the molecular surface of BfAChE showing the key BfAChE residues (italicized labels; encircled Ser74) and Fab410 CDR residues (same orientation and color codes as in A with red oxygen and blue nitrogen atoms). The arrows point to the BfAChE active site gorge entrance.
FIGURE 6.

Comparison of the Fab410-BfAChE complex with other peptidic inhibitor-AChE complexes. A, overall views of the crystalline Fas2-mAChE complex (left) (PDB code 1KU6) and the 1029-Å2 buried interface at the mAChE surface in this complex (center) (left and right mAChE molecules are oriented 90° from each other). Fas2 is displayed in light green, and mAChE Trp286 is in light blue (other color codes as in Fig. 3A). Right, spatial alignment of the key interacting aromatic and positively charged side chains in BfAChE-bound Fab410 (plain labels; color codes as in Fig. 5) and mAChE-bound Fas2 (italicized labels; green side chains) overlaid onto the Fas2 backbone (light green ribbon). Their similar positioning along with the absence of a Fab410 residue mimicking Fas2 Met33 in its interaction with Trp279 (displayed in light blue on the right) is evident. B, overall views of the theoretical model of the Fab410-EeAChE complex (left) and the 1017-Å2 buried interface at the EeAChE surface in this complex (right) (left and right EeAChE molecules are oriented 90° from each other). In the right panel, the buried EeAChE surface is colored according to the L chain and H chain CDRs in bound Fab410 (see Fig. 5A), and EeAChE Ser75, Leu282, and Asn345 are color-coded as in Fig. 5.
Theoretical Modeling of the Fab410-EeAChE Complex
A previously designed model of the EeAChE subunit devoid of the EeAChE-specific insert Ile418–Gln446 and C-terminal tailed peptide Ala576–Leu610 (32) and the present, randomly oriented Fab410 structure were used as templates. The model of the Fab410-EeAChE complex was generated by flexible docking using HADDOCK 2.1 (66) with default parameters and as possible interfacial active residues five residues of the EeAChE PAS (Tyr71, Pro77, Trp281, Ser287, and Gly337) close to those residues shown by mutagenesis to be important for Elec410 binding (45) (see Fig. 2A) and five residues of the Fab410 CDRs (H chain Arg52 and Tyr104 and L chain Ser30, Arg49, and His93 (see Fig. 2B) likely to be important for interaction with EeAChE (32). Neighboring solvent-accessible residues (four for EeAChE and five for Fab410) suitably positioned for being indirectly involved in the binding were defined as passive residues. For each run, the top 200 complexes generated after rigid body energy minimization were subjected to flexible simulated annealing in torsion angle space and to flexible water refinement in Cartesian space, and the three energetically best models scored by HADDOCK were comparatively analyzed. Nine ensembles of four energetically best models of a Fab410-EeAChE complex were obtained of which the cluster with the best HADDOCK scores closely resembles our experimental structure of the Fab410-BfAChE complex.
As a control, a “reference” model of the Fab410-BfAChE complex was generated using the same procedure and a randomly oriented Fab410 molecule relative to the BfAChE subunit, both extracted from the structure of the complex. Ambiguous interaction restraints similar to those for the Fab410-EeAChE complex were defined from each partner using the corresponding five active residues from the interfacial regions of BfAChE (PAS region) and Fab410 (CDRs), respectively, and four and five surrounding passive residues from each partner. For each of the five ensembles of models obtained, the best solution generated by HADDOCK matches our experimental structure of the Fab410-BfAChE complex (root mean square deviation value of 0.65 Å).
RESULTS AND DISCUSSION
Biochemical and Functional Characterization of the Fab410-BfAChE Complex
Preparation, purification, and biochemical analysis of Fab410, which is made of several charge isoforms, and its functional characterization toward its EeAChE antigen were described previously (32) (Table 1). Natural BfAChE was purified from the B. fasciatus venom using standard affinity chromatography and gel filtration procedures (38). Comprehensive electrophoretic analysis showed molecular homogeneity in mass but not in charge, consistent with variations in the composition of the glycan chains linked to several (or all) of the five consensus sites for N-glycosylation (Figs. 1, A and B, and 2A, black closed circles) (38, 41, 67). However, all Fab410 and BfAChE isoforms have the capacity to form complexes as assessed from the native PAGE patterns (Fig. 1C) and analytical gel filtration profiles (data not shown) obtained upon analysis of the complex stoichiometry.
Elec410 was reported to inhibit BfAChE “with an apparent Ki in the nm range” (31). Curves for BfAChE inhibition by Elec410 and Fab410 spanning 2 orders of magnitude again ascertained formation of equivalent complexes despite the charge heterogeneity of both partners, and validated the use of Fab410 for the structural study (Fig. 1D). In turn, the slight difference in the mean IC50 and residual activity values may denote the respective avidities of the IgG and Fab relatives, and/or experimental variation since identical values were found for the EeAChE antigen (Table 1). Compared with the EeAChE antigen, the lower inhibition potency of Elec410 and Fab410 for BfAChE, denoted by the ∼20–80-fold greater mean IC50 values (Table 1), reflects the amino acid sequence and possible N-glycosylation differences found in the PAS regions of the two AChE species and likely to modulate the interaction network at the two complex interfaces (see below). In turn, the ∼3–6-fold greater residual activity of bound BfAChE over EeAChE recorded at saturating Elec410 and Fab410 concentrations may indicate slightly different mechanisms for substrate hydrolysis or for enzyme inhibition by the bound antibody, associated with architectural differences in the gorge paths or active centers of the two AChEs.
Overall View of the Fab410-BfAChE Complex
The 2.7-Å resolution structure of the Fab410-BfAChE complex reveals the same homodimeric assembly of AChE subunits as observed previously in crystal structures of the recombinant mAChE monomer and the natural covalent TcAChE dimer (Table 2 and Fig. 3A). In fact, comparative analysis of the dimer interfaces in BfAChE and mAChE, which share a buried surface area of ∼920 A2, points to subtle differences in the nature of the contributing residues (e.g. BfAChE Gln374 that replaces mAChE His381 at the center of the interface and BfAChE residues Gln379, Pro500, Pro514, and His526 that replace mAChE residues Leu386, Gln508, Arg522, and Arg534 at its periphery (Fig. 2A)) that significantly alter the pattern of polar interactions found in the mAChE dimer and may affect dimer stability. This observation is consistent with the monomeric versus dimeric states of BfAChE in low versus high concentration solutions (34, 37, 38) (see “Experimental Procedures”) and, compared with mAChE, its higher concentration dependence for dimer formation (data not shown). The same features can be predicted for Naja naja oxiana AChE (NnAChE), which was shown to behave as a monomer at 0.2 mg/ml and a dimer at 2 mg/ml (68) and whose single sequence difference with BfAChE in the dimerization domain is a His substitution to Pro514.5
The BfAChE subunit shows the canonical α/β-hydrolase fold of all cholinesterases (Figs. 3, A and B; 4A; and 5A), consistent with their high sequence identity (Fig. 2A). Of the five potential N-glycosylation sites predicted from the BfAChE sequence (and conserved in NnAChE), two, located near the gorge rim at Asn343 and Asn453, are occupied with a well ordered pentasaccharide, whereas no electron density is associated with Asn457, which is located near the Fab410 L chain and is occupied in most AChEs (TcAChE Asn458; mAChE Asn464) (Figs. 2A, 3A, 4A, and 5A). Whether Asn457 is occupied with a highly disordered glycan chain or whether glycosylation at Asn453 “compensates” for the absence of glycosylation at Asn457 is unknown. The fourth and fifth sites, located far from the gorge entrance at Asn258 and Asn533, might be occupied as well because additional electron density is observed at each of these positions, although not sufficiently resolved to permit the building of glycan molecules.
The BfAChE active site gorge, with key functional residues lining its walls and the Ser200/Glu327/His447 catalytic triad at its bottom, adopts the same shape as in other crystallized AChE species. At the gorge entrance, the PAS is characterized by the non-conserved Met70 and Lys285 residues (Figs. 3B and 5A), which respectively replace an invariant Tyr residue and an acidic residue in the long Ω loop (Cys67–Cys94 in BfAChE) and at the tip of the α36,7-α46,7 loop of other AChEs (Fig. 2A) and mainly contribute to the low sensitivity of the snake enzymes to PAS inhibitors (27, 38). Compared with TcAChE or mAChE, the Met substitution in BfAChE slightly enlarges the gorge entrance; however, this feature does not seem to be associated with a greater turnover number (27) (data not shown). Incidentally, BLAST searches using the BfAChE sequence as a template identified close orthologs sharing 86–94% sequence identity along with the two PAS mutations in the genome of the snakes Suta fasciata, Echiopsis curta, and Denisonia devisi, suggesting evolutionary relationships.
At the PAS, bound Fab410, which displays the canonical β-sandwich Ig fold, is trapped between the glycan chains linked to Asn343 and Asn453 on each side of the gorge entrance (see below) with its long axis tilted by ∼30° from the main axis of the gorge (Figs. 3A, 4A, and 5A). At the interface between the VH and VL domains, the pocket-shaped combining site, 12 Å long and 5 Å wide, is delimitated by the protruding CDRs H2 and H3 and the emerging although shorter CDRs H1 and L3 (Fig. 2B). The paratope surface is markedly electropositive and confers a clear anisotropic distribution of charges on the Fab410 variable domains (Fig. 3B) with a dipole moment of 780 debyes that points toward the H chain and emerges near Lys67 in the VH domain (not shown). This feature is consistent with the calculated high pI value (8.1) of Fab410 and its retention close to the cathode in native PAGE (32) (see Fig. 1C, lane 2). It is also consistent with interaction of Fab410 with the PAS of BfAChE competitively with the cationic ligands Fas2 and propidium and bisquaternary ligands edrophonium and decamethonium (27). Compared with the theoretical model of unbound Fab410 (32), the experimental structure of bound Fab410 displays overall good overlap (see the root mean square deviation value under “Experimental procedures”) except for the flexible tip of the long CDR H3, whose inverted curvature better accommodates the long Ω loop at the BfAChE surface.
Evidence for Coexisting Open and Closed States of a Back Door Channel in All BfAChE Subunits
At the bottom of the BfAChE active site, the phenol ring of Tyr442, which is located in loop β8-α18,9 in the back door region (Fig. 2A), adopts two alternate conformations equally represented in each BfAChE subunit (Fig. 4). In one conformation, the hydroxyl group of Tyr442 is tightly bound to the indole nitrogen atoms of Trp84 and Trp442 and to the carbonyl of Gly80 as observed in most AChE structures. In the second conformation, the Tyr442 phenol ring is rotated by ∼110° and points toward the subunit surface, to occupy a hydrophobic pocket lined with Phe423, Leu430, Val438, and Phe446, where the hydroxyl establishes weak interactions with Ala427 in the β8-α18,9 (Ala427–Gly437) surface loop associated with an outward displacement (up to 2 Å) at the loop tip (Fig. 4, A and B). The large rotational gating motion of the Tyr442 phenol ring opens a narrow channel, 15 Å long and 3–4 Å wide (i.e. a radius of approximately one-half that of the constricted region located midway in the active site gorge), roughly perpendicular to the main axis of the gorge path (Fig. 4, C and D). The channel begins at the anionic subsite within the active site pocket, and then it curves around the edge of Trp84 to insert deeper between the αβ3,2 and α18,9 helices, exemplified by Met83 and Glu445, respectively, to finally emerge as a surface dimple lined by side chains from the Ala427–Tyr442 and Leu456–Tyr458 loops in the back door region.
Most of the residues that line this channel (e.g. Met83, Trp84, Trp432, and Glu445) are conserved in most AChE species (Fig. 2A), suggesting that channel opening is a constitutive functional event. In fact, in the aflatoxin-bound TcAChE dimer, a similarly rotated Tyr442 phenol ring associated with an open channel is observed in one subunit, whereas the usual Tyr442 conformation with no channel is retained in the second subunit (26) (Fig. 4, C and D). In unliganded DmAChE, a genuine back door channel exists due to Ile and Asp substitutions to Met83 and the bulky Tyr442, respectively, that destabilize Trp84 (25) (Fig. 4C). However, in both Fab410-bound BfAChE and aflatoxin-bound TcAChE (but not apo-DmAChE), the channel displays a significant bottleneck, dictated by the orientation of the facing Trp84 and Trp432 side chains and narrower than the constricted region in the active site gorge (Fig. 4, C and D). Hence, even with a high Tyr442 rotation frequency as denoted in the two structures, additional conformational changes leading to substantial bottleneck enlargement in these liganded AChEs would be required to permit substantial trafficking of molecules through the back door channel. In this context, species-dependent substitutions (Pro434 in BfAChE versus Glu434 in TcAChE) in the β8-α18,9 loop region bearing Tyr442 may cause local conformational rearrangements and dynamic enlargement of the back door channel. These observations, along with the fact that none of these three AChEs display particularly high catalytic activity compared with the average activity of most AChEs, would support the previously proposed absence of functional relevance of a back door in apo-AChE (24, 69) in contrast with the role of gated tunnels to regulate other enzymes or ion channels (70).
Compared with the various Fas2-AChE complexes, which show tightly bound Fas2 at the PAS with a fully occluded gorge entrance and no back door channel in the crystal state (14, 23, 71–74) and display residual activities not exceeding a few percent in solution (15, 75–77), the semi-occluding Fab410 position at the BfAChE PAS and presumed higher opening frequency of the back door channel in the complex are likely to account for the greater residual activity of the Fab410-BfAChE complex (Table 1). However, the presence of bound Fab410 at the BfAChE PAS raises a question as to whether both conformations of Tyr442 preexist in unliganded BfAChE or whether one of them is correlated with (i.e. associated with or induced by) Fab410 binding and, if so, which one is the apo conformation. In fact, close proximity of residues at the tip of CDR L1 in Fab410 to residues in helix αβ3,2 of BfAChE, e.g. Gln79 (Glu in mAChE and Ser in TcAChE) that lines the exit of the back door channel, suggests that bound Fab410 could interfere with the trafficking of small molecules through the channel. In the absence of a structure of unliganded BfAChE, close examination of the complex interface and structural comparison with other AChEs in the unliganded form (below) are a suitable way to explore this hypothesis. Whether this alternative route is efficiently used during AChE catalysis for substrate/product trafficking and how channel opening is regulated remain to be investigated, in focusing on the dynamic aspects of the enzyme during ligand binding.
The Fab410-BfAChE Complex Interface and Implications of the Structure for BfAChE Inhibition by Fab410
At the complex interface, the concave combining site of Fab410 sequesters the tip of the long Ω loop located on one side of the gorge rim (Figs. 3 and 4A). As a result, the Ω loop tip, whose conformation is already constrained by Pro76, is further rigidified within the Fab410 combining site. In fact, comparison of the Fab410-bound BfAChE and apo-mAChE structures points to a slightly more open (by as much as 2 Å) Ser74–Gln79 segment at the loop tip, suggesting the occurrence of a rigid body movement upon Fab410 binding.
An average surface area of ∼900 Å2 is buried on each partner of the complex with the Fab410 H and L chains contributing ∼500 and ∼400 Å2, respectively, and the binding interface is dominated by electrostatic interactions (Fig. 5 and Table 3). In BfAChE, 12 residues of the long Ω loop segment Tyr68–Met90 contribute ∼70% of the binding interface, whereas neighboring Asp342 in the α17,8-α27,8 loop and Gln272 and Asp276 in the facing helix α36,7 across the gorge entrance provide additional contact points at the interface periphery. The Asn343-linked glycan chain contributes an additional interface area of ∼350 Å2, thereby providing additional stability to bound Fab410.
TABLE 3.
Electrostatic interactions at the BfAChE-Fab410 complex interface
BfAChE residues not conserved in other AChEs are italicized. The corresponding EeAChE residues are indicated in parentheses.
| BfAChE |
Fab410 |
|||
|---|---|---|---|---|
| Residue | Atom | Residue | Atom | CDR |
| Asp72 | O | Tyr109 | Oη | H3 |
| Thr73 | Oγ1 | Arg52 | Nη1 | H2 |
| Oγ1 | Asn56 | Nδ2 | H2 | |
| Ser74 (Ser75) | O | Arg50a | Nη1 | (H) |
| Tyr75 | Oη | His93 | Nδ1 | L3 |
| Pro76 | O | Arg49 | Nη2 | L2 |
| O | Tyr33a | Oη | (L) | |
| Gly77 | O | Tyr31 | N | L1 |
| Gln79 (Ser80) | Oϵ1 | Tyr31 | Oη | L1 |
| N | Ser30 | Oγ | L1 | |
| Glu82 | Oϵ1 | Tyr104 | Oη | H3 |
| Oϵ1 | Arg49 | Nη2 | L2 | |
| Oϵ2 | Tyr109 | Oη | H3 | |
| Pro86 | O | Tyr104 | Oη | H3 |
| Gln272 | Oϵ1 | Tyr105 | Oη | H3 |
| Asp276 | O | Ser55 | Oγ | H2 |
| Lys341 | Nζ | Asn91 | O | L3 |
| Nζ | Ser92 | Oγ | L3 | |
| Asp342 | Oδ2 | His93 | Nδ1 | L3 |
a Fab410 residues not in a CDR.
In Fab410, the central position of the H chain places the strongly electropositive CDR H2 (32) close to the gorge entrance with the side chains of Arg52, Asn56, and Lys57 pointing within the gorge (Fig. 5 and Table 3). The tip of the extended CDR H3, which encompasses the aromatic Tyr104/Tyr105/Tyr109 triplet, comfortably anchors Fab410 at the PAS periphery where it contributes 55% of the area buried by the H chain. In contrast, CDR H1 only weakly contributes to the interface as only Thr31 at its tip interacts with Asp276 in helix α36,7 of BfAChE. Unexpectedly, BfAChE Met70 and Lys285, which are located at the base of the long Ω loop and in the α36,7-α46,7 loop, respectively, and which distinguish the PAS of BfAChE and NnAChE from other AChE species, weakly contribute to Fab410 binding as only Met70 establishes long range van der Waals contacts with Ser55 at the tip of CDR H2. In more detail, the non-CDR residue Arg50 interacts with BfAChE Ser75 in the long Ω loop, whereas Ser55 at the tip of CDR H2 is bound to Asp276 (Ala in NoAChE) across the gorge entrance. In CDH H3, Tyr104, which adopts two alternate conformations related by a 45° arc motion, abuts on the external face of the long Ω loop where it interacts with the Pro86 carbonyl and the Met90 side chain; Tyr105 extends the network of interactions with BfAChE Tyr68 and Met90 in the long Ω loop and with Gln272 in helix α36,7; and Tyr109 is bound to BfAChE Glu82 and in van der Waals contact with Thr73 and Pro76 all in the long Ω loop.
For its part, the Fab410 L chain is positioned near the tip of the long Ω loop in BfAChE, away from the gorge entrance, with CDR L3 contributing most of the binding interface (Fig. 5 and Table 3). In the Naja AChEs, substitution of the Asp276-Glu277 pair in helix α36,7 by an Ala-Lys pair5 may confer electrostatic repulsion with the positively charged CDR H2 and be responsible for the weak inhibition by Elec410 (27). In more detail, the Ser30-Tyr31 pair at the CDR L1 tip on the interior face of the Fab molecule interacts with BfAChE Gln79. In CDR L2, only the Arg49 guanidinium, in stacking interaction with CDR L1 residue Tyr31, interacts with the BfAChE Pro76 carbonyl and the Glu82 carboxylate. In CDR L3, Tyr90, which is located at the L chain/H chain interface, establishes stacking interactions with BfAChE Pro76; Asn91 interacts with BfAChE Phe78; Ser92 interacts with BfAChE Lys341 and Asp342 in the α17,8-α27,8 loop, which is remote from the gorge entrance; and His93, which is in contact with H chain residues Arg50 and His61, interacts with BfAChE Tyr75.
In contrast to bound Fas2 that occludes the AChE gorge entrance in crystalline complexes (see above), bound Fab410 only partially occludes it so that the PAS region near loop Tyr334–Gly335, which faces the long Ω loop across the gorge rim, remains solvent-accessible (Fig. 6A). Moreover, Fab410 lacks an equivalent to Fas2 Met33, which protrudes at the tip of loop II to interact with Trp279 and act as a central plug. Hence, both these features and the open back door are likely to account for the greater residual activity of the Fab410 complex compared with Fas2 complexes (Table 1 and see above). However, the nature and spatial arrangement of key positively charged or aromatic side chains from other Fas2 loops and Fab410 CDRs surrounding the gorge rim are remarkably conserved despite the very moderate overlap of their backbone traces, an observation consistent with the cationic nature of both ligands (Fig. 6A). Indeed, compared with the Fas2-mAChE complex (14), CDR H2 residue Lys57 mimics Fas2 Arg27 near the gorge entrance, and CDR H2 Arg52 and Lys54 correspond to Fas2 Arg24 and Lys25, respectively; the neighboring Fab410 Arg50 coincides with Fas2 Arg37; and CDR L3 Tyr90 and CDR L2 Arg49, respectively, coincide with Fas2 Tyr4 and Arg11 at the gorge periphery, thereby providing still another example of exquisite mimicry of the molecular determinants of organic and peptidic PAS ligands (18).
Implications of the Structure for EeAChE Inhibition by Fab410
All three Elec antibodies were raised against EeAChE, and Elec403, Elec408 and their Fab derivatives inhibit EeAChE only. In contrast, Elec410 and Fab410 inhibit both EeAChE and BfAChE although with distinctive dissociation constant and residual activity values (Table 1), suggesting slightly different Fab410 positioning and/or interaction networks at the respective gorge entrances. In fact, six non-conservative substitutions are found in the PAS regions of these two AChEs of which three (Tyr71, Ser80, and Ser287 in EeAChE in place of Met70, Gln79, and Lys285 in BfAChE) are located within the Fab410 binding site at the BfAChE surface (see Fig. 2A). To explore the possible mode of Fab410 binding onto EeAChE, we performed a software-assisted docking of the crystalline Fab410 molecule onto the PAS of a modeled EeAChE subunit and validated this procedure by redocking Fab410 onto the PAS of BfAChE using the same parameters (see “Experimental Procedures”).
The modeled Fab410-EeAChE complex (Fig. 6B) shows a similarly bound and oriented Fab410 on one side of the gorge rim of EeAChE as observed for BfAChE, consistent with the weak contribution of BfAChE Met70 and Lys285, which are replaced by Tyr71 and Ser287 in EeAChE, to Fab410 binding (Figs. 2A and 6B). The model also highlights those three substitutions in the PAS of EeAChE that are likely to contribute to the greater affinity for Fab410 (Table 1). First, the phenol ring of EeAChE Tyr71, which replaces BfAChE Met70, is well positioned on the solvent-exposed face of the long Ω loop to establish polar interactions with Arg52 and Asn56 in CDR H2. Second, the polar side chain of neighboring EeAChE Ser80, which is smaller than the corresponding BfAChE Gln79, is better suited to closely accommodate the tip of CDR L1. Third, EeAChE Ser287 in place of BfAChE Lys285 in the short Ω loop presents suitable electrostatic complementarity with the facing Lys54 in CDR H2. In turn, EeAChE Ser75 at the tip of the long Ω loop and conserved in BfAChE abuts on Arg50 and Arg52 in CDR H2, a position consistent with its critical contribution to Fab410 binding, as shown by loss of inhibition upon substitution of this small polar side chain by the Leu side chain found in mammalian AChEs (45). As well, the absence of Fab410 interaction with EeAChE Leu282, which corresponds to BfAChE Ser280, reflects the limited reduction in Fab410 affinity observed upon substitution of this residue by the mammalian His counterpart (45). Hence, this experimentally based model of the Fab410-EeAChE complex largely accounts for the greater affinity of EeAChE for Fab410 and the absence of Elec410 effect on human, bovine, Torpedo, and Naja AChEs (27, 31) and mAChE (data not shown).
Of the three N-glycosylation sites located near the BfAChE gorge entrance, those at Asn343 (occupied in the structure) and Asn457 (non-occupied) correspond to EeAChE Asn345 and Asn488, whereas the third site at Asn453 (occupied) corresponds to EeAChE Glu484 (Fig. 2A). Whether EeAChE Asn345 and Asn488 are both occupied is unknown, but at least one is because deglycosylation of EeAChE results in ∼10-fold greater sensitivity to inhibition by Fab403, despite unaltered inhibition by Fab410 (32). Hence, distinctive distribution and/or nature of the glycan chains in the PAS regions of EeAChE and BfAChE may provide distinctive constraints onto Fab410 positioning and contribute to the ∼80-fold difference in the affinities of the two enzymes (Table 1).
Finally, this predicted mode of Fab410 binding at the EeAChE PAS is also consistent with its competition with the large peptidic inhibitor Fas2 but absence of competition with the small organic inhibitors propidium, decamethonium, BW284C51, and the substrate acetylcholine when in excess (31). In fact, structural comparison of the crystalline Fab410-BfAChE and modeled Fab410-EeAChE complexes with crystalline mAChE or TcAChE complexes with acetylcholine (8, 9) and propidium (18) and with the bifunctional ligands BW284C51 (63) and decamethonium (62) points to only one critical steric clash occurring between the propidium alkyl chain and Lys57 in Fab410 CDR H2 (data not shown). This observation along with the greater improvement of propidium binding observed for the M70Y mutant of BfAChE compared with its K285D mutant (38) also largely supports our model of the Fab410-EeAChE complex.
In summary, this biochemical and structural analysis of the Fab410-BfAChE complex illustrates the capacity of the snake venom enzyme to form canonical dimers of subunits despite its unusual, non-amphiphilic C terminus; documents the mode of binding of one of the largest peptidic inhibitors targeting the PAS of an AChE; and reveals the molecular determinants associated with inhibition of BfAChE and EeAChE, but not other AChEs, by this antibody. Most particularly, our data provide a precise picture of the antibody pose on one side of the active site gorge entrance and unveil an equilibrium of closed and open states of a back door channel not observed in previous AChE structures, two features likely to be correlated with the significant residual activity of this particular complex. Hence, this study provides a novel AChE template to explore alternative binding perimeters and topographies at the PAS surface for non-competitive modulators of catalysis along with new determinants and mechanisms associated with the dynamics of back door opening. The availability of crystal structures of AChE complexes with Fab403 and Fab408, which target the PAS and back door region, respectively, would complete the picturing of the molecular determinants and mechanisms involved in regulation of AChE catalysis by the “Elec” mAbs.
Acknowledgments
We are grateful to Cassian Bon (deceased) (Institut Pasteur, Paris, France) and Jacques Grassi, Didier Boquet, and Christophe Créminon (Commissariat à l'Energie Atomique Saclay, Gif-sur-Yvette, France) for the kind gift of biological materials, unpublished data, and enthusiastic support; Bernard Saliou (Institut Pasteur, Paris) and Marianick Juin and Sandrine Conrod (CNRS/Aix-Marseille Université (AMU), Marseille, France) for assistance in protein preparation and analysis; Silvia Spinelli (Architecture et Fonction des Macromolécules Biologiques, Marseille) and the ID14-EH3 staff (European Synchrotron Radiation Facility, Grenoble, France) for crystallographic data collection; Christopher Weise (Freie Universität, Berlin, Germany) and Xavier Cousin (Institut National de la Recherche Agronomique, Rennes, France) for sharing the unpublished complete sequence of N. oxiana AChE; and Jean Massoulié (deceased) and Stéphanie Simon (École Normale Supérieure, Paris) and Igor Fabrichny (CNRS/AMU, Marseille) for fruitful discussions.
This work was supported by the Association Française contre les Myopathies (a Ph.D. grant to L. R.) and (research fees to P. M.).
This paper is dedicated to the memory of Dr. Cassian Bon (Institut Pasteur, CNRS, Muséum National d'Histoire Naturelle, Paris, France), deceased March 2008.
The atomic coordinates and structure factors (code 4QWW) have been deposited in the Protein Data Bank (http://wwpdb.org/).
C. Weise, X. Cousin, and C. Bon, personal communication.
- AChE
- acetylcholinesterase (BfAChE, from B. fasciatus venom
- EeAChE
- from E. electricus electric organ
- mAChE
- recombinant from mouse
- TcAChE
- from T. californica electric organ
- DmAChE
- from Drosophila
- NnAChE
- from N. naja oxiana venom)
- CDR
- complementarity-determining region
- Fas2
- fasciculin 2
- PAS
- peripheral anionic site
- VH
- CH, VL, CL, variable (V) and constant (C) regions of the heavy (H) and light (L) chains, respectively.
REFERENCES
- 1. Massoulié J., Pezzementi L., Bon S., Krejci E., Vallette F. M. (1993) Molecular and cellular biology of cholinesterases. Prog. Neurobiol. 41, 31–91 [DOI] [PubMed] [Google Scholar]
- 2. Taylor P., Radić Z. (1994) The cholinesterases: from genes to proteins. Annu. Rev. Pharmacol. Toxicol. 34, 281–320 [DOI] [PubMed] [Google Scholar]
- 3. Silman I., Sussman J. L. (2008) Acetylcholinesterase: how is structure related to function? Chem. Biol. Interact. 175, 3–10 [DOI] [PubMed] [Google Scholar]
- 4. Sussman J. L., Harel M., Frolow F., Oefner C., Goldman A., Toker L., Silman I. (1991) Atomic structure of acetylcholinesterase from Torpedo californica: a prototypic acetylcholine-binding protein. Science 253, 872–889 [DOI] [PubMed] [Google Scholar]
- 5. Quinn D. M. (1987) Acetylcholinesterase: enzyme structure, reaction dynamics, and virtual transition states. Chem. Rev. 87, 955–979 [Google Scholar]
- 6. Taylor P., Lappi S. (1975) Interaction of fluorescence probes with acetylcholinesterase. The site and specificity of propidium binding. Biochemistry 14, 1989–1997 [DOI] [PubMed] [Google Scholar]
- 7. Radić Z., Pickering N. A., Vellom D. C., Camp S., Taylor P. (1993) Three distinct domains in the cholinesterase molecule confer selectivity for acetyl- and butyrylcholinesterase inhibitors. Biochemistry 32, 12074–12084 [DOI] [PubMed] [Google Scholar]
- 8. Bourne Y., Radić Z., Sulzenbacher G., Kim E., Taylor P., Marchot P. (2006) Substrate and product trafficking through the active center gorge of acetylcholinesterase analyzed by crystallography and equilibrium binding. J. Biol. Chem. 281, 29256–29267 [DOI] [PubMed] [Google Scholar]
- 9. Colletier J. P., Fournier D., Greenblatt H. M., Stojan J., Sussman J. L., Zaccai G., Silman I., Weik M. (2006) Structural insights into substrate traffic and inhibition in acetylcholinesterase. EMBO J. 25, 2746–2756 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Radić Z., Reiner E., Taylor P. (1991) Role of the peripheral anionic site on acetylcholinesterase: inhibition by substrates and coumarin derivatives. Mol. Pharmacol. 39, 98–104 [PubMed] [Google Scholar]
- 11. Szegletes T., Mallender W. D., Thomas P. J., Rosenberry T. L. (1999) Substrate binding to the peripheral site of acetylcholinesterase initiates enzymatic catalysis. Substrate inhibition arises as a secondary effect. Biochemistry 38, 122–133 [DOI] [PubMed] [Google Scholar]
- 12. Mallender W. D., Szegletes T., Rosenberry T. L. (2000) Acetylthiocholine binds to Asp74 at the peripheral site of human acetylcholinesterase as the first step in the catalytic pathway. Biochemistry 39, 7753–7763 [DOI] [PubMed] [Google Scholar]
- 13. Johnson J. L., Cusack B., Hughes T. F., McCullough E. H., Fauq A., Romanovskis P., Spatola A. F., Rosenberry T. L. (2003) Inhibitors tethered near the acetylcholinesterase active site serve as molecular rulers of the peripheral and acylation sites. J. Biol. Chem. 278, 38948–38955 [DOI] [PubMed] [Google Scholar]
- 14. Bourne Y., Taylor P., Marchot P. (1995) Acetylcholinesterase inhibition by fasciculin: crystal structure of the complex. Cell 83, 503–512 [DOI] [PubMed] [Google Scholar]
- 15. Radić Z., Quinn D. M., Vellom D. C., Camp S., Taylor P. (1995) Allosteric control of acetylcholinesterase catalysis by fasciculin. J. Biol. Chem. 270, 20391–20399 [DOI] [PubMed] [Google Scholar]
- 16. Bourne Y., Taylor P., Bougis P. E., Marchot P. (1999) Crystal structure of mouse acetylcholinesterase. A peripheral site-occluding loop in a tetrameric assembly. J. Biol. Chem. 274, 2963–2970 [DOI] [PubMed] [Google Scholar]
- 17. Bourne Y., Marchot P. (2002) in Toxines et Recherches Biomédicales (Goudey-Perrière F., Bon C., Puiseux-Dao S., Sauviat M. P., eds) pp. 287–296, Editions Scientifiques et Médicales Elsevier, Paris [Google Scholar]
- 18. Bourne Y., Taylor P., Radić Z., Marchot P. (2003) Structural insights into ligand interactions at the acetylcholinesterase peripheral anionic site. EMBO J. 22, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bourne Y., Kolb H. C., Radić Z., Sharpless K. B., Taylor P., Marchot P. (2004) Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proc. Natl. Acad. Sci. U.S.A. 101, 1449–1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Colletier J. P., Sanson B., Nachon F., Gabellieri E., Fattorusso C., Campiani G., Weik M. (2006) Conformational flexibility in the peripheral site of Torpedo californica acetylcholinesterase revealed by the complex structure with a bifunctional inhibitor. J. Am. Chem. Soc. 128, 4526–4527 [DOI] [PubMed] [Google Scholar]
- 21. Ripoll D. R., Faerman C. H., Axelsen P. H., Silman I., Sussman J. L. (1993) An electrostatic mechanism for substrate guidance down the aromatic gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 90, 5128–5132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gilson M. K., Straatsma T. P., McCammon J. A., Ripoll D. R., Faerman C. H., Axelsen P. H., Silman I., Sussman J. L. (1994) Open “back door” in a molecular dynamics simulation of acetylcholinesterase. Science 263, 1276–1278 [DOI] [PubMed] [Google Scholar]
- 23. Harel M., Kleywegt G. J., Ravelli R. B., Silman I., Sussman J. L. (1995) Crystal structure of an acetylcholinesterase-fasciculin complex: interaction of a three-fingered toxin from snake venom with its target. Structure 3, 1355–1366 [DOI] [PubMed] [Google Scholar]
- 24. Xu Y., Colletier J. P., Weik M., Qin G., Jiang H., Silman I., Sussman J. L. (2010) Long route or shortcut? A molecular dynamics study of traffic of thiocholine within the active-site gorge of acetylcholinesterase. Biophys. J. 99, 4003–4011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nachon F., Stojan J., Fournier D. (2008) Insights into substrate and product traffic in the Drosophila melanogaster acetylcholinesterase active site gorge by enlarging a back channel. FEBS J. 275, 2659–2664 [DOI] [PubMed] [Google Scholar]
- 26. Sanson B., Colletier J. P., Xu Y., Lang P. T., Jiang H., Silman I., Sussman J. L., Weik M. (2011) Backdoor opening mechanism in acetylcholinesterase based on x-ray crystallography and molecular dynamics simulations. Protein Sci. 20, 1114–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Frobert Y., Créminon C., Cousin X., Rémy M. H., Chatel J. M., Bon S., Bon C., Grassi J. (1997) Acetylcholinesterases from Elapidae snake venoms: biochemical, immunological and enzymatic characterization. Biochim. Biophys. Acta 1339, 253–267 [DOI] [PubMed] [Google Scholar]
- 28. Duhaiman A. S., Alhomida A. S., Rabbani N., Kamal M. A., al-Jafari A. A. (1996) Purification and characterization of acetylcholinesterase from desert cobra (Walterinnesia aegyptia) venom. Biochimie 78, 46–50 [DOI] [PubMed] [Google Scholar]
- 29. Malih I., Ahmad rusmili M. R., Tee T. Y., Saile R., Ghalim N., Othman I. (2014) Proteomic analysis of Moroccan cobra Naja haje legionis venom using tandem mass spectrometry. J. Proteomics 96, 240–252 [DOI] [PubMed] [Google Scholar]
- 30. Vonk F. J., Casewell N. R., Henkel C. V., Heimberg A. M., Jansen H. J., McCleary R. J., Kerkkamp H. M., Vos R. A., Guerreiro I., Calvete J. J., Wüster W., Woods A. E., Logan J. M., Harrison R. A., Castoe T. A., de Koning A. P., Pollock D. D., Yandell M., Calderon D., Renjifo C., Currier R. B., Salgado D., Pla D., Sanz L., Hyder A. S., Ribeiro J. M., Arntzen J. W., van den Thillart G. E., Boetzer M., Pirovano W., Dirks R. P., Spaink H. P., Duboule D., McGlinn E., Kini R. M., Richardson M. K. (2013) The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. U.S.A. 110, 20651–20656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Remy M. H., Frobert Y., Grassi J. (1995) Characterization of monoclonal antibodies that strongly inhibit Electrophorus electricus acetylcholinesterase. Eur. J. Biochem. 231, 651–658 [DOI] [PubMed] [Google Scholar]
- 32. Bourne Y., Renault L., Essono S., Mondielli G., Lamourette P., Boquet D., Grassi J., Marchot P. (2013) Molecular characterization of monoclonal antibodies that inhibit acetylcholinesterase by targeting the peripheral site and backdoor region. PLoS One 8, e77226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ahmed M., Latif N., Khan R. A., Ahmad A., Rocha J. B. T., Mazzanti C. M., Bagatini M. D., Morsch V. M., Schetinger M. R. C. (2012) Enzymatic and biochemical characterization of Bungarus sindanus snake venom acetylcholinesterase. J. Venom. Anim. Toxins Incl. Trop. Dis. 18, 236–243 [Google Scholar]
- 34. Kumar V., Elliott W. B. (1973) The acetylcholinesterase of Bungarus fasciatus venom. Eur. J. Biochem. 34, 586–592 [DOI] [PubMed] [Google Scholar]
- 35. Kumar V., Elliott W. B. (1975) Acetylcholinesterase from Bungarus fasciatus venom-I. Substrate specificity. Comp. Biochem. Physiol. C 51, 249–253 [DOI] [PubMed] [Google Scholar]
- 36. Kumar V., Elliott W. B. (1975) Acetylcholinesterase from Bungarus fasciatus venom-II. Effects of pH and temperature. Comp. Biochem. Physiol. C 51, 255–258 [DOI] [PubMed] [Google Scholar]
- 37. Nickerson P. A., Kumar V. (1974) Electron microscopic studies of acetylcholinesterase from Bungarus fasciatus venom. Toxicon 12, 83–84 [DOI] [PubMed] [Google Scholar]
- 38. Cousin X., Bon S., Duval N., Massoulié J., Bon C. (1996) Cloning and expression of acetylcholinesterase from Bungarus fasciatus venom. A new type of COOH-terminal domain; involvement of a positively charged residue in the peripheral site. J. Biol. Chem. 271, 15099–15108 [DOI] [PubMed] [Google Scholar]
- 39. Porschke D., Créminon C., Cousin X., Bon C., Sussman J., Silman I. (1996) Electrooptical measurements demonstrate a large permanent dipole moment associated with acetylcholinesterase. Biophys. J. 70, 1603–1608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Masson P., Schopfer L. M., Bartels C. F., Froment M. T., Ribes F., Nachon F., Lockridge O. (2002) Substrate activation in acetylcholinesterase induced by low pH or mutation in the π-cation subsite. Biochim. Biophys. Acta 1594, 313–324 [DOI] [PubMed] [Google Scholar]
- 41. Cousin X., Bon C. (1997) Acetylcholinesterase from snake venoms. C. R. Seances Soc. Biol. Fil. 191, 381–400 [PubMed] [Google Scholar]
- 42. Cousin X., Bon C. (1999) Acetylcholinesterase from snake venom as a model for its nerve and muscle counterpart. J. Nat. Toxins 8, 285–294 [PubMed] [Google Scholar]
- 43. Massoulié J. (2002) The origin of the molecular diversity and functional anchoring of cholinesterases. Neurosignals 11, 130–143 [DOI] [PubMed] [Google Scholar]
- 44. Simon S., Massoulié J. (1997) Cloning and expression of acetylcholinesterase from Electrophorus. Splicing pattern of the 3′ exons in vivo and in transfected mammalian cells. J. Biol. Chem. 272, 33045–33055 [DOI] [PubMed] [Google Scholar]
- 45. Simon S., Le Goff A., Frobert Y., Grassi J., Massoulié J. (1999) The binding sites of inhibitory monoclonal antibodies on acetylcholinesterase. Identification of a novel regulatory site at the putative “back door.” J. Biol. Chem. 274, 27740–27746 [DOI] [PubMed] [Google Scholar]
- 46. Renault L., Essono S., Juin M., Boquet D., Grassi J., Bourne Y., Marchot P. (2005) Structural insights into AChE inhibition by monoclonal antibodies. Chem. Biol. Interact. 157–158, 397–400 [DOI] [PubMed] [Google Scholar]
- 47. Massoulié J., Bon S. (1976) Affinity chromatography of acetylcholinesterase. The importance of hydrophobic interactions. Eur. J. Biochem. 68, 531–539 [DOI] [PubMed] [Google Scholar]
- 48. Ellman G. L., Courtney K. D., Andres V., Jr., Feather-Stone R. M. (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem. Pharmacol. 7, 88–95 [DOI] [PubMed] [Google Scholar]
- 49. Leslie A. G. W. (1992) Recent changes to the MOSFLM package for processing film and image plate data. Joint CCP4 and ESF-EAMCB Newsletter on Protein Crystallography, Number 26, Daresbury Laboratory, Warrington, UK [Google Scholar]
- 50. Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 50, 760–763 [DOI] [PubMed] [Google Scholar]
- 51. Navaza J. (1994) AMoRe: an automated package for molecular replacement. Acta Crystallogr. A 50, 157–163 [Google Scholar]
- 52. Ito S., Fujimori T., Hayashizaki Y., Nabeshima Y. (2002) Identification of a novel mouse membrane-bound family 1 glycosidase-like protein, which carries an atypical active site structure. Biochim. Biophys. Acta 1576, 341–345 [DOI] [PubMed] [Google Scholar]
- 53. Suhre K., Sanejouand Y. H. (2004) ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 32, W610–W614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 56. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 [DOI] [PubMed] [Google Scholar]
- 57. Bricogne G., Blanc E., Brandl M., Flensburg C., Keller P., Paciorek W., Roversi P., Sharff A., Smart O. S., Vonrhein C., Womack T. O. (2011) BUSTER, version 2.11.2, Global Phasing Ltd., Cambridge, UK [Google Scholar]
- 58. Davis I. W., Leaver-Fay A., Chen V. B., Block J. N., Kapral G. J., Wang X., Murray L. W., Arendall W. B., 3rd, Snoeyink J., Richardson J. S., Richardson D. C. (2007) MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 35, W375–W383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Baker N. A., Sept D., Joseph S., Holst M. J., McCammon J. A. (2001) Electrostatics of nanosystems: application to microtubules and the ribosome. Proc. Natl. Acad. Sci. U.S.A. 98, 10037–10041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Krissinel E., Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 [DOI] [PubMed] [Google Scholar]
- 61. Chovancova E., Pavelka A., Benes P., Strnad O., Brezovsky J., Kozlikova B., Gora A., Sustr V., Klvana M., Medek P., Biedermannova L., Sochor J., Damborsky J. (2012) CAVER 3.0: a tool for the analysis of transport pathways in dynamic protein structures. PLoS Comput. Biol. 8, e1002708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Harel M., Schalk I., Ehret-Sabatier L., Bouet F., Goeldner M., Hirth C., Axelsen P. H., Silman I., Sussman J. L. (1993) Quaternary ligand binding to aromatic residues in the active-site gorge of acetylcholinesterase. Proc. Natl. Acad. Sci. U.S.A. 90, 9031–9035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Felder C. E., Harel M., Silman I., Sussman J. L. (2002) Structure of a complex of the potent and specific inhibitor BW284C51 with Torpedo californica acetylcholinesterase. Acta Crystallogr. D. Biol. Crystallogr. 58, 1765–1771 [DOI] [PubMed] [Google Scholar]
- 64. Harel M., Kryger G., Rosenberry T. L., Mallender W. D., Lewis T., Fletcher R. J., Guss J. M., Silman I., Sussman J. L. (2000) Three-dimensional structures of Drosophila melanogaster acetylcholinesterase and of its complexes with two potent inhibitors. Protein Sci. 9, 1063–1072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. DeLano W. L. (2010) The PyMOL Molecular Graphics System, version 1.5.0.4 Schrödinger, LLC, New York [Google Scholar]
- 66. Dominguez C., Boelens R., Bonvin A. M. (2003) HADDOCK: a protein-protein docking approach based on biochemical or biophysical information. J. Am. Chem. Soc. 125, 1731–1737 [DOI] [PubMed] [Google Scholar]
- 67. Cousin X., Créminon C., Grassi J., Méflah K., Cornu G., Saliou B., Bon S., Massoulié J., Bon C. (1996) Acetylcholinesterase from Bungarus venom: a monomeric species. FEBS Lett. 387, 196–200 [DOI] [PubMed] [Google Scholar]
- 68. Raba R., Aaviksaar A., Raba M., Siigur J. (1979) Cobra venom acetylcholinesterase. Purification and molecular properties. Eur. J. Biochem. 96, 151–158 [DOI] [PubMed] [Google Scholar]
- 69. Kronman C., Ordentlich A., Barak D., Velan B., Shafferman A. (1994) The “back door” hypothesis for product clearance in acetylcholinesterase challenged by site-directed mutagenesis. J. Biol. Chem. 269, 27819–27822 [PubMed] [Google Scholar]
- 70. Zhou H. X., McCammon J. A. (2010) The gates of ion channels and enzymes. Trends Biochem. Sci. 35, 179–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Kryger G., Harel M., Giles K., Toker L., Velan B., Lazar A., Kronman C., Barak D., Ariel N., Shafferman A., Silman I., Sussman J. L. (2000) Structures of recombinant native and E202Q mutant human acetylcholinesterase complexed with the snake-venom toxin fasciculin-II. Acta Crystallogr. D Biol. Crystallogr. 56, 1385–1394 [DOI] [PubMed] [Google Scholar]
- 72. Carletti E., Colletier J. P., Dupeux F., Trovaslet M., Masson P., Nachon F. (2010) Structural evidence that human acetylcholinesterase inhibited by tabun ages through O-dealkylation. J. Med. Chem. 53, 4002–4008 [DOI] [PubMed] [Google Scholar]
- 73. Cheung J., Rudolph M. J., Burshteyn F., Cassidy M. S., Gary E. N., Love J., Franklin M. C., Height J. J. (2012) Structures of human acetylcholinesterase in complex with pharmacologically important ligands. J. Med. Chem. 55, 10282–10286 [DOI] [PubMed] [Google Scholar]
- 74. Nachon F., Carletti E., Ronco C., Trovaslet M., Nicolet Y., Jean L., Renard P. Y. (2013) Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer's drugs targeting acetyl- and butyryl-cholinesterase. Biochem. J. 453, 393–399 [DOI] [PubMed] [Google Scholar]
- 75. Marchot P., Khélif A., Ji Y. H., Mansuelle P., Bougis P. E. (1993) Binding of 125I-fasciculin to rat brain acetylcholinesterase. The complex still binds diisopropyl fluorophosphate. J. Biol. Chem. 268, 12458–12467 [PubMed] [Google Scholar]
- 76. Radić Z., Duran R., Vellom D. C., Li Y., Cervenansky C., Taylor P. (1994) Site of fasciculin interaction with acetylcholinesterase. J. Biol. Chem. 269, 11233–11239 [PubMed] [Google Scholar]
- 77. Eastman J., Wilson E. J., Cerveñansky C., Rosenberry T. L. (1995) Fasciculin 2 binds to the peripheral site on acetylcholinesterase and inhibits substrate hydrolysis by slowing a step involving proton transfer during enzyme acylation. J. Biol. Chem. 270, 19694–19701 [DOI] [PubMed] [Google Scholar]
- 78. Sikorav J. L., Krejci E., Massoulié J. (1987) cDNA sequences of Torpedo marmorata acetylcholinesterase: primary structure of the precursor of a catalytic subunit; existence of multiple 5′-untranslated regions. EMBO J. 6, 1865–1873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Rachinsky T. L., Camp S., Li Y., Ekström T. J., Newton M., Taylor P. (1990) Molecular cloning of mouse acetylcholinesterase: tissue distribution of alternatively spliced mRNA species. Neuron 5, 317–327 [DOI] [PubMed] [Google Scholar]