

Background: Elevating NAD throughout the body improves oxidative metabolism in skeletal muscle and counteracts effects of high fat feeding.
Results: Boosting NAD synthesis specifically in muscle does not recapitulate these effects.
Conclusion: NAD content does not limit oxidative metabolism in young, healthy muscles.
Significance: Contrary to the presumption of direct, tissue-autonomous effects, the primary sites of action for NAD precursors remain unknown.
Keywords: Mitochondrial Metabolism, NAD Biosynthesis, Nicotinamide, Nicotinamide Adenine Dinucleotide (NAD), Nicotinamide Adenine Dinucleotide (NADH), Redox Regulation, Skeletal Muscle Metabolism, NMN, NAMPT, Nicotinamide Riboside
Abstract
The NAD biosynthetic precursors nicotinamide mononucleotide and nicotinamide riboside are reported to confer resistance to metabolic defects induced by high fat feeding in part by promoting oxidative metabolism in skeletal muscle. Similar effects are obtained by germ line deletion of major NAD-consuming enzymes, suggesting that the bioavailability of NAD is limiting for maximal oxidative capacity. However, because of their systemic nature, the degree to which these interventions exert cell- or tissue-autonomous effects is unclear. Here, we report a tissue-specific approach to increase NAD biosynthesis only in muscle by overexpressing nicotinamide phosphoribosyltransferase, the rate-limiting enzyme in the salvage pathway that converts nicotinamide to NAD (mNAMPT mice). These mice display a ∼50% increase in skeletal muscle NAD levels, comparable with the effects of dietary NAD precursors, exercise regimens, or loss of poly(ADP-ribose) polymerases yet surprisingly do not exhibit changes in muscle mitochondrial biogenesis or mitochondrial function and are equally susceptible to the metabolic consequences of high fat feeding. We further report that chronic elevation of muscle NAD in vivo does not perturb the NAD/NADH redox ratio. These studies reveal for the first time the metabolic effects of tissue-specific increases in NAD synthesis and suggest that critical sites of action for supplemental NAD precursors reside outside of the heart and skeletal muscle.
Introduction
Nicotinamide adenine dinucleotide (NAD) is a ubiquitous pyridine nucleotide that functions as an essential reduction-oxidation (redox) cofactor in cellular metabolism. Recent years have seen growing interest in the expanded roles for NAD as a co-substrate in a myriad of signaling contexts, spurred in part by the discoveries that in yeast enhancing flux through the NAD salvage pathway is necessary and sufficient for lifespan extension by calorie restriction (1, 2) and that in higher eukaryotes NAD levels fluctuate in response to circadian cues, exercise, and nutritional status (3–5). In mammalian cells, NAD is continuously consumed as a substrate in adenosine diphosphate (ADP)-ribosylation, cyclization, and deacylation reactions that influence a host of physiological processes (6). These functions highlight the necessity for NAD biosynthetic pathways in the maintenance of cellular homeostasis and suggest that modulation of the NAD pool might have therapeutic applications.
Although the de novo synthesis of NAD from dietary tryptophan, nicotinic acid, or nicotinamide-containing nucleotides can contribute to the intracellular NAD pool in mammals, the constitutive degradation of NAD to nicotinamide (NAM)2 by endogenous enzymes renders a functional NAD salvage pathway necessary for life (7). The rate-limiting enzyme in the NAD salvage pathway, nicotinamide phosphoribosyltransferase (NAMPT/NamPRTase/PBEF/Visfatin), catalyzes the condensation of NAM and phosphoribosyl pyrophosphate to nicotinamide mononucleotide (NMN), which is subsequently converted to NAD by NMN adenylyltransferases (NMNAT1–3) (8).
NAD salvage is at least partly regulated at the level of NAMPT protein expression, which increases in response to various stresses and disease states, including inflammation, hypoxia, and oncogenesis. NAMPT overexpression is sufficient to elevate NAD concentration and confer cytoprotective effects upon cells in culture (8–10) and in vivo (11). In skeletal muscle, NAMPT protein levels have been observed to more than double in response to chronic exercise (12, 13) or calorie restriction (14, 15). Importantly, administration of the specific NAMPT inhibitor FK866 to calorie-restricted rats can partially or fully revert several beneficial metabolic phenotypes to the ad libitum conditions (15). Although NAMPT maintains a critical co-substrate of the sirtuin family of deacylases and ADP-ribosyltransferases (16), the range of mechanisms through which it can influence mammalian physiology remains largely unresolved.
The observations that global increases in NAD availability appear beneficial to mammalian metabolic health, especially in the context of nutrient excess, do not reveal the particular cells or tissues responsible for the effects. Whole body deletion of major NAD consumers, including CD38 (17), PARP-1 (18), and PARP-2 (19), provides protection against diet-induced obesity and metabolic disease, although it is difficult to determine whether these phenotypes derive from the increased availability of NAD versus the loss of signals that would otherwise arise from the deleted enzymes. Studies targeting NAD synthesis via the direct provision of the NAMPT product, NMN, as well as its unphosphorylated relative, nicotinamide riboside (NR), have bolstered the case that higher intratissue NAD levels improve oxidative metabolism and ameliorate deleterious consequences of a high fat diet (20, 21). Such NAD-boosting strategies have been associated with enhanced mitochondrial biogenesis and function in tissues where elevated NAD was observed, including liver (21), brown adipose (20), and skeletal muscle (17). These findings coincide with enhancements in energy expenditure, thermogenesis, and endurance (19, 20) as well as correction of respiratory and ultrastructural defects in mouse models of mitochondrial disorders (22, 23). However, NAD-modulating interventions involving either germ line genetic mutations or transient, systemically administered treatments cannot conclusively resolve the primary site of action for NAD or the contributions of individual tissues to the whole animal metabolic phenotype. Thus, a conditional transgenic approach is needed to specifically and chronically modulate NAD homeostasis in metabolically significant tissues.
In this study, we investigated the degree to which increasing NAD concentration exerts tissue-autonomous effects on oxidative metabolism in otherwise healthy muscle. By specifically overexpressing NAMPT in muscle, we provide evidence that a modest increase in the steady-state level of NAD is insufficient to improve muscle oxidative function or ameliorate consequences of high fat diet feeding and does not alter the NAD/NADH ratio. The lack of phenotype in our transgenic model suggests that NAD-boosting treatments may benefit muscle via signals that originate from outside the tissue.
EXPERIMENTAL PROCEDURES
Generation of Muscle-specific Nampt Transgenic Mice
A Cre-inducible Nampt construct was generated by placing a floxed transcriptional stop element between a CAGGS promoter and the Nampt cDNA. This construct was then targeted to the Col1A1 locus as described previously (24). Nampt+/WT mice were generated by crossing Namptfl/WT or Namptfl/fl mice to muscle creatine kinase (MCK)-Cre transgenic mice on the C57BL6/J background obtained from The Jackson Laboratory. Nampt+/+ mice were subsequently generated by crossing Nampt+/WT to Namptfl/WT or Namptfl/fl mice. Littermates lacking either MCK-Cre or the floxed allele were used as controls. Mice were studied between the ages of 3 and 9 months as indicated. The NAM supplementation experiment in Fig. 1I was performed in 15-month-old animals.
FIGURE 1.

Overexpression of NAMPT increases NAD concentration in muscle. A, proportion of Cre-recombined genomes in total genomic DNA of quadriceps muscle from mice carrying MCK-Cre in addition to one (NC) or two (NNC) copies of the Nampt transgene as compared with littermates carrying the same number of copies but lacking MCK-Cre (n = 4–9 per group). B, mRNA expression in quadriceps of NAD salvage pathway enzymes measured by quantitative RT-PCR: total nicotinamide phosphoribosyltransferase (Nampt), 3′-untranslated region of endogenous Nampt transcript (Nampt3UTR), nicotinamide mononucleotide adenylyltransferases (NMNAT1–3), and nicotinamide N-methyltransferase (NNMT) (n = 3–6 per group). A Western blot (C) and quantification (D) of NAMPT protein in gastrocnemius muscle (Gas) and heart of each genotype (n = 3 per group) are shown. E, NAD content of quadriceps muscle (n = 4–11 per group). F, representative histological sections of extensor digitorum longus (EDL) and soleus muscles stained with H&E or antibody against myosin heavy chain I (MyHCI). Scale bars represent 50 μm. NAD content in whole hearts (G) and heart weight (H) normalized to tibia length (n = 8–9 per group) are shown. I, NAD content of quadriceps muscle from 15-month-old mice supplied with 400 mg/kg/day NAM in the drinking water for 2 weeks. Error bars represent S.E. *, p < 0.05; **, p < 0.01; ***, p < 0.001 by Student's t test (D and G) or one-way analysis of variance with Bonferroni's multiple comparisons test (B and E). Mice were aged 5–10 months and fed a standard diet unless otherwise noted.
Animal Housing and Diets
All mice were housed according to approved Institutional Animal Care and Use Committee protocols and sacrificed after an overnight fast. Animals were kept in a specific pathogen-free barrier facility with controlled temperature and humidity and a 12-h/12-h light/dark cycle with lights on at 7 a.m. Mice were fed standard chow diet containing 120 ppm niacin (Rodent Diet 5010, LabDiet) unless otherwise noted. Obesity was induced with high fat diet (HFD) (D12492, Research Diets) containing 60% calories from fat. For NAM and NR supplementation experiments, compounds were dissolved weekly in the drinking water at 2.67 mg/ml in light-protected bottles.
Immunoblotting
Tissues were lysed with a motorized homogenizer in lysis buffer (25 mm Tris-HCl, pH 7.9, 100 mm KCl, 5 mm MgCl2, 1% (v/v) Nonidet P-40, 10% (v/v) glycerol) supplemented with Complete protease inhibitor mixture (Roche Applied Science). SDS-PAGE was performed on 4–15% Tris-HCl gels (Bio-Rad) using 20 μg of cleared lysate boiled for 5 min in 1× Laemmli buffer. Gels were transferred to PVDF membranes (Millipore) and blocked with 5% nonfat milk. Primary antibodies were diluted 1:1000 in TBS-Tween 20 according to the manufacturer's instructions. PBEF (A300-372A) was from Bethyl Laboratories, Mitoprofile Total OXPHOS antibody mixture (ab110413) was from MitoSciences, and β-actin-HRP (ab49900) was from Abcam. Antibody binding was detected using chemiluminescent horseradish peroxidase substrate (PerkinElmer Life Sciences) and quantified using NIH ImageJ software.
RNA and DNA Extraction
RNA was isolated from ∼25 mg of muscle tissue using 1 ml of TRIzol (Qiagen) reagent according to the manufacturer's instructions. For total DNA isolation, ∼25 mg of tissue was incubated in digestion buffer (100 mm Tris-HCl, pH 8.5, 200 mm NaCl, 5 mm EDTA, 0.2% (w/v) SDS, 1 mm β-mercaptoethanol) supplemented with 0.3 mg/ml proteinase K (Sigma) overnight at 55 °C followed by an additional 0.3 mg/ml proteinase K for 1 h at 55 °C. Nucleic acids were precipitated by addition of 1.25 m NaCl and ethanol, washed in 70% (v/v) ethanol, and resuspended in Tris-EDTA buffer supplemented with 80 μg/ml RNase A (Roche Applied Science).
Quantitative Real Time PCR and Recombination Analysis
Total RNA (1 μg) was reverse transcribed using a High Capacity cDNA Reverse Transcription kit (Applied Biosystems). Quantitative real time PCR for gene expression was performed on an Applied Biosystems 7900HT system using 15 ng of cDNA/reaction and a standard curve method relative to reference gene 36B4. One control sample was excluded as an outlier according to Grubb's test with α = 0.05 due to low reference gene expression. For LoxP recombination analysis, 15 ng of total DNA was analyzed by a custom TaqMan Copy Number Assay (Invitrogen) normalized to reference gene Trfc with custom primers flanking the 3′ LoxP site of the transgene. For determination of mitochondrial DNA (mtDNA)/nDNA content, 15 ng of total DNA was analyzed with primers specific to mitochondrial gene ND1 (forward, 5-TAA CCG GGC CCC CTT CGA CC-3; reverse, 5-TAA CGC GAA TGG GCC GGC TG-3) and nuclear gene TBP (forward, 5-CCC CTT GTA CCC TTC ACC AAT-3; reverse, 5-GAA GCT GCG GTA CAA TTC CAG-3).
Glucose Tolerance Test
Mice were injected intraperitoneally with d-glucose solution at a dose of 1.5 g/kg of body weight after an overnight fast. Injected mice were allowed free locomotion and access to water. Tail vein blood glucose was measured with a handheld glucometer (Abbott) for the next 2 h.
Citrate Synthase Activity
Citrate synthase activity was assayed as described previously with modifications (25). 200 μl of reaction mixture (125 mm Tris-HCl, pH 8, 0.3 mm acetyl-CoA, 0.1% (v/v) Triton X-100, 0.1 mm 5′,5′-dithiobis(2-nitrobenzoic acid)) was incubated with 5 μg of skeletal muscle protein lysate or 10 μg of isolated skeletal muscle mitochondrial protein at 30 °C for 10 min. The reaction was initiated by the addition of freshly prepared 0.5 mm oxaloacetate, and the reduction of 5′,5′-dithiobis(2-nitrobenzoic acid) was continuously monitored by absorbance at 412 nm for 3 min.
NAD Metabolite Extraction from Muscle and Mitochondria
NAD, NAM, and NMN were extracted from ∼50 mg of muscle tissue in 0.6 m perchloric acid at 4 °C using a TissueLyzer (Qiagen) set to 20 Hz for 6 min. Lysates were pelleted at 20,000 × g for 10 min at 4 °C and diluted 1:200 in ice-cold 100 mm phosphate buffer, pH 8. For determination of NMN, the supernatant was further neutralized with 1 m potassium carbonate and centrifuged to remove insoluble material. NADH was similarly extracted from ∼50 mg of muscle tissue in alkaline extraction buffer (25 mm NH4Ac, 25 mm NaOH, 50% (v/v) acetonitrile) flushed with nitrogen gas. For measurement by cycling assay, alkaline lysates were mixed 1:1 (v/v) with ethanol extraction buffer (250 mm KOH, 50% (v/v) EtOH) and heated at 55 °C for 10 min. Lysate supernatants were diluted 1:50 in ice-cold 100 mm phosphate buffer, pH 8. NAD was extracted from 100 μg of isolated mitochondria by vortexing vigorously in 100 μl of 0.6 m perchloric.
NAD and NADH Measurement
NAD and NADH were immediately measured after extraction by an enzymatic cycling assay in a 96-well format as described previously with modifications (26). Briefly, 5 μl of NAD standards or diluted tissue extracts was combined with 95 μl of cycling mixture (2% ethanol, 100 μg/ml alcohol dehydrogenase, 10 μg/ml diaphorase, 20 μm resazurin, 10 μm flavin mononucleotide, 10 mm nicotinamide, 0.1% BSA in 100 mm phosphate buffer, pH 8.0). The cycling reaction proceeded for 30 min at room temperature, and resorufin accumulation was measured by fluorescence excitation at 544 nm and emission at 590 nm. NAD and NADH values were confirmed by HPLC analysis of the same extracts.
NAD Metabolite Measurement by HPLC
Separation of NAD, NAM, and NADH was performed on non-diluted extracts as described (27) with modifications using an Adsorbosphere XL ODS column (5 μm, 4.6 × 250 mm) preceded by a guard column at 50 °C. Flow rate was set at 0.4 ml/min. The mobile phase was initially 100% mobile phase A (0.1 m potassium phosphate buffer, pH 6.0 containing 3.75% methanol). Methanol was linearly increased with mobile phase B (0.1 m potassium phosphate buffer, pH 6.0 containing 30% methanol) to 50% over 15 min. The column was washed after each separation by increasing mobile phase B to 100% for 5 min. UV absorbance was monitored at 260 and 340 nm with a Shimadzu SPD-M20A. Separation of NMN was carried out on an YMC-Pack ODS-A column (5 μm, 4.6 × 250 mm) at 30 °C. Flow rate was set at 0.4 ml/min. The mobile phase was initially 100% mobile phase A (0.1 m potassium phosphate buffer, pH 6.0) for the first 8 min. Methanol was linearly increased with mobile phase B (0.1 m potassium phosphate buffer, pH 6.0 containing 30% methanol) to 50% over 7 min. The column was washed after each separation by increasing mobile phase B to 100% for 3 min, and UV absorbance was similarly monitored. Pertinent peak areas were integrated by the LabSolution software from Shimadzu, quantified using standard curves, and normalized to weights of frozen tissues.
Mitochondrial Isolation from Skeletal Muscle
Mice were euthanized by cervical dislocation, and their hind limb muscles dissected and placed immediately in ice-cold muscle homogenization buffer (50 mm Tris-HCl, pH 7.4, 100 mm KCl, 5 mm MgCl2, 1 mm EDTA, pH 8.0, 1.8 mm ATP) at pH 7.2. The entire procedure was performed at 4 °C. The fat and connective tissues were removed, and minced muscles were incubated for 2 min in 2.5 ml of homogenization buffer supplemented with 60 units/ml protease from Bacillus licheniformis (Sigma). Muscle pieces were washed twice with 5 ml of unsupplemented homogenization buffer and subjected to an ice-cold motorized homogenizer for 10 min at 150 rpm. A small aliquot of the homogenate was removed and stored at −80 °C for further analysis. The remaining homogenate was centrifuged at 720 × g for 5 min at 4 °C. The pellet was resuspended in homogenization buffer and centrifuged for an additional 5 min at 720 × g. The supernatants were combined and centrifuged at 10,000 × g for 20 min at 4 °C. The supernatant was discarded, and the pellet was resuspended in homogenization buffer and further centrifuged for 10 min at 10,000 × g. The final mitochondrial pellet was resuspended in resuspension buffer (225 mm sucrose, 44 mm KH2PO4, 12.5 mm MgOAc, 6 mm EDTA, pH 7.4) at 4 μl of buffer/mg of muscle tissue.
Mitochondrial Oxygen Consumption
Oxygen consumption was measured using the Oroboros High-Resolution Respirometry System (Innsbruck, Austria). Two 2-ml chambers were loaded with 150 μg of isolated skeletal muscle mitochondrial protein in 37 °C MiRO5 respiration medium (110 mm sucrose, 20 mm HEPES, 10 mm KH2PO4, 20 mm taurine, 60 mm potassium lactobionate, 3 mm MgCl2·6H20, 0.5 mm EGTA, 1 g/liter defatted BSA, pH 7.1). 10 mm pyruvate and 5 mm malate or 20 μm palmitoyl carnitine and 5 mm malate were added as oxidative substrates followed by 4 μmol of ADP to stimulate maximal respiration. Glutamate and cytochrome c were added to replenish TCA cycle intermediates and confirm the integrity of mitochondrial membranes, respectively, following the manufacturer's instructions. To interrogate complex II (succinate dehydrogenase), 1 μm rotenone and 10 mm succinate were added. The assay was ended by addition of 5 μm antimycin A for calculation of residual oxygen consumption.
Histology and Immunohistochemistry
Histological analysis was performed on 4-μm-thick paraffin sections and deparaffinized in Histoclear (National Diagnostics) followed by rehydration through a graded ethanol series. Selected sections were stained with Gill 3 hematoxylin (Thermo Scientific) and eosin (Sigma). For immunohistochemistry, antigens were retrieved following rehydration by microwave heating in EDTA buffer, pH 7.5 for 10 min followed by cooling for 20 min and blocking of endogenous peroxidase with hydrogen peroxide. Slides were incubated overnight at 4 °C with the primary antibody against slow skeletal myosin heavy chain (Abcam, ab11083) diluted 1:3000. Following washing with PBS, slides were incubated with biotinylated secondary antibody (Vector Laboratories, BA-2001) at 37 °C for 1 h, washed three times with PBS, and incubated with streptavidin-conjugated HRP (Vector Laboratories, PK6101) for 1 h at 37 °C. Staining was detected with diaminobenzidine (Dako) chromogen, and slides were counterstained with hematoxylin.
Treadmill, Voluntary Wheel Running, and Grip Strength
Mice were habituated to the Exer3-6 treadmill system with shock detector (Columbus Instruments, Columbus, OH) by running at 10 m/min for 15 min at 0° incline 1 day before the test. To test exercise tolerance, mice were fasted with access to water for 3 h midday before simultaneously running a protocol described in Fig. 3D of 0–25 m/min at 0° incline. Exhaustion was defined as 50 cumulative electrical stimuli (73 V, 0.97 mA, 1 Hz). Exhausted mice were removed from the treadmill and allowed access to food and water. To assess voluntary running, mice were individually housed and allowed access to a computer-monitored running wheel (Columbus Instruments) continuously for 3 weeks. Data were compiled from the 2nd and 3rd weeks after habituation. Grip strength was measured with a rodent grip meter (TSE, Hamburg, Germany) as described previously (28).
FIGURE 3.
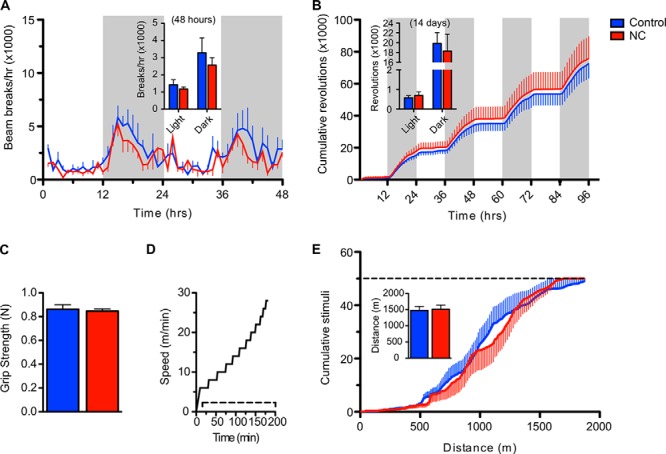
Overexpression of NAMPT in muscle does not change voluntary movement or endurance. A, voluntary ambulatory activity assessed by counting infrared beam breaks. The time course represents the 48-h period following habituation. Shaded panels indicate periods of darkness. Inset, average activity during light and dark hours of the time course (n = 5 mice per group). B, voluntary wheel running activity. The time course represents the first 96-h period following habituation. Inset, average wheel revolutions during light and dark hours over the entire 14-day period (n = 8 per group). C, maximum forelimb grip strength (n = 9–12 per group). N, newtons. D, treadmill protocol for exercise tolerance test at 0° incline. The dashed line indicates the presence of electrical stimuli. E, exercise-induced fatigability. The dashed line indicates the exhaustion threshold. Inset, average total distance run at exhaustion (n = 10–11 per group). Blue and red bars represent control and NC groups, respectively. Error bars represent S.E. No significant differences were observed at any time point by Student's t test. Mice were aged 9 (A) or 4–5 months (B–E).
Indirect Calorimetry
VO2, VCO2, and activity were measured in individually housed mice using an open circuit calorimeter (Columbus Instruments Comprehensive Lab Animal Monitoring System). Room air was supplied at 0.6 liter/min, and exhaust air was sampled at 1-h intervals for 96 h. The respiratory exchange ratio was calculated as the ratio of VCO2/VO2 with lower values indicating oxidation of fatty acids. Activity was monitored by infrared beam interruption. Data were compiled from hours 48 to 96 after habituation.
Serum Non-esterified Fatty Acids and Cholesterol
Plasma samples were collected from the tail veins of awake mice after a 5-h fast. Plasma lipids were analyzed individually by analytical chemistry with an automated Cobas Mira Autoanalyzer (Roche Diagnostics).
Lactate, Pyruvate, and Ketone Measurements
The concentrations of lactate, pyruvate, β-hydroxybutyric acid, and acetoacetate were assayed by GC/MS as their acetylated pentafluorobenzyl derivatives as described (29) with modifications. The derivatized analytes were quantified against an internal standard of β-hydroxypentanoate-d5. Frozen tissues (∼40 mg) were spiked with 20 nmol of β-hydroxypentanoate-d5 and 20 μmol of NaB2H4 and homogenized on ice for 2 min in 0.5 ml of a solvent mixture containing equal parts acetonitrile, methanol, and water as described (30). The cold homogenate was then centrifuged at 3000 × g for 10 min at 4 °C. The supernatants were incubated with 20 μmol of pentafluorobenzyl bromide in acetone solution at 60 °C for 1 h. The pentafluorobenzyl bromide-derivatized analytes were extracted with 1 ml of chloroform, dried with nitrogen gas, and further derivatized by acetylation using 30 μl of acetic anhydride and 30 μl of pyridine for 1 h at 65 °C. GC/MS analysis was carried out on an Agilent 5973 mass spectrometer linked to a Model 6890 gas chromatograph equipped with an autosampler and an Agilent OV-225 capillary column (30 m, 0.32-mm inner diameter). The injection volume was 1 μl with a 30:1 split ratio, and the carrier gas was helium at 2 ml/min. The injector temperature was set at 200 °C, and the transfer line was set at 250 °C. The GC temperature program was as follows: 100 °C for 1 min, increase by 10 °C/min to 230 °C followed by 50 °C/min to 300 °C, and hold for 5 min. Chemical ionization GC/MS was monitored for lactate, pyruvate, β-hydroxybutyric acid, and acetoacetate at m/z ratios of 131, 132, 145, and 146, respectively.
Estimation of Compartmental Free NAD/NADH Ratios
Estimates of NAD/NADH ratios in cytosol and mitochondria were calculated from metabolites presented in Table 1 (extracted from whole tissue) according to the equilibrium constants for NAD-linked lactate dehydrogenase and β-hydroxybutyrate dehydrogenase, respectively, as determined previously (31). Calculations according to the following equations assumed enzymatic equilibrium at 38 °C and pH 7.0 in each compartment.
 |
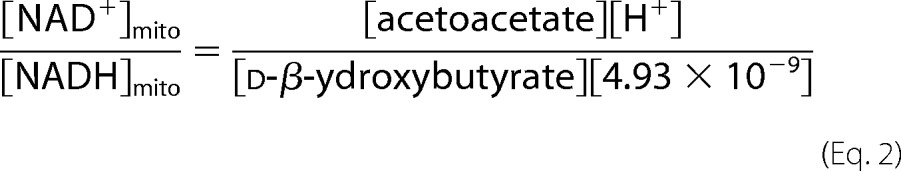 |
TABLE 1.
Concentrations of redox-sensitive metabolites in quadriceps muscle of 5-month-old mice
Concentrations are presented as mean nmol/mg of tissue (S.E.). β-HB, β-hydroxybutyrate. No significant differences were found between genotypes by one-way analysis of variance.
| Group | Observations | [Lactate] | [Pyruvate] | [β-HB] | [Acetoacetate] |
|---|---|---|---|---|---|
| Control | 7 | 14.3 (1.1) | 0.289 (0.029) | 0.068 (0.007) | 0.131 (0.015) |
| NC | 6 | 12.7 (0.9) | 0.313 (0.042) | 0.058 (0.005) | 0.133 (0.020) |
| NNC | 3 | 12.5 (1.7) | 0.285 (0.042) | 0.058 (0.006) | 0.118 (0.015) |
Statistics
Results were analyzed by two-tailed unpaired Student's t test or one-way analysis of variance with Bonferroni's multiple comparisons post hoc test as indicated. p < 0.05 was taken to be significant. Analysis was conducted with GraphPad Prism 5 software.
RESULTS
Generation of mNAMPT Mice
To investigate the role of NAD in muscle, we generated mice carrying an exogenous copy of the Nampt cDNA separated from the CAGGS promoter by a floxed stop cassette. Expression of Cre recombinase under control of the muscle creatine kinase promoter (MCK-Cre) results in excision of the stop cassette and overexpression of Nampt in skeletal and cardiac muscle (mNAMPT mice). An important advantage of targeting muscle is that it avoids complications associated with the reported production of extracellular NAMPT (eNAMPT/PBEF/Visfatin) by adipose tissue (32), immune cells (33), and hepatocytes (34). Deletion of the stop cassette was detected in ∼40% of muscle nuclei using a probe specific to the intact allele (Fig. 1A), consistent with efficient recombination of the genomes contained within myofibers (35). Expression of Nampt mRNA increased ∼10- and 20-fold in the quadriceps of animals carrying one (NC) or two (NNC) copies of the transgene, respectively, whereas expression of the 3′-UTR of endogenous Nampt was unaffected (Fig. 1B). Similar increases in Nampt expression were confirmed in gastrocnemius and the more oxidative soleus muscles (data not shown). Transcripts of enzymes downstream in the NAD salvage pathway were unchanged, although expression of both NMNAT1 and NMNAT3 trended higher with increasing Nampt gene dosage (Fig. 1B). Expression of the transcript for nicotinamide N-methyltransferase, which competes with NAMPT to metabolize NAM, was low in skeletal muscle and unaffected by genotype. In accordance with Nampt transcript levels, we observed a robust increase in NAMPT protein in both skeletal (∼10-fold) and cardiac (∼4-fold) muscles of NC mice (n = 11 and 3, respectively) and an ∼50% increase in intramuscular NAD content (Fig. 1, C–E). Because only a small number of NNC mice were available, we focused primarily on the NC mice for subsequent experiments.
Histological analysis of hind limb muscles containing primarily glycolytic type IIB (extensor digitorum longus) and oxidative type I and IIA fibers (soleus) showed normal polygonal cross-sectional morphology and diameter as well as equivalent staining for type I myosin heavy chain in NC mice as compared with littermate controls (Fig. 1F). NAD levels in heart were also significantly elevated albeit to a lesser degree than in skeletal muscle, consistent with the activity of MCK-Cre in these tissues (36) and the relative expression of NAMPT (Fig. 1G). Gross examination of myocardium did not reveal any major changes in heart tissue, nor was there a change in organ weight (Fig. 1H). To test whether NAD generation in mNAMPT mice is limited by NAM availability, we administered 400 mg/kg NAM in the drinking water for 1 week and examined skeletal muscle NAD levels. No further elevation of NAD was observed in mNAMPT mice under these conditions, suggesting that NAM availability does not limit NAMPT activity in muscle under standard dietary conditions (Fig. 1I).
Characterization of Muscle Oxidative Function
Based on earlier work using NAD-boosting strategies in mice, we hypothesized that skeletal muscle of mNAMPT mice would exhibit enhanced oxidative function. To test this, we examined several markers of mitochondrial content in gastrocnemius and quadriceps muscles. We found protein expression of subunits from four different complexes of the electron transport chain to be similar in control and mNAMPT mice (Fig. 2A). We further observed similar levels of citrate synthase activity (Fig. 2B) and mtDNA content (Fig. 2C) between the two groups. Consistent with these findings, the Nampt transgene had no significant effect on the expression of genes associated with mitochondrial biogenesis (TFAM, PGC1a, mfsn2, and Sirt1), oxidative phosphorylation (CytC, Cox5b, and Ndufs8), coupling (UCP2), or substrate selection (PDK4, MCAD, and Sirt3) in quadriceps or soleus muscles (Fig. 2E).
FIGURE 2.

NAMPT overexpression does not improve muscle oxidative function. A, Western blot of representative components of the electron transport chain complexes in gastrocnemius muscle. B, citrate synthase activity in whole tissue lysates of quadriceps muscle (n = 3–4 mice per group; A.U., arbitrary units). C, relative mtDNA/nDNA ratio in quadriceps muscle (n = 7 per group). D, NAD content in mitochondria isolated from pooled hind limb muscles (n = 4 per group). E, mRNA expression of transcripts related to oxidative function in soleus and quadriceps (quad) muscle measured by quantitative RT-PCR (n = 6 per group). F, representative trace of oxygen consumption rate (OCR; left axis) and chamber oxygen concentration (right axis) of mitochondria isolated from control skeletal muscle. Arrows indicate sequential addition of malate (Mal), pyruvate (Pyr), ADP, glutamate (Glu), cytochrome c (CyC), rotenone (Rot), succinate (Suc), and antimycin A (AnA) as well as opening and closing of the chamber to atmospheric oxygen. G, oxygen consumption rate of mitochondria isolated from skeletal muscle and provided with respective substrates and inhibitors (n = 4 per group). White and gray bars represent control (Ctrl) and NC groups, respectively. Error bars represent S.E. No changes are significant by Student's t test. Mice were aged 5–7 months.
Interventions that alter NAD in cultured cells or the liver have been reported to concomitantly alter the mitochondrial NAD pool (10, 20), which may directly influence oxidative metabolism (37). We thus hypothesized that mitochondria isolated from the muscles of mNAMPT mice would exhibit an increase in NAD content proportional to the effect in whole muscle tissue. Surprisingly, we found that the NAD content of muscle mitochondria was similar between groups (Fig. 2D). Accordingly, mitochondria did not exhibit any alterations in oxidative capacity that persisted through isolation as detected using high resolution respirometry (Fig. 2, F and G). Mitochondria isolated from control and mNAMPT muscle showed similar patterns of oxygen consumption across all complexes on both pyruvate and fatty acid substrates. These results suggest that NAMPT overexpression is not sufficient to promote the biogenesis or respiratory capacity of muscle mitochondria.
We next sought to test the influence of NAMPT on muscle performance in vivo. We found voluntary ambulatory and wheel running activity across the light/dark cycle to be indistinguishable between control and NC mice (Fig. 3, A and B). Forelimb strength and treadmill running capacity were also comparable (Fig. 3, C–E). Together, these data indicate that NAD is unlikely to limit muscle metabolic function in healthy wild type mice.
Muscle NAMPT Does Not Protect against Metabolic Consequences of High Fat Diet
Genetic and pharmacological interventions that elevate NAD levels have been reported to ameliorate many of the negative consequences of high fat diet feeding, including insulin resistance and weight gain. To investigate whether NAMPT-mediated NAD synthesis in muscle conferred similar protective effects, we placed mNAMPT mice on an HFD for 24 weeks. No significant differences in weight gain, plasma free fatty acids, or total cholesterol were observed between control and NC mice (Fig. 4, A–C). After 10 weeks on respective diets, HFD-fed NC and control mice exhibited similar declines in glucose tolerance, VO2, and respiratory exchange ratio as compared with chow-fed littermates (Fig. 4, D, E, and G–J). Interestingly, HFD caused a slight but significant decline in intramuscular NAD in control mice that was completely rescued by the transgene (Fig. 4F). Thus, intramuscular NAD levels do not appear to have a major influence on the general metabolic deterioration caused by HFD.
FIGURE 4.

Overexpression of NAMPT in muscle does not protect against metabolic consequences of high fat diet. A, body weights of mice provided a standard chow diet (CD) or HFD for 24 weeks starting at 3 months of age (n = 6 mice per group). B and C, plasma non-esterified fatty acids (NEFA) and total cholesterol after 24 weeks on the indicated diets (n = 7–10 per group). D and E, glucose tolerance after 20 weeks on the indicated diets (n = 6 per group). AUC, area under the curve. F, NAD content of gastrocnemius muscles after 24 weeks on the indicated diets (n = 5–6 per group). G, oxygen consumption (VO2) normalized to body weight after 24 weeks on the indicated diets. The time course represents the 48-h period following habituation. Shaded panels indicate periods of darkness (n = 5 per group). H, average VO2 during light and dark periods shown in G. I, respiratory exchange ratios (RER) over the same time course shown in G. J, average respiratory exchange ratio during light and dark periods shown in I. Error bars represent S.E. *, p < 0.05; **, p < 0.01; ns, not significant by Student's t test.
NAMPT Overexpression in Muscle Does Not Significantly Affect Steady-state Levels of NAM or NMN
NAM, a substrate of NAMPT, is also a non-competitive inhibitor of sirtuins and a competitive inhibitor of PARPs. Thus, one mechanism by which NAMPT overexpression might influence such enzymes is through the depletion of NAM rather than through the generation of NAD per se. Contrary to our initial expectation, we did not observe a significant depletion of NAM in quadriceps muscles of mNAMPT mice (Fig. 5A), potentially indicating that this metabolite is rapidly replenished. We also found no significant accumulation of NMN, the direct product of NAMPT, in mNAMPT muscle (Fig. 5B), supporting the view that NAMPT catalyzes the rate-limiting step in the NAD salvage pathway.
FIGURE 5.

Chronic elevation of NAD in muscle does not alter the NAD/NADH ratio. NAM (A), NMN (B), and NADH (C) content of quadriceps muscle in 5-month-old mice (n = 4–11 mice per group). D, measured ratio of total NAD/NADH in quadriceps muscle. NAD values were derived from Fig. 1D. Shown are the measured ratio of lactate/pyruvate (E) and β-hydroxybutyrate/acetoacetate (BHB/Acac) (F) in quadriceps (n = 3–7 per group). Shown is the calculated free NAD/NADH ratio in the nucleocytosolic (G) and mitochondrial (H) compartments. Error bars represent S.E. *, p < 0.05; ***, p < 0.001 by one-way analysis of variance with Bonferroni's multiple comparisons test.
Muscle Redox State Is Independent of NAMPT-mediated NAD Synthesis
The reduced form of NAD, NADH, acts as a key electron donor and allosteric regulator of mitochondrial dehydrogenases, and the ratio of oxidized to reduced nucleotides can influence the rates of many reactions within the cell. Although overexpression of NAMPT in cells and treatment of mice with NR have both been reported to increase the NAD/NADH redox ratio (20, 23, 38), it remains unresolved whether changes in absolute NAD concentration or the redox ratio play a greater role in mediating the effects of NAD-boosting interventions. Interestingly, total concentrations of NADH were significantly elevated in the muscles of mNAMPT mice, reflecting an apparent equilibration between the oxidized and reduced forms of NAD to maintain the NAD/NADH ratio across a range of absolute concentrations (Fig. 5, C and D).
A large fraction of the intracellular NADH pool is protein-bound and therefore inaccessible to enzymatic reactions that would lead to its oxidation. NAD(H) is further compartmentalized into mitochondrial and nucleocytosolic pools, suggesting that significant changes in the abundance of free nucleotides within a given compartment could be missed by measuring only total tissue levels. To confirm the maintenance of redox state in the nucleocytosolic compartment of mNAMPT muscle, we determined the concentrations of lactate and pyruvate, which can be used to estimate the ratio of free NAD/NADH in the cytosol via the equilibrium constant for lactate dehydrogenase (Table 1 and Fig. 5G) (31). To test whether redox was altered in the mitochondrial compartment, we determined the concentrations of the ketone bodies β-hydroxybutyrate and acetoacetate and estimated the free NAD/NADH ratio via the equilibrium constant for β-hydroxybutyrate dehydrogenase (Table 1 and Fig. 5H). Consistent with measurements of total metabolite pools, NAMPT overexpression was not found to significantly affect the redox state of either compartment.
DISCUSSION
Chronic deficiency of dietary NAD precursors is known to cause the progressive and lethal disease pellagra (39), but the more recent notion that increasing NAD availability might suffice to enhance the metabolic fitness of an otherwise healthy tissue is just beginning to be explored. We report that targeting the NAD salvage pathway in muscle via overexpression of NAMPT increases the NAD content of skeletal muscles by ∼50% but does not overtly influence muscle physiology or whole body energy balance in the context of standard or high fat diets. Our findings represent the first investigation of these phenotypes using a tissue-specific approach to enhance NAD synthesis and stand in stark contrast to the beneficial effects that have been observed using a variety of genetic and pharmacological approaches to elevate NAD throughout the whole body.
It is noteworthy that our model achieved a relative elevation of skeletal muscle NAD greater than or equal to those reported for small molecule precursor supplementation (20, 40), exercise (3, 4), or deletion of either PARP-1 (18) or PARP-2 (19). Because increased NAD was achieved via supraphysiological levels of NAMPT protein expression and was not further enhanced by supplementing NAM, it is possible that negative feedback maintains the steady-state NAD concentration in our model at or near a theoretical limit for salvage-mediated synthesis. These observations are consistent with reports that the Km of NAMPT for NAM in the presence of ATP is well below our observed tissue concentrations of NAM and that physiological levels of pyridine nucleotides can inhibit NAMPT activity in rat liver extracts (41, 42). Despite the elevated steady-state concentration of muscle NAD that was achieved, mNAMPT mice failed to display phenotypes suggesting any alteration of mitochondrial content (Fig. 2, A–C) or transcriptional adaptations (Fig. 2E), consistent with their unchanged strength and exercise capacity (Fig. 3, B–E). In addition, mNAMPT mice gained weight normally on a high fat diet and did not display changes in glucose tolerance or oxygen consumption (Fig. 4, E and H). Although obese control animals exhibited diminished metabolic health coincident with a small but significant decline in skeletal muscle NAD, this decline was completely rescued by the Nampt transgene to no effect (Fig. 4F). Thus, the slight decrease in muscle NAD does not contribute meaningfully to the detrimental consequences of obesity, nor does elevating NAD have a protective effect.
Although little is known about the dynamics of pyridine nucleotide transport across the mitochondrial membranes, it is appreciated that mitochondria maintain bioenergetics via a distinct NAD pool that is reduced to NADH in a much higher proportion than the nucleocytosolic pool (31) and is shielded from the activity of nuclear and cytosolic enzymes (10, 43, 44). Although distinct, the mitochondrial NAD pool tends to mirror expansion or contraction of the total NAD pool in cultured cells and in the liver where NAD deficiency can limit mitochondrial oxidative metabolism (20, 37). Although we observed a slight trend toward an increase in mitochondrial NAD content in muscle (Fig. 2D), the effect was clearly dampened compared with the increase in whole tissue. Moreover, mitochondria isolated from mNAMPT muscle consumed pyruvate and palmitoyl carnitine at rates equivalent to those of control littermates (Fig. 2G), consistent with the VO2 and respiratory exchange ratio measurements from whole animals (Fig. 4, H and J). Because supplemented NAD precursors can theoretically bypass any compartmentalization of NAMPT activity, it is plausible that their beneficial effects reflect a more pronounced influence on the mitochondrial pool. However, we found that administration of NR at 400 mg/kg, either as a single intraperitoneal bolus or a daily dose in the drinking water for 1 week, significantly increased neither mitochondrial nor whole tissue NAD levels.3 Although an increase in whole muscle NAD was previously reported following NR supplementation in the chow (20), the discrepancy is unlikely to be caused by the route of administration because a more recent study using NR-supplemented chow for up to 16 weeks also found non-significant changes in muscle NAD (22). Thus, the available evidence suggests that NR exerts only a mild influence on skeletal muscle NAD content in young, healthy animals, and it remains to be determined whether specific expansion of the mitochondrial or cytosolic NAD pools directly influences oxidative metabolism in this tissue.
We found intramuscular NMN content to be ∼50–100-fold lower than that of NAD across genotypes and relatively unaffected by transgene expression, consistent with the rate-limiting nature of NAMPT in the NAD salvage pathway (Fig. 5B). However, NAMPT also failed to have a prominent effect on the intramuscular concentrations of its substrate, NAM (Fig. 5A). This is of interest because it has been proposed that NAM acts as a feedback inhibitor of NAD-dependent enzymes in the concentration range that we have detected (1, 45, 46). Indeed, evidence indicates that depletion of NAM by the NAMPT analog Pnc1 is a key mechanism regulating sirtuins in yeast (1). In mouse muscle, however, our findings suggest that homeostatic mechanisms maintain approximately normal tissue NAM levels as was observed in the context of PARP-1 deletion (18). Our results may indicate that (a) flux through the salvage pathway is unchanged, (b) increased NAM consumption is counterbalanced by a corresponding increase in NAM generation by NAD-dependent enzymes, or (c) NAM exchanges continuously with the plasma to maintain a stable tissue concentration irrespective of NAMPT activity.
The cellular NAD/NADH ratio is sensitive to metabolic flux and extrinsic factors and can modify enzymatic activity independently from changes in NAD concentration per se (38, 47). For example, the transcriptional co-repressor C-terminal binding protein is reciprocally regulated by the oxidized and reduced nucleotides (48), and it is debated whether the sirtuins are competitively inhibited by NADH in vivo (14, 46, 47). Thus, any changes in the NAD/NADH ratio could mediate effects of NAD-boosting interventions. Contrary to our expectation that NAMPT overexpression would increase the skeletal muscle NAD/NADH ratio as reported in isolated myoblasts (38) and heart (11) as well as muscles of NR-fed mice (20), we observed equilibration between the oxidized and reduced nucleotides to maintain the redox state in mNAMPT muscle (Fig. 5D). Several key differences distinguish our experiment from previous studies. First, experiments in cultured myoblasts may not accurately model the changes in nutrient availability, glycolytic flux, or ATP demand experienced by muscle fibers in vivo; all of these affect the redox state of NAD. Moreover, the redox states of liver and other tissues may influence muscle in vivo via circulating redox-active metabolites, including lactate, pyruvate, and ketone bodies. Second, in the cardiac model of Hsu et al. (11), NAMPT overexpression shifted the redox ratio in concert with a larger increase in NAD than has been reported for any other intervention (∼5-fold), yet a partial equilibration was still apparent. Lastly, the dietary delivery of NR is distinct from our genetic model in that it amounts to a series of discrete systemic administrations, which may be responsible for its effect on muscle redox state. Our finding that redox state was not chronically affected in mNAMPT mice indicates that the metabolic processes responsible for establishing the NAD/NADH ratio are rapid relative to the basal rate of NAD synthesis. Analogous observations were made following deletion of the circadian regulator Rev-erbα in which the muscle NAD/NADH ratio was maintained in the context of a diminished NAD pool (50). Our data indicating that lactate/pyruvate and β-hydroxybutyrate/acetoacetate ratios were unchanged in the muscles of transgenic mice (Fig. 5, G and H) provide additional evidence that NAD equilibrates with NADH and, insofar as the associated enzymes can be presumed to be in equilibrium, adds compartment-specific resolution to both nucleocytosolic and mitochondrial pools of the free nucleotides. Thus, homeostatic mechanisms appear to maintain the NAD/NADH ratio in muscle despite a chronic 50% increase in the size of the NAD pool.
It is interesting to consider why increased NAD availability did not have clear effects on skeletal muscle physiology. Unlike yeast where sirtuins are the most prominent consumers of NAD (51), mammalian genomes encode several additional classes of NAD-consuming enzymes that play important roles in signaling and metabolism and depend on the same NAD pool (52). The evolution of different classes of NAD-dependent enzymes was likely accompanied by additional layers of regulation responsible for modifying the enzyme activities in different contexts. In support of this view, the best studied mammalian sirtuin, SIRT1, is now appreciated to have numerous binding partners and post-translational modifications (53) at least some of which can regulate deacetylase activity independently from changes in NAD (54). Therefore, it will be important to identify the specific tissues or cell types that are capable of responding directly to changes in NAD availability.
Overall, our findings using a conditional Nampt allele support a model in which the effects of globally increasing NAD availability on muscle physiology are not muscle-autonomous. It remains possible that more profound muscle-autonomous effects would emerge following larger increases in NAD, concurrent with energetic stress, or in a diseased state. However, we note that none of these conditions were required for NR supplementation or PARP-1 deletion to benefit muscle (18, 20). Interestingly, overexpression of the NAD-dependent deacetylase SIRT1 in the brain has recently been shown to improve muscle physiology (55, 56), whereas overexpression of the enzyme in the muscle itself was ineffective (57). Moreover, NMN can promote insulin secretion in cultured islets and in vivo, consistent with the stimulation of SIRT1 in pancreatic β cells (7, 21), and knockdown of nicotinamide N-methyltransferase was recently shown to elevate adipose tissue NAD levels in concert with increased whole body energy expenditure (49). Thus, it is possible that improvements in muscle physiology during NAD precursor supplementation occur via pleiotropic effects of NAD-dependent enzymes in tissues other than skeletal muscle. It will be important to determine in future studies whether the central nervous system, hormones, circulating metabolites, or a combination thereof mediates the effects of global NAD availability on muscle physiology. It will also be interesting to determine the tissue-autonomous impact of enhanced NAD salvage capacity under conditions known to deplete the metabolite, including genotoxic stress (10) and the course of natural aging (21, 40).
In summary, our muscle-specific transgenic approach provides an important step toward dissecting the mechanisms by which NAD availability influences mammalian physiology. A deeper understanding of the tissues and signals that mediate these effects will greatly facilitate the development of NAD-boosting therapeutics to combat comorbidities of obesity and aging.
Acknowledgments
We are indebted to D. Sinclair for valuable reagents and advice as well as D. Cromley and D. Rader for the analysis of plasma lipids. We also thank R. Ahima and R. Dhir of the Mouse Phenotyping, Physiology, and Metabolism Core and H. Collins of the Radioimmunoassay and Biomarkers Core, both provided by the University of Pennsylvania Diabetes Research Center (National Institutes of Health Grant P30-DK19525), as well as the Case Western Reserve Mouse Metabolic Phenotyping Center (National Institutes of Health Grant U24-DK76174). We thank R. Dellinger of ChromaDex for constructive discussions and the provision of NR and all members of the Baur laboratory for support and constructive feedback.
This work was supported, in whole or in part, by National Institutes of Health Grants R01-DK098656 (to J. A. B.) and R01-GM097409 (to E. O.-N.). This work was also supported by American Heart Association Grant 11SDG5560001 (to E. O.-N.) and an Ellison Medical Foundation New Scholar Award (to J. A. B.).
D. W. Frederick, A. Dávila, Jr., and J. A. Baur, unpublished results.
- NAM
- nicotinamide
- PBEF
- pre-B cell-enhancing factor
- NMN
- nicotinamide mononucleotide
- NMNAT
- NMN adenylyltransferase
- PARP
- poly(ADP-ribose) polymerase
- NR
- nicotinamide riboside
- MCK
- muscle creatine kinase
- HFD
- high fat diet
- NC
- one-copy transgenic
- NNC
- two-copy transgenic.
REFERENCES
- 1. Anderson R. M., Bitterman K. J., Wood J. G., Medvedik O., Sinclair D. A. (2003) Nicotinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 423, 181–185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lin S. J., Defossez P. A., Guarente L. (2000) Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 289, 2126–2128 [DOI] [PubMed] [Google Scholar]
- 3. Cantó C., Gerhart-Hines Z., Feige J. N., Lagouge M., Noriega L., Milne J. C., Elliott P. J., Puigserver P., Auwerx J. (2009) AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 458, 1056–1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cantó C., Jiang L. Q., Deshmukh A. S., Mataki C., Coste A., Lagouge M., Zierath J. R., Auwerx J. (2010) Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell Metab. 11, 213–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nakahata Y., Sahar S., Astarita G., Kaluzova M., Sassone-Corsi P. (2009) Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 324, 654–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belenky P., Bogan K. L., Brenner C. (2007) NAD+ metabolism in health and disease. Trends Biochem. Sci. 32, 12–19 [DOI] [PubMed] [Google Scholar]
- 7. Revollo J. R., Körner A., Mills K. F., Satoh A., Wang T., Garten A., Dasgupta B., Sasaki Y., Wolberger C., Townsend R. R., Milbrandt J., Kiess W., Imai S. (2007) Nampt/PBEF/Visfatin regulates insulin secretion in β cells as a systemic NAD biosynthetic enzyme. Cell Metab. 6, 363–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Revollo J. R., Grimm A. A., Imai S. (2004) The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 279, 50754–50763 [DOI] [PubMed] [Google Scholar]
- 9. Rongvaux A., Galli M., Denanglaire S., Van Gool F., Dreze P. L., Szpirer C., Bureau F., Andris F., Leo O. (2008) Nicotinamide phosphoribosyl transferase/pre-B cell colony-enhancing factor/visfatin is required for lymphocyte development and cellular resistance to genotoxic stress. J. Immunol. 181, 4685–4695 [DOI] [PubMed] [Google Scholar]
- 10. Yang H., Yang T., Baur J. A., Perez E., Matsui T., Carmona J. J., Lamming D. W., Souza-Pinto N. C., Bohr V. A., Rosenzweig A., de Cabo R., Sauve A. A., Sinclair D. A. (2007) Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 130, 1095–1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hsu C. P., Oka S., Shao D., Hariharan N., Sadoshima J. (2009) Nicotinamide phosphoribosyltransferase regulates cell survival through NAD+ synthesis in cardiac myocytes. Circ. Res. 105, 481–491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Costford S. R., Bajpeyi S., Pasarica M., Albarado D. C., Thomas S. C., Xie H., Church T. S., Jubrias S. A., Conley K. E., Smith S. R. (2010) Skeletal muscle NAMPT is induced by exercise in humans. Am. J. Physiol. Endocrinol. Metab. 298, E117–E126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koltai E., Szabo Z., Atalay M., Boldogh I., Naito H., Goto S., Nyakas C., Radak Z. (2010) Exercise alters SIRT1, SIRT6, NAD and NAMPT levels in skeletal muscle of aged rats. Mech. Ageing Dev. 131, 21–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen D., Bruno J., Easlon E., Lin S.-J., Cheng H.-L., Alt F. W., Guarente L. (2008) Tissue-specific regulation of SIRT1 by calorie restriction. Genes Dev. 22, 1753–1757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Song J., Ke S. F., Zhou C. C., Zhang S. L., Guan Y. F., Xu T. Y., Sheng C. Q., Wang P., Miao C. Y. (2014) Nicotinamide phosphoribosyltransferase is required for the calorie restriction-mediated improvements in oxidative stress, mitochondrial biogenesis, and metabolic adaptation. J. Gerontol. A Biol. Sci. Med. Sci. 69, 44–57 [DOI] [PubMed] [Google Scholar]
- 16. Imai S., Guarente L. (2014) NAD and sirtuins in aging and disease. Trends Cell Biol. 24, 464–471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Barbosa M. T., Soares S. M., Novak C. M., Sinclair D., Levine J. A., Aksoy P., Chini E. N. (2007) The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 21, 3629–3639 [DOI] [PubMed] [Google Scholar]
- 18. Bai P., Cantó C., Oudart H., Brunyánszki A., Cen Y., Thomas C., Yamamoto H., Huber A., Kiss B., Houtkooper R. H., Schoonjans K., Schreiber V., Sauve A. A., Menissier-de Murcia J., Auwerx J. (2011) PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 13, 461–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bai P., Canto C., Brunyánszki A., Huber A., Szántó M., Cen Y., Yamamoto H., Houten S. M., Kiss B., Oudart H., Gergely P., Menissier-de Murcia J., Schreiber V., Sauve A. A., Auwerx J. (2011) PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 13, 450–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cantó C., Houtkooper R. H., Pirinen E., Youn D. Y., Oosterveer M. H., Cen Y., Fernandez-Marcos P. J., Yamamoto H., Andreux P. A., Cettour-Rose P., Gademann K., Rinsch C., Schoonjans K., Sauve A. A., Auwerx J. (2012) The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 15, 838–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yoshino J., Mills K. F., Yoon M. J., Imai S. (2011) Nicotinamide mononucleotide, a key NAD+ intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 14, 528–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Khan N. A., Auranen M., Paetau I., Pirinen E., Euro L., Forsström S., Pasila L., Velagapudi V., Carroll C. J., Auwerx J., Suomalainen A. (2014) Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol. Med. 6, 721–731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cerutti R., Pirinen E., Lamperti C., Marchet S., Sauve A. A., Li W., Leoni V., Schon E. A., Dantzer F., Auwerx J., Viscomi C., Zeviani M. (2014) NAD+-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 19, 1042–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Firestein R., Blander G., Michan S., Oberdoerffer P., Ogino S., Campbell J., Bhimavarapu A., Luikenhuis S., de Cabo R., Fuchs C., Hahn W. C., Guarente L. P., Sinclair D. A. (2008) The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One 3, e2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Trounce I. A., Kim Y. L., Jun A. S., Wallace D. C. (1996) Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 264, 484–509 [DOI] [PubMed] [Google Scholar]
- 26. Graeff R., Lee H. C. (2002) A novel cycling assay for cellular cADP-ribose with nanomolar sensitivity. Biochem. J. 361, 379–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Litt M. R., Potter J. J., Mezey E., Mitchell M. C. (1989) Analysis of pyridine dinucleotides in cultured rat hepatocytes by high-performance liquid chromatography. Anal. Biochem. 179, 34–36 [DOI] [PubMed] [Google Scholar]
- 28. Akpan I., Goncalves M. D., Dhir R., Yin X., Pistilli E. E., Bogdanovich S., Khurana T. S., Ucran J., Lachey J., Ahima R. S. (2009) The effects of a soluble activin type IIB receptor on obesity and insulin sensitivity. Int. J. Obes. 33, 1265–1273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tomcik K., Ibarra R. A., Sadhukhan S., Han Y., Tochtrop G. P., Zhang G. F. (2011) Isotopomer enrichment assay for very short chain fatty acids and its metabolic applications. Anal. Biochem. 410, 110–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Des Rosiers C., Montgomery J. A., Desrochers S., Garneau M., David F., Mamer O. A., Brunengraber H. (1988) Interference of 3-hydroxyisobutyrate with measurements of ketone-body concentration and isotopic enrichment by gas-chromatography mass spectrometry. Anal. Biochem. 173, 96–105 [DOI] [PubMed] [Google Scholar]
- 31. Williamson D. H., Lund P., Krebs H. A. (1967) The redox state of free nicotinamide-adenine dinucleotide in the cytoplasm and mitochondria of rat liver. Biochem. J. 103, 514–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Romacho T., Sánchez-Ferrer C. F., Peiró C. (2013) Visfatin/Nampt: an adipokine with cardiovascular impact. Mediators Inflamm. 2013, 946427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friebe D., Neef M., Kratzsch J., Erbs S., Dittrich K., Garten A., Petzold-Quinque S., Blüher S., Reinehr T., Stumvoll M., Blüher M., Kiess W., Körner A. (2011) Leucocytes are a major source of circulating nicotinamide phosphoribosyltransferase (NAMPT)/pre-B cell colony (PBEF)/visfatin linking obesity and inflammation in humans. Diabetologia 54, 1200–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garten A., Petzold S., Barnikol-Oettler A., Körner A., Thasler W. E., Kratzsch J., Kiess W., Gebhardt R. (2010) Nicotinamide phosphoribosyltransferase (NAMPT/PBEF/visfatin) is constitutively released from human hepatocytes. Biochem. Biophys. Res. Commun. 391, 376–381 [DOI] [PubMed] [Google Scholar]
- 35. Bothe G. W., Haspel J. A., Smith C. L., Wiener H. H., Burden S. J. (2000) Selective expression of Cre recombinase in skeletal muscle fibers. Genesis 26, 165–166 [PubMed] [Google Scholar]
- 36. Brüning J. C., Michael M. D., Winnay J. N., Hayashi T., Hörsch D., Accili D., Goodyear L. J., Kahn C. R. (1998) A muscle-specific insulin receptor knockout exhibits features of the metabolic syndrome of NIDDM without altering glucose tolerance. Mol. Cell 2, 559–569 [DOI] [PubMed] [Google Scholar]
- 37. Peek C. B., Affinati A. H., Ramsey K. M., Kuo H. Y., Yu W., Sena L. A., Ilkayeva O., Marcheva B., Kobayashi Y., Omura C., Levine D. C., Bacsik D. J., Gius D., Newgard C. B., Goetzman E., Chandel N. S., Denu J. M., Mrksich M., Bass J. (2013) Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 342, 1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fulco M., Cen Y., Zhao P., Hoffman E. P., McBurney M. W., Sauve A. A., Sartorelli V. (2008) Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev. Cell 14, 661–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bogan K. L., Brenner C. (2008) Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 28, 115–130 [DOI] [PubMed] [Google Scholar]
- 40. Gomes A. P., Price N. L., Ling A. J., Moslehi J. J., Montgomery M. K., Rajman L., White J. P., Teodoro J. S., Wrann C. D., Hubbard B. P., Mercken E. M., Palmeira C. M., de Cabo R., Rolo A. P., Turner N., Bell E. L., Sinclair D. A. (2013) Declining NAD+ induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Burgos E. S., Schramm V. L. (2008) Weak coupling of ATP hydrolysis to the chemical equilibrium of human nicotinamide phosphoribosyltransferase. Biochemistry 47, 11086–11096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dietrich L. S., Muniz O., Powanda M. (1968) NAD synthesis in animal tissues. J. Vitaminol. 14, (suppl.) 123–129 [DOI] [PubMed] [Google Scholar]
- 43. Di Lisa F., Ziegler M. (2001) Pathophysiological relevance of mitochondria in NAD+ metabolism. FEBS Letters 492, 4–8 [DOI] [PubMed] [Google Scholar]
- 44. Pittelli M., Formentini L., Faraco G., Lapucci A., Rapizzi E., Cialdai F., Romano G., Moneti G., Moroni F., Chiarugi A. (2010) Inhibition of nicotinamide phosphoribosyltransferase: cellular bioenergetics reveals a mitochondrial insensitive NAD pool. J. Biol. Chem. 285, 34106–34114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Bitterman K. J., Anderson R. M., Cohen H. Y., Latorre-Esteves M., Sinclair D. A. (2002) Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast Sir2 and human SIRT1. J. Biol. Chem. 277, 45099–45107 [DOI] [PubMed] [Google Scholar]
- 46. Schmidt M. T., Smith B. C., Jackson M. D., Denu J. M. (2004) Coenzyme specificity of Sir2 protein deacetylases: implications for physiological regulation. J. Biol. Chem. 279, 40122–40129 [DOI] [PubMed] [Google Scholar]
- 47. Lin S.-J., Ford E., Haigis M., Liszt G., Guarente L. (2004) Calorie restriction extends yeast life span by lowering the level of NADH. Genes Dev. 18, 12–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Zhang Q., Wang S. Y., Fleuriel C., Leprince D., Rocheleau J. V., Piston D. W., Goodman R. H. (2007) Metabolic regulation of SIRT1 transcription via a HIC1:CtBP corepressor complex. Proc. Natl. Acad. Sci. U.S.A. 104, 829–833 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49. Kraus D., Yang Q., Kong D., Banks A. S., Zhang L., Rodgers J. T., Pirinen E., Pulinilkunnil T. C., Gong F., Wang Y. C., Cen Y., Sauve A. A., Asara J. M., Peroni O. D., Monia B. P., Bhanot S., Alhonen L., Puigserver P., Kahn B. B. (2014) Nicotinamide N-methyltransferase knockdown protects against diet-induced obesity. Nature 508, 258–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Woldt E., Sebti Y., Solt L. A., Duhem C., Lancel S., Eeckhoute J., Hesselink M. K., Paquet C., Delhaye S., Shin Y., Kamenecka T. M., Schaart G., Lefebvre P., Nevière R., Burris T. P., Schrauwen P., Staels B., Duez H. (2013) Rev-erb-α modulates skeletal muscle oxidative capacity by regulating mitochondrial biogenesis and autophagy. Nat. Med. 19, 1039–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. de Figueiredo L. F., Gossmann T. I., Ziegler M., Schuster S. (2011) Pathway analysis of NAD+ metabolism. Biochem. J. 439, 341–348 [DOI] [PubMed] [Google Scholar]
- 52. Berger F. (2004) The new life of a centenarian: signalling functions of NAD(P). Trends Biochem. Sci. 29, 111–118 [DOI] [PubMed] [Google Scholar]
- 53. Revollo J. R., Li X. (2013) The ways and means that fine tune Sirt1 activity. Trends Biochem. Sci. 38, 160–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gerhart-Hines Z., Dominy J. E., Jr., Blättler S. M., Jedrychowski M. P., Banks A. S., Lim J.-H., Chim H., Gygi S. P., Puigserver P. (2011) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD+. Mol. Cell 44, 851–863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Satoh A., Brace C. S., Rensing N., Cliften P., Wozniak D. F., Herzog E. D., Yamada K. A, Imai S. (2013) Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 18, 416–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ramadori G., Fujikawa T., Anderson J., Berglund E. D., Frazao R., Michán S., Vianna C. R., Sinclair D. A., Elias C. F., Coppari R. (2011) SIRT1 deacetylase in SF1 neurons protects against metabolic imbalance. Cell Metab. 14, 301–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. White A. T., McCurdy C. E., Philp A., Hamilton D. L., Johnson C. D., Schenk S. (2013) Skeletal muscle-specific overexpression of SIRT1 does not enhance whole-body energy expenditure or insulin sensitivity in young mice. Diabetologia 56, 1629–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]