

Background: An asymmetric dimer of ErbB kinases in which one kinase activates the other is essential for ErbB activity.
Results: A hierarchy of ErbB kinase activator-receiver preferences is mediated by intracellular regions.
Conclusion: Intracellular ErbB asymmetry shapes signaling output from ErbB heterodimers but is not coupled to extracellular asymmetry.
Significance: The signaling output from an ErbB varies with different partners and ligands.
Keywords: Cell-surface Receptor, Epidermal Growth Factor (EGF), Epidermal Growth Factor Receptor (EGFR), Phosphotyrosine signaling, Receptor Tyrosine Kinase
Abstract
The EGF receptor (EGFR) family comprises four homologs in humans collectively known as the ErbB or HER proteins. ErbB proteins are receptor tyrosine kinases that become activated when ligands bind to their extracellular regions and promote formation of specific homo- and heterodimers with enhanced tyrosine kinase activity. An essential feature of ErbB activation is formation of an asymmetric kinase dimer in which the C-terminal lobe of one kinase serves as the activator or donor kinase by binding the N-terminal lobe of a receiver or acceptor kinase and stabilizing its active conformation. ErbB extracellular regions are also thought to form active asymmetric dimers in which only one subunit binds ligand. The observation that the unliganded ErbB2 kinase preferentially serves as the activator kinase when paired with EGFR/ErbB1 implied that extracellular asymmetry in ErbB proteins might be coupled to intracellular asymmetry with unliganded partners favoring the activator kinase position. Using cell-based stimulation assays and chimeric ErbB proteins, we show that extracellular asymmetry is not coupled to intracellular asymmetry and that ErbB intracellular regions are sufficient to determine relative kinase activator-receiver orientation. We further show a hierarchy of activator-receiver preferences among ErbB proteins, with EGFR/ErbB1 being the strongest receiver, followed by ErbB2 and then ErbB4, and that cis-phosphorylation of EGFR and ErbB2 appears to be negligible. This hierarchy shapes the nature of signaling responses to different ligands in cells expressing multiple ErbB proteins.
Introduction
The EGF receptor (EGFR)2 is the founding member of the class I family of receptor tyrosine kinases (1, 2) and is composed of an extracellular ligand-binding region, a single membrane-spanning helix, a cytoplasmic tyrosine kinase domain, and a C-terminal ∼230-amino acid non-globular tail that contains several sites of tyrosine phosphorylation (3, 4). The EGFR family comprises four homologs in humans: EGFR (ErbB1/HER1), ErbB2 (HER2), ErbB3 (HER3), and ErbB4 (HER4). Each ErbB is essential for normal development, and abnormal ErbB activity is associated with several cancers (3, 5, 6). Eleven EGF-like ligands are known to bind and activate one or more ErbB proteins by promoting formation of specific ErbB homo- or heterodimers (6, 7). In addition to active ErbB1 and ErbB4 homodimers, evidence exists for active forms of all possible ErbB heterodimers (6, 8–10).
ErbB2 and ErbB3 are atypical in that ErbB2 has no known ligand and ErbB3 has negligible kinase activity (6). ErbB2 is the preferred dimerization partner of each of the other ErbB proteins, however, and the ErbB2/ErbB3 heterodimer in particular mediates highly potent cell growth signals (8, 11–13). Crystal structures of ErbB extracellular regions rationalized the unique properties of ErbB2 (14, 15). In the absence of ligand, the ErbB1, ErbB3, and ErbB4 extracellular regions adopt a tethered conformation in which a loop from domain II contacts a pocket in domain IV (16–18). In this tethered conformation, ligand-binding surfaces on domains I and III are held apart. Ligand binding requires breaking the domain II/IV contact to bring domains I and III together and stabilize an extended conformation in which the loop from domain II is exposed and free to mediate receptor dimerization (Fig. 1). The ErbB2 extracellular region adopts a constitutively extended conformation with a direct contact between domains I and III and an exposed domain II loop (19). ErbB2 is thus capable of dimerizing with other ErbB proteins without binding ligand itself. Previous structural analyses of Drosophila EGFR and human ErbB proteins have uncovered the significance of an ∼30° difference in the relative orientation of domains I and III in ErbB2 versus ligand-bound ErbB proteins (19–22), which rationalizes the absence of ErbB2 homodimers and indicates the presence of conformational asymmetry in the extracellular regions of ErbB2-containing ErbB heterodimers. Mutagenesis studies imply the presence of similar active asymmetric homodimers of ErbB1 and ErbB4 in which only one ErbB subunit binds ligand (22).
FIGURE 1.
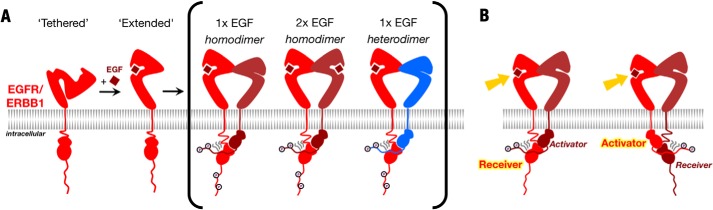
Schematic representation of ErbB activation. A, in the absence of ligand, EGFR/ErbB1 adopts a compact tethered state. Ligand binding promotes a conformational change in the extracellular regions to an extended conformation that exposes surfaces that mediate dimerization with liganded or unliganded partners. The intracellular kinases within these dimers form an asymmetric dimer contact essential for stimulating kinase activity. B, extracellular asymmetry in ErbB dimers with a single ligand bound may be coupled to the intracellular asymmetric kinase dimer.
Asymmetric dimerization of ErbB intracellular juxtamembrane and kinase domains is also an essential feature of active ErbB proteins (23–25). In the asymmetric kinase dimer, the C-terminal lobe of one kinase contacts the N-terminal lobe of the other kinase and stabilizes it in an active conformation. The kinase contributing the C-terminal lobe contact is thus known as the “donor” or “activator” kinase, and the kinase contributing the N-terminal lobe contact is known as the “acceptor” or “receiver” kinase. Only the receiver kinase is believed to be active within asymmetric dimers, and it phosphorylates the C-terminal tail of the activator kinase in trans owing to its proximity to the receiver kinase active site (25). The nomenclature can be confusing, as the C-terminal tail of the activator kinase is a substrate of the receiver kinase, and its phosphorylation is often used as an indicator of receiver kinase activity. To minimize confusion, we will use the activator-receiver versus donor-acceptor terminology. Although much is known about the conformation of isolated ErbB extracellular, transmembrane, and kinase regions in the active state, precisely how extracellular dimerization promotes formation of active asymmetric kinase dimers (or how asymmetric kinase dimers are prevented in inactive states) is not well understood.
It was recently reported that ErbB2 fails to phosphorylate kinase-deficient ErbB1 in ErbB1/ErbB2 heterodimers (26). This observation implies that ErbB2 prefers the activator position when paired with ErbB1, and it was speculated that the distinct unliganded conformation of the ErbB2 extracellular region could be coupled to this preferential kinase conformation. Owing to the presence of unliganded ErbB proteins in active asymmetric homodimers, coupling of extracellular and intracellular asymmetry could be a general feature of ErbB dimers. To test whether extracellular asymmetry is indeed coupled to intracellular asymmetry in ErbB dimers (Fig. 1B) or whether the intrinsic asymmetry of the ErbB1 and ErbB2 kinases is sufficient to drive the preference of the ErbB2 kinase for the activator position, we created chimeric ErbB proteins in which the extracellular region of one ErbB was spliced onto the transmembrane and intracellular regions of another ErbB. Coexpressing these chimeric ErbB proteins with native and kinase-deficient ErbB variants demonstrated that the intracellular regions alone are sufficient to determine relative preferences for the activator or receiver position and that intracellular ErbB asymmetry is not coupled to extracellular asymmetry. Furthermore, we uncovered a hierarchy of activator-receiver preference in which ErbB1 is the strongest receiver, followed by ErbB2 and then ErbB4, which appears to function as a receiver when paired only with itself in homodimers or with ErbB3. As this hierarchy determines which kinases will become active and which C-terminal tails will become phosphorylated within specific ErbB heterodimers, the extent of ErbB cross-talk and the nature of signaling output from a given ErbB will vary depending on the stimulating ligand and coexpressed ErbB proteins.
EXPERIMENTAL PROCEDURES
Materials and Plasmids
Plasmids encoding full-length human ErbB1, ErbB2, and ErbB4 were generated by cloning between XbaI and BssHII sites in pACTIN-SV, a vector that drives expression through use of an insect actin promoter (27). Kinase-deficient mutations (ErbB1-D837N, ErbB2-D845N, and ErbB4-D843N) and N- and C-terminal lobe activator/receiver interface mutations (ErbB2-I714Q and ErbB2-V956R) were generated using the Phusion site-directed mutagenesis kit protocol (Thermo Scientific). The sequences of all expressed genes were determined and validated. To generate the chimeric ErbB1/ErbB2 receptors, an MfeI restriction site was introduced in the nucleotide sequence encoding the ErbB1 amino acid sequence SIA (residues 645–647). The B1b2 chimera included amino acids 1–644 of ErbB1 and amino acids 653–1255 of ErbB2. The B2b1 chimera included amino acids 1–652 of ErbB2 and amino acids 645–1210 of ErbB1. Numbering begins with the first amino acid of the signal sequence. EGF and neuregulin-1β (NRG1β) were expressed and purified from Escherichia coli as described previously (22).
Cell Culture, Transfection, and Expression
S2R+ cells were grown at 25 °C in homemade protein-free insect medium (28). 12 h prior to transfection, 1 × 106 cells were plated in each well of a 6-well plate and allowed to adhere. 0.5 μg of each indicated expression plasmid was then transiently transfected into the cells in each well using Effectene (Qiagen) according to the manufacturer's protocol. Transfected cells were allowed to grow for 36 h prior to stimulation.
ErbB Stimulation Assays
For each ErbB combination, S2R+ cells were transfected in duplicate (2 wells) or triplicate (3 wells) for EGF or NRG1β stimulation or unstimulated control. For stimulation, the indicated ligand was added directly to insect cell medium at a final concentration of 100 ng/ml EGF or 1000 ng/ml NRG1β and incubated at room temperature for 5 min. Cells were then washed with phosphate-buffered saline and lysed in radioimmune precipitation assay buffer (25 mm Tris (pH 7.5), 150 mm NaCl, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, and 1 mm EDTA) supplemented with 5 μm lapatinib, 0.5 mm activated Na3VO4, 1 mm PMSF, and 250 units Benzonase nuclease (Sigma). Lapatinib and vanadate were included to prevent kinase and phosphatase activity, respectively, during lysis.
Immunoblotting and Detection
Whole cell lysates were normalized for total protein concentration using the BCA protein assay kit (Thermo Scientific). Lysates were separated on 7.5% polyacrylamide gels containing SDS and transferred to PVDF membranes for immunoblotting. Blots were treated in order with the indicated primary antibody and a horseradish peroxidase-conjugated secondary antibody and developed by chemiluminescence exposure to film.
RESULTS
Functional Human ErbB Proteins Can Be Expressed in S2R+ Cells
To investigate the behavior of human ErbB proteins in a cell line devoid of cross-reactive ErbB proteins, we transiently expressed wild-type and variant forms of these receptors in Drosophila melanogaster S2R+ cells. All human ErbB proteins except ErbB3 expressed well in these cells and became phosphorylated in response to appropriate ligands (Fig. 2B). Transfection conditions were adjusted to maintain expression levels below ∼100,000 receptors/cell to minimize complications from receptor overexpression (Fig. 2A). Phosphorylation of transfected ErbB proteins was assessed by Western blotting with ErbB site-specific anti-phosphotyrosine antibodies. Low-level ligand-independent ErbB phosphorylation was observed in some experiments. These signals were generally at least 20-fold lower than ligand-stimulated phosphorylation responses in comparable experiments.
FIGURE 2.
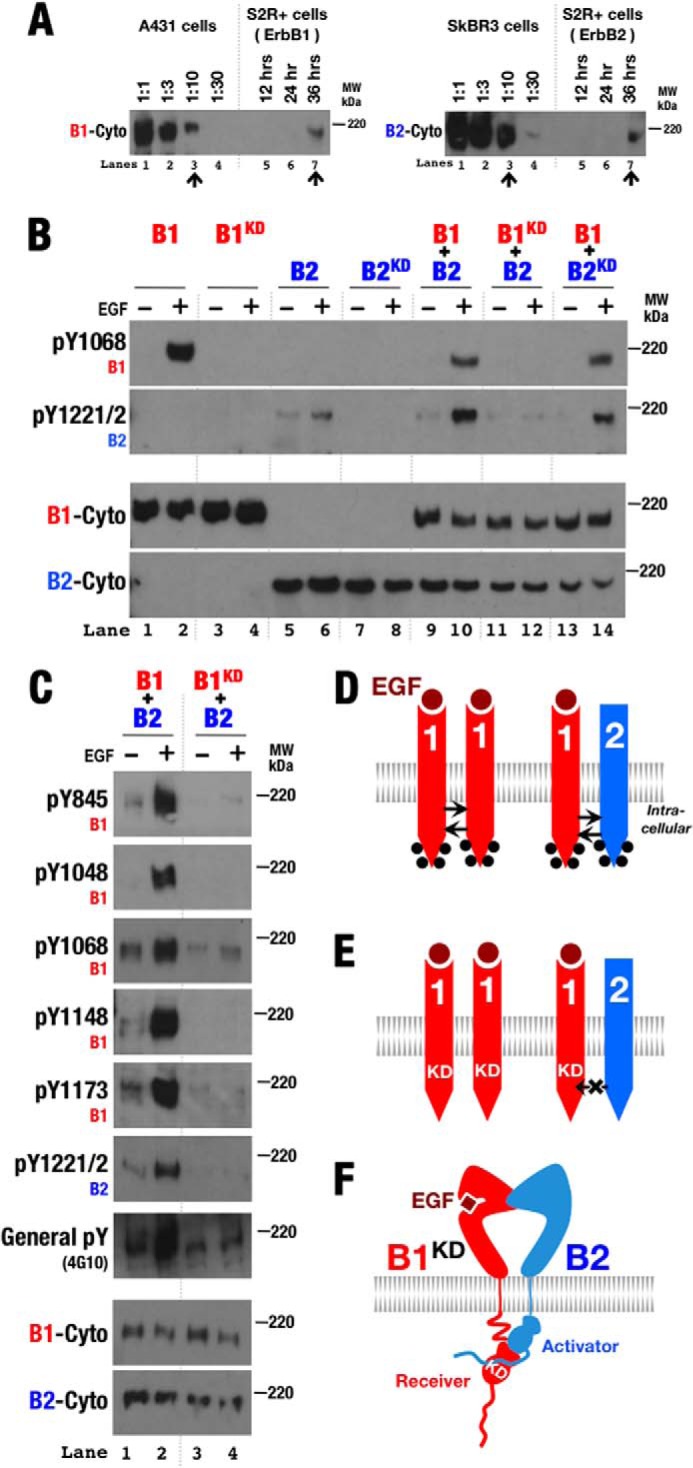
ErbB2 preferentially serves as the activator kinase when paired with ErbB1. A, to estimate receptor expression levels in S2R+ cells by Western blot analysis, cell lysates of ErbB-expressing S2R+ cells were compared against dilution standards of A431 cells (1.55 × 106 ErbB1 receptors/cell) (49) and SkBR3 cells (1.26 × 106 ErbB2 receptors/cell) (50). B, Western blots of whole cell lysates from S2R+ cells transiently transfected with wild-type and kinase-deficient (KD) forms of ErbB1 (B1) and ErbB2 (B2) and then treated with EGF (+) or left untreated (−). Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1221/1222 (pY1221/2; ErbB2), and the ErbB1 (B1-Cyto) and ErbB2 (B2-Cyto) cytoplasmic regions were used as indicated. C, similar Western blots as in B but utilizing antibodies specific for the indicated phosphotyrosines in ErbB1 as well as a general anti-phosphotyrosine antibody (4G10). D and E, schematic representations of phosphorylation patterns observed in A. Phosphorylation is indicated by black dots. F, schematic showing the preference of ErbB2 to serve as the activator kinase when paired with ErbB1.
ErbB1 Kinase Preferentially Serves as the Receiver Kinase When Paired with ErbB2
Wild-type and kinase-deficient variants of ErbB1 and ErbB2 expressed independently behaved as expected when expressed in S2R+ cells: only ErbB1 became phosphorylated in the presence of EGF (Fig. 2B), but both ErbB1 and ErbB2 became phosphorylated in the presence of EGF when coexpressed (Fig. 2B). Curiously, as observed previously (26), loss of ErbB1 (but not ErbB2) kinase activity resulted in loss of ligand-dependent phosphorylation of both ErbB1 and ErbB2 in coexpression experiments (Fig. 2B, lanes 11 and 12). This loss was observed for all five sites in the ErbB1 tail assessed with site-specific antibodies (Fig. 2C). ErbB1 thus appears able to phosphorylate kinase-deficient ErbB2, but not vice versa (Fig. 2, D and E). As ErbB phosphorylation occurs in trans (29), this confirms that the ErbB1 kinase preferentially adopts the receiver position and ErbB2 adopts the activator position in ErbB1/ErbB2 heterodimers in this system (Fig. 2F).
ErbB1 and ErbB2 Intracellular Regions Determine Kinase Activator-Receiver Preference
To test whether the unliganded ErbB2 extracellular region directs the preference of the ErbB2 kinase for the activator position when paired with ErbB1 (26), chimeric ErbB variants with the ErbB1 extracellular and ErbB2 transmembrane and intracellular regions (B1b2) or ErbB2 extracellular and ErbB1 transmembrane and intracellular regions (B2b1) were created (Fig. 3A). Both chimeras expressed, and B1b2 (but not B2b1) became phosphorylated in response to ligand when expressed alone (Fig. 3B). B1b2 phosphorylation in response to ligand demonstrated that the ErbB2 intracellular region is capable of serving as both the receiver and activator kinase when paired with itself; B2b1 presumably failed to become phosphorylated owing to its inability to bind ligand (Fig. 3C). Introduction of either N-terminal lobe receiver surface or C-terminal lobe activator surface mutations into B1b2 disrupted signaling, but coexpression of these two B1b2 variants restored activity (Fig. 3, D and E), indicating that B1b2 relies on the classical ErbB1 asymmetric kinase dimer for signaling (25). Also of note, the basal phosphorylation level of the B2b1 chimera was slightly elevated compared with wild-type ErbB1 (Fig. 3B), hinting that the ErbB2 extracellular region may be less able to autoinhibit the ErbB1 kinase. Surface biotinylation experiments (data not shown) showed that a substantial fraction of B2b1 was expressed on the cell surface, and B2b1 migrated on SDS-PAGE at a position consistent with normal glycosylation. These observations suggest that misfolding of B2b1 is unlikely to explain its basal activity.
FIGURE 3.

Chimeric ErbB proteins preserve ligand responsiveness and function through the canonical kinase dimer interface. A, schematic diagram of chimeric ErbB proteins with the extracellular region of one ErbB and the transmembrane and intracellular regions of another. B, Western blots of whole cell lysates from S2R+ cells transiently transfected with normal and kinase-deficient (KD) variants of ErbB1 (B1), ErbB2 (B2), and the ErbB chimera depicted in A. Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1221/1222 (pY1221/2; ErbB2), and the ErbB1 (B1-Cyto) and ErbB2 (B2-Cyto) cytoplasmic regions were used as indicated. C, schematic representation of phosphorylation patterns observed in B. Phosphorylation is indicated by black dots. D, Western blots of whole cell lysates from S2R+ cells transiently transfected with wild-type and variant forms of B1b2 were treated with EGF (+) or left untreated (−). Antibodies against phospho-Tyr1221/1222 (ErbB2) and ErbB2 cytoplasmic regions were used as indicated. B1b2N harbors the N-terminal lobe mutation I714Q, which disrupts ErbB2 receiver function; B1b2C harbors the C-terminal lobe mutation V956R, which disrupts ErbB2 activator function (25). E, schematic representation of phosphorylation observed in D.
Combined expression of ErbB1, ErbB2, B1b2, B2b1, and their kinase-deficient variants demonstrated that the preference of the ErbB2 kinase to adopt the activator position when paired with ErbB1 is intrinsic to the ErbB1 and ErbB2 transmembrane and intracellular regions and is not coupled to the extracellular regions (Fig. 4). First, unlike ErbB2, the B2b1 kinase was capable of serving as the receiver kinase and phosphorylating kinase-deficient ErbB1 (Fig. 4A, lanes 11 and 12). The ErbB2 extracellular region thus does not confer a preference for the activator position to its associated kinase. Conversely, the ErbB2 kinase became able to serve as the receiver kinase and phosphorylate its partner when paired with kinase-deficient B1b2 (Fig. 4A, lanes 13 and 14). Thus, removing intracellular differences within an ErbB1/ErbB2 heterodimer by making intracellular regions homodimeric removes the preference of one partner to be the activator or receiver kinase. Furthermore, kinase-deficient B1b2 was phosphorylated when coexpressed with B2b1 (Fig. 4A, lanes 7 and 8), demonstrating that the B2b1 kinase is capable of serving as the receiver kinase despite the inability of its extracellular region to bind ligand.
FIGURE 4.
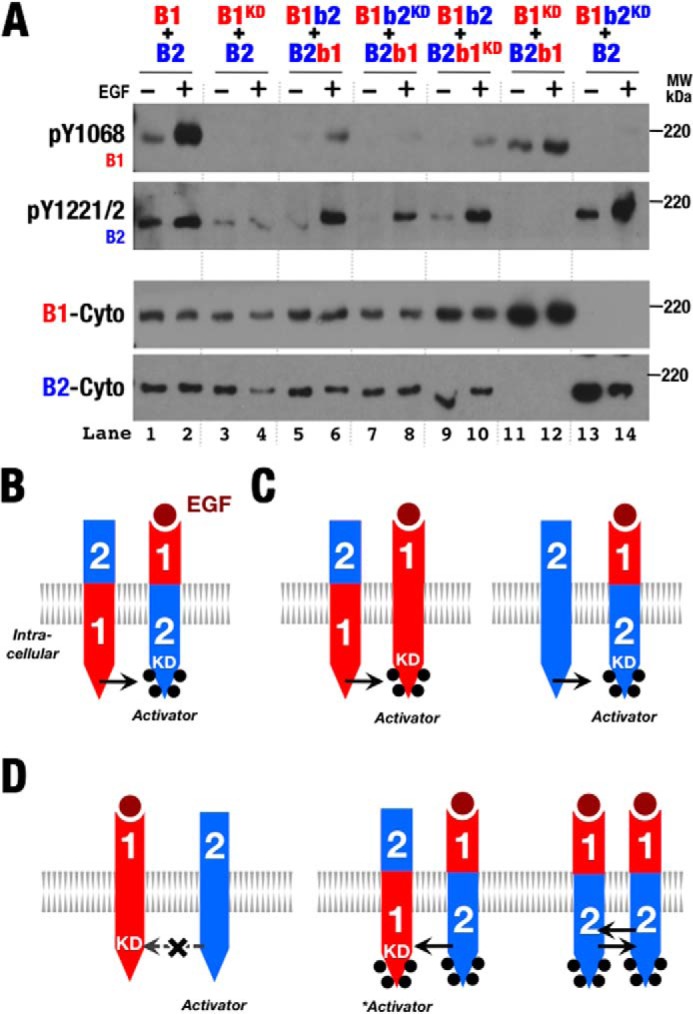
ErbB2 intracellular regions are sufficient to confer kinase activator role relative to ErbB1. A, Western blots of whole cell lysates from S2R+ cells transiently transfected with normal and kinase-deficient (KD) variants of ErbB1 (B1), ErbB2 (B2), and the ErbB chimera depicted in Fig. 3A. Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1221/1222 (pY1221/2; ErbB2), and the ErbB1 (B1-Cyto) and ErbB2 (B2-Cyto) cytoplasmic regions were used as indicated. B–D, schematic representations of phosphorylation patterns observed in A. Phosphorylation is indicated by black dots.
Contrary to expectations based on a model in which the ErbB2 kinase strictly adopts the activator position when paired with an ErbB1 kinase, weak ligand-dependent phosphorylation of either B2b1 or kinase-deficient B2b1 was observed when coexpressed with B1b2 (Fig. 4A, lanes 5, 6, 9, and 10). The B1b2 kinase was able to serve as the receiver kinase when paired with the ErbB1 intracellular region in these instances (Fig. 4B). A possible explanation for this apparent role reversal relative to non-chimeric ErbB1 and ErbB2 pairings is that, in this case, the B1b2 intracellular region was able to autophosphorylate through homodimerization owing to its ability to bind ligand (Fig. 3B, lanes 3 and 4). A difference between ErbB2 and B1b2 is that B1b2 is capable of autophosphorylation, and this phosphorylation and/or the consequent interaction with cytoplasmic factors may influence the relative ability of the B1b2 kinase (or ErbB kinases in general) to serve as the activator or receiver kinase (Fig. 4D).
To test whether C-terminal tail phosphorylation enables the ErbB2 kinase to serve as the receiver when paired with ErbB1, we examined the activity of a B1b2 variant in which five C-terminal tail phosphorylation sites were substituted with phenylalanine (30). These mutations did not prevent B1b2 from being phosphorylated at alternative sites and had no effect on the chimeric B1b2 kinase receiver activity when paired with B2b1 (Fig. 5), however.
FIGURE 5.
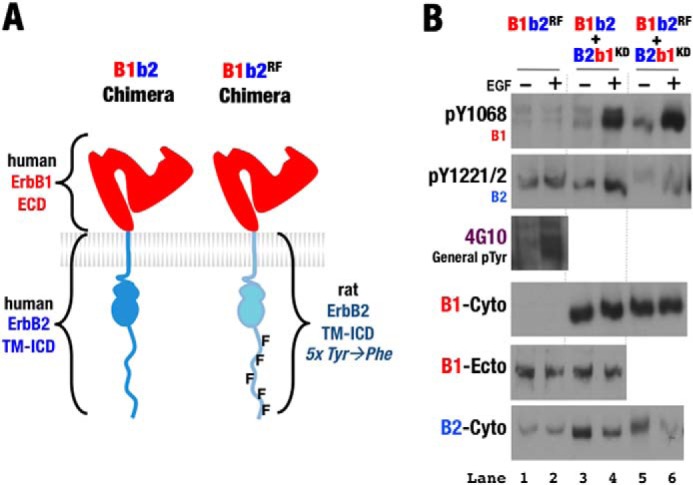
Phe variant C-terminal tail ErbB2 does not disrupt chimeric heterodimer activation. A, schematic representation of the human B1b2 chimera containing the extracellular domain (ECD) of human ErbB1 and the transmembrane and intracellular domains (TM-ICD) of either human ErbB2 or rat ErbB2 harboring five Tyr-to-Phe mutations (30). B, Western blots of whole cell lysates from S2R+ cells transiently transfected with wild-type, kinase-deficient (KD), and Phe variant (RF) forms of the indicated ErbB chimeras. Cotransfected cells were treated with EGF (+) or left untreated (−). Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1221/1222 (pY1221/2; ErbB2), general phospho-Tyr, the ErbB1 ectodomain (B1-Ecto), and the ErbB1 (B1-Cyto) and ErbB2 (B2-Cyto) cytoplasmic regions were used as indicated. Western blots show that Phe variant B1b2 retains receiver activity relative to B2b1 (lanes 5 and 6).
ErbB2 Kinase Preferentially Serves as the Receiver Kinase When Paired with ErbB4
In contrast to kinase-deficient ErbB1, when kinase-deficient ErbB4 was coexpressed with ErbB2, it became phosphorylated in a ligand-dependent manner (Fig. 6, A, lanes 11 and 12; and B). When ErbB4 was coexpressed with wild-type or kinase-deficient ErbB2, however, ErbB2 was not phosphorylated in the presence of ligand (Fig. 6A, lanes 7–10). These observations are consistent with ErbB2 preferring to serve as the receiver kinase when paired with ErbB4. This result was at first surprising owing to its contrast with the behavior of ErbB1 when paired with ErbB2. However, ErbB4 is most closely related to the pseudokinase ErbB3 (Fig. 6C), which must function predominantly as an activator due to its weak or nonexistent kinase activity (31–33). This result may also seem surprising, as the ErbB4 ligand NRG was originally purified based on its ability to stimulate ErbB2 phosphorylation (34, 35). The cells used in these assays, MDA-453 or MCF-7 cells, have subsequently been shown to express ErbB3 (36, 37), however, and cotransfection of ErbB3 into COS-7 cells with ErbB2 results in NRG-dependent ErbB2 phosphorylation (38), likely through ErbB3-dependent higher order oligomers of ErbB2 and ErbB3 (39). The NRG-dependent ErbB2 phosphorylation observed in earlier experiments thus likely stems from the presence of ErbB3 or some other factor.
FIGURE 6.

ErbB4 preferentially serves as the activator kinase when paired with ErbB2. A, Western blots of whole cell lysates from S2R+ cells transiently transfected with wild-type and kinase-deficient (KD) forms of ErbB1 (B1), ErbB2 (B2), and ErbB4 (B4) and treated with EGF or NRG1β (+) or left untreated (−). Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1221/1222 (pY1221/2; ErbB2), phospho-Tyr1284 (pY1284; ErbB4), and the ErbB1 (B1-Cyto), ErbB2 (B2-Cyto), and ErbB4 (BCyto) cytoplasmic regions were used as indicated. B, schematic representation of phosphorylation patterns observed in A. Phosphorylation is indicated by black dots. C, phylogenetic tree of human ErbB proteins based on sequence conservation generated by the maximum likelihood method (51). The scale bar indicates probability of amino acid change as an indicator of divergence time.
Attempts to show that the preference of the ErbB4 kinase to serve as the activator kinase relative to ErbB2 is independent of the extracellular regions were hindered by the constitutive activity of the ErbB4 intracellular region when appended to the ErbB2 extracellular region (data not shown). This activity is reminiscent of the higher basal activity of B2b1 and hints again that the ErbB2 extracellular region may be less able to inhibit the intrinsic kinase activity of other ErbB proteins.
This preference of the ErbB4 kinase to serve as the activator when paired with ErbB2 was also surprising, as we presumed that phosphorylating the ErbB2 C-terminal tail and engaging its downstream effectors would be needed to distinguish the response of cells expressing ErbB2 and ErbB4 from those expressing ErbB4 only and provide selective pressure for coexpression. We note, however, that although ErbB2 is not strongly phosphorylated when paired with ErbB4, it is functioning as a receiver kinase and likely phosphorylating non-ErbB substrates (40). This activity would diversify the ligand-dependent responses of ErbB4 only-expressing cells from cells expressing ErbB4 and ErbB2 and provide basis for selection of coexpression of ErbB2 and ErbB4.
ErbB1 Kinase Preferentially Serves as the Receiver Kinase When Paired with ErbB4
ErbB1 and ErbB4 are able to heterodimerize as evidenced by EGF-dependent phosphorylation of ErbB4 and NRG1β-dependent phosphorylation of kinase-deficient ErbB4 when coexpressed with ErbB1 (Fig. 7A, lanes 5–10). Neither ErbB1 nor kinase-deficient ErbB1 was phosphorylated in response to the ErbB4 ligand NRG1β when coexpressed with ErbB4 (Fig. 7A, lanes 5–10), however, suggesting that ErbB1 preferentially serves as the receiver kinase in ErbB1/ErbB4 heterodimers. Consistent with ErbB1 preferring the receiver role when paired with ErbB4, kinase-deficient ErbB4 was phosphorylated in response to EGF or NRG1β when coexpressed with ErbB1. Phosphorylation of ErbB1 in response to EGF when coexpressed with ErbB4 likely stems from ErbB1 homodimers (Fig. 7B), and the inability of phosphorylated ErbB4 to serve as the receiver kinase when paired with ErbB1 distinguishes ErbB4 from ErbB2.
FIGURE 7.
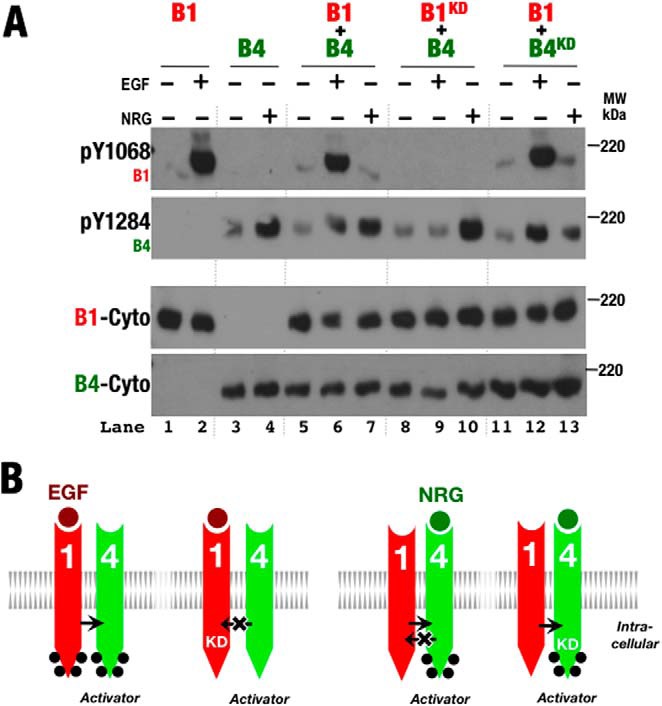
ErbB4 preferentially serves as the activator kinase when paired with ErbB1. A, Western blots of whole cell lysates from S2R+ cells transiently transfected with wild-type and kinase-deficient (KD) forms of ErbB1 (B1) and ErbB4 (B4) and treated with EGF or NRG1β (+) or left untreated (−). Antibodies against phospho-Tyr1068 (pY1068; ErbB1), phospho-Tyr1284 (pY1284; ErbB4), and the ErbB1 (B1-Cyto) and ErbB4 (B4-Cyto) cytoplasmic regions were used as indicated. B, schematic representation of phosphorylation patterns observed in A. Phosphorylation is indicated by black dots.
DISCUSSION
It has recently been recognized that ErbB2-containing heterodimers and active ErbB homodimers with a single ligand bound likely resemble asymmetric Drosophila EGFR dimers with a single high-affinity ligand bound (21, 22). In addition to an asymmetric dimer of ErbB kinases (23–25), ErbB extracellular regions in active, singly ligated ErbB heterodimers are also conformationally distinct. The observation by Pike and co-workers (26) that ErbB2 fails to phosphorylate kinase-deficient ErbB1 implies that the ErbB2 kinase preferentially serves as the activator kinase in ErbB1/ErbB2 heterodimers, and it was suggested that that the distinct unliganded conformation of the ErbB2 extracellular region may couple to the intracellular region and drive its preference for being the activator kinase. To test whether extracellular asymmetry in active ErbB proteins is indeed coupled to intracellular asymmetry (or perhaps determined by the intrinsic variation of ErbB1 and ErbB2 kinase regions), we created chimeric ErbB proteins with the extracellular region from one ErbB and the transmembrane and intracellular regions from another.
Coexpression of wild-type and kinase-deficient forms of native and chimeric ErbB proteins showed that ErbB transmembrane and intracellular regions are sufficient to determine the preference of specific ErbB kinases to serve as the activator or receiver kinase. Extracellular asymmetry is thus not coupled to intracellular asymmetry, consistent with mutagenesis results showing a loose coupling between ErbB1 extracellular and intracellular regions (41). In addition to confirming that the ErbB2 kinase preferentially serves as the activator kinase when paired with ErbB1, we have shown that the ErbB2 and ErbB1 kinases preferentially serve as the receiver kinase when paired with ErbB4. These results uncover a consistent pattern in which ErbB1 has the strongest propensity to serve as the receiver kinase, followed by ErbB2 and then ErbB4. The ErbB1, ErbB2, and ErbB4 kinases are each capable of functioning as the receiver and activator kinase as evidenced by active ErbB1 and ErbB4 homodimers and the activity of the B1b2 chimera. Our inability to express ErbB3 in S2R+ cells regrettably precluded including it in our studies, but its weak or inactive kinase (31–33), divergent N-terminal lobe receiver site sequence (25, 42, 43), and closer relationship to ErbB4 strongly suggest that its intracellular region has evolved to serve solely if not preferentially in the activator role.
Two physical bases likely underlie specific activator-receiver preferences among ErbB proteins. The first is sequence variability. Although amino acids present in the ErbB asymmetric dimer interface are highly conserved, more variability is seen in N-terminal lobe and juxtamembrane latch regions (receiver kinase) than in C-terminal lobe regions (activator kinase) (Fig. 8). Although it is difficult to parse the energetic contributions of individual amino acid substitutions to interaction interfaces, N-terminal lobe and juxtamembrane latch differences thus appear to be more likely sources of activator-receiver preference variability. A second potential factor influencing kinase activator-receiver preferences is the intrinsic balance between active and inactive kinase conformations. Receiver kinases are stabilized in the active state (25), and any relative preference for the active state among ErbB kinases will enhance their ability to serve in the receiver role. An example of this phenomenon is apparent in the recent observation that ErbB1 kinase-activating mutations associated with lung cancer greatly enhance the ability of ErbB1 to function as a receiver kinase (44). We are not aware of any evidence suggesting that the ErbB1 kinase more strongly favors an active conformation than the ErbB4 kinase under basal conditions, however, and only note this mechanism as a possibility.
FIGURE 8.

Alignment of ErbB intracellular domain sequences. A sequence alignment was generated using ClustalW2 (52). N-terminal lobe (receiver) interface residues are colored orange, C-terminal lobe (activator) interface residues are colored red, juxtamembrane residues are colored green, and C-terminal latch residues are colored blue. Purple marks indicate sequence variation in highlighted regions. Variation occurs in only 16 of the 57 residues found in activator/receiver interfaces. Hs, Homo sapiens.
We note that we have considered the activity of coexpressed ErbB proteins and ErbB variants in the context of homo- and heterodimers but cannot rule out the possibility of higher order ErbB oligomers. In particular, heteromeric ErbB complexes may consist of tetramers formed by dimerization of distinct homodimers (45). Whatever the stoichiometry of ErbB heteromers, however, our results indicate that the canonical asymmetric kinase dimer (25) and the activator-receiver preferences uncovered here are operative within any higher order oligomer. We have also considered cis-phosphorylation to be negligible, as wild-type ErbB1 and ErbB2 phosphorylated only in trans when paired with an activating ErbB (Figs. 6 and 7), but we cannot exclude the possibility that cis-phosphorylation may account for some of our observations.
We were intrigued by the apparent ability of the ErbB2 kinase in the context of the B1b2 chimera to switch roles and serve as the receiver kinase relative to the B2b1 chimera (Fig. 4D). The ability of the B1b2 C-terminal tail to become autophosphorylated through homodimers suggested that the phosphorylation state of ErbB2 might influence its preference for serving as the activator or receiver kinase. Substitution of five ErbB2 C-terminal tail tyrosines with phenylalanine within the B1b2 chimera did not affect its ability to switch roles and serve as the receiver kinase when paired with B2b1, however, and the basis for this role reversal needs further investigation.
In addition to functional ErbB1 and ErbB4 homodimers, earlier studies of coexpressed ErbB proteins found evidence for all possible ErbB heterodimers and identified a hierarchy of ErbB interactions in which ErbB2 appears to be the preferred partner of all ErbB proteins (8–10, 46). Our identification of a distinct hierarchy of ErbB kinase interactions is essentially an independent phenomenon, however, as ErbB kinase activator-receiver preferences contribute only partially, if at all, to overall receptor interaction hierarchies. Rather, kinase activator-receiver preferences determine which kinases become active and which C-terminal tails become phosphorylated within specific ErbB heterodimers. Assuming nonequivalent (i) kinase substrate specificities and (ii) sets of downstream effectors that interact with phosphorylated C-terminal tails (3, 40), which kinases and/or C-terminal tails become activated will determine the signaling output from a given input. We presume that activating an ErbB kinase independently of phosphorylating its C-terminal tail or vice versa, which is a consequence of kinase activator-receiver preferences, will thus result in qualitatively different signaling outputs.
In cells expressing both ErbB1 and ErbB4, for example, ErbB1/ErbB4 heterodimers appear to form in response to both EGF (an ErbB1-specific ligand) and NRG1β (an ErbB3- and ErbB4-specific ligand). Due to the preference of the ErbB1 kinase for the receiver position when paired with ErbB4, however, the signaling response to EGF and NRG1β will be different. Following EGF treatment, the ErbB1 (but not ErbB4) kinase will be activated, and both the ErbB1 and ErbB4 C-terminal tails will become phosphorylated. In response to NRG1β, the ErbB4 and ErbB1 kinases will be activated, but only the ErbB4 C-terminal tail will be phosphorylated. This receptor activation and phosphorylation pattern rationalizes previous reports that EGF, but not NRG1β, stimulates recruitment of the ErbB1 C-terminal tail effectors Shc and Grb2 (8, 47). Furthermore, the ErbB2-dependent responses of cells coexpressing ErbB1 and ErbB2 will likely differ from those of cells coexpressing ErbB4 and ErbB2 (Fig. 9B). In the former case, the ErbB2 C-terminal tail will become phosphorylated, but the ErbB2 kinase will not be activated. In the latter case, the ErbB2 kinase will be activated, but its C-terminal tail will not be phosphorylated. Consistent with this scheme, a previous study has shown that anchorage-independent growth of T47D cells expressing ErbB4 requires ErbB2 kinase activity, but not ErbB2 C-terminal tail phosphorylation (Fig. 9C) (48).
FIGURE 9.

Kinase activator-receiver hierarchy in ErbB heterodimers. A, schematic diagrams of ErbB heterodimers showing kinase activator-receiver preferences and C-terminal tail phosphorylation patterns (black dots). B, the ErbB2 kinase is the dominant activator when paired with ErbB1 but the dominant receiver when paired with ErbB4. C, due to kinase activator-receiver preferences, ErbB1/ErbB4 heterodimers will result in different patterns of kinase activation and C-terminal tail phosphorylation depending on whether EGF or NRG1β is the stimulating ligand.
The four human ErbB proteins constitute an intertwined signaling network tuned to respond to at least 11 ligands with differing specificities and affinities for individual ErbB proteins. We have shown here that the roles of kinases in asymmetric kinase dimers characteristic of active ErbB heterodimers are not coupled to extracellular asymmetry. Within specific ErbB heterodimers, however, individual ErbB kinases show a hierarchy of preference for the receiver or activator position depending on their partner. This preference hierarchy will influence the nature of the signaling responses of cells expressing multiple ErbB proteins and adds an additional layer of diversity to ErbB signaling.
Acknowledgments
We thank Jennifer Kavran, Patrick Byrne, and Lily Raines for helpful discussions. We also thank William Papavasiliou and William Muller (McGill University) for generously providing rat erbB2 cDNA clones.
This work was supported, in whole or in part, by National Institutes of Health Grant R01GM099321 (to D. J. L.).
- EGFR
- EGF receptor
- NRG1β
- neuregulin-1β.
REFERENCES
- 1. Blume-Jensen P., Hunter T. (2001) Oncogenic kinase signalling. Nature 411, 355–365 [DOI] [PubMed] [Google Scholar]
- 2. Lemmon M. A., Schlessinger J. (2010) Cell signaling by receptor tyrosine kinases. Cell 141, 1117–1134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Olayioye M. A., Neve R. M., Lane H. A., Hynes N. E. (2000) The ErbB signaling network: receptor heterodimerization in development and cancer. EMBO J. 19, 3159–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Downward J., Yarden Y., Mayes E., Scrace G., Totty N., Stockwell P., Ullrich A., Schlessinger J., Waterfield M. D. (1984) Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 307, 521–527 [DOI] [PubMed] [Google Scholar]
- 5. Holbro T., Civenni G., Hynes N. E. (2003) The ErbB receptors and their role in cancer progression. Exp. Cell Res. 284, 99–110 [DOI] [PubMed] [Google Scholar]
- 6. Yarden Y., Sliwkowski M. X. (2001) Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2, 127–137 [DOI] [PubMed] [Google Scholar]
- 7. Jorissen R. N., Walker F., Pouliot N., Garrett T. P., Ward C. W., Burgess A. W. (2003) Epidermal growth factor receptor: mechanisms of activation and signalling. Exp. Cell Res. 284, 31–53 [DOI] [PubMed] [Google Scholar]
- 8. Graus-Porta D., Beerli R. R., Daly J. M., Hynes N. E. (1997) ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 16, 1647–1655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tzahar E., Waterman H., Chen X., Levkowitz G., Karunagaran D., Lavi S., Ratzkin B. J., Yarden Y. (1996) A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 16, 5276–5287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Riese D. J., 2nd, van Raaij T. M., Plowman G. D., Andrews G. C., Stern D. F. (1995) The cellular response to neuregulins is governed by complex interactions of the ErbB receptor family. Mol. Cell. Biol. 15, 5770–5776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Alimandi M., Romano A., Curia M. C., Muraro R., Fedi P., Aaronson S. A., Di Fiore P. P., Kraus M. H. (1995) Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 10, 1813–1821 [PubMed] [Google Scholar]
- 12. Karunagaran D., Tzahar E., Beerli R. R., Chen X., Graus-Porta D., Ratzkin B. J., Seger R., Hynes N. E., Yarden Y. (1996) ErbB-2 is a common auxiliary subunit of NDF and EGF receptors: implications for breast cancer. EMBO J. 15, 254–264 [PMC free article] [PubMed] [Google Scholar]
- 13. Pinkas-Kramarski R., Shelly M., Glathe S., Ratzkin B. J., Yarden Y. (1996) Neu differentiation factor/neuregulin isoforms activate distinct receptor combinations. J. Biol. Chem. 271, 19029–19032 [DOI] [PubMed] [Google Scholar]
- 14. Burgess A. W., Cho H. S., Eigenbrot C., Ferguson K. M., Garrett T. P., Leahy D. J., Lemmon M. A., Sliwkowski M. X., Ward C. W., Yokoyama S. (2003) An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 12, 541–552 [DOI] [PubMed] [Google Scholar]
- 15. Leahy D. J. (2004) Structure and function of the epidermal growth factor (EGF/ErbB) family of receptors. Adv. Protein Chem. 68, 1–27 [DOI] [PubMed] [Google Scholar]
- 16. Bouyain S., Longo P. A., Li S., Ferguson K. M., Leahy D. J. (2005) The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. U.S.A. 102, 15024–15029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cho H. S., Leahy D. J. (2002) Structure of the extracellular region of HER3 reveals an interdomain tether. Science 297, 1330–1333 [DOI] [PubMed] [Google Scholar]
- 18. Ferguson K. M., Berger M. B., Mendrola J. M., Cho H. S., Leahy D. J., Lemmon M. A. (2003) EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 11, 507–517 [DOI] [PubMed] [Google Scholar]
- 19. Cho H. S., Mason K., Ramyar K. X., Stanley A. M., Gabelli S. B., Denney D. W., Jr., Leahy D. J. (2003) Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 421, 756–760 [DOI] [PubMed] [Google Scholar]
- 20. Alvarado D., Klein D. E., Lemmon M. A. (2009) ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 461, 287–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Alvarado D., Klein D. E., Lemmon M. A. (2010) Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 142, 568–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu P., Cleveland T. E., 4th, Bouyain S., Byrne P. O., Longo P. A., Leahy D. J. (2012) A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. U.S.A. 109, 10861–10866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Aertgeerts K., Skene R., Yano J., Sang B. C., Zou H., Snell G., Jennings A., Iwamoto K., Habuka N., Hirokawa A., Ishikawa T., Tanaka T., Miki H., Ohta Y., Sogabe S. (2011) Structural analysis of the mechanism of inhibition and allosteric activation of the kinase domain of HER2 protein. J. Biol. Chem. 286, 18756–18765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qiu C., Tarrant M. K., Choi S. H., Sathyamurthy A., Bose R., Banjade S., Pal A., Bornmann W. G., Lemmon M. A., Cole P. A., Leahy D. J. (2008) Mechanism of activation and inhibition of the HER4/ErbB4 kinase. Structure 16, 460–467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang X., Gureasko J., Shen K., Cole P. A., Kuriyan J. (2006) An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 125, 1137–1149 [DOI] [PubMed] [Google Scholar]
- 26. Macdonald-Obermann J. L., Piwnica-Worms D., Pike L. J. (2012) Mechanics of EGF receptor/ErbB2 kinase activation revealed by luciferase fragment complementation imaging. Proc. Natl. Acad. Sci. U.S.A. 109, 137–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Huynh C. Q., Zieler H. (1999) Construction of modular and versatile plasmid vectors for the high-level expression of single or multiple genes in insects and insect cell lines. J. Mol. Biol. 288, 13–20 [DOI] [PubMed] [Google Scholar]
- 28. Inlow D., Shauger A., Maiorella B. (1989) Insect cell culture and baculovirus propagation in protein-free culture. J. Tissue Culture Methods 12, 13–16 [Google Scholar]
- 29. Lammers R., Van Obberghen E., Ballotti R., Schlessinger J., Ullrich A. (1990) Transphosphorylation as a possible mechanism for insulin and epidermal growth factor receptor activation. J. Biol. Chem. 265, 16886–16890 [PubMed] [Google Scholar]
- 30. Dankort D. L., Wang Z., Blackmore V., Moran M. F., Muller W. J. (1997) Distinct tyrosine autophosphorylation sites negatively and positively modulate Neu-mediated transformation. Mol. Cell. Biol. 17, 5410–5425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guy P. M., Platko J. V., Cantley L. C., Cerione R. A., Carraway K. L., 3rd (1994) Insect cell-expressed p180erbB3 possesses an impaired tyrosine kinase activity. Proc. Natl. Acad. Sci. U.S.A. 91, 8132–8136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jura N., Shan Y., Cao X., Shaw D. E., Kuriyan J. (2009) Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. U.S.A. 106, 21608–21613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mendrola J. M., Shi F., Park J. H., Lemmon M. A. (2013) Receptor tyrosine kinases with intracellular pseudokinase domains. Biochem. Soc. Trans. 41, 1029–1036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Holmes W. E., Sliwkowski M. X., Akita R. W., Henzel W. J., Lee J., Park J. W., Yansura D., Abadi N., Raab H., Lewis G. D. (1992) Identification of heregulin, a specific activator of p185erbB2. Science 256, 1205–1210 [DOI] [PubMed] [Google Scholar]
- 35. Peles E., Bacus S. S., Koski R. A., Lu H. S., Wen D., Ogden S. G., Levy R. B., Yarden Y. (1992) Isolation of the Neu/HER-2 stimulatory ligand: a 44 kd glycoprotein that induces differentiation of mammary tumor cells. Cell 69, 205–216 [DOI] [PubMed] [Google Scholar]
- 36. Knowlden J. M., Hutcheson I. R., Jones H. E., Madden T., Gee J. M., Harper M. E., Barrow D., Wakeling A. E., Nicholson R. I. (2003) Elevated levels of epidermal growth factor receptor/c-erbB2 heterodimers mediate an autocrine growth regulatory pathway in tamoxifen-resistant MCF-7 cells. Endocrinology 144, 1032–1044 [DOI] [PubMed] [Google Scholar]
- 37. Wang S., Huang J., Lyu H., Lee C. K., Tan J., Wang J., Liu B. (2013) Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 4, e556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sliwkowski M. X., Schaefer G., Akita R. W., Lofgren J. A., Fitzpatrick V. D., Nuijens A., Fendly B. M., Cerione R. A., Vandlen R. L., Carraway K. L., 3rd (1994) Coexpression of ErbB2 and ErbB3 proteins reconstitutes a high affinity receptor for heregulin. J. Biol. Chem. 269, 14661–14665 [PubMed] [Google Scholar]
- 39. Zhang Q., Park E., Kani K., Landgraf R. (2012) Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc. Natl. Acad. Sci. U.S.A. 109, 13237–13242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Carpenter G. (1992) Receptor tyrosine kinase substrates: Src homology domains and signal transduction. FASEB J. 6, 3283–3289 [DOI] [PubMed] [Google Scholar]
- 41. Lu C., Mi L. Z., Grey M. J., Zhu J., Graef E., Yokoyama S., Springer T. A. (2010) Structural evidence for loose linkage between ligand binding and kinase activation in the epidermal growth factor receptor. Mol. Cell. Biol. 30, 5432–5443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jura N., Endres N. F., Engel K., Deindl S., Das R., Lamers M. H., Wemmer D. E., Zhang X., Kuriyan J. (2009) Mechanism for activation of the EGF receptor catalytic domain by the juxtamembrane segment. Cell 137, 1293–1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Red Brewer M., Choi S. H., Alvarado D., Moravcevic K., Pozzi A., Lemmon M. A., Carpenter G. (2009) The juxtamembrane region of the EGF receptor functions as an activation domain. Mol. Cell 34, 641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Red Brewer M., Yun C. H., Lai D., Lemmon M. A., Eck M. J., Pao W. (2013) Mechanism for activation of mutated epidermal growth factor receptors in lung cancer. Proc. Natl. Acad. Sci. U.S.A. 110, E3595–E3604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schlessinger J. (2000) Cell signaling by receptor tyrosine kinases. Cell 103, 211–225 [DOI] [PubMed] [Google Scholar]
- 46. Riese D. J., 2nd, Stern D. F. (1998) Specificity within the EGF family/ErbB receptor family signaling network. BioEssays 20, 41–48 [DOI] [PubMed] [Google Scholar]
- 47. Olayioye M. A., Graus-Porta D., Beerli R. R., Rohrer J., Gay B., Hynes N. E. (1998) ErbB-1 and ErbB-2 acquire distinct signaling properties dependent upon their dimerization partner. Mol. Cell. Biol. 18, 5042–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mill C. P., Zordan M. D., Rothenberg S. M., Settleman J., Leary J. F., Riese D. J., 2nd (2011) ErbB2 is necessary for ErbB4 ligands to stimulate oncogenic activities in models of human breast cancer. Genes Cancer 2, 792–804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Panke C., Weininger D., Haas A., Schelter F., Schlothauer T., Bader S., Sircar R., Josel H. P., Baer U., Burtscher H., Mundigl O., Grote M., Brinkmann U., Sustmann C. (2013) Quantification of cell surface proteins with bispecific antibodies. Protein Eng. Des. Sel. 26, 645–654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Costantini D. L., Bateman K., McLarty K., Vallis K. A., Reilly R. M. (2008) Trastuzumab-resistant breast cancer cells remain sensitive to the auger electron-emitting radiotherapeutic agent 111In-NLS-trastuzumab and are radiosensitized by methotrexate. J. Nucl. Med. 49, 1498–1505 [DOI] [PubMed] [Google Scholar]
- 51. Dereeper A., Guignon V., Blanc G., Audic S., Buffet S., Chevenet F., Dufayard J. F., Guindon S., Lefort V., Lescot M., Claverie J. M., Gascuel O. (2008) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res. 36, W465–W469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Goujon M., McWilliam H., Li W., Valentin F., Squizzato S., Paern J., Lopez R. (2010) A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38, W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]