

Background: Plant cryptochromes are blue light sensors forming an exceptionally stable flavin neutral radical as a signaling state.
Results: Blockage of proton transfer to flavin severely reduces the lifetime of the radical.
Conclusion: The proton donor aspartic acid acts as an intrinsic stabilizer of the signaling state.
Significance: The role of a key structural element is identified, distinguishing plant cryptochromes from other members of the family.
Keywords: Chlamydomonas, cryptochrome, flavoprotein, IR Spectroscopy, Photoreceptor, Radical
Abstract
Plant cryptochromes regulate the circadian rhythm, flowering time, and photomorphogenesis in higher plants as responses to blue light. In the dark, these photoreceptors bind oxidized FAD in the photolyase homology region (PHR). Upon blue light absorption, FAD is converted to the neutral radical state, the likely signaling state, by electron transfer via a conserved tryptophan triad and proton transfer from a nearby aspartic acid. Here we demonstrate, by infrared and time-resolved UV-visible spectroscopy on the PHR domain, that replacement of the aspartic acid Asp-396 with cysteine prevents proton transfer. The lifetime of the radical is decreased by 6 orders of magnitude. This short lifetime does not permit to drive conformational changes in the C-terminal extension that have been associated with signal transduction. Only in the presence of ATP do both the wild type and mutant form a long-lived radical state. However, in the mutant, an anion radical is formed instead of the neutral radical, as found previously in animal type I cryptochromes. Infrared spectroscopic experiments demonstrate that the light-induced conformational changes of the PHR domain are conserved in the mutant despite the lack of proton transfer. These changes are not detected in the photoreduction of the non-photosensory d-amino acid oxidase to the anion radical. In conclusion, formation of the anion radical is sufficient to generate a protein response in plant cryptochromes. Moreover, the intrinsic proton transfer is required for stabilization of the signaling state in the absence of ATP.
Introduction
Plant cryptochromes form a subfamily of the large and diverse cryptochrome/photolyase family and play a vital role as photoreceptors in the responses of plants to light conditions in the environment (1). These blue light receptors regulate several aspects of photomorphogenesis (2, 3) and interact directly with the phytochromes as red/far-red light receptors (4). Cryptochromes synchronize the circadian clock of plants with the external light-dark cycle (5) and play a major role in the initiation of flowering by the change in day length (6). Furthermore, cryptochromes are involved in the regulation of stomatal opening (7) and trigger programmed cell death induced by singlet oxygen (8).
Plant cryptochromes bind FAD as a chromophore in the photolyase homology region (PHR)2 (9), which comprises about 500 N-terminal amino acids. Close to the FAD binding pocket, the PHR may additionally bind ATP (Fig. 1) (10, 11). The long C-terminal extension (CCT) of plant cryptochromes is sufficient for constitutive activation of the signaling cascade (12). This effect could be narrowed down in sequence to an 80-residue motif that includes a few residues of the PHR and the adjacent region of the CCT (13). Binding of the PHR to the CCT in the dark is considered to act as a repressor of this motif.
FIGURE 1.
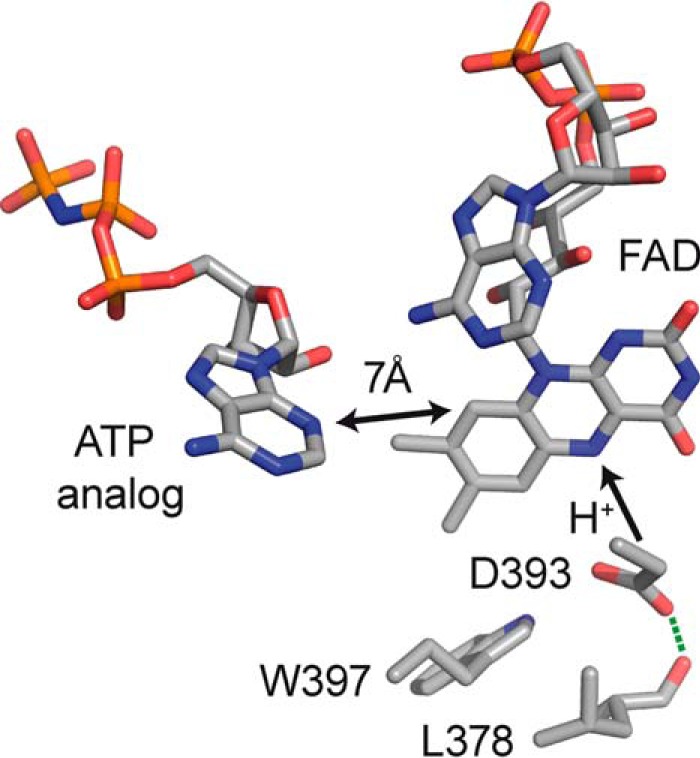
Detail of the flavin-binding pocket in the plant cryptochrome CPH1 modeled on the crystal structure of AtCRY1-PHR (PDB code 1U3D). Trp-397 acts as electron donor to flavin after excitation by blue light. The proton donor Asp-393 (Asp-396 in AtCRY1) was replaced with a cysteine in this study. The position of the ATP analog was determined in AtCRY1-PHR after soaking the crystal.
Upon illumination of plant cryptochromes, the oxidized FAD is reduced to the anion radical by a nearby tryptophan residue with a time constant of 400 femtoseconds (14) and subsequently protonated within a few microseconds (15, 16) to form the neutral radical of FAD (17) (Fig. 1). Several experiments point to the fact that this neutral radical constitutes the signaling state of the receptor in vivo (18–20), but this model of activation is under debate (21, 22). Illumination of plant cryptochrome leads to conformational changes in the PHR (23) and in the CCT (24). The latter are associated with a significant change in the diffusion coefficient with a time constant of 400 ms (25).
Many recent studies have dealt with the electron transfer cascade via a tryptophan triad in cryptochromes and its role in activation of the sensor by photoreduction (14, 17, 26–30). Much less is known about the role of the subsequent step of proton transfer to flavin. In plant cryptochromes, this step is unusually separated in time from the initiating electron transfer by 6 orders of magnitude (14, 15). A much faster proton transfer has been postulated in theoretical studies (31), but indications for such a competing process have been found only to some extent above the physiological pH (pH 7.4) and in the absence of ATP (32). In plant cryptochromes, proton transfer is required for the antagonizing effect of green light found in vivo (18, 19, 33) because only the protonated radical absorbs green light to a significant extent. Furthermore, proton transfer is a prerequisite for a further photoreduction of the radical to the fully reduced state. This photoreaction finally leads to a regeneration of the oxidized state by reaction with oxygen (34).
Interestingly, only in plant cryptochromes is an intrinsic proton donor, aspartic acid 396, conserved close to flavin (11) (in the numbering of Arabidopsis cryptochrome 1 (AtCRY1)), whereas it is replaced with a cysteine in animal type I cryptochromes and with an asparagine in all other cryptochromes (Fig. 1). Deprotonation of this residue accompanies flavin reduction, as has been deduced from FTIR spectroscopic experiments and structural considerations (23, 35, 36). In support of this finding, a high pKa value of this aspartic acid has been indirectly determined by its impact on the absorption spectrum of the flavin (32). According to this study, it shifts strongly from 3.7 in solution to 7.4 in AtCRY1 and to above 9 upon addition of ATP. An exchange of this residue by asparagine leads to a change in the redox potential of flavin to more negative values (37) and to some photolyase-like activity of DNA repair by the plant cryptochrome (36).
Therefore, we investigated the role of proton transfer in the PHR of a plant cryptochrome from the unicellular green alga Chlamydomonas reinhardtii. It has been suggested that this cryptochrome, Chlamydomonas photolyase homologue 1 (CPH1), plays a role as a negative modulator of the clock because its level correlates negatively with the sensitivity in clock resetting by blue light in vivo (38). CPH1 groups together with cryptochromes from higher plants, mosses, and ferns to the subfamily plant cryptochrome (1). Accordingly, CPH1-PHR exhibits all characteristics of PHRs from higher plant cryptochromes (14, 15, 23, 39). We exchanged aspartic acid with cysteine (D393C) and found that proton transfer is completely blocked in this mutant but that conformational changes are retained. The consequences of this blockage for the signaling of plant cryptochrome are discussed.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
Wild-type protein was prepared as described previously (39). The mutation of aspartic acid 393 to cysteine was introduced by overlapping PCR into a sequence coding for six histidines and the N-terminal 504 amino acids of CPH1 from C. reinhardtii (39). The mutation was confirmed by sequence analysis. The construct was cloned into a modified pET11a vector (Novagen) coding for a Strep-tag II at the C terminus of the protein (39). Escherichia coli BL21 (DE 3) pLysE (Invitrogen) containing the expression construct was grown in double yeast tryptone broth supplemented with 0.57 mm ampicillin at 37 °C in the dark. When the optical density (OD) at 600 nm reached 0.5, the temperature was lowered to 18 °C. At an OD of 0.8, isopropyl 1-thio-β-d-galactopyranoside was added to a final concentration of 10 μm. After 20 h, the cells were harvested by centrifugation (5000 × g, 15 min). The cell pellet was resuspended in 50 mm sodium phosphate buffer (pH 7.9), 0.1 m NaCl, 20% glycerol, a tablet of Complete EDTA-free (Roche Applied Science), and DNase. Before cell disruption via French press (2 × 1000 p.s.i.g.), the resuspension of the cells was titrated with NaOH to a pH of 8.3. The lysate was centrifuged (108,000 × g, 1 h) and loaded onto a Strep-Tactin-Sepharose column (IBA). After washing with 50 mm sodium phosphate buffer (pH 7.9), 0.1 m NaCl, and 20% glycerol, the protein was eluted in the same buffer containing 2.5 mm d-desthiobiotin. The eluted protein was washed twice with the 10-fold volume of sodium phosphate buffer without d-desthiobiotin using an Amicon Ultra 4 filter device (Millipore) with a 50-kDa cutoff.
UV-visible Spectroscopy
Spectra were recorded with a Shimadzu 2450 PC spectrophotometer. For photoreduction experiments, an integration sphere was used. The samples were kept at 10 °C during the measurements. MgCl2 was added to a final concentration of 17 mm. The influence of ATP was investigated by adding ATP to a final concentration of 3 mm. For illumination, a light-emitting diode (Luxeon Star, Philips Lumileds) with an emission maximum at 455 nm (20 nm full width at half-maximum) was used. Between two illuminations, a dark time of 3 min was applied. The kinetics of the dark recovery were recorded at 20 °C and 450 nm after illumination with the light-emitting diode with an intensity of 16 milliwatt/cm2 for 10 s. The time windows of recording were chosen to cover at least three times the slowest time constant of the biexponential fit. Both the D393C mutant and the wild type were diluted to the same A450 = 0.28 before the experiments.
Time-resolved UV-visible Spectroscopy
The flash photolysis setup with an intensified charge-coupled device camera has been described previously (40). Here a tunable optical parametric oscillator (Opta) pumped by the third harmonic of an Nd:YAG laser (Spectra Physics, GCR-12) was used to generate 450-nm pulses with a duration of 10 ns and an energy density of 2 mJ/cm2 at a repetition rate of (1.6 s)−1 as selected by a shutter. Horizontal windows of 4 × 10 mm limited the exposure of the sample for excitation and probe light, respectively. To ensure a homogeneous excitation of the sample, a mirror was placed behind the cuvette. For each experiment, an alternating series of reference spectra with probe light only and signal spectra with additional laser excitation was recorded. A dark spectrum without excitation or probe light was subtracted from both spectra. The opening time of the probe light shutter was limited to 200 μs or 1 ms depending on the time window of detection. The concentration of the sample was adjusted to an A450 = 0.3. A sample volume of 2.5 ml was placed in a 10 × 10 mm quartz cuvette and was stirred for 750 ms after each excitation with a 5-mm stirrer bar (Thermo Scientific, Variomag Mini) to minimize multiple excitations. Fifteen experiments were averaged per time point at 20 °C. The time window of detection was set to 10% of the delay time for the experiments at 2–200 μs. Spectra from 2–40 ms were acquired with a window of detection of 100 μs.
FTIR Spectroscopy
All steps were performed in the dark. For experiments with CPH1-PHR-D393C, the glycerol content was reduced to ≤ 1% by a buffer exchange. The protein was concentrated by ultrafiltration to A450 ≥ 5. ATP was added to a final concentration of 2.5 mm. A droplet of 5 μl of protein solution was applied to a BaF2 window, and the water content was gently reduced for 7 min at a pressure of 500 millibar. The sample was sealed by a second window. For experiments with d-amino acid oxidase (DAO), lyophilized protein (Sigma-Aldrich) was dissolved in 10 mm potassium phosphate buffer (pH 8), 50 mm NaCl, and 70 mm 2-mercaptoethanol. The solution was washed twice with the 10-fold volume of the same buffer by ultrafiltration using an Amicon Ultra 4 filter device (Millipore) with a 30-kDa cutoff and was finally concentrated to A450 = 3. A droplet of 10–15 μl of the protein solution was applied to a BaF2 window, and the sample was gently deoxygenated by several cycles of applying a vacuum of 60 millibar and flushing with nitrogen.
FTIR spectra were recorded on Bruker IFS/66v and 66v/S spectrometers with a spectral resolution of 2 cm−1. The temperature of the sample was maintained ambient for DAO and adjusted to 10 °C for CHP1-PHR-D393C. For light-induced difference spectroscopy, a long-wave pass filter (OCLI) was placed between the sample and detector. The filter blocked stray light and restricted the recording range to <2000 cm−1 to improve the signal-to-noise ratio. CPH1-PHR-D393C was illuminated for 4 s with a 455-nm light-emitting diode (Luxeon Star, Philips Lumileds) with an intensity of ∼20 milliwatt/cm2 at the sample. DAO was illuminated with the same light source for 30 s. Light-minus-dark difference spectra were collected after illumination. For CPH1-PHR-D393C and DAO, representative difference spectra of nine and six independent preparations were selected and averaged to a total of 9216 and 20,480 scans, respectively.
RESULTS
Changes in the Photoreaction of the D393C Mutant Compared with the Wild Type
In plant cryptochromes, a light-induced formation of the flavin neutral radical from the oxidized state was observed. The postulated proton donor Asp-393 in CPH1-PHR (Asp-396 in AtCRY1) was replaced with a cysteine by site-directed mutagenesis to investigate its influence on the photoresponse of the plant cryptochrome. The D393C mutant was expressed and purified similarly as the wild type. The absorption spectrum shows maxima at 364, 376, and 451 nm, close to those at 364, 375, and 449 nm of the wild type (39) (Fig. 2). The position of the absorption band in the UVA range is a sensitive indicator for the hydrogen bonding interactions in the flavin binding pocket (41, 42) and its polarity (43). The replacement of aspartic acid with cysteine did not lead to significant changes in the spectrum, which points to the presence of an aspartic acid instead of a charged aspartate in the wild type, in agreement with previous findings (23, 35).
FIGURE 2.
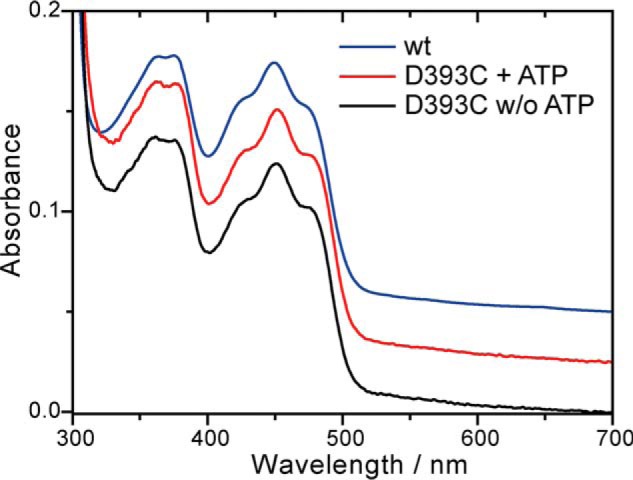
Absorption spectra of the D393C mutant of CPH1-PHR in the absence and the presence of ATP. Comparison to the spectrum of the wild type shows that, despite the mutation, the polarity and hydrogen bonding network of the flavin binding pocket is fully preserved.
Illumination with blue light induced only small changes in the spectrum of the D393C mutant. Even after prolonged illumination of 10 s, the changes were too small to allow an identification of the reaction product (Fig. 3A). In contrast, a significant amount of neutral radical was formed under identical conditions in the wild type (Fig. 3A). Therefore, the replacement of the aspartic acid led to the absence of any long-lived product in the photoreaction of the plant cryptochrome.
FIGURE 3.

Absorption difference spectra and decay kinetics after illumination of the D393C mutant and wild-type CPH1-PHR with blue light for the indicated time intervals. A and B, difference spectra were obtained in the absence (A) and presence (B) of ATP under otherwise identical conditions. Binding of ATP stabilizes a flavin anion radical as the photoproduct in the mutant, whereas the neutral radical is detected in the wild type under both conditions. w/o, without. C, representative decay kinetics of the radical photoproduct in the presence of ATP. The biexponential fit yields a main decay component with 90% amplitude with a time constant of 6100 s for the wild type and 300 s for the mutant. The neutral radical is significantly more stabilized by the protein environment than the anion radical. D, absorption difference spectra of the D393C mutant at different pH values. Formation of a flavin anion radical is observed independently of the pH value.
It has been observed that the presence of ATP stabilizes the flavin neutral radical in the plant cryptochrome (39, 44) and increases its yield (32). Therefore, the influence of ATP on the photoreaction of the mutant was investigated. The absorption spectrum was not significantly affected by the presence of ATP (Fig. 2). After illumination, the formation of a long-lived reaction product was observed (Fig. 3B). However, the spectral features differed from those of the neutral radical. The product is assigned to the flavin anion radical with maxima at 372, 400, and 511 nm because of the high similarity of its characteristic difference spectrum to those of photoreduced Drosophila cryptochrome and glucose oxidase (45). Therefore, in the presence of ATP, photoreduction of the D393C mutant of the plant cryptochrome leads to the same product as that found in animal type I cryptochromes, such as the Drosophila cryptochrome (45, 46), despite the low homology in amino acid sequence.
To determine whether the D393C replacement has an impact on the efficiency of radical formation or on the stability of the photoproduct, decay kinetics at 450 nm were recorded under identical conditions for the wild type and the D393C mutant in the presence of ATP (Fig. 3C). The conversion efficiency directly after illumination was determined on the basis of the similar absorption coefficients of the neutral radical and the anion radical at 450 nm (22). As a result, the efficiency was only moderately affected, with a loss of oxidized flavin of 39% in the wild type and of 26% in the mutant, respectively. The time constants of the decay were obtained by a biexponential fit and yielded 6100 s (90%) and 720 s (10%) for the wild type and 300 s (90%) and 3800 s (10%) for the D393C mutant. Accordingly, most of the radical product decayed ∼20 faster in the mutant compared with the wild type. In summary, the removal of the proton donor significantly decreased the radical lifetime but had a less pronounced effect on the efficiency of its formation. The stabilizing effect of ATP binding is independent of the presence of Asp-393.
Role of Cysteine in the Blockage of Proton Transfer
Cysteine does not act as a substitute for aspartic acid as the proton donor to flavin, despite its pKa of 8.1 in solution compared with that of the flavin radical of 8.3 (47). Therefore, the formation of the flavin anion radical in the mutant might either be the result of a blockage of proton transfer to flavin by the cysteine or of an increase in accessibility of the binding pocket to the basic bulk solution. The experiments were conducted at a pH of 7.9, which might be sufficient for a deprotonation of the flavin radical, considering a pKa in glucose oxidase of 7.5 (48). However, lowering of the external pH to 6.8 and 6.1 did not lead to a change in the major product of the reaction (Fig. 3D). Further experiments at even lower pH values were prevented by a strong increase in scattering upon illumination. We conclude that the formation of the flavin anion radical as the photoproduct is not a consequence of the basic pH in the bulk but, rather, of a blockage of proton transfer by cysteine.
Microsecond Lifetime of the Flavin Anion Radical in the Absence of ATP
Time-resolved UV-visible spectra of the D393C mutant were recorded to analyze the identity and stability of the photoproduct in the absence of ATP. The spectra show the presence of the anion radical at 2 μs after the laser pulse and its decay within 10 ms (Fig. 4A). A kinetic analysis revealed a heterogeneous decay that was represented by a biexponential function with time constants of 9 μs (40%) and 400 μs (60%) (Fig. 4B). Such structural heterogeneity has been found previously for plant cryptochromes, as reflected in the biphasic decay of the neutral radical (17, 39). The lifetime of the radical state in the D393C mutant is reduced by about 6 orders of magnitude compared with that of the wild type, with a time constant of decay of 200 s (39). Therefore, a blockage of proton transfer leads to a severe reduction of the radical lifetime. An additional effect on the efficiency of radical formation cannot be excluded.
FIGURE 4.
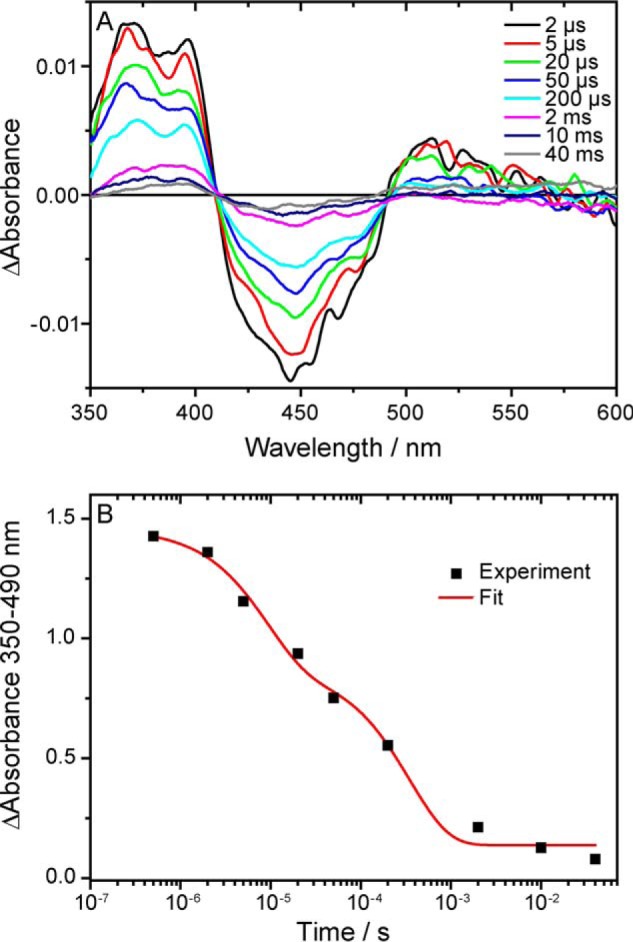
Time-resolved UV-visible absorption difference spectroscopy on the D393C mutant of CPH1-PHR in the absence of ATP. A, spectra were recorded at the indicated time points after the laser pulse and show the decay of the flavin anion radical within 10 ms. B, the integral difference absorbance in the spectral range of 350–490 nm was plotted against time on a logarithmic scale. A biexponential fit of the decay yields two time constants of 9 and 400 μs.
Preservation of Light-induced Conformational Changes in the D393C Mutant
To characterize the response of the protein moiety to light in the D393C mutant, FTIR difference spectroscopy was applied in the presence of ATP. Blue light induces changes in the vibrational signature that are detected as negative bands originating from the dark form and positive bands from the photoproduct. Prominent bands were recorded after illumination of the D393C mutant for 4 s (Fig. 5A). Negative bands at 1707 and 1692 cm−1 as well as at 1577 and 1547 cm−1 have been found previously in the difference spectrum of the wild type (23) and are correspondingly assigned to contain contributions from carbonyl and CN/CC stretches of oxidized flavin, respectively.
FIGURE 5.
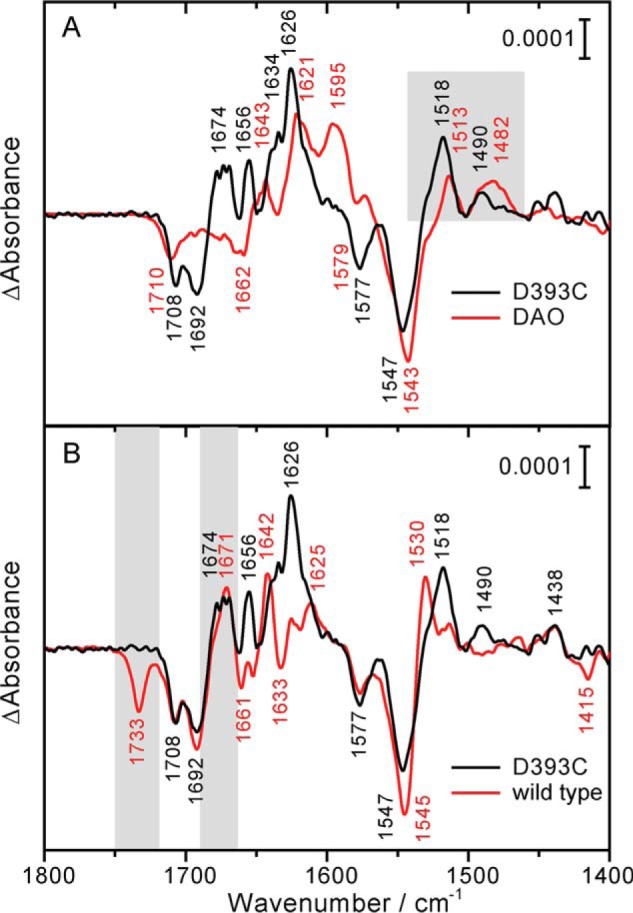
FTIR difference spectra after illumination of the D393C mutant of CPH1-PHR, of the wild type, and of DAO. A, difference spectrum of the D393C mutant. For comparison, DAO was photoreduced to the flavin anion radical and scaled to the area of the C4 = O band of oxidized flavin at 1708 cm−1. Similarities in band pattern are obtained from the comparison (highlighted in gray), including a marker band for the anion radical at 1490 and 1482 cm−1, respectively. B, difference spectra of the D393C mutant and the wild type (taken from Ref. 23). Contributions from the protein moiety are highlighted in gray. The mutation led to a loss of the band at 1733 cm−1, which was assigned to deprotonation of Asp-393 in the wild type. Light-induced secondary structural changes were preserved, as indicated by the positive band at near 1671 cm−1. Differences in the positive band pattern were caused by the formation of the flavin anion radical and neutral radical in the mutant and the wild type, respectively.
Positive bands represent the formation of the flavin anion radical. For an unequivocal assignment, DAO was used as a reference and photoreduced in the presence of the electron donor 2-mercaptoethanol and in the absence of oxygen at pH 8. Under similar conditions, DAO has been found by UV-visible spectroscopy to selectively form the flavin anion radical (48). The difference spectrum of DAO shows a positive band at 1482 cm−1 that is not present in the neutral radical (23) or fully reduced state (49) of flavoproteins. Therefore, in combination with the band at 1513 cm−1, this signal is considered to represent a marker band of the anion radical, in agreement with previous findings on the N392C mutant of bacterial DASH cryptochrome (50) and a trapping of the anion radical in (6–4) photolyase at 200 K (51). The D393C mutant of CPH1-PHR shows similar characteristic bands at 1490 and 1518 cm−1 (Fig. 5A), which confirms the formation of the anion radical in this plant cryptochrome. A further comparison of the infrared spectra of the D393C mutant and of DAO reveals a similar band pattern. Bands at 1656, 1634, and 1626 cm−1 are detected in the D393C mutant that correspond to those at 1643, 1621, and 1595 cm−1 in the spectrum of DAO, although significantly downshifted in frequency. The presence of similar bands in the spectra of two completely different flavoproteins implies that they originate from the flavin chromophore.
After the identification of all contributions from flavin to the difference spectrum of the D393C mutant, a single positive band at 1674 cm−1 remains. This band is accordingly assigned to the protein moiety of the cryptochrome. The amplitude of the signal argues against a contribution by a side chain. The spectral region at around 1674 cm−1 is associated (52) and strongly correlated (53) with the presence of turn elements, which points to some light-induced rearrangement in the tertiary structure of the PHR. The same band has been detected previously and similarly assigned in the spectrum of the wild type (Fig. 5B) (23). There its assignment was supported by including the analysis of a sample, in which this light-induced conformational change was suppressed by lyophilizing and redissolving the PHR. It can be concluded that the D393C mutation does not interfere with this protein response despite the change in the radical photoproduct and its lifetime. In contrast, the prominent band found at 1733 cm−1 in the spectrum of the wild type is not detected in the mutant (Fig. 5B). This band has been assigned to the deprotonation of Asp-393 (23, 35), which is further confirmed by comparison with the D393C mutant.
DISCUSSION
The Role of the Flavin Anion Radical in Plant and Animal Type I Cryptochromes
The decisive steps of signal generation in the plant cryptochrome between the initial ultrafast electron transfer to FAD (14) and the structural change in the CCT (24, 25) are unclear. In particular, the role of the proton transfer to the flavin radical needs to be elucidated considering the fact that the aspartic acid close to flavin is exclusively present in the plant cryptochrome. The loss of the IR signal of the aspartic acid in the D393C mutant further supports its role as proton donor in the wild type (Fig. 5B).
It has been postulated that the secondary structural change in a turn motif upon photoreduction is part of the signal generation in the plant cryptochrome (23). Therefore, the corresponding band near 1671 cm−1 might be considered as a marker band for signaling. This view is supported by the finding that, in the non-photosensory DAO, this structural change is absent (Fig. 5A). In contrast, the signature of this structural change is preserved in the mutant D393C (Fig. 5B). Accordingly, protonation of the anion radical might not be required for the signal generation in the plant cryptochrome. It is likely that further light-induced conformational changes accompany signaling, which have not been analyzed here with respect to their preservation upon anion radical formation. The corresponding signals might overlap with those from the chromophore, or these processes might not lead to prominent contributions in the infrared spectral range. Particularly, the impact of the D393C replacement on the light-induced structural change of the CCT was not studied because purification of full-length CPH1 comprising its exceptionally long and unconserved CCT of ∼500 amino acids (54) has not been successful to date.
A prominent role of the anion radical in the mechanism of the plant cryptochrome has been proposed before. It has been suggested to act as the dark, resting form of the receptor (22). However, the lifetime of the anion radical in the plant cryptochrome is limited to a few microseconds in vitro (15, 16) because of the fast protonation by the inherent proton donor aspartic acid. This time interval is too short to allow the anion radical to absorb sufficient light for generation of a signal, which argues against a role of the anion radical as a dark form of the receptor. In contrast, the proposal that formation of the anion radical is the main initiator of signaling would reconcile a fundamental difference between the photoactivation of plant and animal type I cryptochromes. In the latter, the neutral radical state is not formed at all, leading to a model in which photoreduction of the oxidized state to the anion radical or even further, to the anionic fully reduced state, drives C-terminal conformational changes (45, 55). However, the inversion of the experiment demonstrated here, i.e. the conversion of animal type I cryptochromes into plant cryptochromes by a corresponding cysteine-to-aspartic acid mutation, was unsuccessful because it did not lead to a light-induced formation of the neutral radical state (46). The latter experiment highlights the fundamental differences in the binding pocket of flavin between animal type I and plant cryptochromes. More precisely, the aspartic acid cannot be stabilized in animal type I cryptochromes, leading to a presence of an aspartate in the dark without any ability to donate the proton for the neutral flavin radical upon illumination.
The Role of Proton Transfer and Formation of the Neutral Radical State
The neutral radical has been demonstrated to represent the signaling state of the plant cryptochrome by comparing the attenuating effects of green light in vitro and in vivo (18, 19). A further important role of proton transfer for signaling is revealed here (Fig. 6) besides the roles mentioned in the Introduction. A strong difference in the stability of the anion and neutral radical was observed in CPH1-PHR of about 6 orders of magnitude in the lifetime. Therefore, proton transfer stabilizes the radical state for the time range later than a few milliseconds even under aerobic conditions. This time range is crucial for the signaling process because of the unfolding of the CCT with a time constant of 400 ms (25). It is conceivable, in general, that such a conformational change occurs after relaxation of the initiating photochemical process in the cofactor. However, the in vivo recordings of AtCRY1 and AtCRY2 point to a connection between the cofactor state and the protein response because of the similar lifetime of several minutes of the radical state in insect cells and the signaling process in planta (20).
FIGURE 6.
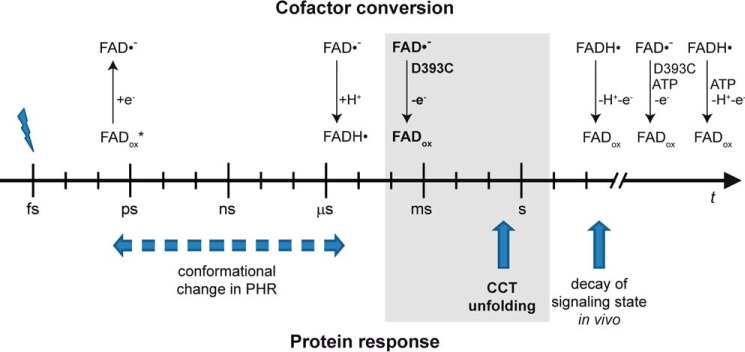
Time line of events in the plant cryptochrome after illumination with blue light. The photochemical processes of the cofactor FAD and subsequent responses of the protein are separated. Only the longest time constants determined for these processes are depicted for clarity. Protonation of the anion radical is an intrinsic process of the receptor that prolongs the lifetime of the radical by 6 orders of magnitude. When this proton transfer is blocked, the lifetime is not sufficient anymore to drive CCT unfolding (highlighted in gray). Alternatively, binding of ATP may stabilize the radical state depending on its current cellular concentration.
Alternatively, the radical state can be stabilized by the presence of ATP (Fig. 3), the presence of reductants (17), or the absence of oxygen (39). The main advantage of proton transfer over the latter effects is that it is an intrinsic property of the plant cryptochrome that is guaranteed by the aspartic acid close to the flavin and is independent of the current conditions in the cell in vivo. Taken together, the extraordinary stability of the neutral radical state under aerobic conditions and its photoreversibility to the oxidized state are two reasons to rationalize the presence of the conserved aspartic acid in the plant cryptochrome even when the anion radical might be sufficient to produce the signal.
Effect of ATP on the Photoreaction of the Mutant
ATP binding leads to an increased yield (32) and stability (39, 44) of the radical photoproduct. It is likely that this effect is also of physiological significance because of the high binding affinity determined in vitro (10). This interpretation is complicated by the fact that the lifetimes of the radical and the signaling state in vivo of a few minutes for AtCRY1 (20) only match well with that of the isolated receptor in the absence of ATP (44) (Fig. 6).
It is evident from the experiments on the D393C mutant that the two effects of ATP binding need to be clearly separated in mechanism. Recent experimental and theoretical studies on AtCRY1 rationalized the higher yield of radical formation in the presence of ATP by an increase in the fraction of protonated Asp-396 in the dark (32, 56). Simulations suggest that Asp-396 then fully adopts the conformation in which it is hydrogen-bonded to the backbone of Met-381 (Leu-378 in CPH1) and allows for an efficient electron transfer pathway from Trp-400 to the flavin (56). In the case of the mutant, cysteine cannot simply attain the same hydrogen-bonded conformation because of the difference in structure. Further theoretical analysis is necessary to investigate whether the small effect of the mutation on the formation efficiency (Fig. 3C) can be reconciled with this molecular model of the ATP effect. Concerning the stabilizing role of ATP, it is demonstrated here that this effect is not dependent on the presence or protonation state of Asp-396/Asp-393. More specifically, the stabilizing effect cannot be linked to the presence of a charged aspartate after illumination or to the accessibility of an external base catalyst. Further unlikely candidates are a change in the redox potential or in the accessibility to oxygen. Its extent seems to be mainly dependent on the final protonation state of the flavin radical.
The Proton Transfer Pathway Is Different from Those in Other Flavoproteins
Formation of the neutral radical state is observed in many flavoproteins (48) in the absence of any amino acid identified to act as a proton donor. This proton transfer is necessary for the photoreduction of photolyases from the oxidized to the fully reduced state as well without stabilization of the neutral radical state (57, 58). Most likely, proton transfer is mediated by bulk water and transferred via structural water and hydrogen bonding pathways to flavin upon photoreduction. This unspecific pathway seems to be slow because, in photolyase from E. coli, protonation is not observed within hundreds of microseconds after anion radical formation (59). A kinetic barrier has been identified for the respective deprotonation step from the neutral radical to the oxidized state, which can be lowered by the N386D mutation close to flavin (60). A role of the aspartate as a proton shuttle in this photolyase mutant has accordingly been suggested.
Aspartic acid in the plant cryptochrome, however, acts as an efficient and fast donor within a few microseconds (15, 16) and not merely as a shuttle for protons from the bulk (23). Its replacement with cysteine efficiently blocks proton transfer to the N(5) position of flavin, similar to the effect of the N392C mutant in the bacterial DASH cryptochrome (50). In contrast, the conservative mutation of aspartic acid to asparagine did not interfere with the unspecific pathway from the outside (36). Most probably, the bifurcated hydrogen bonding ability of asparagine in other cryptochromes as well as in cyclobutane pyrimidine dimer and (6-4) photolyases as opposed to the unidirectional hydrogen bonding of cysteine in the D393C mutant and animal type I cryptochrome makes the decisive difference for the shuttling of protons to flavin. In summary, cysteine acts as a gatekeeper for proton transfer irrespective of the general tendency of the flavoprotein to stabilize either the neutral or anion radical state.
Conclusions
Cryptochromes are a very diverse protein family for which new members with photosensory function are constantly being uncovered. It is striking that only members of the plant cryptochrome subfamily carry an aspartic acid close to flavin with its intrinsic property of proton donation. A role of the flavin neutral radical in these cryptochromes as initiator of signaling is extended by our finding that the initially formed anion radical is capable of inducing the same conformational change in the PHR domain as the neutral radical. Despite the clear differences in the flavin binding pocket between plant and animal type I cryptochromes, this finding might point to a common origin of the signaling process. However, we have to conclude from the timeline of events that the formation of the anion radical in the plant cryptochrome is not sufficient to drive signaling because it decays before the onset of the response of the CCT (Fig. 6). Therefore, the lifetime of the radical is prolonged by the intrinsic proton transfer as one prerequisite for the functionality of this blue light sensor.
Acknowledgments
We thank Joachim Heberle and Thomas Hellweg for support, Dominik Immeln for performing the mutagenesis, and Meike Spexard for assistance with the expression.
This work was supported by Deutsche Forschungsgemeinschaft Grant KO3580/2-1. Part of these results were presented at the 16th International Congress on Photobiology held in Córdoba, Argentina, in 2014.
- PHR
- photolyase homology region
- CCT
- C-terminal extension
- OD
- optical density
- DAO
- d-amino acid oxidase.
REFERENCES
- 1. Chaves I., Pokorny R., Byrdin M., Hoang N., Ritz T., Brettel K., Essen L. O., van der Horst G. T., Batschauer A., Ahmad M. (2011) The cryptochromes: blue light photoreceptors in plants and animals. Annu. Rev. Plant Biol. 62, 335–364 [DOI] [PubMed] [Google Scholar]
- 2. Ahmad M., Lin C., Cashmore A. R. (1995) Mutations throughout an Arabidopsis blue-light photoreceptor impair blue-light-responsive anthocyanin accumulation and inhibition of hypocotyl elongation. Plant J. 8, 653–658 [DOI] [PubMed] [Google Scholar]
- 3. Lin C., Yang H., Guo H., Mockler T., Chen J., Cashmore A. R. (1998) Enhancement of blue-light sensitivity of Arabidopsis seedlings by a blue light receptor cryptochrome 2. Proc. Natl. Acad. Sci. U.S.A. 95, 2686–2690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hughes R. M., Vrana J. D., Song J., Tucker C. L. (2012) Light-dependent, dark-promoted interaction between Arabidopsis cryptochrome 1 and phytochrome B proteins. J. Biol. Chem. 287, 22165–22172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Somers D. E., Devlin P. F., Kay S. A. (1998) Phytochromes and cryptochromes in the entrainment of the Arabidopsis circadian clock. Science 282, 1488–1490 [DOI] [PubMed] [Google Scholar]
- 6. Guo H., Yang H., Mockler T. C., Lin C. (1998) Regulation of flowering time by Arabidopsis photoreceptors. Science 279, 1360–1363 [DOI] [PubMed] [Google Scholar]
- 7. Mao J., Zhang Y. C., Sang Y., Li Q. H., Yang H. Q. (2005) A role for Arabidopsis cryptochromes and COP1 in the regulation of stomatal opening. Proc. Natl. Acad. Sci. U.S.A. 102, 12270–12275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Danon A., Coll N. S., Apel K. (2006) Cryptochrome-1-dependent execution of programmed cell death induced by singlet oxygen in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 103, 17036–17041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lin C., Robertson D. E., Ahmad M., Raibekas A. A., Jorns M. S., Dutton P. L., Cashmore A. R. (1995) Association of flavin adenine dinucleotide with the Arabidopsis blue light receptor CRY1. Science 269, 968–970 [DOI] [PubMed] [Google Scholar]
- 10. Bouly J. P., Giovani B., Djamei A., Mueller M., Zeugner A., Dudkin E. A., Batschauer A., Ahmad M. (2003) Novel ATP-binding and autophosphorylation activity associated with Arabidopsis and human cryptochrome-1. Eur. J. Biochem. 270, 2921–2928 [DOI] [PubMed] [Google Scholar]
- 11. Brautigam C. A., Smith B. S., Ma Z., Palnitkar M., Tomchick D. R., Machius M., Deisenhofer J. (2004) Structure of the photolyase-like domain of cryptochrome 1 from Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 101, 12142–12147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yang H. Q., Wu Y. J., Tang R. H., Liu D., Liu Y., Cashmore A. R. (2000) The C termini of Arabidopsis cryptochromes mediate a constitutive light response. Cell 103, 815–827 [DOI] [PubMed] [Google Scholar]
- 13. Yu X., Shalitin D., Liu X., Maymon M., Klejnot J., Yang H., Lopez J., Zhao X., Bendehakkalu K. T., Lin C. (2007) Derepression of the NC80 motif is critical for the photoactivation of Arabidopsis CRY2. Proc. Natl. Acad. Sci. U.S.A. 104, 7289–7294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Immeln D., Weigel A., Kottke T., Pérez Lustres J. L. (2012) Primary events in the blue light sensor plant cryptochrome: intraprotein electron and proton transfer revealed by femtosecond spectroscopy. J. Am. Chem. Soc. 134, 12536–12546 [DOI] [PubMed] [Google Scholar]
- 15. Langenbacher T., Immeln D., Dick B., Kottke T. (2009) Microsecond light-induced proton transfer to flavin in the blue light sensor plant cryptochrome. J. Am. Chem. Soc. 131, 14274–14280 [DOI] [PubMed] [Google Scholar]
- 16. Maeda K., Robinson A. J., Henbest K. B., Hogben H. J., Biskup T., Ahmad M., Schleicher E., Weber S., Timmel C. R., Hore P. J. (2012) Magnetically sensitive light-induced reactions in cryptochrome are consistent with its proposed role as a magnetoreceptor. Proc. Natl. Acad. Sci. U.S.A. 109, 4774–4779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giovani B., Byrdin M., Ahmad M., Brettel K. (2003) Light-induced electron transfer in a cryptochrome blue-light photoreceptor. Nat. Struct. Biol. 10, 489–490 [DOI] [PubMed] [Google Scholar]
- 18. Banerjee R., Schleicher E., Meier S., Viana R. M., Pokorny R., Ahmad M., Bittl R., Batschauer A. (2007) The signaling state of Arabidopsis cryptochrome 2 contains flavin semiquinone. J. Biol. Chem. 282, 14916–14922 [DOI] [PubMed] [Google Scholar]
- 19. Bouly J. P., Schleicher E., Dionisio-Sese M., Vandenbussche F., Van Der Straeten D., Bakrim N., Meier S., Batschauer A., Galland P., Bittl R., Ahmad M. (2007) Cryptochrome blue light photoreceptors are activated through interconversion of flavin redox states. J. Biol. Chem. 282, 9383–9391 [DOI] [PubMed] [Google Scholar]
- 20. Herbel V., Orth C., Wenzel R., Ahmad M., Bittl R., Batschauer A. (2013) Lifetimes of Arabidopsis cryptochrome signaling states in vivo. Plant J. 74, 583–592 [DOI] [PubMed] [Google Scholar]
- 21. Li X., Wang Q., Yu X., Liu H., Yang H., Zhao C., Liu X., Tan C., Klejnot J., Zhong D., Lin C. (2011) Arabidopsis cryptochrome 2 (CRY2) functions by the photoactivation mechanism distinct from the tryptophan (Trp) triad-dependent photoreduction. Proc. Natl. Acad. Sci. U.S.A. 108, 20844–20849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu B., Liu H., Zhong D., Lin C. (2010) Searching for a photocycle of the cryptochrome photoreceptors. Curr. Opin. Plant Biol. 13, 578–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Immeln D., Pokorny R., Herman E., Moldt J., Batschauer A., Kottke T. (2010) Photoreaction of plant and DASH Cryptochromes probed by infrared spectroscopy: the neutral radical state of flavoproteins. J. Phys. Chem. B 114, 17155–17161 [DOI] [PubMed] [Google Scholar]
- 24. Partch C. L., Clarkson M. W., Ozgür S., Lee A. L., Sancar A. (2005) Role of structural plasticity in signal transduction by the cryptochrome blue-light photoreceptor. Biochemistry 44, 3795–3805 [DOI] [PubMed] [Google Scholar]
- 25. Kondoh M., Shiraishi C., Müller P., Ahmad M., Hitomi K., Getzoff E. D., Terazima M. (2011) Light-induced conformational changes in full-length Arabidopsis thaliana cryptochrome. J. Mol. Biol. 413, 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Biskup T., Hitomi K., Getzoff E. D., Krapf S., Koslowski T., Schleicher E., Weber S. (2011) Unexpected electron transfer in cryptochrome identified by time-resolved EPR spectroscopy. Angew. Chem. Int. Ed. Engl. 50, 12647–12651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biskup T., Paulus B., Okafuji A., Hitomi K., Getzoff E. D., Weber S., Schleicher E. (2013) Variable electron transfer pathways in an amphibian cryptochrome: tryptophan versus tyrosine-based radical pairs. J. Biol. Chem. 288, 9249–9260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Brazard J., Usman A., Lacombat F., Ley C., Martin M. M., Plaza P., Mony L., Heijde M., Zabulon G., Bowler C. (2010) Spectro-temporal characterization of the photoactivation mechanism of two new oxidized cryptochrome/photolyase photoreceptors. J. Am. Chem. Soc. 132, 4935–4945 [DOI] [PubMed] [Google Scholar]
- 29. Weber S., Biskup T., Okafuji A., Marino A. R., Berthold T., Link G., Hitomi K., Getzoff E. D., Schleicher E., Norris J. R. (2010) Origin of light-induced spin-correlated radical pairs in cryptochrome. J. Phys. Chem. B 114, 14745–14754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zeugner A., Byrdin M., Bouly J. P., Bakrim N., Giovani B., Brettel K., Ahmad M. (2005) Light-induced electron transfer in Arabidopsis cryptochrome-1 correlates with in vivo function. J. Biol. Chem. 280, 19437–19440 [DOI] [PubMed] [Google Scholar]
- 31. Solov'yov I. A., Domratcheva T., Moughal Shahi A. R., Schulten K. (2012) Decrypting cryptochrome: revealing the molecular identity of the photoactivation reaction. J. Am. Chem. Soc. 134, 18046–18052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Müller P., Bouly J. P., Hitomi K., Balland V., Getzoff E. D., Ritz T., Brettel K. (2014) ATP binding turns plant cryptochrome into an efficient natural photoswitch. Sci. Rep. 4, 5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sellaro R., Crepy M., Trupkin S. A., Karayekov E., Buchovsky A. S., Rossi C., Casal J. J. (2010) Cryptochrome as a sensor of the blue/green ratio of natural radiation in Arabidopsis. Plant Physiol. 154, 401–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Müller P., Ahmad M. (2011) Light-activated cryptochrome reacts with molecular oxygen to form a flavin-superoxide radical pair consistent with magnetoreception. J. Biol. Chem. 286, 21033–21040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Kottke T., Batschauer A., Ahmad M., Heberle J. (2006) Blue-light-induced changes in Arabidopsis cryptochrome 1 probed by FTIR difference spectroscopy. Biochemistry 45, 2472–2479 [DOI] [PubMed] [Google Scholar]
- 36. Burney S., Wenzel R., Kottke T., Roussel T., Hoang N., Bouly J. P., Bittl R., Heberle J., Ahmad M. (2012) Single amino acid substitution reveals latent photolyase activity in Arabidopsis cry1. Angew. Chem. Int. Ed. Engl. 51, 9356–9360 [DOI] [PubMed] [Google Scholar]
- 37. Balland V., Byrdin M., Eker A. P., Ahmad M., Brettel K. (2009) What makes the difference between a cryptochrome and DNA photolyase? A spectroelectrochemical comparison of the flavin redox transitions. J. Am. Chem. Soc. 131, 426–427 [DOI] [PubMed] [Google Scholar]
- 38. Forbes-Stovall J., Howton J., Young M., Davis G., Chandler T., Kessler B., Rinehart C. A., Jacobshagen S. (2014) Chlamydomonas reinhardtii strain CC-124 is highly sensitive to blue light in addition to green and red light in resetting its circadian clock, with the blue-light photoreceptor plant cryptochrome likely acting as negative modulator. Plant Physiol. Biochem. 75, 14–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Immeln D., Schlesinger R., Heberle J., Kottke T. (2007) Blue light induces radical formation and autophosphorylation in the light-sensitive domain of Chlamydomonas cryptochrome. J. Biol. Chem. 282, 21720–21728 [DOI] [PubMed] [Google Scholar]
- 40. Bauer C., Rabl C. R., Heberle J., Kottke T. (2011) Indication for a radical intermediate preceding the signaling state in the LOV domain photocycle. Photochem. Photobiol. 87, 548–553 [DOI] [PubMed] [Google Scholar]
- 41. Salzmann S., Tatchen J., Marian C. M. (2008) The photophysics of flavins: What makes the difference between gas phase and aqueous solution? J. Photochem. Photobiol. A 198, 221–231 [Google Scholar]
- 42. Kotaki A., Naoi M., Yagi K. (1970) Effect of proton donors on the absorption spectrum of flavin compounds in apolar media. J. Biochem. 68, 287–292 [DOI] [PubMed] [Google Scholar]
- 43. Raffelberg S., Gutt A., Gärtner W., Mandalari C., Abbruzzetti S., Viappiani C., Losi A. (2013) The amino acids surrounding the flavin 7a-methyl group determine the UVA spectral features of a LOV protein. Biol. Chem. 394, 1517–1528 [DOI] [PubMed] [Google Scholar]
- 44. Burney S., Hoang N., Caruso M., Dudkin E. A., Ahmad M., Bouly J. P. (2009) Conformational change induced by ATP binding correlates with enhanced biological function of Arabidopsis cryptochrome. FEBS Lett. 583, 1427–1433 [DOI] [PubMed] [Google Scholar]
- 45. Berndt A., Kottke T., Breitkreuz H., Dvorsky R., Hennig S., Alexander M., Wolf E. (2007) A novel photoreaction mechanism for the circadian blue light photoreceptor Drosophila cryptochrome. J. Biol. Chem. 282, 13011–13021 [DOI] [PubMed] [Google Scholar]
- 46. Oztürk N., Song S. H., Selby C. P., Sancar A. (2008) Animal type 1 cryptochromes. Analysis of the redox state of the flavin cofactor by site-directed mutagenesis. J. Biol. Chem. 283, 3256–3263 [DOI] [PubMed] [Google Scholar]
- 47. Müller F. (1987) Flavin radicals: chemistry and biochemistry. Free Radic. Biol. Med. 3, 215–230 [DOI] [PubMed] [Google Scholar]
- 48. Massey V., Palmer G. (1966) On the existence of spectrally distinct classes of flavoprotein semiquinones: a new method for the quantitative production of flavoprotein semiquinones. Biochemistry 5, 3181–3189 [DOI] [PubMed] [Google Scholar]
- 49. Wille G., Ritter M., Friedemann R., Mäntele W., Hübner G. (2003) Redox-triggered FTIR difference spectra of FAD in aqueous solution and bound to flavoproteins. Biochemistry 42, 14814–14821 [DOI] [PubMed] [Google Scholar]
- 50. Iwata T., Zhang Y., Hitomi K., Getzoff E. D., Kandori H. (2010) Key dynamics of conserved asparagine in a cryptochrome/photolyase family protein by Fourier transform infrared spectroscopy. Biochemistry 49, 8882–8891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamada D., Zhang Y., Iwata T., Hitomi K., Getzoff E. D., Kandori H. (2012) Fourier-transform infrared study of the photoactivation process of Xenopus (6-4) photolyase. Biochemistry 51, 5774–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Barth A. (2007) Infrared spectroscopy of proteins. Biochim. Biophys. Acta 1767, 1073–1101 [DOI] [PubMed] [Google Scholar]
- 53. Goormaghtigh E., Ruysschaert J. M., Raussens V. (2006) Evaluation of the information content in infrared spectra for protein secondary structure determination. Biophys. J. 90, 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Reisdorph N. A., Small G. D. (2004) The CPH1 gene of Chlamydomonas reinhardtii encodes two forms of cryptochrome whose levels are controlled by light-induced proteolysis. Plant Physiol. 134, 1546–1554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Vaidya A. T., Top D., Manahan C. C., Tokuda J. M., Zhang S., Pollack L., Young M. W., Crane B. R. (2013) Flavin reduction activates Drosophila cryptochrome. Proc. Natl. Acad. Sci. U.S.A. 110, 20455–20460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Cailliez F., Müller P., Gallois M., de la Lande A. (2014) ATP binding and aspartate protonation enhance photoinduced electron transfer in plant cryptochrome. J. Am. Chem. Soc. 136, 12974–12986 [DOI] [PubMed] [Google Scholar]
- 57. Jorns M. S., Wang B. Y., Jordan S. P., Chanderkar L. P. (1990) Chromophore function and interaction in Escherichia coli DNA photolyase: reconstitution of the apoenzyme with pterin and/or flavin derivatives. Biochemistry 29, 552–561 [DOI] [PubMed] [Google Scholar]
- 58. Schleicher E., Hitomi K., Kay C. W., Getzoff E. D., Todo T., Weber S. (2007) Electron nuclear double resonance differentiates complementary roles for active site histidines in (6-4) photolyase. J. Biol. Chem. 282, 4738–4747 [DOI] [PubMed] [Google Scholar]
- 59. Henbest K. B., Maeda K., Hore P. J., Joshi M., Bacher A., Bittl R., Weber S., Timmel C. R., Schleicher E. (2008) Magnetic-field effect on the photoactivation reaction of Escherichia coli DNA photolyase. Proc. Natl. Acad. Sci. U.S.A. 105, 14395–14399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Damiani M. J., Nostedt J. J., O'Neill M. A. (2011) Impact of the N5-proximal Asn on the thermodynamic and kinetic stability of the semiquinone radical in photolyase. J. Biol. Chem. 286, 4382–4391 [DOI] [PMC free article] [PubMed] [Google Scholar]