

Background: The glycolytic enzyme GAPDH is a non-canonical RNA-binding protein.
Results: A dimer interface mutation impairs the two-step binding of GAPDH to AU-rich elements from TNF-α mRNA and leads to RNA structural changes.
Conclusion: Dimer and tetramer interface residues are important for GAPDH-RNA binding.
Significance: We propose a novel model for GAPDH binding to AU-rich RNA via the oligomeric interfaces.
Keywords: Conformational Change, Crystallography, Fluorescence Anisotropy, Fluorescence Resonance Energy Transfer (FRET), Hydrogen Exchange Mass Spectrometry, RNA, Tumor Necrosis Factor (TNF), AU-rich Elements, GAPDH, Oligomerization
Abstract
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) is an enzyme best known for its role in glycolysis. However, extra-glycolytic functions of GAPDH have been described, including regulation of protein expression via RNA binding. GAPDH binds to numerous adenine-uridine rich elements (AREs) from various mRNA 3′-untranslated regions in vitro and in vivo despite its lack of a canonical RNA binding motif. How GAPDH binds to these AREs is still unknown. Here we discovered that GAPDH binds with high affinity to the core ARE from tumor necrosis factor-α mRNA via a two-step binding mechanism. We demonstrate that a mutation at the GAPDH dimer interface impairs formation of the second RNA-GAPDH complex and leads to changes in the RNA structure. We investigated the effect of this interfacial mutation on GAPDH oligomerization by crystallography, small-angle x-ray scattering, nano-electrospray ionization native mass spectrometry, and hydrogen-deuterium exchange mass spectrometry. We show that the mutation does not significantly affect GAPDH tetramerization as previously proposed. Instead, the mutation promotes short-range and long-range dynamic changes in regions located at the dimer and tetramer interface and in the NAD+ binding site. These dynamic changes are localized along the P axis of the GAPDH tetramer, suggesting that this region is important for RNA binding. Based on our results, we propose a model for sequential GAPDH binding to RNA via residues located at the dimer and tetramer interfaces.
Introduction
To respond to external stimuli, cells rapidly alter protein expression via several mechanisms acting at the levels of transcription, mRNA degradation, translation, and protein degradation. Posttranscriptional regulation of mRNA decay rates offers multiple points of control in regard to cellular homeostasis, including viral defense, gene expression flexibility, and control of aberrant mRNA biogenesis (1–3). For many regulatory genes, a discrete family of cis-acting sequences termed AU-rich elements (AREs),6 located in the mRNA 3′-UTRs, recruit trans-acting factors or ARE-binding proteins that alter mRNA decay rates in response to specific stimuli (4, 5). AREs generally span 50–150 nucleotides and are characterized by the presence of multiple copies of the AUUUA pentanucleotide as well as poly-A or poly-U regions (6). Several ARE-binding proteins have been identified, including the Hu family of proteins (7, 8), tristetraprolin (9), AUF1 (10–12), and the glycolytic enzyme GAPDH (13).
GAPDH is a homotetramer best described as a dimer of dimers with known regions of dimerization (14). Each 335-amino acid polypeptide contains two domains: an NAD+ binding/Rossmann-fold domain (residues 1–150 and 317–335) and a catalytic domain (residues 151–316). GAPDH is a multifunctional enzyme implicated in a variety of cellular processes including nucleic acid binding (13, 15–19), DNA replication and repair (20), nuclear tRNA transport (18), apoptosis (21, 22), microtubule bundling (23), membrane fusion (24), neurodegenerative disorders (25, 26), and heme incorporation (27). The mechanism by which GAPDH switches among these various functions in the cell is unknown but may depend on the cell status and GAPDH posttranslational modifications (for reviews, see Refs. 28 and 29).
GAPDH binding to AREs within the 3′-UTR of various mRNAs has been shown in vitro and in vivo (13, 30–37). GAPDH plays key roles in vascular homeostasis and cancer cell proliferation by posttranscriptionally regulating expression of endothelin-1 and colony-stimulating factor-1 via binding to the AREs of their mRNA 3′-UTR (33, 36). The lack of a canonical RNA binding sequence (38) raises the question of the exact localization of the RNA binding site(s). Earlier studies suggested that the RNA binding site may be localized to the positively charged substrate binding groove (39) or the N-terminal domain of GAPDH (13). The latter hypothesis was supported by results showing inhibition of RNA binding by ATP, NAD+, and NADH (13, 31). However, other studies have suggested that regions outside the Rossmann fold may also contribute to RNA binding and sequence specificity (30, 40). Clearly more studies are needed to elucidate the GAPDH binding site for RNA.
Here we characterize for the first time GAPDH binding to an RNA oligonucleotide corresponding to the core ARE region from the TNF-α mRNA 3′-UTR via electrophoretic mobility shift assay (EMSA), fluorescence anisotropy, and fluorescence resonance energy transfer (FRET) studies. We identified a mutation at the GAPDH dimer interface that impacts complex formation with RNA and the global structure of the RNA ligand. We further characterized the effect of the mutation by x-ray crystallography, biophysical assays, and mass spectrometry. We show that the mutation does not affect GAPDH oligomerization but promotes dynamic changes within protein regions clustered along the P axis of the GAPDH tetramer. Our results allow us to propose a new model for RNA binding to GAPDH.
EXPERIMENTAL PROCEDURES
Materials
2′-OH-de-protected and purified RNA oligonucleotides were purchased from IDT or Dharmacon Research. Oligonucleotides incorporating 5′- or 3′-terminal cyanine 3 (Cy3) or fluorescein (Fl) dyes are indicated by relevant prefixes or suffixes, respectively. Optima (LC/MS) grade water and acetonitrile were obtained from Fisher, and formic acid was obtained from Sigma. D2O, DCl, and NaOD were obtained from Cambridge Isotopes.
Molecular Biology, Protein Expression, and Purification
Untagged wild-type human GAPDH cDNA (Gene accession number NM_002046) was cloned into the pET21a vector. Mutants were generated via site-directed mutagenesis (Stratagene). For the T229K GAPDH mutant, the following primer pairs were used: forward primer (5′-CTGAACGGGAAGCTCAAAGGCATG-3′) and reverse primer (5′-CATGCCTTTGAGCTTCCCGTTCAG-3′). The mutation was confirmed by DNA sequencing (Genewiz).
Wild-type and mutant GAPDH proteins were expressed in BL21(DE3)pRIPL Escherichia coli cells (Life Technologies). Cells were cultured overnight at 37 °C. Protein expression was induced with 0.4 mm isopropyl 1-thio-β-d-galactopyranoside when the A600 reached 0.6–0.8. Cells were grown for 4–5 h at 37 °C, then pelleted and frozen at −80 °C until further use. To purify GAPDH proteins, cell pellets were lysed in buffer A (20 mm Tris-HCl, pH 8.0, 30 mm NaCl, 1 mm DTT), 15 units/ml benzonase (Sigma), 0.5 mg/ml lysozyme, protease inhibitor mixture (Roche Applied Science) via sonication. The first chromatography step was an anion-exchange column (GE Healthcare) equilibrated in buffer A. GAPDH did not bind to the column and was directly loaded onto an Affi-Blue-Sepharose affinity column (Bio-Rad) equilibrated in buffer A. GAPDH was eluted in 100% buffer B (20 mm Tris-HCl pH 8.0, 1.4 m NaCl, 1 mm DTT). SDS-PAGE was used to analyze the purity of the elution fractions. Fractions containing GAPDH were pooled, dialyzed in buffer A using 10-kDa Amicon concentrators (Millipore), aliquoted, and stored at −80 °C. The protein was further purified via size-exclusion chromatography on a Superdex 200 column (GE Healthcare) equilibrated in buffer C (20 mm Tris-HCl, pH 8.0, 30 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine). Fractions containing GAPDH were pooled and concentrated to ∼10 mg/ml, aliquoted, and stored at −80 °C. The final protein yields were ∼70 mg and ∼20 mg per liter of cell culture for wild-type and T229K GAPDH, respectively.
RNA Electrophoretic Mobility Shift Assays
For our experiments, we used the AU-rich RNA derived from TNF-α (ARE38; 5′-GUGAUUAUUUAUUAUUUAUUUAUUAUUUAUUUAUUUAG-3′) and control RNA derived from rabbit β-globin (Rβ31; 5′-UGGCCAAUGCCCUGGCUCACAAAUACCACUG-3′). Both oligonucleotides were resuspended in 10 mm Tris-HCl, pH 8.0, before use. RNA concentrations and efficiency of fluorophore-labeling were assessed by absorbance spectroscopy, as described (41). 5′-Hydroxyl RNA oligos were radiolabeled to a specific activity of ∼3–5 × 103 cpm/fmol using [γ-32P]ATP and T4 polynucleotide kinase as described (42). RNA-EMSAs were used to determine the minimum number of GAPDH-RNA complexes formed. Briefly, we incubated 0.2 nm 32P-labeled RNA oligos (ARE38 or Rβ31) with increasing molar concentrations of GAPDH in binding buffer (10 mm Tris-HCl, pH 8.0, 50 mm KCl, 2 mm DTT, 0.5 mm EDTA, 0.1 mg/ml acetylated bovine serum albumin, 0.1 mg/ml heparin, 10% (v/v) RNase/DNase-free glycerol) for 20 min at room temperature. The mixture was run on 6% native acrylamide gels in 0.5× Tris borate EDTA buffer at 200 V, 4 °C. After 180 min, gels were dried and visualized using a Typhoon FLA-9500 Phosphorimager (GE Healthcare).
Fluorescence Anisotropy Assays
GAPDH-RNA binding equilibria were quantitatively assessed by fluorescence anisotropy (42, 43). Unlike EMSA, this technique does not require fractionation of reaction products and thus permits resolution of binding constants under true solution equilibrium conditions. Briefly, we incubated limiting concentrations (0.2 nm) of fluorescein-labeled RNA oligos (Fl-ARE38 or Fl-Rβ31) with increasing concentrations of GAPDH in binding buffer without glycerol in a final volume of 100 μl. After incubating reactions at room temperature for 1 min, fluorescence intensity and total reaction anisotropy (At) were recorded using a Beacon 2000 Fluorescence Polarization System (Panvera) equipped with a 490-nm excitation filter and a 535-nm emission filter. Preliminary on-rate analysis indicated that equilibrium was achieved in <1 min (data not shown). Equilibrium constants for RNA-GAPDH complexes were determined by plotting anisotropy values (At) as a function of GAPDH protein concentration ([P]) and fitting using nonlinear regression. Because protein binding did not alter the fluorescence quantum yield of any RNA substrate tested, data sets were fit using binary and sequential two-step binding models described by Equations 1 and 2, respectively.
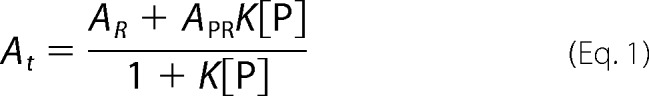 |
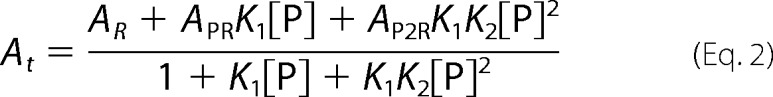 |
AR, APR, and AP2R represent the intrinsic anisotropies of unbound RNA (R) or RNA following one (PR) or two (P2R) protein binding events, respectively, whereas K1 and K2 are the equilibrium association constants (K = 1/KD) associated with the first and second binding events.
The appropriateness of each fit was assessed by the coefficient of determination (R2) and a residual runs test for residual nonrandomness (binding model discarded if p < 0.05). Pairwise comparisons between binding models were performed using the F test with the simpler model (greater degrees of freedom) preferred unless p < 0.05. We performed three independent anisotropy experiments for each oligonucleotide and GAPDH protein. PRISM v3.03 software (GraphPad) was used to perform nonlinear regression and statistical analyses.
Intramolecular FRET Experiments
Structural changes in RNA induced by GAPDH proteins were monitored by changes in the distance between 3′-Fl (donor) and 5′-Cy3 (acceptor) groups attached to the termini of RNA substrates using FRET as described (12, 44). Briefly, FRET efficiency (EFRET) varies inversely with the scalar distance (r) between a fluorescent donor and an appropriate acceptor according to the equation,
 |
The Förster distance (R0) can be calculated from the fluorophore spectral data and represents the distance between donor and acceptor yielding EFRET = 0.5 (45). For FRET between Fl and Cy3 dyes attached to single-stranded nucleic acids, R0 is 55.7 Å (46).
Varying concentrations of GAPDH proteins were incubated with RNA substrates ARE38-Fl (donor alone) or Cy3-ARE38-Fl (donor-acceptor pair) as described for anisotropy analyses (see above), with final RNA concentrations of 2 nm. Fluorescence from the FRET donor (Fl) was measured using a Cary Eclipse fluorometer (Varian) equipped with a sub-microcell cuvette (λex = 485 nm, λem = 518 nm, 10-nm slit widths). Background fluorescence was quantified from samples lacking RNA substrates. Inner filter effects and photobleaching were insignificant in these experiments (data not shown). EFRET between donor and acceptor fluorophores was calculated using the equation,
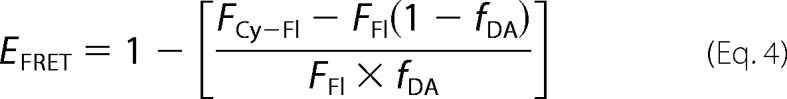 |
where FCy-Fl is the background-corrected fluorescence of the donor in the presence of the acceptor (measured from the Cy-ARE38-Fl substrate), FFl is the background-corrected donor fluorescence from reactions lacking the acceptor (using the ARE38-Fl substrate), and fDA is the fractional labeling of the Cy3 acceptor on the Cy-ARE38-Fl RNA.
To correlate protein-dependent changes in RNA conformation with formation of specific GAPDH-RNA complexes, we estimated the relative concentration of each complex with varying protein concentrations. These fractional distributions were calculated using the sequential two-step association model and affinity constants resolved by anisotropy analyses (described above).
Intermolecular FRET Experiments
To determine the number of RNA molecules bound to GAPDH, we used single-labeled Cy-ARE38 and ARE38-Fl substrates and measured EFRET as above. To maximize the potential for detection of intermolecular FRET, we used 50 nm ARE38-Fl and 450 nm Cy3-ARE38. Because FRET was measured by looking for changes in donor (Fl) emission when acceptors (Cy3) are present, 90% of any detectable complexes between GAPDH and two RNAs containing at least one ARE38-Fl would also contain one Cy3-ARE38 RNA.
Crystallization, X-ray Data Collection, and Refinement
Wild-type and T229K GAPDH were crystallized using a Phoenix crystallization robot (Art Robbins) and PEG/Ion (Hampton) and PACT (Qiagen) commercial screens. The drops consisted of 0.1 μl of GAPDH protein (5–10 mg/ml) and 0.1 μl of screen solution. Crystals of T229K GAPDH were obtained in 20% (w/v) PEG3350, 0.02 m ZnCl2. Crystals of wild-type GAPDH were obtained in 20% (w/v) PEG6000, 0.1 m sodium acetate, pH 5.0, 0.01 m ZnCl2. After growing for 1 week, crystals were flash-cooled in liquid N2.
X-ray diffraction data were collected on beamline 12.3.1 at the Advanced Light Source at Lawrence Berkeley National Laboratory for wild-type GAPDH and beamline 7–1 at the Stanford Synchrotron Radiation Laboratory for T229K GAPDH. For both data sets, data processing and reduction were done with HKL2000 (47). Phasing by molecular replacement was done with PHASER (48) using the human GAPDH structure (PDB code 1U8F) as a starting model. The final models were obtained with iterative cycles of refinement with PHENIX (49) and rebuilding with COOT (50). The final models and structure factors were deposited with the PDB (PDB entries 4WNC and 4WNI for wild-type and T229K GAPDH, respectively).
Small-angle X-ray Scattering (SAXS)
SAXS data were collected at the Advanced Light Source 12.3.1 beamline (SIBYLS). The wavelength was 1 Å, and the sample-to-detector distance was 1.5 m, resulting in scattering vectors, q, ranging from 0.01 to 0.32 Å−1 (q is defined as 4πsinθ/λ, where 2θ is the scattering angle). All experiments were performed on 20-μl samples at 20 °C using a Hamilton robot for loading samples from the 96-well plate. The buffer used for all samples was 20 mm Tris, pH 8.0, 30 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine. Wild-type and T229K GAPDH were purified by size-exclusion chromatography before SAXS experiments using a 24-ml S200 column (GE Healthcare). SAXS data were collected on concentrated pooled fractions (ranging from 0.25 to 8 mg/ml). Fractions eluting before the column void volume were used for buffer subtraction. For each sample, sequential exposures were taken (0.5, 0.5, 1, 2, and 5s), and data were monitored for radiation damage. Data collected at protein concentrations of 0.25 and 0.5 mg/ml were noisy and showed signs of aggregation at low q and were not processed further. Data in the range of 1–8 mg/ml were analyzed with PRIMUS (51), SCATTER (LBNL BL12.3.1 software), and GNOM (52) to generate Guinier, Kratky, and P(r) plots and determine Rg (radius of gyration), I(0), and Dmax (maximum dimension of the particle). The experimental SAXS data and the calculated SAXS curve obtained from high resolution structures of tetrameric GAPDH were compared with CRYSOL (53). We determined the overall χ2 parameter, which reflects the fit of the experimental curve to the calculated curve.
Hydrogen Deuterium Exchange Mass Spectrometry
The coverage maps for GAPDH were obtained from undeuterated controls as follows; 1 μl of wild-type GAPDH (8.5 mg/ml) or T229K GAPDH (7.1 mg/ml) in size-exclusion chromatography (SEC) buffer (20 mm Tris, pH 8.0, 28 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine) was diluted with 9 μl of SEC buffer at room temperature followed with 50 μl of ice-cold quench buffer (0.1 m phosphate buffer, 1.5 m guanidine-HCl, pH 2.4). The samples were immediately injected into a Waters HDX nanoAcquity UPLC (Waters, Milford, MA) with in-line pepsin digestion (Poroszyme immobilized pepsin cartridge from Applied Biosystems). The resulting peptides were trapped on an Acquity UPLC BEH C18 peptide trap and separated on an Acquity UPLC BEH C18 column using a 7-min, 5 to 35% acetonitrile (0.1% formic acid) gradient directly into a Waters Synapt G2 mass spectrometer (Waters, Milford, MA). MSE data were acquired with a 20–30-V ramp trap collision energy for high energy acquisition of product ions as well as continuous lock mass (leucine enkephalin) for mass accuracy correction. Peptides were identified using the ProteinLynx Global Server 2.5.1 (PLGS) from Waters.
H/D exchange reactions were performed as follows: 1 μl of wild-type or T229K GAPDH was incubated in 9 μl of SEC buffer, 99.99% D2O, pD 8.0, at room temperature for various lengths of time (10 s, 1 min, 10 min, 1h, and 2h) and quenched immediately prior to injection. Back exchange correction was performed against fully deuterated controls acquired by incubating 1 μl of wild-type or T229K GAPDH in 9 μl of 20 mm Tris, 28 mm NaCl, 1 mm Tris(2-carboxyethyl)phosphine, 3.8 m guanidine-DCl, 99.99% D2O, pD 8.0, for 2 h at room temperature before quenching and injection. The various deuteration time points and controls were acquired in triplicate.
The deuterium uptake by the peptides identified by ProteinLynx Global Server 2.5.1 through increasing deuteration time and fully deuterated control was determined using Water's DynamX 2.0 software. The normalized percentage of deuterium uptake (%Deuteration) at an incubation time t for a given peptide was calculated as,
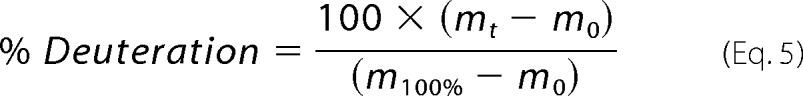 |
with mt the centroid mass at incubation time t, m0 the centroid mass of the undeuterated control, and m100% the centroid mass of the fully deuterated control.
Peptides displaying significant differences in conformation as detected by exchange mass spectrometry (HDX-MS) were determined using the criteria described by Houde et al. (54). For each peptide, the difference in back-exchange-corrected deuterium incorporation was determined at each time point. The threshold for significance in the difference in conformation between two states as detected by HDX-MS is based on two conditions; (i) at least one time point with at least a 0.5-Da difference and (ii) a sum of differences over all 5 time points (in triplicates) superior to 1.1 Da.
Nano-electrospray Ionization Native Mass Spectrometry (Nano-ESI/MS)
The purified wild-type and T229K GAPDH samples were dialyzed into 0.1 m ammonium acetate, pH 7.4, 1 mm DTT using Amicon Ultra-4 centrifugal filter units (EMD Millipore). The concentration of the buffer-exchanged protein was calculated by measuring A280 absorbance. In the analysis of the oligomers, various protein concentrations were obtained by serial dilution starting from the stock solution of buffer exchanged protein sample.
Nano-ESI/MS analysis was carried out utilizing a modified quadrupole-ion mobility-time of flight (Q-IM-TOF) instrument (Synapt G2, Waters Corp., Manchester, UK) with a customized surface-induced dissociation device installed before the IM chamber as previously described (55). All experiments were carried out using a nanoelectrospray source using a capillary voltage of 1.0–1.3 kV and a cone voltage of 50 V. No heating was applied to the cone. Procedures of nano-ESI glass capillary and surface preparation can be found elsewhere (56). Instrumental conditions used were 5 mbar for the source/backing pressure, 2 mbar for nitrogen gas pressure in the IM cell, 120 ml·min−1 gas flow into the helium cell, ∼6 × 10−7 mbar in the TOF analyzer, and a wave velocity and height of 300 ms−1 and 20 V respectively for IM experiments. Nano-ESI/MS is a gentle method of ionization where salts and solvent are often retained, giving higher m/z values than those calculated from sequence, especially for oligomers. A more accurate determination of the molecular weight can be obtained from tandem nano-ESI MS (MS/MS) measurements of fragments, as this MS/MS approach results in the loss of salts and other nonspecific adducts. In surface-induced dissociation experiments, ions were steered toward the surface, and fragments were collected and separated in the IM chamber. In collision-induced dissociation (CID) experiments, ions experienced multiple collisions with argon gas in the region before the IM chamber. CID energy-resolved mass spectrometry analysis was performed by plotting the relative abundance of fragments present over a range of collision energies. By calculating the fraction of tetramer precursor remaining at similar CID collision energy, one can infer similar resistance to dissociation for the T229K tetramer versus the wild-type GAPDH tetramer.
RESULTS
Wild-type GAPDH Binds Tightly to the ARE from TNF-α mRNA
We first tested GAPDH binding to an RNA oligonucleotide derived from TNF-α 3′-UTR. We used a 38-mer containing five AUUUA motifs (hereafter referred to as ARE38 or ARE probe). This probe is very similar to other ARE-RNAs shown to bind GAPDH in vivo and in vitro (13, 30, 33, 36). Our EMSA experiments with wild-type GAPDH and ARE38 showed two major GAPDH-RNA complexes (Fig. 1a). A higher mobility complex (complex 1) formed at low GAPDH concentrations and a second complex of slower mobility (complex 2) formed at higher GAPDH concentrations. Binding to the ARE probe was specific, as no binding was observed with the control RNA Rβ31 lacking the AUUUA motifs, even at high GAPDH concentrations (Fig. 1a).
FIGURE 1.
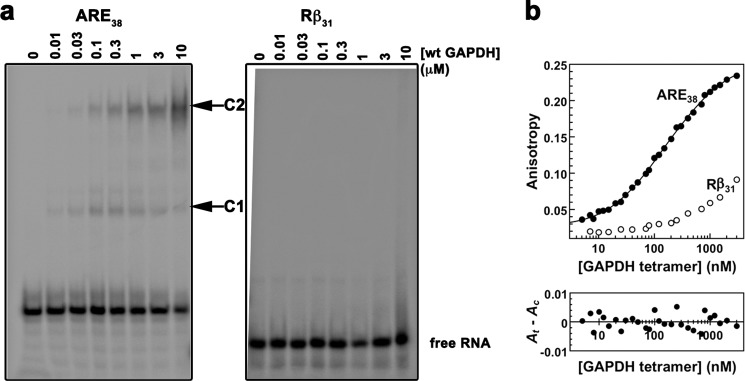
In vitro binding of ARE38 RNA substrate to wild-type GAPDH. a, RNA-EMSA for wild-type GAPDH with ARE38 and control Rβ31. Binding reactions containing a 5′-32P-labeled ARE38 or control Rβ31 RNA substrate and wild-type GAPDH protein were assembled as described under “Experimental Procedures.” The arrows indicate two distinct GAPDH-RNA complexes (labeled C1 and C2). No complex formation was observed with control RNA. b, fluorescence anisotropy for wild-type GAPDH with ARE38 and control Rβ31. The fluorescence anisotropy of reactions containing indicated Fl-labeled RNA substrates (0.2 nm) and increasing concentrations of wild-type GAPDH were measured as described under “Experimental Procedures.” GAPDH binding to the Fl-ARE38 RNA was resolved by the sequential two-step binding algorithm. A residuals plot showing random deviation of the preferred binding model (Ac) from observed data (At) is shown in the lower part of the figure.
To determine the binding affinity of GAPDH for the ARE substrate, we performed fluorescence anisotropy measurements at equilibrium using conditions similar to those used for the EMSA experiments. Anisotropy of the fluorescent ARE probe was dramatically enhanced by GAPDH, whereas that of the control Rβ31 RNA increased only slightly at the highest GAPDH concentrations used in the assay (Fig. 1b). The data were best fitted to a sequential two-step binding model and yielded dissociation constants KD1 of 97 ± 21 nm and KD2 of 1.4 ± 0.4 μm, indicating a tight binding event followed by a weaker binding event. This binding model was validated by the random distribution of residuals (Fig. 1b) and high coefficients of determination for all experiments (R2 > 0.99).
A Mutation at the Dimer Interface Impairs GAPDH Binding to the ARE Probe
The RNA binding site in GAPDH is unknown. To test the hypothesis that RNA may bind in the positively charged substrate binding region of GAPDH, we mutated residues located either in the positively charged groove or at the GAPDH dimer interface. Several mutants showed impaired expression and solubility and could not be used for our studies. In contrast, the T229K GAPDH mutant was soluble and purified for subsequent characterizations. T229K GAPDH showed impaired RNA binding compared with wild-type GAPDH by EMSA (Fig. 2a). In particular, higher T229K GAPDH concentrations were necessary to form the slow mobility complex 2. We verified this observation quantitatively by fluorescence anisotropy (Fig. 2b). Similar to wild-type GAPDH, the data for the T229K mutant were fitted to a sequential two-step binding model (Fig. 2b) yielding dissociation constants KD1 of 131 ± 22 nm and KD2 of 3.5 ± 0.8 μm. Although the tight binding event (KD1) was not affected, the second dissociation constant (KD2) was significantly increased, indicating that the T229K mutation impaired formation of complex 2.
FIGURE 2.
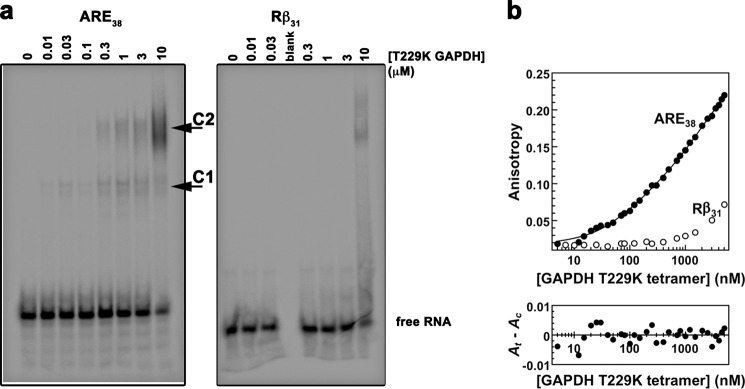
In vitro binding of ARE38 RNA substrate to T229K GAPDH. a, RNA-EMSA for T229K GAPDH with ARE38 and control Rβ31. Binding reactions containing a 5′-32P-labeled ARE38 or control Rβ31 RNA substrate and T229K GAPDH protein were assembled as described under “Experimental Procedures.” The arrows indicate two distinct T229K GAPDH-RNA complexes (labeled C1 and C2). No complex formation was observed with control RNA until the highest protein concentration. b, fluorescence anisotropy for T229K GAPDH with ARE38 and control Rβ31. The fluorescence anisotropy of reactions containing the indicated Fl-labeled RNA substrates (0.2 nm) and increasing concentrations of T229K GAPDH were measured as described under “Experimental Procedures.” T229K GAPDH binding to the Fl-ARE38 RNA was resolved by the sequential two-step binding algorithm. A residuals plot showing random deviation of the preferred binding model (Ac) from observed data (At) is shown in the lower part of the figure.
T229K GAPDH Assembles as a Homotetramer
Our results revealed that the GAPDH T229K mutation impairs RNA binding. The Thr-229 residue is mostly invariant among mammalian GAPDH proteins (99.2% identity among 250 sequences) and is located on strand β11 at the dimer interface (Fig. 3). This mutation was shown previously via gel filtration to disrupt GAPDH oligomerization (57). Therefore, we hypothesized that this mutation may decrease RNA binding via disruption of GAPDH quaternary assembly. To test this hypothesis, we performed SEC, SAXS, and x-ray crystallography studies of wild-type and T229K GAPDH. First, SEC profiles of wild-type and T229K GAPDH revealed that both proteins eluted as a single peak corresponding to tetrameric GAPDH, even at protein concentrations as low as 0.5 mg/ml (data not shown).
FIGURE 3.

T229K induces subtle conformational changes at the dimer interface. a, T229K GAPDH is a dimer of dimers. T229K GAPDH is composed of four identical subunits (R, Q, P, O) arranged in a tetrameric quaternary assembly. The subunits are related by three 2-fold symmetry axes (P, Q, R; Q is not shown). The dimer interface is located along the P axis (between monomers R and Q or between P and O). The tetramer interface (between dimers R/Q and P/O) is located along the R axis. Each subunit is constituted of an NAD+ binding domain (cyan) and a catalytic domain (dark blue). Three NAD+ molecules are bound (shown as sticks). The Lys-229 mutation (magenta sticks) is located at the dimer interface. b, superimposition of the x-ray structures of wild-type and T229K GAPDH. The wild-type (magenta) and T229K GAPDH (blue) tetrameric structures superimpose well. The T229K mutation (green rectangle) is located at the dimer interface. c, stereo view of the region surrounding the T229K mutation. Both structures are represented as sticks colored as in b. The T229K promotes a cascade of subtle but significant conformational changes at the dimer interface in region 298–303 of the adjacent monomer and regions 229–231 and 210–216 of the same monomer.
Second, we crystallized wild-type and T229K GAPDH and determined their x-ray structures at 1.99 Å and 2.3 Å, respectively (Table 1). For wild-type GAPDH, the final model includes 8 monomers in the asymmetric unit, 2399 water molecules, 6 zinc atoms, 2 acetate molecules, and 6 NAD+ cofactors. For T229K GAPDH, the final model includes 4 monomers in the asymmetric unit with protein residues 3–335, 710 water molecules, 3 zinc atoms, and 3 NAD+ cofactors (Fig. 3a). Both proteins are tetramers composed of a dimer of dimers. The identical subunits are related by three 2-fold symmetry axes (P, Q, and R). The dimer interface is located along the P axis (between monomers R and Q or P and O), whereas the tetramer interface (between dimers R/Q and P/O) is located along the R axis (Fig. 3a). Importantly, several residues at or near the dimer interface can be posttranslationally modified. Acetylation of Lys-227 and Lys-251 (58), phosphorylation of Thr-246 (59) and Thr-237 (60), O-GlcNacylation of Thr-229 (57), and S-nitrosation of Cys-247 (61) have been shown to affect GAPDH interactions with other proteins or GAPDH localization in the cell. Therefore, the dimer interface is a key region for regulation of GAPDH activities by posttranslational modifications.
TABLE 1.
X-ray data collection and refinement statistics
| Wild-type GAPDH | T229K GAPDH | |
|---|---|---|
| Data collection | ||
| Space group | P21 | P212121 |
| Wavelength (Å) | 1.116 | 1.127 |
| Resolution (Å) | 50.0-1.99 (2.02-1.99)a | 49.5-2.3 (2.38-2.30)a |
| Unit cell parameters (Å) | a = 132.5 b = 81.4, c = 147.8, β = 90.058 | a = 81.4 b = 133.1, c = 147.8 |
| Measurements | 422,327 (20,829) | 117,182 (3,352) |
| Unique reflections | 216,349 (10,672) | 63,669 (3,951) |
| Redundancy | 3.7 (3.3) | 4.8 (3.1) |
| Completeness (%) | 99.9 (99.9) | 88.5 (55.4) |
| 〈I/σ(I)〉 | 15.6 (18.7) | 10.7 (12.1) |
| Rmerge (%)b | 9.0 (34.2) | 6.2 (23.5) |
| Refinement | ||
| Resolution range (Å) | 49.5-1.99 (2.06-1.99) | 49.5-2.3 (2.38-2.30) |
| No. of protein atoms | 20,208 | 10,100 |
| No. of water atoms | 2,399 | 710 |
| No. of hetero atoms | 278 | 135 |
| r.m.s.d. bond lengths (Å) | 0.009 | 0.003 |
| r.m.s.d. bond angle (°) | 1.18 | 0.78 |
| Rwork (%)c | 13.9 (16.3) | 16.9 (19.6) |
| Rfree (%)d | 17.9 (19.0) | 19.8 (24.0) |
| Wilson B-factor (Å2) | 17.3 | 28.6 |
| Average B-factor (Å2) | 22.3 | 34 |
| Ramachandran plot (%) | ||
| Favored | 96.4 | 96 |
| Outliers | 0.34 | 0.3 |
| Molprobity clashscoree | 0.83 | 1.23 |
| Protein Data Bank code | 4WNC | 4WNI |
a Values in parentheses apply to the highest resolution shell.
b Rmerge = ΣhΣi|I(h,i) − 〈I(h)〉|/ΣhΣiI(h,i), where I(h,i) is the intensity of the ith observation of reflection h, and 〈I(h)〉 is the average intensity of redundant measurements of reflection h.
c Rwork = Σh‖Fobs| − |Fcalc‖/Σh|Fobs|.
d Rfree = Σh‖ Fobs| − |Fcalc‖/Σh|Fobs| for 5% of the reserved reflections.
e Values were from the Molprobity server.
To identify potential differences between wild-type and T229K GAPDH, we superimposed their x-ray structures (PDB entries 4WNC and 4WNI for wild-type and T229K GAPDH, respectively). The overall root mean square deviation (r.m.s.d.) between the mutant and wild-type GAPDH structures is 0.272 Å for 1329 atoms superimposed. For comparison, we superimposed each monomer to each other in the wild-type and mutant structures. The average r.m.s.d. ranged from 0.041 Å to 0.363 Å and from 0.167 Å to 0.492 Å for all 333 amino acids superimposed for wild-type GAPDH and T229K GAPDH, respectively. For the mutant structure, the higher intersubunit r.m.s.d. is mostly due to the C subunit, which displays a much higher average B-factor (55.4 Å2 versus 35 Å2) and less-defined electron density than the other subunits. Regardless, this result confirmed that the wild-type and mutant structures are very similar and that the mutation did not induce large structural rearrangements (Fig. 3b).
Interestingly, the regions in the vicinity of T229K exhibited a cascade of subtle but significant conformational changes in the mutant structure. First, the backbone of residues 299–305 located at the dimer interface in the adjacent subunit was displaced by 0.3–0.6 Å in the mutant compared with the wild-type structure (Fig. 3c). This conformational change was likely necessary to accommodate the bulkier side chain of Lys-229 compared with Thr-229. In addition, Ile-301 adopted a different side-chain conformation to avoid energetically unfavorable contacts with the Lys-229 side chain. Second, the backbones of residues 230 and 231, directly following the mutation, were shifted by 0.5 Å to accommodate the bulky side chain of Ile-301. Finally, the backbones of residues 210–212 were shifted by 0.4–0.7 Å to accommodate the displacement of the 229–231 backbones. Thus, our structural determinations showed that T229K induced subtle conformational changes at the dimer interface while leaving the tetramer mostly unchanged.
To rule out artifacts due to crystallization conditions at high GAPDH concentration, we measured SAXS data for wild-type and T229K GAPDH proteins at several concentrations. Data collected in the range of 1–8 mg/ml (6.9–55 μm tetramer) showed no sign of aggregation (Fig. 4, a and b). The Guinier analysis yielded a Rg of 32.6 ± 0 Å and 32.9 ± 0.1 Å for wild-type and T229K GAPDH, respectively. The maximum distance (Dmax) and Rg values derived from the pair distribution function P(r) were 85 Å and 32.5 ± 0 Å for wild-type GAPDH and 86 Å and 32.8 ± 0.1 Å for T229K GAPDH, respectively. The comparison of the experimental SAXS data with the calculated SAXS curves for tetrameric wild-type and T229K GAPDH structures (Fig. 4, c and d) showed good agreement for both wild-type and T229K GAPDH data (χ2 of 1.02 and 1.06, respectively). These results confirmed that wild-type and mutant GAPDH share similar overall dimensions, and both assemble as tetramers in solution.
FIGURE 4.

Wild-type GAPDH and T229K assemble as tetramers in solution. a, experimental scattering profiles collected for concentrated SEC fractions for wild-type GAPDH (2 mg/ml) and T229K GAPDH (1 mg/ml). b, pair distribution function (P(r)) for wild-type and T229K GAPDH. The P(r) functions were not normalized to unity (individual y scales for wild-type and T229K GAPDH). c and d, experimental and theoretical scattering profiles for wild-type and T229K GAPDH. The theoretical scattering calculated for the tetrameric GAPDH structures (PDB entries 4WNC and 4WNI for wild-type and T229K GAPDH, respectively; solid line) was fitted with the program CRYSOL.
Collectively, our biophysical and structural characterizations confirmed that both wild-type and T229K GAPDH assemble as tetramers in the crystal and in solution. In addition, our crystallographic data indicated that Lys-229 induced small conformational changes mainly at the dimer interface. This is in contrast to previous studies which showed that the T229K mutation disrupted GAPDH oligomerization and yielded predominantly monomeric GAPDH (57). The reasons for this discrepancy are not clear, but our extensive biophysical and structural characterizations unambiguously show that this mutation does not significantly disrupt GAPDH tetramers at concentrations above 0.5 mg/ml.
The T229K Mutation Induces Conformational Changes in Interfacial Regions of the Tetramer
To elucidate the role played by Thr-229 in RNA binding, we performed hydrogen-deuterium HDX-MS studies for wild-type and T229K GAPDH. For both proteins, the peptide coverage was 98.5%. Comparison of deuterium exchange rates in wild-type and T229K GAPDH revealed differences in three main regions (Figs. 5 and 6). The first region was located at the dimer interface directly across from the T229K mutation. The C-terminal domain peptide 301–307 is located on β-strand β15, which is part of the 8-strand β-sheet that forms the dimer interface. In wild-type GAPDH, this peptide exhibited low deuterium uptake at early time points (<10% at 10 s), indicative of peptide with amide hydrogens that were protected from exchange (Fig. 5a). In the mutant, there was a significant increase in deuterium incorporation starting at 10 s (>30%), indicative of structural changes that exposed amide hydrogens to exchange. Furthermore, this peptide showed significantly increased deuterium uptake at later time points in the mutant, suggesting that the backbone of this region was not only more structurally exposed to solvent deuterium but also more conformationally dynamic. This result is in agreement with our crystallographic data showing conformational changes of this region in the T229K x-ray structure compared with wild-type GAPDH.
FIGURE 5.

T229K promotes dynamical changes in three main regions of the GAPDH tetramer. a, regions at the dimer interface that show significant differences in deuterium incorporation between wild-type and T229K GAPDH are colored in shades of orange in the wild-type GAPDH structure (middle). Representative traces of deuterium incorporation as a function of time are shown for peptides 301–307 (top; increased deuterium incorporation) and 78–93 (bottom; no difference in deuterium incorporation). The color coding of the difference in deuteration corresponds to that of Fig. 6a. b, regions at the tetramer interface that display significant differences in deuterium incorporation between wild-type and T229K GAPDH are highlighted in shades of orange in the structure of wild-type GAPDH (middle). Representative traces of deuterium incorporation as a function of time are shown for peptides 177–182 (top) and 196–204 (bottom), which both show increased deuterium incorporation. The color coding of the difference in deuteration corresponds to that of Fig. 6a. c, regions in the NAD+ binding site that display significant differences in deuterium incorporation between wild-type and T229K GAPDH are highlighted in shades of orange in the structure of wild-type GAPDH (middle). Representative traces of deuterium incorporation as a function of time are shown for peptides 94–102 (top) and 183–195 (bottom), which both show increased deuterium incorporation. The color coding of the difference in deuteration corresponds to that of Fig. 6a.
FIGURE 6.

T229K promotes conformational changes along the P axis of the GAPDH tetramer. a, differences in percentage deuteration (Δ%D = %D T229K − %D wild type) for the different time points (0, 10 s, 10 min, 1 h, 2 h) are mapped on the amino acid sequence of wild-type GAPDH and colored according to %D (see the scale). The secondary structure elements and residues involved in the dimer interface (#), the tetramer interface (dot), or NAD+ binding (*) are indicated above the sequence. b, regions that present significant difference in percentage deuteration (shown in a) are mapped to the surface of the T229K tetrameric structure. Molecules P, O, Q, and R of the tetramer are shown. The 2-fold symmetry axis P, Q, and R are indicated. Peptides are colored as in a. The T229K mutation is shown in magenta. Differences in % deuteration between wild-type and T229K GAPDH are localized along the P axis of the tetramer, at the dimeric and tetrameric interfaces, and near the NAD+ binding site. The region 251–260 (black oval) was recently shown to cross-link to AU-RNA in yeast GAPDH (71) and is strategically located near the dimer interface along the P axis.
The second region was located at the tetramer interface. The contiguous peptides 177–182, 183–195, and 196–204 all participate in the tetramer interface (Fig. 5, b and c). In addition, peptides 177–182 and 196–204 also contribute to the dimer interface. Peptide 183–195 adopts an extended loop conformation in our crystal structure of wild-type GAPDH, is relatively solvent-exposed, and only makes three hydrogen bonds between its amides and surrounding regions. This peptide showed increased deuterium uptake in the mutant at almost all time points, indicating both structural changes and increased dynamics of this peptide. In contrast, the shorter flanking regions 177–182 and 196–204 partially fold into a β-strand and an α-helix, respectively, are less solvent-exposed, and make five and three hydrogen bonds between their amides and neighboring regions, respectively. Accordingly, these peptides displayed low deuterium uptake and no significant difference between wild-type and mutant GAPDH at early time points. However, at later time points, these peptides exhibited significantly increased deuterium incorporation in the mutant (Fig. 5b), which suggests that these peptides are more dynamic.
The third region affected by the mutation was the NAD+ binding site. We observed increased deuterium uptake in the regions 33–41, 94–102, 144–152, and 182–184 in the mutant compared with wild-type GAPDH at early time points that became negligible at the latest time points (Fig. 5c). This result was surprising as these peptides, which participate in NAD+ binding, were far from the mutation (∼24 Å). Although NAD+ does not seem to participate extensively in the tetramer interface, it is located very close to peptide 183–195 that is significantly affected by the mutation (see above). These results suggest that the mutation may allosterically perturb NAD+ binding.
Taken together, our results showed that the T229K mutation induced conformational changes at the dimeric and tetrameric interfaces of GAPDH as well as in the NAD+ binding region (Fig. 6a). We showed that a single mutation promoted both short-range and long-range effects on the structure and dynamics of regions important for the integrity of the tetramer assembly. Interestingly, all these changes mapped to regions of the GAPDH tetramer located along the P symmetry axis. No change was observed on the other faces of the tetramer (Fig. 6b). Although the increased conformational flexibility of the interfacial regions does not necessarily equate to a decrease in tetramer stability, these changes could potentially lead to destabilization of the T229K tetramer compared with wild-type GAPDH. We did not observe any change in the oligomeric state of T229K GAPDH at the concentrations used in our biophysical characterizations (>3.5 μm tetramer). However, from these experiments alone, we cannot exclude that the mutation may disrupt the tetramer at the lower concentrations used in the EMSA and fluorescence studies.
T229K Assembles as a Tetramer over a Wide Range of Concentrations
Our HDX-MS studies revealed changes in the conformation of protein regions located at the dimer and tetramer interfaces and suggested that the T229K tetramer may be less stable than the wild-type GAPDH tetramer. To further elucidate the effect of the T229K mutation on GAPDH oligomerization, we used nano-ESI/MS/MS. First, nano-ESI surface-induced dissociation MS/MS spectra for wild-type and T229K GAPDH showed a mixture of NAD+-free and NAD+-bound species (Fig. 7a). In the absence of NAD+, the measured masses (36,053 ± 14 Da and 36,084 ± 38 Da for wild-type and T229K GAPDH, respectively) closely matched their calculated masses (36,053 and 36,080, respectively). The proportions of NAD+-free to NAD+-bound species determined by area under the curve analysis were 57:43 and 69:31 for wild-type and T229K GAPDH, respectively. These results suggested that NAD+ binding was weaker in the mutant protein. Second, we measured nano-ESI/MS spectra for T229K GAPDH at concentrations ranging from 0.24 μm to 61 μm (Fig. 7b). We showed that the mutant protein was present solely as a tetramer at all concentrations tested. Third, we performed CID energy-resolved mass spectrometry analysis, which showed no significant difference in the depletion of the tetramer between wild-type and T229K GAPDH (Fig. 7c).
FIGURE 7.

Nano-ESI/MS characterization of wild-type and T229K GAPDH. a, nano-ESI/MS and MS/MS spectra were obtained by spraying 10 μm protein sample (wild-type and T229K GAPDH) in 0.1 m NH4OAc, pH 7.4. Nano-ESI surface-induced dissociation MS/MS spectra of tetramer revealed the presence of two monomer charge state distributions with measured masses of 36,053 ± 14 Da and 36,581 ±19 Da for wild-type GAPDH and 36,084 ± 38 Da and 36,611 ± 27 Da for T229K GAPDH. These two charge states distributions correspond to NAD+-free and NAD+-bound GAPDH monomers. Wild-type and T229K GAPDH both exist as mixtures of NAD+-free and NAD+-bound mixture (ratios of 57:43 and 69:31 for wild-type and T229K GAPDH, respectively). b, nano-ESI/MS spectra for T229K GAPDH were obtained by spraying the protein sample in 0.1 m NH4OAc, pH 7.4. The concentration of T229K GAPDH was varied by performing serial dilutions from an initial stock solution of 60.9 μm over a range of 0.2–60.9 μm. The nano-ESI/MS spectra show the presence of tetramers only (purple) at all concentrations. c, CID energy-resolved mass spectrometry plot showing no significant difference in the tetramer population of wild-type GAPDH and T229K as a function of the collision energy.
In summary, our ESI/MS results showed that the T229K mutation does not lead to significant changes in oligomerization across a wide range of concentrations. Thus, we can postulate that both wild-type and T229K GAPDH are mostly tetrameric in the conditions used for our EMSA, fluorescence anisotropy, and FRET assays. In addition, the mutant enzyme showed decreased NAD+ binding compared with wild-type GAPDH, consistent with the HDX-MS results showing increased dynamics of protein regions involved in NAD+ binding.
The T229K Mutation Affects the Conformation of Bound RNA Ligands
To determine whether GAPDH binding affects the structure of the Cy3-ARE38-Fl probe, we performed intramolecular FRET experiments (Fig. 8, a–c). In the absence of protein and Mg2+ ions, the average FRET efficiency (EFRET) of the labeled RNA probe was 0.40–0.44, consistent with the dynamic population of folded RNA structures described previously (44). After adding wild-type GAPDH, we observed no significant change in EFRET across a range of protein concentrations (EFRET < 0.5; Fig. 8c), suggesting that wild-type GAPDH binding did not alter the global structure of the RNA ligand. In contrast, titration of the T229K GAPDH protein yielded a significant increase in FRET efficiency to 0.65 (p = 0.004; Fig. 8c), indicating a shortening of the distance between the two fluorescent labels within the RNA probe. We calculated the average distances between the two labels based on EFRET as follows: 61 Å, 58 Å, and 49 Å for RNA alone, RNA with wild-type GAPDH, and RNA with T229K GAPDH, respectively. Furthermore, comparing protein-dependent changes in EFRET with the fractional concentration of each species present in solution (Fig. 8c) showed that the RNA structural condensation correlated well with the first high affinity T229K binding event (KD1). No further significant change was observed with the second binding event.
FIGURE 8.

T229K binding leads to structural changes of the ARE38 RNA. a, schematic of RNA folding measured by FRET. Both fluorophores are excited at 490 nm, but Fl is the only one producing significant quantum emission at 518 nm. The loss of energy from Fl to Cy3 due to FRET leads to a decrease in fluorescence emission at 518 nm. b, fluorescence emission spectra (λex = 490 nm) of the Cy3-ARE38-Fl RNA substrate incubated without protein or with 500 nm wild-type GAPDH (green) or T229K GAPDH (magenta). Decreased emission at 518 nm in the presence of the T229K mutant indicates an increase in EFRET. c, intramolecular FRET. EFRET of the Cy3-ARE38-Fl RNA substrate (solid black circles) was monitored in solution as a function of tetrameric GAPDH protein concentration as described under “Experimental Procedures.” Each experiment was done in triplicate. The fractional concentrations of free RNA (solid line), complex 1 (C1, dotted line), and complex 2 (C2, dashed line) are shown as a function of tetrameric GAPDH concentration (left, wild type; right, T229K mutant). Product concentrations are based on values of K1 and K2 determined from anisotropy experiments in Figs. 1b and 2b, with the total RNA concentration set to 2 nm. d, intermolecular FRET was measured between independently labeled RNA substrates Cy3-ARE38 and ARE38-Fl (9:1 ratio) without protein and with wild-type or T229K GAPDH as described under “Experimental Procedures.”
GAPDH Binds a Single RNA Ligand
To determine whether GAPDH ribonucleoprotein complexes contained single or multiple RNA ligands, we measured the FRET efficiency of mixtures of RNA substrates labeled individually with 5′-Cy3 or 3′-Fl in the absence or presence of GAPDH proteins. 5′-Cy3 and 3′-Fl RNAs were mixed in a 9:1 ratio so that, should GAPDH bind multiple RNA targets, any complex containing an Fl-labeled RNA (from which EFRET is measured) would most likely contain a Cy3-labeled substrate as well. The results of these experiments showed no detectable FRET either in the absence or presence of wild-type or T229K GAPDH (Fig. 8d). These data suggest that GAPDH ribonucleoprotein complexes contained a single RNA molecule and that the changes in intramolecular FRET efficiency observed upon T299K binding to the double-labeled RNA (see above) were not the result of multiple RNA binding events but reflected intramolecular conformational changes in individual RNA molecules.
DISCUSSION
Here we present biochemical, biophysical, structural, and mass spectrometry results that provide new mechanistic information regarding GAPDH binding to RNA. Although GAPDH was shown to bind several AU-rich RNA targets in vitro and in vivo (13, 30–37), we showed for the first time that GAPDH binds with high affinity to the ARE responsible for posttranscriptional regulation of TNF-α mRNA (62, 63). Previous studies relied solely on EMSA to determine an apparent KD for GAPDH binding to ARE-RNA (36). Here we have used fluorescence anisotropy to quantitatively determine the binding affinity of GAPDH to TNF-α ARE38. We showed that GAPDH binding to this RNA substrate proceeded via a sequential two-step binding model, in agreement with our RNA-EMSA results. The first binding event (complex 1) occurred with mid-nm affinity, whereas the second binding event (complex 2), seen as a slower migrating species, occurred with low μm affinity. This is the first time that precise quantification of binding affinity has been performed for a GAPDH-RNA complex. Although the two-step sequential protein model may be specific to the ARE38 probe used here, this RNA probe shares a number of features with other oligoribonucleotides shown to bind GAPDH previously (13, 30, 33, 36). Indeed AREs from the 3′-UTRs of IFN-γ, CSF-1, ET-1, IL-2, GM-CSF, and c-myc mRNAs also contain three to eight AUUUA motifs and are similar in length to ARE38 studied here. Thus, we reason that this behavior may be a general feature of GAPDH binding to AU-rich RNAs. Further studies to confirm this hypothesis are currently under way.
The stoichiometry of complex 1 and complex 2 remains to be determined, but our biophysical and mass spectrometry characterizations strongly suggest that each binding event involves a GAPDH tetramer (see below). We further performed FRET experiments to determine the number of RNA molecules bound to GAPDH. Although we did not observe any significant change in FRET efficiency upon the addition of GAPDH, we cannot rule out that more than one RNA molecule may be bound to GAPDH. The GAPDH tetramer is rather large with overall dimensions spanning 90 × 90 × 84 Å3. Thus, if two RNA molecules were to bind on opposite sides of a GAPDH tetramer, the distance between the probes would likely be too large to observe any significant FRET. Further experiments are needed to determine the exact stoichiometry of these GAPDH-RNA complexes. Regardless, our results extend the repertoire of ARE-containing RNAs that are specifically targeted by GAPDH, although the biological significance of GAPDH binding to TNF-α mRNA in cells remains to be determined.
Although GAPDH is undoubtedly an ARE-binding protein, it lacks the well known sequence motifs shared by many canonical RNA-binding proteins (38). As a result, the GAPDH binding site for RNA remains unknown. Conflicting studies suggested that RNA binding is mediated by either the NAD+ binding domain alone (13, 30, 64) or by the entire GAPDH protein (36). Others proposed that RNA binds to the tetramer groove (39). To identify residues important for RNA binding, we mutated several GAPDH residues located in the positively charged groove or at the dimer interface. Of the mutants made, only T229K could be expressed and purified in quantity and quality suitable for our biophysical and structural characterizations. As seen for wild-type GAPDH, two complexes were observed with T229K GAPDH. However, the mutant displayed lower affinity at the second binding step (KD2). Furthermore, the mutant promoted structural changes in the bound RNA ligand that were not observed with wild-type GAPDH.
The effects of the T229K mutation on the formation of complex 2 and on the structure of the associated RNA ligand were surprising as the mutation is located far from the NAD+ binding site. Previous studies suggested that this mutation led to GAPDH monomerization (57). We determined via biophysical and structural analyses that wild-type and T229K GAPDH presented no significant difference in terms of overall structure and oligomerization at concentrations above 0.5 mg/ml (3.5 μm tetramer). In addition, ESI-MS revealed that T229K GAPDH was tetrameric at concentrations between 0.2 and 60 μm, in contrast with previous studies. In our hands, we observed a mixture of oligomeric species for wild-type GAPDH only after repeated freezing-thawing cycles, suggesting that the oligomeric integrity of the protein had been compromised (data not shown). This is in agreement with previous studies showing that tetrameric GAPDH could be easily dissociated into monomers at low temperature (65, 66) and that freeze-thaw cycles significantly affected protein interfaces (67–69). Based on our results, we concluded that the wild-type and T229K GAPDH proteins were mostly tetrameric in the concentration range used for EMSA, fluorescence anisotropy, and FRET.
The crystal structure of the T229K mutant showed a cascade of conformational changes in regions neighboring the mutation at the dimer interface. This result was supported by HDX-MS experiments showing that the regions surrounding the mutation were more dynamic in the mutant protein. HDX-MS also allowed us to observe changes in regions participating in the tetramer interface and NAD+ binding region and that were not observed in the crystal structure. This difference is likely due to the fact that the protein conformation is somewhat more constrained in the tightly packed crystal structure compared with the protein in solution. Regardless, our results demonstrated that (i) RNA binding may be affected by dynamic changes in key interfacial regions and (ii) the mutation did not affect RNA binding via disruption of oligomerization as previously thought. In light of our results, we propose several scenarios to explain the effect of the mutation on RNA binding.
Direct Effect
if Thr-229 is part of the RNA binding site. On the one hand, we reason that mutation of Thr to the positively charged Lys would likely increase RNA binding via electrostatic interactions. On the other hand, because T229K induces a cascade of conformational changes at the dimer and tetramer interfaces, we propose that these regions directly participate in RNA binding.
Indirect Effect Linked to GAPDH Oligomerization
T229K affects regions at the dimer and tetramer interfaces, suggesting that the oligomerization state of GAPDH may be important for RNA binding. Although we do not know yet whether RNA binding disrupts GAPDH oligomerization, our results unambiguously demonstrate that the T229K mutation alone does not disrupt oligomerization.
Indirect Effect Linked to the NAD+ Binding Site
T229K promotes concerted long range structural and dynamic changes in regions located close to the NAD+ binding site and in the positively charged groove. Furthermore, the mutant enzyme displayed decreased NAD+ binding compared with wild-type GAPDH. NAD+ is strategically positioned close to the tetramer interface, and previous studies have shown that the cofactor favors GAPDH tetramerization (65, 70). It is, therefore, possible that RNA binds in or close to the NAD+ binding site, as proposed by others (13, 30). However some of these experiments may have been biased by technical limitations, notably, that the UV-cross-linking experiments used to detect GAPDH-RNA complexes would be inhibited by UV absorbance from the high concentrations of adenosine nucleotides (ATP, NAD+, NADH) used to demonstrate competition for the NAD+ binding site. Quantitative assays to unequivocally determine whether NAD+ inhibits RNA binding to GAPDH are necessary.
Regardless, our studies reveal that the T229K mutation promotes long-range dynamic changes in the conformation of protein regions involved in NAD+ binding leading to the disruption of the integrity of interfacial protein loops. Our combined mass spectrometry results showed that the mutation at the dimer interface allosterically perturbed the tetramer interface and the NAD+ binding site, thus linking GAPDH interfaces and cofactor binding. Because the T229K mutation promotes short range and long range conformational changes along the P axis of the GAPDH tetramer, we hypothesize that these regions play a key role in RNA binding (Fig. 6). Based on our combined results, we propose a model for RNA binding to wild-type and T229K GAPDH (Fig. 9). In the absence of protein and Mg2+ ions and in the presence of 50 mm KCl, the ARE38 probe adopts a partially folded structure with a distance separating the probes estimated to be 61 Å (44). Complex 1 would correspond to RNA bound to one GAPDH tetramer via the dimer and tetramer interfaces and along the GAPDH P axis. This binding mode for RNA is strongly supported by recent studies showing that the conserved region 250–258 in yeast GAPDH (region 251–260 in human GAPDH) is involved in AU-RNA binding (71). This region is strategically located adjacent to the dimer interface along the P axis and directly across from the T229K mutation (Fig. 6b). Thus, we propose that the dimer interface acts as a platform for RNA binding. Our intramolecular FRET experiments showed that only the T229K mutant significantly altered the structure of the bound ARE38 via condensation of the RNA interterminal distance by 11 Å. The mechanism of RNA remodeling by T229K is not known, but we propose that the mutation affected the conformation of the bound RNA without significantly affecting the binding affinity of complex 1. The resulting T229K-RNA complex 1 would then represent a suboptimal substrate for the second binding event compared with wild-type GAPDH-RNA. Further studies are needed to confirm this model.
FIGURE 9.
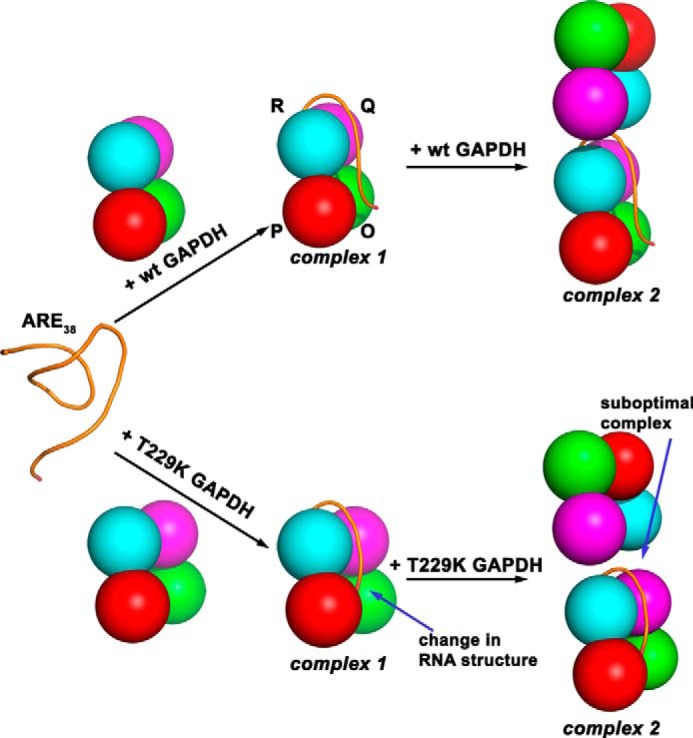
Models for RNA binding to GAPDH. We propose that ARE38 substrate derived from TNF-α mRNA 3′-UTR binds at the dimer interface and along the GAPDH tetramer P axis close to the NAD+ binding site to form complex 1 for both wild-type and T229K GAPDH. However, the T229K mutation promotes dynamic changes in protein regions at the dimer and tetramer interfaces and induces a structural change in the RNA conformation (condensation) and shown in our model as a different conformation of RNA in complex 1. Increasing concentrations of wild-type GAPDH lead to formation of complex 2. For T229K GAPDH, the change in RNA conformation and dynamic changes of protein regions impair similar formation of complex 2 (differences with wild-type GAPDH are indicated by arrows for T229K complexes 1 and 2).
Overall, our results suggested that a single interfacial mutation had a significant effect on RNA binding to GAPDH. This mutation was proposed to mimic the posttranslational O-GlcNacylation of Thr-229 by O-GlcNac transferase (57), but it is not known whether this GAPDH modification regulates RNA binding. Other posttranslational modifications of GAPDH have been identified near the dimer interface, including acetylation (58, 72), phosphorylation (59, 60), and O-GlcNacylation by other glycosyltransferases (73). In some cases, these modifications have been shown to alter GAPDH localization, interaction with other proteins, and possibly its oligomerization state. Therefore, we speculate that posttranslational modifications may regulate the RNA binding function of GAPDH in the cell. Studies are under way to confirm this hypothesis. Because GAPDH is involved in so many distinct cellular functions, posttranslational modifications in response to specific cell stimuli would provide an elegant switch to turn on/off specific GAPDH functions to regulate cellular processes.
In conclusion, our multidisciplinary results have extended the repertoire of AU-rich RNAs targeted by GAPDH and have revealed new aspects of the mostly uncharacterized molecular details of GAPDH-RNA interactions. In addition, our findings have extended our understanding of the complex interplay between the NAD+ binding site and the various protein interfaces in GAPDH.
Acknowledgments
We thank Franziska Seeger and Regina Ulis for cloning of the human GAPDH gene in the pET21 vector and Jacob P. Neal and John Chavis III for help with GAPDH expression and purification. We thank the University of Maryland School of Pharmacy Mass Spectrometry Center. We thank Prof. Richard Karpel for insightful discussions and critical reading of the manuscript.
This work was supported in part by National Institutes of Health Grant R01 CA102428 (to G. M. W.). This work was also supported in part by National Science Foundation Grant DBI 0923551 (to V. H. W.) and the American Heart Association Grant PRE6900008 (to B. E. Z.).
The atomic coordinates and structure factors (codes 4WNC and 4WNI) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- ARE
- adenine-uridine rich element
- HDX
- hydrogen/deuterium exchange
- ESI
- electrospray mass spectrometry
- CID
- collision-induced dissociation
- SAXS
- small-angle x-ray scattering
- SEC
- size-exclusion chromatography
- Cy3
- cyanine 3
- Fl
- fluorescein
- IM
- ion mobility
- r.m.s.d.
- root mean square deviation.
REFERENCES
- 1. White E. J., Brewer G., Wilson G. M. (2013) Post-transcriptional control of gene expression by AUF1: mechanisms, physiological targets, and regulation. Biochim. Biophys. Acta 1829, 680–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Parker R., Song H. (2004) The enzymes and control of eukaryotic mRNA turnover. Nat. Struct. Mol. Biol. 11, 121–127 [DOI] [PubMed] [Google Scholar]
- 3. Wilusz C. J., Wormington M., Peltz S. W. (2001) The cap-to-tail guide to mRNA turnover. Nat. Rev. Mol. Cell Biol. 2, 237–246 [DOI] [PubMed] [Google Scholar]
- 4. Wilson G. M., Brewer G. (1999) The search for trans-acting factors controlling messenger RNA decay. Prog. Nucleic Acid Res. Mol. Biol. 62, 257–291 [DOI] [PubMed] [Google Scholar]
- 5. Shim J., Karin M. (2002) The control of mRNA stability in response to extracellular. Mol. Cells 14, 323–331 [PubMed] [Google Scholar]
- 6. Barreau C. (2005) AU-rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 33, 7138–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Myer V. E. (1997) Identification of HuR as a protein implicated in AUUUA-mediated mRNA decay. EMBO J. 16, 2130–2139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Peng S. S.-Y., Chen C. Y., Xu N., Shyu A. B. (1998) RNA stabilization by the AU-rich element binding protein, HuR, an ELAV protein. EMBO J. 17, 3461–3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lai W. S., Carballo E., Strum J. R., Kennington E. A., Phillips R. S., Blackshear P. J. (1999) Evidence that tristetraprolin binds to AU-rich elements and promotes the deadenylation and destabilization of tumor necrosis factor α mRNA. Mol. Cell. Biol. 19, 4311–4323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brewer G., Saccani S., Sarkar S., Lewis A., Pestka S. (2003) Increased interleukin-10 mRNA stability in melanoma cells is associated with decreased levels of A + U-rich element binding factor AUF1. J. Interferon Cytokine Res. 23, 553–564 [DOI] [PubMed] [Google Scholar]
- 11. Sarkar B., Xi Q., He C., Schneider R. J. (2003) Selective degradation of AU-rich mRNAs promoted by the p37 AUF1 protein isoform. Mol. Cell. Biol. 23, 6685–6693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zucconi B. E., Ballin J. D., Brewer B. Y., Ross C. R., Huang J., Toth E. A., Wilson G. M. (2010) Alternatively expressed domains of AU-rich element RNA-binding protein 1 (AUF1) regulate RNA-binding affinity, RNA-induced protein oligomerization, and the local conformation of bound RNA ligands. J. Biol. Chem. 285, 39127–39139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Nagy E., Rigby W. (1995) Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD(+)-binding region (Rossmann fold). J. Biol. Chem. 270, 2755–2763 [DOI] [PubMed] [Google Scholar]
- 14. Jenkins J. L., Tanner J. J. (2006) High-resolution structure of human d-glyceraldehyde-3-phosphate dehydrogenase. Acta Crystallogr. D Biol. Crystallogr. 62, 290–301 [DOI] [PubMed] [Google Scholar]
- 15. Karpel R. L., Burchard A. C. (1981) A basic isozyme of yeast glyceraldehyde-3-phosphate dehydrogenase with nucleic acid helix-destabilizing activity. Biochim. Biophys. Acta 654, 256–267 [DOI] [PubMed] [Google Scholar]
- 16. Perucho M., Salas J., Salas M. (1977) Identification of the mammalian DNA-binding protein P8 as glyceraldehyde 3-phosphate dehydrogenase. Eur. J. Biochem. 81, 557–562 [DOI] [PubMed] [Google Scholar]
- 17. Perucho M., Salas J., Salas M. (1980) Study of the interaction of glyceraldehyde 3-phosphate dehydrogenase with DNA. Biochim. Biophys. Acta 606, 181–195 [DOI] [PubMed] [Google Scholar]
- 18. Singh R., Green M. R. (1993) Sequence-specific binding of transfer RNA by glyceraldehyde-3-phosphate dehydrogenase. Science 259, 365–368 [DOI] [PubMed] [Google Scholar]
- 19. Schultz D. E., Hardin C. C., Lemon S. M. (1996) Specific interaction of glyceraldehyde 3-phosphate dehydrogenase with the 5′-nontranslated RNA of hepatitis A virus. J. Biol. Chem. 271, 14134–14142 [DOI] [PubMed] [Google Scholar]
- 20. Baxi M. D., Vishwanatha J. K. (1995) Uracil DNA-glycosylase/glyceraldehyde-3-phosphate dehydrogenase is an Ap4A binding protein. Biochemistry 34, 9700–9707 [DOI] [PubMed] [Google Scholar]
- 21. Hara M. R., Agrawal N., Kim S. F., Cascio M. B., Fujimuro M., Ozeki Y., Takahashi M., Cheah J. H., Tankou S. K., Hester L. D., Ferris C. D., Hayward S. D., Snyder S. H., Sawa A. (2005) S-Nitrosylated GAPDH initiates apoptotic cell death by nuclear translocation following Siah1 binding. Nat. Cell Biol. 7, 665–674 [DOI] [PubMed] [Google Scholar]
- 22. Sen N., Hara M. R., Kornberg M. D., Cascio M. B., Bae B. I., Shahani N., Thomas B., Dawson T. M., Dawson V. L., Snyder S. H., Sawa A. (2008) Nitric oxide-induced nuclear GAPDH activates p300/CBP and mediates apoptosis. Nat. Cell Biol. 10, 866–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Muronetz V. I., Wang Z. X., Keith T. J., Knull H. R., Srivastava D. K. (1994) Binding constants and stoichiometries of glyceraldehyde 3-phosphate dehydrogenase-tubulin complexes. Arch. Biochem. Biophys. 313, 253–260 [DOI] [PubMed] [Google Scholar]
- 24. Glaser P. E., Gross R. W. (1995) Rapid Plasmenylethanolamine-selective fusion of membrane bilayers catalyzed by an isoform of glyceraldehyde-3-phosphate dehydrogenase: discrimination between glycolytic and fusogenic roles of individual isoforms. Biochemistry 34, 12193–12203 [DOI] [PubMed] [Google Scholar]
- 25. Nakajima H., Amano W., Fujita A., Fukuhara A., Azuma Y. T., Hata F., Inui T., Takeuchi T. (2007) The active site cysteine of the proapoptotic protein glyceraldehyde-3-phosphate dehydrogenase is essential in oxidative stress-induced aggregation and cell death. J. Biol. Chem. 282, 26562–26574 [DOI] [PubMed] [Google Scholar]
- 26. Mazzola J. L., Sirover M. A. (2002) Alteration of nuclear glyceraldehyde-3-phosphate dehydrogenase structure in Huntington's disease fibroblasts. Mol. Brain Res. 100, 95–101 [DOI] [PubMed] [Google Scholar]
- 27. Chakravarti R., Aulak K. S., Fox P. L., Stuehr D. J. (2010) GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc. Natl. Acad. Sci. U.S.A. 107, 18004–18009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sirover M. A. (1999) New insights into an old protein: the functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys. Acta 1432, 159–184 [DOI] [PubMed] [Google Scholar]
- 29. Sirover M. A. (2011) On the functional diversity of glyceraldehyde-3-phosphate dehydrogenase: Biochemical mechanisms and regulatory control. Biochim. Biophys. Acta 1810, 741–751 [DOI] [PubMed] [Google Scholar]
- 30. Nagy E., Henics T., Eckert M., Miseta A., Lightowlers R. N., Kellermayer M. (2000) Identification of the NAD+-binding fold of glyceraldehyde-3-phosphate dehydrogenase as a novel RNA-binding domain. Biochem. Biophys. Res. Commun. 275, 253–260 [DOI] [PubMed] [Google Scholar]
- 31. De B. P., Gupta S., Zhao H., Drazba J. A., Banerjee A. K. (1996) Specific interaction in vitro and in vivo of glyceraldehyde-3-phosphate dehydrogenase and la protein with cis-acting rnas of human parainfluenza virus type 3. J. Biol. Chem. 271, 24728–24735 [DOI] [PubMed] [Google Scholar]
- 32. Bonafé N., Gilmore-Hebert M., Folk N. L., Azodi M., Zhou Y., Chambers S. K. (2005) Glyceraldehyde-3-phosphate dehydrogenase binds to the AU-rich 3′-untranslated region of colony-stimulating factor-1 (CSF-1) messenger RNA in human ovarian cancer cells: possible role in CSF-1 posttranscriptional regulation and tumor phenotype. Cancer Res. 65, 3762–3771 [DOI] [PubMed] [Google Scholar]
- 33. Zhou Y., Yi X., Stoffer J. B., Bonafe N., Gilmore-Hebert M., McAlpine J., Chambers S. K. (2008) The multifunctional protein glyceraldehyde-3-phosphate dehydrogenase is both regulated and controls colony-stimulating factor-1 messenger RNA stability in ovarian cancer. Mol Cancer Res. 6, 1375–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chang C.-H., Curtis J. D., Maggi L. B., Jr., Faubert B., Villarino A. V., O'Sullivan D., Huang S. C.-C., van der Windt G. J., Blagih J., Qiu J., Weber J. D., Pearce E. J., Jones R. G., Pearce E. L. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Wang R. Y., Nagy P. D. (2008) Tomato bushy stunt virus co-opts the RNA-binding function of a host metabolic enzyme for viral genomic RNA synthesis. Cell Host Microbe 3, 178–187 [DOI] [PubMed] [Google Scholar]
- 36. Rodríguez-Pascual F., Redondo-Horcajo M., Magán-Marchal N., Lagares D., Martínez-Ruiz A., Kleinert H., Lamas S. (2008) Glyceraldehyde-3-phosphate dehydrogenase regulates endothelin-1 expression by a novel, redox-sensitive mechanism involving mRNA stability. Mol. Cell. Biol. 28, 7139–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Backlund M., Paukku K., Daviet L., De Boer R. A., Valo E., Hautaniemi S., Kalkkinen N., Ehsan A., Kontula K. K., Lehtonen J. Y. (2009) Posttranscriptional regulation of angiotensin II type 1 receptor expression by glyceraldehyde 3-phosphate dehydrogenase. Nucleic Acids Res. 37, 2346–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Burd C. G., Dreyfuss G. (1994) Conserved structures and diversity of functions of RNA binding proteins. Science 265, 615–621 [DOI] [PubMed] [Google Scholar]
- 39. Carmona P., Rodríguez-Casado A., Molina M. (1999) Conformational structure and binding mode of glyceraldehyde-3-phosphate dehydrogenase to tRNA studied by Raman and CD spectroscopy. Biochim. Biophys. Acta 1432, 222–233 [DOI] [PubMed] [Google Scholar]
- 40. Evguenieva-Hackenberg E., Schiltz E., Klug G. (2002) Dehydrogenases from all three domains of life cleave RNA. J. Biol. Chem. 277, 46145–46150 [DOI] [PubMed] [Google Scholar]
- 41. Wilson G. M., Sutphen K., Moutafis M., Sinha S., Brewer G. (2001) Structural remodeling of an A + U-rich RNA element by cation or AUF1 binding. J. Biol. Chem. 276, 38400–38409 [DOI] [PubMed] [Google Scholar]
- 42. Wilson G. M., Sun Y., Lu H., Brewer G. (1999) Assembly of AUF1 Oligomers on U-rich RNA targets by sequential dimer association. J. Biol. Chem. 274, 33374–33381 [DOI] [PubMed] [Google Scholar]
- 43. Wilson G. M., Sutphen K., Chuang K.-y., Brewer G. (2001) Folding of A+U-rich RNA elements modulates AUF1 binding: potential roles in regulation of mRNA turnover. J. Biol. Chem. 276, 8695–8704 [DOI] [PubMed] [Google Scholar]
- 44. Fialcowitz E. J., Brewer B. Y., Keenan B. P., Wilson G. M. (2005) A hairpin-like structure within an AU-rich mRNA-destabilizing element regulates trans-factor binding selectivity and mRNA decay kinetics. J. Biol. Chem. 280, 22406–22417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lakowicz J. (1999) in Principles of Fluorescence Spectroscopy, 2nd Ed., pp. 367–394, Kluwer Academic/Plenum, New York [Google Scholar]
- 46. Norman D. G., Grainger R. J., Uhrín D., Lilley D. M. (2000) Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry 39, 6317–6324 [DOI] [PubMed] [Google Scholar]
- 47. Otwinowski Z., Minor W. (1997) Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 [DOI] [PubMed] [Google Scholar]
- 48. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Adams P. D., Grosse-Kunstleve R. W., Hung L. W., Ioerger T. R., McCoy A. J., Moriarty N. W., Read R. J., Sacchettini J. C., Sauter N. K., Terwilliger T. C. (2002) PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr. D Biol. Crystallogr. 58, 1948–1954 [DOI] [PubMed] [Google Scholar]
- 50. Emsley P., Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 [DOI] [PubMed] [Google Scholar]
- 51. Konarev P. V., Volkov V. V., Sokolova A. V., Koch M. H. J., Svergun D. I. (2003) PRIMUS: a Windows PC-based system for small-angle scattering data analysis. J. Appl. Crystallogr. 36, 1277–1282 [Google Scholar]
- 52. Svergun D. I. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 495–503 [Google Scholar]
- 53. Kozin M. B., Svergun D. I. (2001) Automated matching of high- and low-resolution structural models. J. Appl. Crystallogr. 34, 33–41 [Google Scholar]
- 54. Houde D., Berkowitz S. A., Engen J. R. (2011) The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 100, 2071–2086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhou M., Dagan S., Wysocki V. (2012) Protein subunits released by surface collisions of noncovalent complexes: native-like compact structures revealed by ion mobility mass spectrometry. Angew. Chem. Int. Ed. Engl. 51, 4336–4339 [DOI] [PubMed] [Google Scholar]
- 56. Galhena A. S., Dagan S., Jones C. M., Beardsley R. L., Wysocki V. H. (2008) Surface-induced dissociation of peptides and protein complexes in a quadrupole/time-of-flight mass spectrometer. Anal. Chem. 80, 1425–1436 [DOI] [PubMed] [Google Scholar]
- 57. Park J., Han D., Kim K., Kang Y., Kim Y. (2009) O-GlcNAcylation disrupts glyceraldehyde-3-phosphate dehydrogenase homo-tetramer formation and mediates its nuclear translocation. Biochim. Biophys. Acta 1794, 254–262 [DOI] [PubMed] [Google Scholar]
- 58. Ventura M., Mateo F., Serratosa J., Salaet I., Carujo S., Bachs O., Pujol M. J. (2010) Nuclear translocation of glyceraldehyde-3-phosphate dehydrogenase is regulated by acetylation. Int. J. Biochem. Cell Biol. 42, 1672–1680 [DOI] [PubMed] [Google Scholar]
- 59. Yogalingam G., Hwang S., Ferreira J. C., Mochly-Rosen D. (2013) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase C δ (δPKC) inhibits mitochondrial elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J. Biol. Chem. 288, 18947–18960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Huang Q., Lan F., Zheng Z., Xie F., Han J., Dong L., Xie Y., Zheng F. (2011) Akt2 kinase suppresses glyceraldehyde-3-phosphate dehydrogenase (GAPDH)-mediated apoptosis in ovarian cancer cells via phosphorylating GAPDH at threonine 237 and decreasing its nuclear translocation. J. Biol. Chem. 286, 42211–42220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jia J., Arif A., Willard B., Smith J. D., Stuehr D. J., Hazen S. L., Fox P. L. (2012) Protection of extraribosomal {RPL13a} by {GAPDH} and dysregulation by S-nitrosylation. Mol. Cell 47, 656–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang T., Kruys V., Huez G., Gueydan C. (2002) AU-rich elements-mediated translational control: complexity and multiple activities of trans-acting factors. Biochem. Soc. Trans. 30, 952–958 [DOI] [PubMed] [Google Scholar]
- 63. Wang E., Ma W.-J., Aghajanian C., Spriggs D. R. (1997) Posttranscriptional regulation of protein expression in human epithelial carcinoma cells by adenine-uridine-rich elements in the 3′-untranslated region of tumor necrosis factor-α messenger RNA. Cancer Res. 57, 5426–5433 [PubMed] [Google Scholar]
- 64. Nicholls C., Pinto A. R., Li H., Li L., Wang L., Simpson R., Liu J.-P. (2012) Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) induces cancer cell senescence by interacting with telomerase RNA component. Proc. Natl. Acad. Sci. 109, 13308–13313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Constantinides S. M., Deal W. C. (1969) Reversible dissociation of tetrameric rabbit muscle glyceraldehyde 3-phosphate dehydrogenase into dimers or monomers by adenosine triphosphate. J. Biol. Chem. 244, 5695–5702 [PubMed] [Google Scholar]
- 66. Stancel G. M., Deal W. C., Jr. (1969) Reversible dissociation of yeast glyceraldehyde 3-phosphate dehydrogenase by adenosie triphosphate. Biochemistry 8, 4005–4011 [DOI] [PubMed] [Google Scholar]
- 67. Griko Y. V., Privalov P. L., Sturtevant J. M., Venyaminov SYu. (1988) Cold denaturation of staphylococcal nuclease. Proc. Natl. Acad. Sci. 85, 3343–3347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chang B. S., Kendrick B. S., Carpenter J. F. (1996) Surface-induced denaturation of proteins during freezing and its inhibition by surfactants. J. Pharm. Sci. 85, 1325–1330 [DOI] [PubMed] [Google Scholar]
- 69. Zhang A., Singh S. K., Shirts M. R., Kumar S., Fernandez E. J. (2012) Distinct aggregation mechanisms of monoclonal antibody under thermal and freeze-thaw stresses revealed by hydrogen exchange. Pharm. Res. 29, 236–250 [DOI] [PubMed] [Google Scholar]
- 70. Hoagland V. D., Jr., Teller D. C. (1969) Influence of substrates on the dissociation of rabbit muscle d-glyceraldehyde-3-phosphate dehydrogenase. Biochemistry 8, 594–602 [DOI] [PubMed] [Google Scholar]
- 71. Kramer K., Sachsenberg T., Beckmann B. M., Qamar S., Boon K.-L., Hentze M. W., Kohlbacher O., Urlaub H. (2014) Photo-cross-linking and high-resolution mass spectrometry for assignment of RNA-binding sites in RNA-binding proteins. Nat. Methods 11, 1064–1070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Seo J., Jeong J., Kim Y. M., Hwang N., Paek E., Lee K.-J. (2008) Strategy for comprehensive identification of post-translational modifications in cellular proteins, including low abundant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J. Proteome Res. 7, 587–602 [DOI] [PubMed] [Google Scholar]
- 73. Gao X., Wang X., Pham T. H., Feuerbacher L. A., Lubos M.-L., Huang M., Olsen R., Mushegian A., Slawson C., Hardwidge P. R. (2013) NleB, a bacterial effector with glycosyltransferase activity, targets GAPDH function to inhibit NF-κB activation. Cell Host Microbe 13, 87–99 [DOI] [PMC free article] [PubMed] [Google Scholar]