

Abstract
Antithrombin III (AT III) is a plasmatic α-glicoprotein formed by a single peptidic chain. AT III inhibits thrombin (first target) and free Xa, IXa ,VIIa plasmatic factors. In plasma AT III is found under two forms: α-antithrombin and β-antithrombin. Deficiency of AT III represents a risk factor for thromboembolic disease. There are known both quantitative and qualitative AT III deficiencies. Incidence of AT III inherited deficiency is relative rare (1:10.000). Acquired deficiency of AT III is more frequent. The transmission of AT III deficiency is autosomal dominant with variable shield factor. Homozygous is incompatible with life (death immediately after birth). Thrombosis appears around the age of twenty years, and in 4-5 decades of life 2/3 of patients are symptomatics. Traumatisms, surgical interventions, estrogenic treatment, precipitated thrombotic complications. Obesity and dyslipidemic syndrome are risk factors. Thrombosis affects the venous system at these patients. Arterial thrombosis are less reported. The most frequent localisations are: the veins of the legs, mesenteric veins, cave veins, superficial periombilical veins. Treatment of AT III deficiency is: administration of AT III concentrates (with a plasmatic level by 80% from normal value) and heparinotherapy. The treatment with AT III concentrates is for patients which faced major surgical interventions, pregnant women with AT III deficiency. The women with AT III deficiency should avoid the utilisation of oral contraceptives.
Keywords: AT III deficiency, venous thrombosis, AT III concentate, heparinotherapy
Introduction
AT III is the most important plasmatic inhibitor for the activated coagulation factors [1 ,2 ]. Primary target is certainly thrombin followed by inhibition of the free factors of coagulation : X a factor, IX a factor, VII a factor.
Deficiency of AT III represents a risk factor for thromboembolic disease[3 ,4 ]. Both qualitative and quantitative deficiencies of AT III are known. The incidence of inherited deficiency is relative rare (1:10.000). Acquired deficiencies are more frequent. A decreased activity of AT III may be resolved by the heparin or low molecular weight heparin administration. Determination of the level of AT III should be performed before and in the time of heparin administration [5 ]. AT III is partially consumed during heparin therapy. In a lot of cases, there is indicated a fresh level of refined AT III and plasma. The substitution treatment with AT III and the normalisation level in the plasma have beneficial effects in sepsis and inflamatory diseases through decreased concentrations of some proinflamatory cytokines ( IL6, IL8)[6, 7 ].
Case report
We present the case of a 28-years old man, with second degree obesity, dyslipidemic syndrome, healthy carrier of HBs antigen. At 26 years old, the patient faced a surgical intervention for thrombosis of the portal vein and at 28 years he was operated for thrombosis of mesenteric veins. The evolution was favourable after treatment with 1 and 1/2 cp of Sintrom /day, with a Quick time between 25-30% and INR value of 2,5-3,5.
The patient went to the Clinic of Hematology from Craiova where the putative diagnosis was thrombophilia.
There was determined AT III concentration, which represented 40% of the normal value; concentrations of protein C, S and Leiden V factor were normal.
Discussion
Deficiency of AT III is autosomally dominant transmitted with variable shield factor. In 1965 Egeberg described the first case of AT III deficiency. Until now, there have been found in literature many other cases. It is estimated that 2-4% through thrombotic episodes, encountered before 50 years are correlated with genetic deficiency of AT III. The majority of the patients are heterozygous with plasmatic levels of AT III approximatively 50% from normal values. Homozygous status is incompatible with life (severe venous thrombosis immediately after birth).
Inherited types of AT III deficiencies are:
Type I of AT III deficiency is quantitative, the concentration and activity of AT III are decreased; is a result of reduced synthesis of biologically normal protease inhibitor molecules.
Type II - deficiency of AT III is qualitative (the substitution of an aminoacid with another aminoacid), resulting a functional deficit of the factor. It is produced by a discrete molecular defect without the protease inhibitor.
Lane classified this type of deficiency in more variants:
a. type II RS (realixe site) included mutations which affect the aminoacids of the clivation zone of the AT III by thrombin (between Arg 393 and Ser 394) or some adjacent aminoacids.
b. type II HBS (heparin binding site)- is independent of the interaction AT III-heparin[8].
c. typel II PE (pleiotropic effect) - multiple mutations result in abnormalities to the reactive site as well as binding sites.
The heterozygous patients with familial deficiency of AT III type I presented recurrent thrombosis; the ones with HBS type deficiency has not had an increased incidence of thrombosis, although there were cases in literature described by the descendants of these patients with thrombosis before 20 years [9].
Deficiency of AT III type II may be segregated by special methods such as the determination of heparin binding properties [10].
Acquired deficiency of AT III are present in: cirrhosis, liver cancer, nephropathy, DIC, sepsis, preeclampsia, treatment with L-asparaginaze or oral contraceptives, poli traumatisms, severe intoxications, heparinotherapy.
In deficiency of AT III anticoagulant activity is ineffectual, fact showed by a defective extention of aPTT or an important increase of the markers: soluble fibrin, 1+2 factor.
AT III activity must be followed in the relation by the activity of procoagulant factors.
Clinical manifestations in the deficiency of AT III: thrombotic manifestations appear around the age of twenty, and in the 4-5 decades of life 2/3 of patients are symptomatics. Thrombotic accidents are precipited by traumatisms, surgery, estrogenic treatment [11]. Obesity and dyslipidemic syndromes are important increasing factors. Thrombosis affect especially the venous system and more the rarely arterial system.. The veins of the legs, mesenteric veins and cave veins are frequently affected. Thrombosis may affect the splenic vein or superficial periombilical veins [12].
AT III determination it is made via activated X factor or activated II factor, in the presence of a normal concentration of heparin (heparin-cofactor method).
Diagnosis of AT III deficiency involves familial study (siblings) and the elimination of a pathological status which induced a decreased level of plasmatic concentration of AT III.: decrease liver synthesis of proteins, proteic deficiencies, chronic nephropathy (nephrotic syndrom with urinary wastage of AT III), abusive intake of AT III (DIC, acute leukemias, hemolytic uremic syndrome, sepsis with Gramm negative).
Treatment of AT III deficiency is made with:
1. AT III concentrates contain 1000 UI of AT III ( KiberninR HS 1000)
Administration of 50 UI of AT III/Kgc increases plasmatic concentration of AT III and reaches approximately a 120% value at a heterozygous patient with a initial plasmatic concentration of 50%. The plasmatic level of AT III must be maintained at levels above 80%.
Treatment with concentrates of AT III is specific for the patients which faced surgical interventions and pregnant women with AT III deficiency (AT III concentrate + heparin prophylaxis)[13].
2. Heparin therapy is for all patients with AT III deficiency, including pregnant women with risk of embriopathy at these preparates. Women with AT III deficiency should avoid administration of oral contraceptives [14].
In the context of frequent thrombotic accidents at a young age and decreased level of AT III the established diagnosis was of deficiency of AT III; familial study showed an inherited nature of deficiency (patient's father was heterozygous but without thrombotic accidents and the descendants of the patient, aged between 2-6 years, presenting a 50% of deficiency of AT IIII, without thrombosis until those ages)(Fig. 1). The prominence of the deficiency of AT III heterozygous form at the patient as well as his descendants is important for the prevention of thrombotic accidents in the future.
Fig.1.
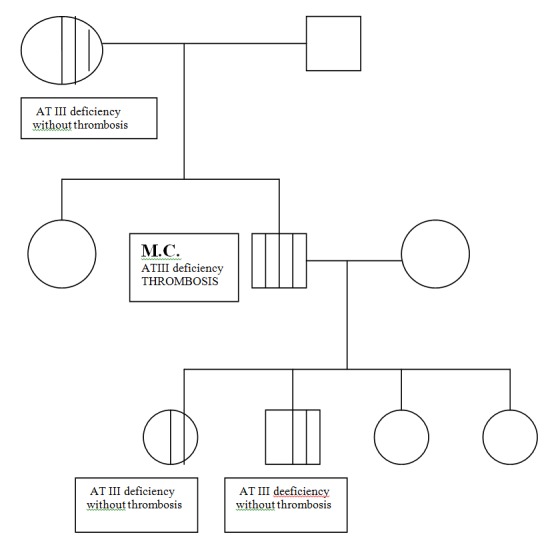
Familial study of patient M.C., 28 years old, with inherited deficiency of AT III heterozygous form and thrombosis
Conclusions
In the presence of spontaneous venous thromboembolism with uncommon localisations (mesenteric veins, portal vein, renal and retinal veins) in young patients inherited antithrombin deficiency must be considered. Obesity, dyslipidemic syndrome, immobilization, surgical procedures, acute or chronic infectious diseases increase risk of thrombosis .
References
- 1.Cucuianu M. Hemostaza. Cluj-Napoca: Ed.Dacia; 1994. pp. 216–222. [Google Scholar]
- 2.Kolde MJ. Haemostasis. Basel: Ed. Pentapharm Ltd; 2004. pp. 37– 39. [Google Scholar]
- 3.Furie B. Pathogenesis of thrombosis. J.Biol.Chem. 2009;267:255–263. doi: 10.1182/asheducation-2009.1.255. [DOI] [PubMed] [Google Scholar]
- 4.Zakai HA, et al. Inflammation and hemostasis biomarkers and cardiovascular risk in the elderly: The cardiovascular health study. J.Thromb.Haemost. 2007;5:1128–1135. doi: 10.1111/j.1538-7836.2007.02528.x. [DOI] [PubMed] [Google Scholar]
- 5.Glynn RJ, et al. Effect of low dose aspirin on the occurence of venous thrombo -embolism. Ann J. Intern. Med. 2007;147:525–533. doi: 10.7326/0003-4819-147-8-200710160-00004. [DOI] [PubMed] [Google Scholar]
- 6.Everett BM, et al. Lipid biomarkers, hormone and the risk of venous thrombo embolism in women. J.Thromb.Haemost. 2009;7:588–596. doi: 10.1111/j.1538-7836.2009.03302.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mant MJ, et al. Severe acute DIC. An . Journ.Med. 1979;67:557–563. doi: 10.1016/0002-9343(79)90225-0. [DOI] [PubMed] [Google Scholar]
- 8.Oven E, et al. Body mass index and the risk of venous thrombosis among post menopausal women. J. Thromb.Haemost. 2006;83:545–548. [Google Scholar]
- 9.Lippi G., et al. Activated partial thromboplastin time : new tricks for an old dogma. Semin Thromb. Hemost. 2008;34:604–611. doi: 10.1055/s-0028-1104539. [DOI] [PubMed] [Google Scholar]
- 10.Brenne KA, et al. Haemostatic changes in pregnancy. Best Pract. Clin. Haematol. 2003;16:153– 168. doi: 10.1016/s1521-6926(03)00021-5. [DOI] [PubMed] [Google Scholar]
- 11.James AH, et al. Thrombosis, thrombophilia and thromboprophylaxis in pregnancy. Clin.Adv.Haematol.Oncol. 2005;3:187–197. [PubMed] [Google Scholar]
- 12.James AH. Venous thromboembolism in pregnancy arterioscler. Thromb.Vasc.Biol. 2009;29:326–331. doi: 10.1161/ATVBAHA.109.184127. [DOI] [PubMed] [Google Scholar]
- 13.Chen YM, et al. Allosteric disulfide bonds in thrombosis and thrombolysis. J.Thromb.Haemost. 2006;4:2533–2541. doi: 10.1111/j.1538-7836.2006.02236.x. [DOI] [PubMed] [Google Scholar]
- 14.Robertson L, et al. hrombophilia in pregnancy : A systematic Review. Br.J.Haematol. 2006;132:171–196. doi: 10.1111/j.1365-2141.2005.05847.x. [DOI] [PubMed] [Google Scholar]