

Abstract
The study of viruses in high containment offers unique challenges for technology-intense approaches. These approaches include high-throughput screening for small-molecule antivirals and genetic perturbation-based screens for host factors required for viral replication. Here, we describe the use of whole-genome scale pooled short hairpin RNA (shRNA) libraries to screen for host factors necessary for viral infection at BSL2, and the transition of this technique into the BSL4 environment. Pooled screening provides a unique way to circumvent many of the technological challenges associated with other high-throughput screening approaches in high containment. Our pooled screening approach identified host factors involved in the replication of orthopoxviruses (Vaccinia and Monkeypox) and filoviruses (Ebola and Marburg) under conditions that enable straightforward screen-to-follow-up approaches.
Introduction
The history of human disease is populated with outbreaks and endemic assaults from highly pathogenic viruses. Smallpox, caused by the poxvirus variola, caused more than 300 million deaths before its eradication from nature in the 1970s.1,2 The list also includes viruses currently causing repeated outbreaks with high mortality such as the Ebola and Marburg viruses.3,4 All of these viruses represent an important type of attack on their host: They are successful obligate parasites. Viruses rely on proteins expressed by the cells they infect to successfully enter the cell, replicate their genome, and exit the cell. Denied the use of these cellular proteins and pathways, viruses are no longer able to replicate.
Understanding what host factors and pathways are used by viral pathogens is an important aspect of virology.5,6 Not only does understanding the cellular proteins used by viruses elucidate important aspects of the viral lifecycle, targeting the function of host proteins that are utilized by viruses can lead to new avenues for antiviral treatment through the application of existing small molecules targeting host factors.7,8 Discovery of the cellular factors required for viral infection has historically been a gradual process. Recent technological advances offer the ability to accelerate this process, mapping virus-host interactions on a genomic scale, but also bring new challenges. Biological safety complications arise when working with highly pathogenic viruses, which limits the ease with which this technology can be utilized.
Identifying host factors exploited by pathogens is difficult in high containment, as the necessary safety precautions pose significant hurdles to assay development.9–11 Genome-scale pooled screening with genetic perturbations, such as RNA interference (RNAi), open reading frames (ORFs), or clustered regularly interspaced short palindromic repeats (CRISPRs), provides the opportunity to identify host factors in high containment for reduced cost of assay development and less manipulation during the assay.12–14 Pooled screens offer the ability to apply genome-scale screening without the equipment usually associated with HTS, which can be a major advantage for high containment research. Here, we describe the development and optimization of genome-scale pooled RNAi screens to identify host factors necessary for viral infection. We detail our recent efforts to apply this technology under intermediate (BSL2) and high (BSL4) biocontainment conditions.
Materials and Methods
Cell Culture and Viruses
A549 cells (CCL-85) were obtained from the ATCC. 293T, 786-O, RKO, and KYSE-30 cells were obtained from the Broad Institute. The vaccinia virus (VACV) used in this study was strain Western Reserve or a derivative thereof.15,16 Monkeypox virus (MPXV) experiments were completed with modified MPXV Zaire 1979 at USAMRIID under appropriate containment conditions.17 Ebola Zaire Kikwit 1995, Marburg Angola, and ebolavirus (EBOV)-enhanced green fluorescent protein (EGFP) experiments were completed at USAMRIID under appropriate containment conditions.18
Fluorescence-Activated Cell Sorting
A549 cells were infected with VACV, using fluorescent reporter virus and multiplicity of infection (MOI) as indicated in the section “Identifying orthopoxvirus host factors: fluorescent reporter pooled screen.” Cells were fixed with 4% formaldehyde, washed, and resuspended in fluorescence-activated cell sorting (FACS) buffer (phosphate-buffered saline, 1% bovine serum albumin, 0.05% sodium azide). Fixed cells were sorted on an MoFlo2 (Beckman Coulter) cell sorter; Venus-negative cells were collected with gates set on a control uninfected cell population.
Cell Viability Endpoint Assay
Cell lines 293T, KYSE-30, 786-O, or RKO were infected with EBOV at MOI 1 or MOI 5, as indicated in the section “Identifying filovirus host factors: cell viability pooled screen.” Cytopathic effects (CPEs) were monitored by light microscopy daily, with cell death caused by EBOV infection noted by day 6 postinfection.
EBOV-EGFP Infection
A549 cells were seeded in 96-well plates the day before transduction. Cells were transduced with lentiviral vectors expressing short hairpin RNAs (shRNAs) at MOI 1. At 24 h post-transduction, cells were selected with puromycin for 4 days. Cells were infected with EBOV-EGFP at MOI 0.5. A SPECTRAMax M5 fluorimeter was used to read EGFP fluorescence (excitation: 485, emission: 515, cutoff: 495) each day for 4 days after infection.
Results
Genome-Scale RNAi Screening: Pooled Versus Arrayed Approaches
Research identifying host factors involved in virus replication often uses RNAi techniques in an arrayed format; this successful approach has been well described elsewhere.19–21 Inherent in this approach is a high level of mechanization and automation of the assay, including the reduction in size of an infection assay to a 386-well or smaller format.22 A pooled screen represents a modification of this approach, involving the transduction of a large population of cells with a library of lentiviral vectors that express multiple individual shRNA constructs targeting each gene in the human genome (Fig. 1).12,13 After selection for integration of the lentiviral vector, each cell in the remaining population will express one shRNA construct designed to inhibit the expression of one host gene. These cells can then be infected with the pathogenic virus of interest without the need for assay miniaturization.
Fig. 1.
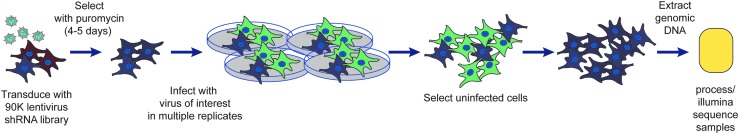
Schematic representing the general pooled shRNA assay evaluating host proteins necessary for viral infection. Cells are transduced with the pooled shRNA lentiviral library at low MOI, selected for 4–5 days with puromycin to ensure lentiviral integration, then infected with the pathogenic virus of interest. The uninfected cells are selected, and the genomic DNA is extracted to process and sequence the integrated lentiviral vectors. Modified from Filone et al.7 MOI, multiplicity of infection; shRNA, short hairpin RNA.
After infection, cells that no longer support viral replication can be collected at an experimental endpoint when infected and uninfected cells are easily separated (e.g., FACS or viability). Genetic analysis allows for the identification of the shRNAs present in the cells that are no longer able to support infection.7,12,13 The shRNA contained in the sorted populations can be quantified by next-generation sequencing (NGS) to determine the hairpins enriched in the uninfected cells compared with the starting population. This information can be used to determine the cell-autonomous host genes important for infection with the pathogenic virus of interest (Table 1). The absence of a host gene may prevent viral infection because that protein is necessary for the viral lifecycle, or the decrease in host gene expression may establish an antiviral state in the cell, preventing infection.23 Secondary screening is necessary to validate the host genes and to determine the mechanism of action of the inhibition.
Table 1.
Example of Pooled Short Hairpin RNA Screen Protocol
| Step | Parameter | Value | Description |
|---|---|---|---|
| 1 | Seed cells | 1 mL | Cells in media/polybrene solution (4 mg/mL final concentration) |
| 2 | Transduce with lentivirus library | 50 μL | Spin infection: 2,000 rpm for 1 h at 37°C |
| 3 | Pool replicates in T225 flasks | 50 mL | Let cells sit at RT for 20 min before placing them in an incubator |
| 4 | Select for infection | 4–5 days | Puromycin selection for integration of lentiviral vector |
| 5 | Infect with virus of interest | 100% infection | Allow to proceed for appropriate length of time for chosen readout |
| 6 | Select uninfected cells | Various readout | FACS or cell viability |
| 7 | Sequence lentiviral vector | NGS | Illumina sequence the processed lentiviral vectors |
Step Notes
1. Want sufficient cell representation to maintain at least 500 lentiviral-transduced cells per perturbagen in the pool. Polybrene concentration predetermined for your cell type.
2. The amount of virus to infect ∼30%–50% of the cells must be predetermined.
4. Add puromycin after cells have settled in flasks; appropriate concentration for your cell line predetermined.
5. For fluorescent sorting or cell viability assay, the length of time predetermined for your virus/cell line combination.
6. FACS: sort for fluorescent-reporter low cells; cell viability: collect live cells after allowing CPE to proceed.
7. Extract genomic DNA, PCR process lentiviral vectors, sequence the shRNA insert by NGS.
CPE, cytopathic effect; FACS, fluorescence activated cell sorting; NGS, next generation sequencing; shRNA, short hairpin RNA.
The pooled approach provides a simple screen setup that can easily be done by a single individual. This contrasts with the need for robotics in the lab to infect 384- or 1,536-well plates for arrayed screening. Furthermore, high containment labs may not have the equipment necessary to read the results in suite; moving plates from one biosafety level (BSL) to another to read the results can complicate assay throughput. A pooled screen requires no additional equipment than what is usually found in a high containment lab (BSC, centrifuges, and incubators), while genomic DNA isolation and preparation can be completed at BSL2 before NGS-based deconvolution. This simplicity of assay setup is a major advantage for enabling screening with highly pathogenic agents.
Optimization of Pooled Screens
During our development of virus-required host factor screens, we identified several important parameters to be optimized for a successful pooled screen: library choice, adequate perturbagen representation in the screen, appropriate screen timing, optimized viral infection conditions, proper choice of assay endpoint, optimal analysis, and experimental follow up. The optimization of the pooled screen parameters (library choice, screen size and timing, and choice of virus) significantly impacts the efficacy of the screen. Proper consideration of the analysis and planning the validation experiments inform the primary screen design (controls, replicates, etc.), to ensure the initial screen provides the desired information.
There are several choices of pooled shRNA libraries, divided into pol III (usually using a human U6 or H1 promoter) or pol II (usually using a miR scaffold) driven shRNA generation approaches.24–28 We utilized the RNAi Consortium (TRC, www.broadinstitute.org/rnai/public/) library that uses a U6-driven method to generate shRNA hairpins. This library has been effectively used in many types of pooled screens.12,13 In all cases, the library should contain five or more nonoverlapping shRNAs targeting each gene. The inclusion of multiple hairpins for each gene provides the opportunity for orthogonal hits on the same gene, which significantly aids in the identification of important genes.
Adequate representation of the pool during each phase of the screen is important. We have found that each peturbagen (shRNA hairpin) should be represented in at least 500 individual cells during the initial infection. If the assay requires sub-sampling, then this minimum representation should be maintained. There is a distribution of penetrance for each perturbagen within a cell (lentivirus integration site, etc.), which contributes to the noise of any assay; the signal can be increased above the noise by performing the screen at adequate representation.
The timing of the screen is also an important consideration. We have obtained our best results when selection of the cells transduced with the shRNA lentiviral vectors is allowed to proceed for 4–5 days. Since mRNA degradation does not immediately reduce the concentration of existing cellular protein, allowing a period of 4–5 days after transduction is prudent so the cells reach a new shRNA-determined steady state. The entire screen should be arranged to keep the length of the screen as brief as possible beyond this initial selection point, to prevent reduced cell proliferation or cell death due to the loss of host proteins. If essential cellular proteins are sufficiently knocked down and cause cell death, then these proteins will not be interrogated during the screen. As cells die due to the knockdown of host proteins, potential information about pathways utilized by the virus of interest will be lost. Furthermore, cells dying due to the absence of an essential host protein will bias the screen to genes not necessary for cell survival.
In contrast to arrayed screens, where there is significant flexibility in infection rates required for a successful screen, for pooled screens it is important that all cells be infected with the pathogenic virus, as noninfected cells will increase the false-positive rate. The MOI, timing of infection, and length of assay must allow for all susceptible cells to be infected by the virus of interest. This enriches for uninfected cells that have acquired resistance to viral infection due to the knockdown of a host protein by the shRNA treatment, and not due to suboptimal infection conditions that allowed a subset of cells to remain uninfected and phenocopy resistant cells. If these conditions are not set, the screen may have a high rate of false positives, with shRNAs called potential hits when those cells were not infected because the virus was not able to infect all of the cells in the pool. Ideally, the stochastic uninfected cells would be determined to be false positives during analysis of the NGS dataset, but optimization of the screening conditions is preferable.
We have found that effective pooled screens require a clean readout for infection. There are many options, including sorting for fluorescent reporters, identifying cell surface markers, or assaying for cell death; for any option, there must be a significant signal-to-noise ratio. The expression of a fluorescent reporter by the virus of interest can be used to effectively separate uninfected (nonfluorescent) cells from infected cells; this requires careful planning, as described next. Optimization for high, consistent levels of fluorescent reporter expression is required to increase screening success. The fluorescent and nonfluorescent cells can be separated by FACS; however, safety protocols and fixation techniques must be established before cell sorting to prevent infectious aerosol formation. Likewise, if the screen endpoint is selection for cells that survive a normally lytic infection, assessment of consistent death due to viral infection is critical before the screen begins.
It is also important that an appropriate method for data analysis be identified. In our screens, we isolated the genomic DNA from uninfected cells, which contains the integrated shRNA in the viral vector. PCR primers against the viral vector allow for amplification of the shRNA, and NGS (such as Illumina HiSeq) provides the information necessary to determine the shRNA sequences (and therefore the host genes) that cause an inhibition of viral infection. The NGS data can be analyzed in several ways to determine the genes over-represented in the uninfected pool of cells.12,13 We chose to categorize the genes using the second best hairpin approach. Using software developed by the Broad Institute, GENE-E (www.broadinstitute.org/cancer/software/GENE-E/), we ordered the hairpins by abundance in the uninfected pool. Each host gene was targeted by at least five shRNAs in our screens; we then ranked the genes based on the hairpin with the second best rank in the overall pool. This analysis ensured that at least two hairpins for each gene were over-represented for each chosen gene, which decreased the chances of attempting to investigate off-target effects.
The analysis data provides a list of genes of interest, which then must be studied further. There are several ways to determine which genes to investigate first. If the screen is performed in replicates, the gene lists can be compared across replicates, with the strongest hits chosen for validation first. The genes can be analyzed to determine common functions using reactome or gene onotology terms in programs such as DAVID (http://david.abcc.ncifcrf.gov/) or Panther (www.pantherdb.org/) and gene networks using a program such as Ingenuity Pathway Analysis (IPA; www.ingenuity.com/products/ipa). The potential hits must then be validated using secondary screening methods. The secondary experiments will confirm which genes are important for viral infection, and those can be investigated for their mechanism of action.
Example Screens
Our lab has completed two whole-genome scale pooled shRNA screens that identified host factors necessary for viral infection. The first screen identified host factors necessary for orthopoxvirus infection using a fluorescent vaccinia virus with cell sorting to identify uninfected cells.7 The second screen, performed in a BSL4 laboratory, identified host proteins necessary for infection with filoviruses, using cell death to identify cells that are capable of supporting Ebolavirus or Marburgvirus (MARV) infection. Next, we describe the optimization approaches taken to enable each screen. While every screen needs to be individually optimized for best results, the principles outlined next highlight some of the major considerations for any pooled screen using shRNA, CRISPR, or ORF perturbations.
Identifying orthopoxvirus host factors: fluorescent reporter pooled screen
To identify host factors necessary for infection with orthopoxviruses, we completed a whole-genome scale pooled shRNA screen utilizing a vaccinia virus that expressed a fluorescent reporter and sorted for uninfected cells. At endpoint, these cells were FACS sorted for low reporter expression and integrated shRNA sequences were quantified by NGS to identify the cells with reduced host proteins that impacted orthopoxvirus infection.7 The critical optimization steps for this screen included developing reporter viruses that allowed appropriate retention of the fluorophore in infected cells after fixation, optimizing the choice of the fluorophores expressed by the virus, and optimizing the sorting protocol.
Our first step for optimization was to develop a panel of viruses that expressed fluorescent reporters to facilitate easy identification of infected versus uninfected cells. Orthopoxvirus gene expression occurs in a cascade of early, intermediate, and late genes throughout the lifecycle of the virus;29,30 therefore, reporter viruses were made to express fluorophores under canonical early (C11R), intermediate (G8R), or late (F17R) promoters, either individually or in combination, allowing for a dissection of the virus lifecycle (Table 2).16 In the screen, we wanted to investigate all steps of the viral lifecycle before egress, allowing us to identify genes necessary for viral entry, early gene expression, DNA replication, and intermediate or late gene expression in one screen. Therefore, we chose to use a recombinant VACV that expressed a late reporter. For secondary and follow-up screening, we were able to use multistage VACV reporter viruses to quickly determine the step in the viral lifecycle inhibited by the RNAi knockdown.
Table 2.
Vaccinia Fluorescent Reporter Viruses
| Category | Virus | Promoter | Fluorophore |
|---|---|---|---|
| Vaccinia fluorescent reporter viruses | |||
| Single-stage reporter | Early Venus (EV) | C11R | Venus |
| Intermediate Venus (IV) | G8R | Venus | |
| Late Venus (LV) | F17R | Venus | |
| Late Red (LR) | F17R | mCherry | |
| A4L-Venus | A4L endogenous | Venus-p39 fusion protein | |
| A4L-mCherry | A4L endogenous | mCherry-p39 fusion protein | |
| Multi-stage reporters | Late Red, Early Venus (LREV) | C11R, F17R | Venus (Early), mCherry (Late) |
| Intermediate Red, Early Venus (IREV) | G8R, F17R | Venus (Early), mCherry (Intermediate) | |
| Triple Virus (TrpV) | C11R, G8R, F17R | Venus (Early), mCherry (Intermediate), TagBFP (Late) | |
| Control | Promoter-less Venus (PLV) | No promoter | Venus (not expressed) |
VACV created to monitor viral infection by fluorescent reporter, with promoter(s) and fluorophore(s) listed.
VACV, vaccinia virus.
The next step in our optimization was to determine the best fluorophore to be used for the reporter virus. The level of fluorescence, the stability of the protein, and the signal-to-noise ratio were considered. We found the GFP derivative Venus had the best characteristics for our purposes: Venus has a long half-life and higher signal-to-noise ratio than EGFP (signal-to-noise ratio of 154.9 for Venus versus 8.1 for GFP).16 Expression of Venus from a canonical late viral promoter provided a clear signal 12 h postinfection. Since sorting can create a dangerous aerosol, infected cells must be fixed before sorting. However, when cells were appropriately fixed to transport for sorting, there was evidence that the fluorophore leaked from a portion of the fixed cells, leading to a mixing of the uninfected and infected population (Fig. 2A). To prevent fluorophore leakage, Venus was fused to a late viral protein, p39 (encoded by A4L), which is a part of the viral core.15 This protein associates with membranes in the cell, preventing the fluorophore signal from diminishing due to the fixation process (Fig. 2B).31 Since A4L-Venus retained fluorescence in fixed cells, maintaining a good signal-to-noise ratio, this virus was used in the pooled shRNA screen.
Fig. 2.

Sorting Vaccinia-infected A549 cells. (A) Uninfected (black) and VACV LV-infected (MOI 10; green) cells were sorted by level of Venus expression. (B) Uninfected (black) and VACV A4L-Venus-infected (MOI 10; green) cells were sorted by level of Venus expression. LV, Late Venus; VACV, vaccinia virus.
Next, we optimized cell type, MOI, and time of infection. We wanted a cell type that was transduced well with the lentiviral vectors used to deliver the shRNA constructs, to ensure that the knockdown library would be accepted. The cell line also needed to demonstrate consistent, near-100% levels of VACV infection, as measured by A4L-Venus expression. We chose A549 cells, which are easily transduced by the lentivirus vectors for the shRNA library, and infected to high levels with VACV. To prevent cell death due to negative effects of host protein knockdown, the duration of the assay should be as short as possible. We optimized the assay to allow VACV infection to progress for 12 h before fixation and sorting. In A549 cells, we found that infection with VACV A4L-Venus at MOI 5 gave near-100% infection levels at 12 hpi as measured by fluorescence.
Finally, we wanted to ensure that the chosen virus and infection conditions allowed for isolation of the uninfected cells via FACS. We optimized sorting conditions by comparing uninfected cells (Fig. 3, green line) and cells infected with VACV A4L-Venus at MOI 5 (optimal infection levels; blue line). Cells infected with MOI 0.05 were tested to represent screening conditions, with a portion of the population no longer supporting infection (red line). These data demonstrate that differentiation of infected and uninfected cells is possible by sorting for A4L-Venus expression levels.
Fig. 3.

Optimization of VACV A4L-Venus sorting conditions. (A) Histogram of cells sorted by Venus expression levels. Uninfected cells (green line), VACV A4L-Venus MOI 5 infected cells (blue line), and VACV A4L-Venus MOI 0.05 infected cells (red line) demonstrate ability to sort by level of Venus expression. (B) Percentage of cells collected in the low-Venus gate, set by the uninfected cell population, and percentage of cells in the high-Venus gate, set by the VACV A4L-Venus MOI 5 infected cells, for each of the three samples.
Using the optimized conditions described earlier, a genome-scale pooled RNAi screen was successfully completed to identify host factors necessary for orthopoxvirus infection. We performed the initial lentivirus transduction at a scale that resulted in >500 cells on average containing each shRNA (12e6 cells/pool). These cells were infected in four replicate pools with VACV A4L-Venus for 12 h. We collected the cells with the lowest 1% of Venus expression, which allowed identification of the shRNAs that prevented VACV A4L-Venus expression. The genomic DNA was isolated from the uninfected cells, and primers against the flanking lentiviral construct were used to amplify the hairpin sequences from the low Venus fraction. The primers also contained a short barcode to enable different samples to be amplified and then pooled onto the same Illumina lane to allow multiplexing (www.broadinstitute.org/rnai/public/resources/protocols). These were sequenced using Illumina technology, and the enriched hairpins within each independent barcoded PCR reaction were quantified. If multiplexing is used to sequence several samples within the same Illumina lane, then data should be normalized to adjust for the variable sequence depth using the following (or similar) calculation: Log2(((shRNA reads/total reads for sample)×1e6)+1). Hairpins were converted into enriched gene sets using the RIGER-weighted second best method.7
A selection of the enriched genes were tested in secondary screening. We tested five nonoverlapping shRNAs per candidate gene in a 96-well plate arrayed format. After transduction with the lentiviral vector expressing the shRNA, cells were selected with puromycin, then infected with VACV Late Red, Early Venus (LREV) to determine the point in the viral lifecycle during which the host genes were utilized. We read the fluorophores each day for 4 days and normalized to control shRNAs; 35% of the genes were confirmed as necessary for VACV infection. These genes were also tested in high containment against monkeypox in a similar arrayed format, with a cross-confirmation rate of ∼50%.
The results from this screen have been published7; we included all of the gene data from the primary screen, and the LREV readout from the secondary screen, so other researchers could independently analyze and draw conclusions about the strength of the data. The paper also includes detailed information about the processing of the gDNA and the analysis of the potential shRNA hits. The orthopoxvirus screen confirmed that pooled genome-scale RNAi screening could identify host factors necessary for viral infection and taught valuable lessons for the requirements that would need to be adapted for a high containment screen.
Identifying filovirus host factors: cell viability pooled screen
We applied the lessons learned in our VACV screen to the experimental design of a second, similar screen completed in the BSL4 laboratory investigating host proteins necessary for filovirus infection. The same optimization considerations needed to be developed, including assay end point, kinetics of the screen, cell line, and virus selection. Although there are fluorescent protein-expressing filoviruses that could be used in an FACS-based screen (similar to the poxvirus assay described earlier), this modification is known to attenuate the virus.32 In addition, there are safety concerns about aerosol formation pertaining to FACS BSL4 agents, and we were unable to obtain the ability to sort cells that were infected in the BSL4 laboratory. Therefore, we modified the assay design to avoid a fluorophore readout.
Cell death as a readout is a simple and robust endpoint of many virus infections, and it does not require manipulation of the virus to express a reporter protein; therefore, we considered a cell viability assay a more applicable design for high containment viruses, especially those without reverse genetics systems or for viruses where modification leads to significant attenuation. Another advantage to screening for cell viability is that we were able to choose the species of virus that we were most interested in screening, rather than ones that were available with a reporter. We chose the highly pathogenic Ebola Zaire and Marburg Angola for our screen identifying host factors necessary for filovirus infection.
Screen optimization again required identification of an appropriate cell line. Here, we needed a cell line that was (1) permissive for lentiviral transduction for delivery of the shRNA, (2) supportive for filovirus infection, and (3) showed strong cytopathogenesis and cell death within a relatively short period after filovirus infection. The cells also needed to be derived from human tissue to ensure the proper targeting of the shRNA library. After an extensive screening process, we chose a subset of cell lines competent for lentiviral transduction to screen for filovirus infection, including A549, 786-O, RKO, KYSE-30, and 293T cells (Fig. 4). As described earlier, we wanted to obtain close to 100% filovirus infection of cells under normal conditions, and also wanted these filovirus-infected cells to die in a relatively short timeframe (<2 weeks). Testing showed that 293T cells filled criteria 1 and 2 and also showed robust CPE and cell death after infection with either Marburg or Ebola virus (Fig. 4). After EBOV infection at MOI 5, the 293T cells showed high levels of filovirus infection. These cells began to die, and within 6 days of infection a majority of the infected cells were no longer viable.
Fig. 4.

Cell line optimization for cell viability Ebola screen. Cell lines 786-O, RKO, KYSE-30, and 293T were infected with EBOV at MOI 5. Cell viability was monitored by measuring CPE at 1 or 5 days postinfection. CPE, cytopathic effect; EBOV, ebolavirus.
After the identification of the appropriate screening cell line, scale-up for screening was relatively simple. The procedure was as follows: lentiviral transduction of 293T cells at an MOI 0.3 (to enrich for cells containing a single shRNA), followed by selection with puromycin for 4 days to ensure lentiviral integration and shRNA expression. The knockdown cells were then infected at MOI ∼1 with either EBOV or MARV in two T225 flasks per pool; the cells were split as appropriate to ensure the uninfected cells were properly maintained during the screen. An important note is that we were able to carry out biological replicates of this screen due to the ease of scale-up. After 6 days, the cells that were still alive were presumed to be uninfected due to the manipulation of a host protein.
These cells were collected and lysed with TRIzol to ensure no viable virus was removed from high containment. The genomic DNA was isolated from the TRIzol in a BSL2 laboratory, then amplified, and sequenced using Illumina technology as described for the orthopoxvirus screen.7 A subset of genes that were enriched in the uninfected cells were selected to test in secondary screening using EBOV-EGFP in a 96-well plate arrayed format with A549 and HeLa cells. The secondary, arrayed screens should have positive and negative control shRNA constructs on each plate to allow for normalization of the data. In this case, we had two negative controls: shRNAs that targeted GFP and wells without lentivirus vectors, so no viable cells remained after puromycin selection. We also selected several host genes that showed no effect on EBOV replication in the primary screen to target for positive controls. For data analysis, we subtracted the background of the negative control wells and normalized them to the positive control wells. We also included control genes known to be necessary for filovirus infection, to ensure the infections and analysis were performed properly.
Here, we include example analysis data from the EBOV-EGFP secondary screen. A control gene, CARS2, did not affect EBOV infection and was one of the genes used to normalize the data (Fig. 5A, left). When GFP is targeted, the EBOV-EGFP reporter mRNA is unable to be translated, and the fluorescence readout is 2% of that seen in virus-infected cells where an irrelevant gene had been silenced. A decrease in protein levels of the EBOV entry receptor, NPC1, inhibits EBOV infection, to 8% of control levels.33,34 Knockdown of cathepsin B, which is necessary for processing of the EBOV glycoprotein before receptor binding,35,36 decreased EGFP expression by more than 50%.34 Other factors, such as components of the homotypic fusion and protein complex, which affects endosome fusion and maturation (VPS16, VPS39, and VPS41),37–39 proteins involved in endosome maturation (FIG4, PIKFYVE),40 and the lysosomal protein BRI3,41 also limited Ebola replication when their protein levels were reduced by shRNA treatment. Knockdown of these genes decreased Ebola-dependent EGFP expression by 60%–85% (Fig. 5A, gray bars). These factors had been identified as necessary for entry of pseudovirions utilizing EBOV glycoprotein;34 here, we confirm that they are necessary for infection with pathogenic EBOV. These data validate the capacity of the secondary screen to identify host proteins necessary for filovirus infection.
Fig. 5.

EBOV-EGFP secondary screening. (A) A549 cells transduced with two independent shRNAs targeting the host proteins indicated were infected with EBOV-EGFP at MOI 0.5 in duplicate. At 68 h postinfection, EGFP was read, and the average normalized RFU graphed with standard error. Black bars represent control genes. Gray bars represent genes not previously tested with pathogenic EBOV. (B) A549 cells were transduced with five independent shRNAs targeting RAB39B. Cells were infected with EBOV-EGFP at MOI 0.5 in duplicate. Fluorescence was read each day for 4 days post EBOV-EGFP infection. Data were normalized to 100% of day 4 control hairpin and graphed with standard error. EGFP, enhanced green fluorescent protein.
We also tested a variety of host proteins that were identified in our primary, pooled filovirus screen, but not previously reported in the literature. One gene, RAB39B, is a small GTPase that regulates trafficking between vesicular compartments.42 We targeted RAB39B with five distinct shRNA constructs in A549 cells; four of the shRNAs inhibited EBOV infection (Fig. 5B). This figure clearly demonstrates the variation in effect of the shRNA sequences, suggesting that the extent of protein knockdown correlates with the level of virus inhibition associated with each shRNA.
As with other RNAi techniques, the level of protein knockdown varies between the shRNA constructs, and not all shRNAs will lead to a sufficient decrease in the target protein level to observe a phenotype. The efficacy of a construct also varies across cells lines, which is why it is important to test several constructs in several cell lines when verifying potential hits. Of the novel genes identified in the primary filovirus screen, more than 60% were confirmed as necessary for EBOV infection. The format for the pooled RNAi screen to identify proteins affecting filovirus infection described here can be developed for many other high containment pathogens as well.
Future Outlook
Pooled screens can be used to identify host proteins and pathways necessary for infection of a wide variety of high-consequence pathogens that require the protection of BSL3 or BSL4 high containment precautions. This information allows us to probe the mechanisms involved in the viral lifecycle, and to better understand the cellular pathways hijacked by the virus. Furthermore, we were able to identify host pathways that may already be well studied, with developed small-molecule inhibitors that could be used as antiviral treatments.
The general optimization protocols for pooled genome-scale shRNA screens described here can be used for many different pathogens at high containment, whether they can be manipulated to express fluorescent reporters or are ideal for cell viability studies. These protocols will also be useful for other pooled screening platforms, such as ORF or CRISPR/Cas9 technology.14 As new pathogenic viruses are identified, or known viruses spread to new areas of the world, it is imperative that we are able to quickly identify the host pathways utilized by the virus and to leverage that information into a better understanding of the virus lifecycle and to development of new therapeutics. A streamlined, genome-scale pooled RNAi screen allows for a unique opportunity to optimize the screen under any containment condition and to rapidly identify candidate host factors necessary for infection.
Abbreviations Used
- BSC
biosafety cabinet
- BSL
biosafety level
- CPE
cytopathic effect
- CRISPRs
clustered regularly interspaced short palindromic repeats
- EBOV
ebolavirus
- EGFP
enhanced green fluorescent protein
- FACS
fluorescence-activated cell sorting
- HTS
high-throughput screens
- LREV
Late Red, Early Venus
- LV
Late Venus
- MARV
marburgvirus
- MOI
multiplicity of infection
- MPXV
monkeypox virus
- NGS
next-generation sequencing
- ORF
open reading frame
- RNAi
RNA interference
- shRNA
short hairpin RNA
- VACV
vaccinia virus
Acknowledgments
The authors would like to thank the Boston University Flow Cytometry Core Facility, especially Anna Belkina, for assistance with the cell sorting experiment analysis. They would also like to acknowledge the cell sorting staff at the MIT Koch Institute for sorting the VACV screen. They would like to thank Monica Ogg and Kenny Lin for training and assistance in high containment. C.M.F. was supported by the Postgraduate Research Participation Program and the U.S. Army Research and Medical Command administered by the Oak Ridge Institute for Science and Education (ORISE) and training grant 5T32AI089673-04. This work was also supported by a SPARC grant from the Broad Institute and in part by R03 MH094169 to J.H.C.
Disclosure Statement
No competing financial interests exist.
References
- 1.Henderson DA, Moss B: Smallpox and Vaccinia. In: Vaccines (Plotkin SA, Orenstein WA, eds.). Saunders, Philadelphia, USA, 1999 [Google Scholar]
- 2.World Health Organization: The Global Eradication of Smallpox : Final Report of the Global Commission for the Certification of Smallpox Eradication, Geneva, December 1979. 122, 1980. Available at: whqlibdoc.who.int/publications/a41438.pdf (last accessed January26, 2015) [Google Scholar]
- 3.Feldmann H, Geisbert TW: Ebola haemorrhagic fever. Lancet 2011;377:849–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Piot P, Muyembe J-J, Edmunds WJ: Ebola in west Africa: from disease outbreak to humanitarian crisis. Lancet Infect Dis 2014;14:1034–1035 [DOI] [PubMed] [Google Scholar]
- 5.Panda D, Cherry S: Cell-based genomic screening: elucidating virus-host interactions. Curr Opin Virol 2012;2:784–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedel CC, Haas J: Virus-host interactomes and global models of virus-infected cells. Trends Microbiol 2011;19:501–508 [DOI] [PubMed] [Google Scholar]
- 7.Filone CM, Caballero IS, Dower K, et al. : The master regulator of the cellular stress response (HSF1) is critical for orthopoxvirus infection. PLoS Pathog 2014;10:e1003904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Karlas A, Machuy N, Shin Y, et al. : Genome-wide RNAi screen identifies human host factors crucial for influenza virus replication. Nature 2010;463:818–822 [DOI] [PubMed] [Google Scholar]
- 9.Panchal RG, Kota KP, Spurgers KB, et al. : Development of high-content imaging assays for lethal viral pathogens. J Biomol Screen 2010;15:755–765 [DOI] [PubMed] [Google Scholar]
- 10.Hoenen T, Groseth A, Callison J, Takada A, Feldmann H: A novel Ebola virus expressing luciferase allows for rapid and quantitative testing of antivirals. Antiviral Res 2013;99:207–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uebelhoer LS, Albariño CG, McMullan LK, et al. : High-throughput, luciferase-based reverse genetics systems for identifying inhibitors of Marburg and Ebola viruses. Antiviral Res 2014;106:86–94 [DOI] [PubMed] [Google Scholar]
- 12.Luo B, Cheung HW, Subramanian A, et al. : Highly parallel identification of essential genes in cancer cells. Proc Natl Acad Sci U S A 2008;105:20380–20385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheung HW, Cowley GS, Weir BA, et al. : Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci U S A 2011;108:12372–12377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang T, Wei JJ, Sabatini DM, Lander ES: Genetic screens in human cells using the CRISPR-Cas9 system. Science 2014;343:80–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dower K, Filone CM, Hodges EN, et al. : Identification of a pyridopyrimidinone inhibitor of orthopoxviruses from a diversity-oriented synthesis library. J Virol 2012;86:2632–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dower K, Rubins KH, Hensley LE, Connor JH: Development of Vaccinia reporter viruses for rapid, high content analysis of viral function at all stages of gene expression. Antiviral Res 2011;91:72–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnston SC, Lin KL, Connor JH, et al. : In vitro inhibition of monkeypox virus production and spread by Interferon-β. Virol J 2012;9:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Towner JS, Paragas J, Dover JE, et al. : Generation of eGFP expressing recombinant Zaire ebolavirus for analysis of early pathogenesis events and high-throughput antiviral drug screening. Virology 2005;332:20–27 [DOI] [PubMed] [Google Scholar]
- 19.Buehler G, Ang KL, Feroze F, et al. : Cell-Based RNAi Assay Development for HTS. In: Assay Guidance Manual [Online] (Sittampalam GS, Coussens NP, Nelson H, et al. eds). Eli Lilly & Company and the National Center for Advancing Translational Sciences, Bethesda, USA, 2013 [PubMed] [Google Scholar]
- 20.Echeverri CJ, Perrimon N: High-throughput RNAi screening in cultured cells: a user's guide. Nat Rev Genet 2006;7:373–384 [DOI] [PubMed] [Google Scholar]
- 21.Snijder B, Sacher R, Rämö P, et al. : Single-cell analysis of population context advances RNAi screening at multiple levels. Mol Syst Biol 2012;8:579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Inglese J, Johnson RL, Simeonov A, et al. : High-throughput screening assays for the identification of chemical probes. Nat Chem Biol 2007;3:466–479 [DOI] [PubMed] [Google Scholar]
- 23.Colina R, Costa-Mattioli M, Dowling RJO, et al. : Translational control of the innate immune response through IRF-7. Nature 2008;452:323–328 [DOI] [PubMed] [Google Scholar]
- 24.Silva JM, Li MZ, Chang K, et al. : Second-generation shRNA libraries covering the mouse and human genomes. Nat Genet 2005;37:1281–1288 [DOI] [PubMed] [Google Scholar]
- 25.Moffat J, Grueneberg DA, Yang X, et al. : A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 2006;124:1283–1298 [DOI] [PubMed] [Google Scholar]
- 26.Berns K, Hijmans EM, Mullenders J, et al. : A large-scale RNAi screen in human cells identifies new components of the p53 pathway. Nature 2004;428:431–437 [DOI] [PubMed] [Google Scholar]
- 27.Li L, Lin X, Khvorova A, Fesik SW, Shen Y: Defining the optimal parameters for hairpin-based knockdown constructs. RNA 2007;13:1765–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kesireddy V, van der Ven PFM, Fürst DO: Multipurpose modular lentiviral vectors for RNA interference and transgene expression. Mol Biol Rep 2010;37:2863–2870 [DOI] [PubMed] [Google Scholar]
- 29.Yang Z, Bruno DP, Martens CA, Porcella SF, Moss B: Simultaneous high-resolution analysis of vaccinia virus and host cell transcriptomes by deep RNA sequencing. Proc Natl Acad Sci U S A 2010;107:11513–11518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang Z, Reynolds SE, Martens CA, et al. : Expression profiling of the intermediate and late stages of poxvirus replication. J Virol 2011;85:9899–9908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cudmore S, Blasco R, Vincentelli R, et al. : A vaccinia virus core protein, p39, is membrane associated. J Virol 1996;70:6909–6921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ebihara H, Theriault S, Neumann G, et al. : In vitro and in vivo characterization of recombinant Ebola viruses expressing enhanced green fluorescent protein. J Infect Dis 2007;196Suppl:S313–S322 [DOI] [PubMed] [Google Scholar]
- 33.Côté M, Misasi J, Ren T, et al. : Small molecule inhibitors reveal Niemann-Pick C1 is essential for Ebola virus infection. Nature 2011;477:344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carette JE, Raaben M, Wong AC, et al. : Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011;477:340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Misasi J, Chandran K, Yang J-Y, et al. : Filoviruses require endosomal cysteine proteases for entry but exhibit distinct protease preferences. J Virol 2012;86:3284–3292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chandran K, Sullivan NJ, Felbor U, Whelan SP, Cunningham JM: Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005;308:1643–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nickerson DP, Brett CL, Merz AJ: Vps-C complexes: gatekeepers of endolysosomal traffic. Curr Opin Cell Biol 2009;21:543–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Poteryaev D, Datta S, Ackema K, Zerial M, Spang A: Identification of the switch in early-to-late endosome transition. Cell 2010;141:497–508 [DOI] [PubMed] [Google Scholar]
- 39.Rink J, Ghigo E, Kalaidzidis Y, Zerial M: Rab conversion as a mechanism of progression from early to late endosomes. Cell 2005;122:735–749 [DOI] [PubMed] [Google Scholar]
- 40.Sbrissa D, Ikonomov OC, Fu Z, et al. : Core protein machinery for mammalian phosphatidylinositol 3,5-bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve-PIKfyve complex. J Biol Chem 2007;282:23878–23891 [DOI] [PubMed] [Google Scholar]
- 41.Wu H, Liu G, Li C, Zhao S: bri3, a novel gene, participates in tumor necrosis factor-alpha-induced cell death. Biochem Biophys Res Commun 2003;311:518–524 [DOI] [PubMed] [Google Scholar]
- 42.Cheng H, Ma Y, Ni X, et al. : Isolation and characterization of a human novel RAB (RAB39B) gene. Cytogenet Genome Res 2002;97:72–75 [DOI] [PubMed] [Google Scholar]