

Abstract
Epithelial injury, alternative macrophage accumulation, and fibroproliferation coexist in the lungs of patients with idiopathic pulmonary fibrosis (IPF). Chitinase 3–like 1 (CHI3L1) is a prototypic chitinase-like protein that has been retained over species and evolutionary time. However, the regulation of CHI3L1 in IPF and its ability to regulate injury and/or fibroproliferative repair have not been fully defined. We demonstrated that CHI3L1 levels were elevated in patients with IPF. High levels of CHI3L1 are associated with progression—as defined by lung transplantation or death—and with scavenger receptor–expressing circulating monocytes in an ambulatory IPF population. In preterminal acute exacerbations of IPF, CHI3L1 levels were reduced and associated with increased levels of apoptosis. We also demonstrated that in bleomycin-treated mice, CHI3L1 expression was acutely and transiently decreased during the injury phase and returned toward and eventually exceeded baseline levels during the fibrotic phase. In this model, CHI3L1 played a protective role in injury by ameliorating inflammation and cell death, and a profibrotic role in the repair phase by augmenting alternative macrophage activation, fibroblast proliferation, and matrix deposition. Using three-dimensional culture system of a human fibroblast cell line, we found that CHI3L1 is sufficient to induce low grade myofibroblast transformation. In combination, these studies demonstrate that CHI3L1 is stimulated in IPF, where it represents an attempt to diminish injury and induce repair. They also demonstrate that high levels of CHI3L1 are associated with disease progression in ambulatory patients and that a failure of the CHI3L1 antiapoptotic response might contribute to preterminal disease exacerbations.
INTRODUCTION
Pulmonary fibrosis is a component of various interstitial pneumonias and can be caused by exposure to fibrosis-inducing agents such as silica (1), coal dust (2), radiation, and certain chemotherapeutic agents (3, 4). It is also a feature found in disorders such as scleroderma (5), sarcoidosis (6), and cystic fibrosis (7). Idiopathic pulmonary fibrosis (IPF) is a particularly deadly form of this disease with an unknown cause and chronic, progressive fibrosis of the lungs. The natural history of IPF is highly variable and unpredictable, and treatment options for this disorder are limited (8). Thus, investigations aimed at dissecting the mechanisms promoting disease progression have the potential to identify novel therapeutic targets and strategies.
Current paradigms of fibrosis include a prominent role for both injury and repair (9) in which chronic or unresolved injury initiates a tissue response characterized by the recruitment of inflammatory cells and the concomitant activation of fibroblasts and myofibroblasts. In the setting of IPF, the simultaneous presence of apoptosis, inflammation, aberrant fibroblast proliferation, and exaggerated extracellular matrix (ECM) deposition accompanies distorted pulmonary architecture and compromised lung function (4, 10, 11). Studies from our laboratory and others have demonstrated that injury and apoptosis are prerequisites for the development of fibrosis and tissue remodeling (12) that are also modulated in part by macrophage-driven inflammatory responses (13, 14). In keeping with this concept, tissue injury and inflammation can also contribute to the severity, rate of progression, and/or reversibility of fibrotic responses in animal models (15–18). Despite the temporally heterogeneous presence of injury, mild inflammation, and fibroblast activation that exists in the IPF lung, only scant data exist regarding the simultaneous regulation of all phases of fibrogenesis in this disease. Therefore, the discovery of a protein with the ability to exert compartment-specific effects on different components of the fibrotic response might importantly advance our knowledge of the mechanism(s) through which tissue remodeling and fibrosis occur.
The 18 glycosyl hydrolase gene family contains true chitinases that enzymatically cleave chitin, and chitinase-like proteins (CLPs) that bind but do not cleave this polysaccharide. These chitinases and CLPs are found across species and are widely expressed from prokaryotes to eukaryotes. Although endogenous chitin or chitin synthases do not exist in mammals, chitinases and CLPs such as acidic mammalian chitinase (AMCase) and chitinase 3–like 1 (CHI3L1) are expressed in the lung and other organs (19–21). CHI3L1, which is also called YKL-40 in man and BRP-39 in the mouse, is the prototypic CLP (22). It can be readily detected in the circulation of normal individuals, and its expression is dysregulated in the circulation and/or tissues from patients with a variety of diseases characterized by inflammation and/or tissue remodeling. To begin to understand the biology of CHI3L1/BRP-39/YKL-40, we generated and characterized mice with null mutations of CHI3L1 (BRP-39−/−) and transgenic mice that use the Clara cell CC10 promoter to overexpress YKL-40 in an inducible and lung-restricted manner (YKL-40 Tg) (19, 23, 24). Studies with these mice and other experimental systems demonstrated that CHI3L1 is a critical regulator of anti-pathogen and adaptive T helper 2 (TH2) responses (19, 25). They also demonstrated that CHI3L1 has impressive proinflammatory and antiapoptotic effects (23, 24) and contributes to interleukin-13 (IL-13)–induced pulmonary inflammation, fibrosis, transforming growth factor–β1 (TGF-β1) elaboration (19), and fibroblast proliferation (26). In patients with IPF, high levels of CHI3L1/YKL-40 can be detected in the lungs and circulation (27). However, the differential regulation of CHI3L1 in the setting of clinically divergent forms of IPF has not been defined, and the roles that CHI3L1 plays in the different facets of this and related disorders have not been elucidated.
On the basis of the studies noted above, we hypothesized that CHI3L1 has distinct roles in the pathogenesis of the injury and repair phases of pulmonary fibrosis in diseases such as IPF. To test this hypothesis, we characterized the regulation of CHI3L1 in ambulatory patients with IPF, patients with acute exacerbations (AEs) of IPF, and age-matched nonfibrotic controls. We also defined the roles of CHI3L1 in the injury and repair phases of bleomycin-induced responses in the murine lung and developed a bioengineering-based culture system based on decellularized lungs to further define CHI3L1’s effect on inflammation and remodeling. These studies demonstrate that CHI3L1 has distinct roles in pulmonary injury and fibroproliferation and that it is stimulated in IPF where it may represent an attempt to diminish injury and induce repair. They also demonstrate that high levels of CHI3L1 reflect disease progression in ambulatory patients and that a failure of the CHI3L1 antiapoptotic protective response is seen in preterminal IPF disease exacerbations.
RESULTS
CHI3L1 expression is enhanced in the IPF lung
To determine the clinical relevance of CHI3L1 in human IPF, we performed immunohistochemistry for CHI3L1 on lung sections obtained from IPF lung biopsies (n = 7) and compared them to the tumor-free margin of wedge resections of metastatic nonprimary lung cancers (n = 4). Here, consistent with previous reports (27), we found a significant increase in both quantities and percentages of CHI3L1-expresssing cells in IPF lungs, which, by morphology and immunofluorescence co-detection, appeared to be epithelial cells and macrophages that were detected both in fibrotic-appearing regions and in more normal areas (Fig. 1, A and B, and figs. S1 to S3). Immunofluorescence evaluation of a small number of cytospins prepared from IPF lung digests revealed that the CHI3L1 macrophages colocalized mainly with CCL-18 and, to a much smaller extent, inducible nitric oxide synthase (iNOS) (fig. S3). These data indicate that CHI3L1 expression is enhanced in the lungs of patients with IPF.
Fig. 1. CHI3L1 levels are increased in IPF.

(A) Immunohistochemical detection of CHI3L1 in control and IPF lung tissue. Scale bars, 200 μm (low-power images) and 100 μm (high-power images). (B) Compared to controls (n = 4), both total quantities (left axis) and percentages (right axis) of CHI3L1+ cells are increased in the IPF lung (n = 7). Left axis: P < 0.0001, unpaired Student’s t test. Right axis: P = 0.008, unpaired Student’s t test. (C) Plasma CHI3L1 is increased in IPF (n = 64) compared to age-matched normal controls (n = 42). P < 0.0001, Mann-Whitney comparison. (D) Compared to stable subjects (n = 41), plasma CHI3L1 is increased in progressive IPF subjects that experienced death by any cause or lung transplantation (n = 23). P = 0.0019, Mann-Whitney comparison.
CHI3L1 is elevated in the circulation of ambulatory patients with IPF, where it is associated with disease progression
We next turned our attention to the detection of CHI3L1 in the peripheral blood. In these experiments, we assembled a prospectively recruited cohort of subjects with IPF (n = 64) and age-matched controls (n = 42) recruited from the Translational Research Program in the Section of Pulmonary, Critical Care, and Sleep Medicine at Yale. Subject characteristics are shown in table S1. Here, we found as previously shown (27), CHI3L1 was increased by more than four-fold in the plasma of IPF patients relative to controls (Fig. 1C). Seventy-two–week longitudinal follow-up of the subjects with IPF revealed that the highest levels of CHI3L1 were seen in ambulatory subjects who went on to experience a disease progression defined by the significant clinical event of lung transplantation or death (Fig. 1D), and further analysis revealed that, when adjusted for covariates such as age, gender, and disease severity, the ideal concentration of CHI3L1 allowing separation of progressive from stable subjects was 190 ng/ml (fig. S4). These data demonstrate that circulating CHI3L1 levels are elevated in IPF in general and in subjects with progressive disease in particular, thereby suggesting a possible role in disease pathogenesis.
CHI3L1 is relatively reduced in AEs of IPF
Because we had found that CHI3L1 levels were elevated in patients experiencing lung transplantation or death, we thought it possible that CHI3L1 levels would be highest in the setting of an “acute exacerbation,” a clinical entity in which the fibrotic lung manifests a superimposed injury response that is often characterized by diffuse alveolar damage (DAD) on histopathology that is associated with an exceedingly high mortality (28). To test this hypothesis, we compared CHI3L1 expression in IPF-AE lung tissue obtained at autopsy to the lung biopsies described above. CHI3L1-expresssing cells in the IPF-AE lungs were decreased compared to non–IPF-AE lungs (Fig. 2A) with sustained expression in macrophages but reduced expression of CHI3L1+ pneumocytes that appeared to be related to lack of expression by epithelial cells (figs. S1 and S2). Reduced detection of CHI3L1 was also found in preterminal plasma specimens obtained from five patients with IPF-AE, where CHI3L1 levels were found to be 60.7% reduced compared to those recruited from the outpatient setting who were not actively suffering an AE (Fig. 2B).
Fig. 2. The levels of CHI3L1 are decreased, and cell death is increased in AEs of IPF.

(A to C) Compared to ambulatory IPF (n = 7), quantities of CHI3L1+ cells are decreased in IPF-AE (n = 6). Overall analysis of variance (ANOVA) P value: 0.0058; comparison of IPF and IPF-AE: P < 0.05 with Bonferroni post-test comparison. (B) Compared to ambulatory IPF (n = 64), plasma CHI3L1 levels are decreased in IPF-AE (n = 5). P = 0.0116, Mann-Whitney comparison. (C) Compared to IPF lung biopsies (n = 4), TUNEL+ cells are increased in IPF-AE (n = 4). Overall P value for ANOVA: P = 0.002; comparison of IPF and IPF-AE: P < 0.05, Bonferroni post-test comparison. (D) TUNEL staining in normal, IPF, and IPF-AE lung tissue. Scale bars, 200 μm (low-power images) and 100 μm (high-power images).
The finding that low levels of CHI3L1 were seen in the context of marked and fatal development of lung injury led us to believe that elevated levels of CHI3L1 in IPF patients might function to control local tissue injury. Previous studies from our laboratory demonstrated that CHI3L1 exerts important antiapoptotic effects on a variety of cells including oxidant-injured epithelium (24, 29). However, a relationship between CHI3L1 and cell death responses in AEs of IPF has not been described. To test this hypothesis, we compared the levels of DNA injury using terminal deoxynucleotidyl transferase–mediated deoxyuridine tri-phosphate nick end labeling (TUNEL) evaluations of tissues from autopsy samples from patients who died from AEs of IPF (30) and biopsies from the out-patient IPF patients described above. These studies revealed increased TUNEL staining in the lungs from IPF-AE patients compared to IPF lung biopsies (Fig. 2, C and D). The TUNEL-positive cells bore the histologic appearance of epithelial cells and inflammatory cells (Fig. 2D). These data indicate that the lungs of patients with fatal IPF-AE contain exaggerated cell death responses, that these findings are accompanied by reduced levels of the tissue and circulating anti-apoptotic protein CHI3L1, and that this finding may in fact be related to this terminal event.
CD206+ macrophages are increased in the IPF lung
Current paradigms of IPF include a potential role for alternatively activated macrophages in the immunomodulation of existing fibrotic responses (9). We have previously demonstrated that CHI3L1 augments expression of the alternative macrophage activation marker CD206 in the presence of IL-13 (19). To determine whether this association also exists in human IPF, we assessed lung tissues from the same subjects shown in Fig. 1 for the expression of CD206 using immunohistochemistry. Semiquantitative analysis revealed a nearly twofold increase in the detection of CD206+ macrophages in the IPF lung (Fig. 3B). These data indicate that the increased expression of intrapulmonary CHI3L1 in human IPF is accompanied by the accumulation of CD206+ macrophages.
Fig. 3.

(A) Immunohistochemical detection of CD206 in normal and IPF lungs. Scale bars, 200 μm (low-power image) and 50 μm (high-power image). (B) Compared to histologically normal lung (n = 4), detection of CD206+ cells is increased in IPF lung biopsies (n = 7). P = 0.0002, unpaired Student’s t test. (C) Circulating CD14hiCD206+ cells are increased in IPF (n = 38) relative to age-matched normal controls (n = 42). P = 0.0342, Mann-Whitney comparison. (D) Schematic of three-dimensional Transwell culture system. Monocyte-derived cell population resides on the plate’s bottom, decellularized lung scaffold slice is placed on the Transwell insert, and fibroblasts are seeded onto the scaffold. (E to G) Fibroblast-seeded lung scaffolds grown in the presence of (E) no monocytes, (F) monocytes derived from the circulation of an IPF subject with low CD206 expression, and (G) monocytes derived from the circulation of an IPF subject with abundant CD206 expression. Scale bars, 200 μm. (H) Compared to cells from subjects with CD14hiCD206+ below the median value, circulating monocytes from IPF subjects with CD14hiCD206+ cells exceeding the median value cause increased fibroblast density in decellularized lungs (P = 0.0065, unpaired Student’s t test; n = 4 per group). (I) Relative to subjects with stable disease (n = 27), CD14hiCD206+ cell concentrations are increased in progressive IPF (n = 11). P = 0.033, Mann-Whitney comparison.
CD206+ monocytes are increased in the IPF blood
Because we had found CD206 expression on macrophages in the IPF lung, we thought it likely that CD206 might be expressed on a related cell population in the peripheral blood. We and others have previously shown that peripheral blood monocytes obtained from subjects with lung fibrosis display enhanced expression of the scavenger receptors that are typically associated with alternatively activated or “M2” macrophages (13, 31). Here, we extended those studies by exploring the relationship between the quantities of M2-related scavenger receptor expression (32) on circulating monocytes and the IPF disease state in a moderately sized subset of the subjects with IPF (n = 38) and demographically matched normal controls (n = 42). Subject characteristics are presented in table S2. Because monocytes are typically distinguished in part based on low or high expression of CD14 (CD14lo versus CD14hi), we first assessed the co-expression of CD14 and CD206 in our subjects. Flow cytometric detection of peripheral blood mononuclear cells (PBMCs) obtained from these individuals revealed that, in comparison to control, CD14hi monocytes of subjects with IPF showed increased quantities of cells coexpressing CD206 (Fig. 3C) that displayed an extremely weak positive correlation with CHI3L1 levels (fig. S3). Little to no CD206 expression was detected on CD14lo and CD14− populations. Therefore, further analysis was conducted on this CD206+CDhi population. The gating strategy is shown in fig. S5.
To determine whether these CD206+ cells might influence the fibrotic responses that define IPF (33), we took advantage of a bioengineering-centered, three-dimensional culture system based on decellularized mammalian lung that we had originally developed as an ex vivo platform for the regeneration of functional lung tissue (34). Lungs processed in this manner retain many of the biochemical and anatomic cues of the intact lung, and as a result, this method has been adapted for the study of fibroblast biology in the setting of fibrosis based on its potential superiority to two-dimensional culture systems in modeling the biochemical and biomechanical properties of the intact lung (34, 35). Here, as shown in Fig. 3D, decellularized scaffolds prepared from adult rat lungs were placed in Transwell plates, seeded with the MRC5 human fibroblast cell line, and grown in the presence of monocytes obtained from IPF subjects with either strong or weak expression of CD206 on CD14hi cells whose data are included in Fig. 3C. These studies found that fibroblast-seeded scaffolds cultured in the presence of cells obtained from the circulation of subjects with CD206-expressing cells above the median value of 5.92 × 104 cells/ml (“high CD206”) displayed higher fibroblast density than those scaffolds grown in the presence of cells from subjects with CD206 expression below this value (“Low CD206,” Fig. 3, D to H). Neither population of cells induced α-SMA (smooth muscle actin) expression in this model, suggesting that CD206 expression identifies a population of monocyte-derived cells capable of stimulating the accumulation, but not myofibroblast transformation, of cultured human fibroblasts.
To determine whether these cells were related to clinical outcomes, we performed longitudinal studies. This analysis revealed that when patients were stratified into “progressive” (defined by the criteria of lung transplantation or all-cause mortality within 72 weeks of study entry) compared with stable (subjects not meeting these endpoints), samples obtained from progressive subjects contained the greatest quantities of CD206+CD14hi cells (Fig. 3I and table S3). PBMC RNA from these subjects also displayed increased expression of the M2-associated transcription factor IRF-4 and reduced expression of the M1-associated transcription factor IRF-5 (fig. S5). These data indicate that the increased concentrations of circulating CHI3L1 present in IPF are accompanied by enhanced detection of fibroblast-stimulating CD206+ monocytes and an M2-skewed gene expression profile in the peripheral blood, thereby suggesting a relationship between these entities.
Bleomycin acutely and transiently decreases the expression of CHI3L1
The human data described above support the contention that CHI3L1 is an important regulator of a variety of potentially pathogenic processes driving IPF. To further understand the role(s) of CHI3L1 in this setting, we used murine modeling. Intratracheal bleomycin was used in these experiments because it causes an early injury response characterized by extensive epithelial cell apoptosis and inflammation and later fibroproliferative repair and scarring. Our studies demonstrated that bleomycin treatment acutely and transiently decreased CHI3L1 mRNA detection compared to saline-treated control mice (Fig. 4A). The peak reduction of CHI3L1 was seen on day 3, and the levels then returned toward and eventually exceeded baseline on days 14 to 21 (Fig. 4A). In a similar manner, the levels of bronchoalveolar lavage (BAL) CHI3L1 were initially suppressed and then normalized (Fig. 4B). Immunofluorescence staining revealed that CHI3L1 was expressed by airway epithelial cells, alveolar type II epithelial cells, and macrophages in the lung (fig. S6). These studies demonstrate that bleomycin regulates CHI3L1 gene expression in a time-dependent manner, with CHI3L1 levels acutely and transiently decreasing during the injury phase and increasing during the later fibroproliferative repair phase of this response.
Fig. 4. Bleomycin acutely and transiently decreases the expression of CHI3L1.
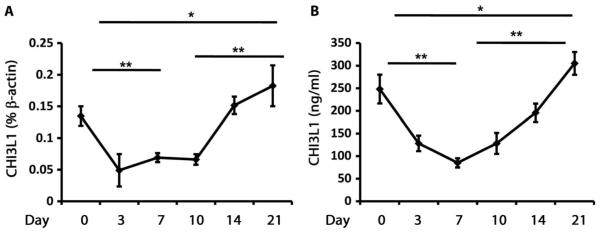
(A) CHI3L1 transcript was measured in whole-lung RNA extracts from wild-type (WT) mice treated with intratracheal bleomycin using quantitative reverse transcription polymerase chain reaction (qRT-PCR). Compared to day 0 (n = 4), CHI3L1 transcript levels are decreased on day 7 (n = 4). P = 0.003, unpaired Student’s t test. Compared to day 0 (n = 4), CHI3L1 transcript levels are increased on day 21 (n = 4). P = 0.03, unpaired Student’s t test. Compared to day 10 (n = 5), CHI3L1 transcript levels are increased on day 21 (n = 4). P = 0.0019, unpaired Student’s t test. (B) CHI3L1 protein in BAL fluid from WT mice treated with intratracheal bleomycin was quantitated using enzyme-linked immunosorbent assay (ELISA). Compared to day 0 (n = 4), CHI3L1 levels are decreased on day 7 (n = 4). P = 0.002, unpaired Student’s t test. Compared to day 0 (n = 4), CHI3L1 levels are increased on day 21 (n = 4). P = 0.05, unpaired Student’s t test. Compared to day 10 (n = 5), CHI3L1 levels are increased on day 21 (n = 4). P = 0.009, unpaired Student’s t test.
Transgenic CHI3L1/YKL-40 regulates injury and repair in a temporally specific manner in vivo
This biphasic regulation of CHI3L1 described above suggests that this protein participates in fibrosis in a temporally heterogeneous, compartment-specific manner. To define the roles of CHI3L1 in the pathogenesis of the tissue effects of bleomycin, we characterized its effects on injury and repair using YKL-40 Tg mice developed in our laboratory, in which YKL-40 is selectively and inducibly targeted to the lung using the CC10 promoter (19). We first employed transgene activation 2 days before the bleomycin was administered and kept the gene “on” for 14 days after bleomycin was administered. In these experiments, transgenic YKL-40 caused a modest but significant increase in collagen accumulation (Fig. 5A and fig. S7), BAL cellularity (Fig. 5B), and macrophage and lymphocyte accumulation (Fig. 5C) as assessed in the 14-day time point.
Fig. 5.

(A to C) Bleomycin was administered 2 days after YKL-40 Tg expression, and the transgene was activated from days 0 to 14. (A) Compared to bleomycin-treated WT mice (n = 4), total lung collagen is increased in YKL-40 Tg mice (n = 4). P = 0.02. (B) On day 7, BAL cells are decreased in bleomycin-treated YKL-40 Tg mice (n = 4) compared to similarly treated WT controls (n = 5). P = 0.019. On day 14, BAL cells are increased in bleomycin-treated YKL-40 Tg mice (n = 4) compared to similarly treated WT controls (n = 6). P = 0.002. (C) Macrophages and lymphocytes are increased in YKL-40 Tg mice (n = 4) compared to controls (n = 6). P = 0.015 and P = 0.03, respectively. (D to E) YKL-40 Tg expression was induced from day 0 to day 5. (D) Compared to similarly treated WT animals (n = 4), total lung collagen is unaltered in bleomycin-treated YKL-40 Tg mice (n = 4). P = 0.09. (E) Compared to WT animals (n = 4), TUNEL+ cells are decreased in bleomycin-treated YKL-40 Tg mice (n = 4). P = 0.019. (F to I) YKL-40 Tg was activated from days 5 to 14. (F) Compared to bleomycin-treated WT mice (n = 4), total lung collagen is increased in bleomycin-treated YKL-40 Tg mice (n = 4). P = 0.0006. In bleomycin-treated YKL-40 Tg mice (n = 6), relative to WT animals (n = 4), (G) Col1-α1 mRNA is increased (P = 0.006), (H) fibronectin mRNA is increased (P = 0.005), and (I) α-SMA mRNA is increased in bleomycin-treated YKL-40 Tg mice (P = 0.002). All comparisons were performed using unpaired Student’s t test. Dox, doxycycline.
These data suggested one of two explanations: either CHI3L1 exerts only small effects on repair and remodeling in this system or, as was suggested by the human data above, CHI3L1 has different time-dependent effects. To test this hypothesis, we next turned on the YKL-40 Tg only during the tissue injury phase (days 0 to 5) at which point the transgene was turned off (fig. S8). In these experiments, the YKL-40 Tg mice had slightly decreased levels of collagen deposition (Fig. 5D), significantly decreased levels of epithelial cell apoptosis (Fig. 5E), and BAL protein leakage (fig. S9) compared to lungs from wild-type controls. In contrast, when the YKL-40 transgene was kept off during the early tissue injury phase and turned on only during tissue repair (days 5 to 14) (fig. S8), collagen content was markedly enhanced (Fig. 5F and fig. S7), matrix molecule gene expression was augmented (Fig. 5, G and H), and α-SMA gene expression was significantly increased (Fig. 5I). In addition, late overexpression of CHI3L1 increased mortality compared to wild-type mice (fig. S10). These studies demonstrate that the early bleomycin-induced decrease in CHI3L1 is associated with significant levels of epithelial apoptosis and that restoration of CHI3L1 at this time point abrogates injury and subsequent tissue fibrosis. They also demonstrate that interventions that increase the levels of CHI3L1 during fibroproliferative repair augment fibrosis and matrix molecule gene expression. When viewed in combination, these studies demonstrate that CHI3L1 shows temporally specific and seemingly opposed effects on repair and remodeling with the ability to decrease injury during the early phase and stimulate fibrosis and matrix gene expression when administered during fibroproliferative repair.
Endogenous CHI3L1 regulates injury, inflammation, and repair
The above data suggest that CHI3L1 suppresses tissue injury while promoting repair. On the basis of these data, we thought it likely that complete removal of CHI3L1 would lead to enhanced cell death responses and a relative lack of repair. To test this hypothesis, we compared the bleomycin responses in wild-type mice and mice with null mutations of CHI3L1. These studies demonstrated that, when compared to the wild-type controls, null mutant mice manifested exaggerated epithelial apoptosis (fig. S11), enhanced accumulation of macrophages and neutrophils (fig. S11), and mortality (fig. S10) in the early days after bleomycin administration. In keeping with the enhanced injury and diminished fibroproliferative responses, in these mice, collagen accumulation was unchanged (fig. S11). To determine whether null mutation of CHI3L1 in the repair phase ameliorates fibrosis, YKL-40 Tg mice were bred onto the CHI3L1 null background and YKL-40 Tg was activated only during tissue injury phase (days 0 to 5) to allow a normal tissue injury response. In these studies, mice lacking CHI3L1 displayed significantly decreased fibrosis development (fig. S11). In addition, bone marrow chimeras were generated to specifically address the role of local and bone marrow–derived CHI3L1 in injury and repair. In these studies, host-derived CHI3L1 was required to prevent cell death in tissue injury (fig. S12). In contrast, CHI3L1 expression by bone marrow–derived cells (BMDCs) was not required for the development of fibrosis in bleomycin-exposed mice (although because not all recipient cells might have been replaced by BMDCs, it is possible that some host cells may have confounded these results), but its presence on BMDCs increased the detection of soluble collagen in CHI3L1 null recipients of wild-type bone marrow (fig. S12). These data show that CHI3L1 in structural cells is required for the maximal cytoprotective effects but that expression by BMDCs is sufficient to amplify existing fibrotic responses in the bleomycin model, findings that support both the human data presented above and our transgenic findings by demonstrating that endogenous CHI3L1 controls injury while augmenting fibroproliferative repair in a compartment-specific manner.
Chronic fibroproliferation in YKL-40 Tg mice is associated with increased CD206+ cells
We were next interested in the BMDC that might be contributing to fibroproliferation in our model. We previously demonstrated that CHI3L1 plays a major role in M2 macrophage differentiation (19) and that M2 macrophage activation is a putative mechanism in pulmonary fibrosis (31). On the basis of this rationale and the human data presented above, we thought it possible that the profibrotic effects of CHI3L1 were enacted at least in part via the accumulation of M2 macrophages (32). To test this hypothesis, we assessed the macrophage differentiation markers in whole-lung RNA extracts of bleomycin-treated YKL-40 Tg mice and wild-type controls. These studies demonstrated that relative to wild-type mice, the expression of molecules associated with M2 differentiation including YM-1, CD206, and FIZZ-1 was increased during the fibroproliferative phase of the bleomycin response in YKL-40 Tg mice (Fig. 6, A to C). Conversely, the expression of moieties that have been associated with M1 differentiation such as iNOS was significantly decreased in these tissues (Fig. 6, A to D). In concordance with these findings, fluorescence-activated cell sorting (FACS) analysis demonstrated that the number of F4/80+CD45+CD11c+CD206+ (hereinafter called “CD206hi”) macrophages was significantly elevated in the lungs of YKL-40 Tg mice compared to wild-type mice (Fig. 6E and fig. S13). When the CD206hi population of macrophages was sorted and compared to CD206lo cells, they were found to express significantly more YM-1 and CD206, and less iNOS (fig. S13). Further, expression of the M2 markers was higher in the M2 cells from YKL-40 Tg mice compared to that from wild-type controls (fig. S13). When total lung macrophages were depleted in this model using liposomal clodronate, we found a significant reduction in total lung collagen and α-SMA expression (Fig. 6, F and G) that was accompanied by a reduction in BAL macrophages (Fig. 6H). These data indicate that eradication of macrophages, which show a predominant shift toward alternative activation in YKL-40 Tg mice, is sufficient to reduce tissue fibrosis in the setting of CHI3L1 overexpression.
Fig. 6. WT and YKL-40 Tg mice were subjected to intratracheal bleomycin administration, the YKL-40 Tg was expressed from days 5 to 14, whole-lung mRNA was extracted, and (A) YM-1, (B) CD206, (C) FIZZ-1, and (D) iNOS expression were evaluated by qRT-PCR.

Compared to controls (n = 4), YM-1 is increased in YKL-40 Tg mice (n = 4). P = 0.016, unpaired Student’s t test. CD206 is increased in YKL-40 Tg mice (n = 4). P = 0.005, unpaired Student’s t test. FIZZ-1 is not significantly altered in YKL-40 Tg mice (n = 4). P = 0.15, unpaired Student’s t test. iNOS is decreased in YKL-40 Tg mice (n = 4). P = 0.029, unpaired Student’s t test. (E) Lungs were digested and FACS analysis was used to analyze CD11c and CD206 expression on CD45+F4/80+ gated cell populations. CD45+F4/80+CD11C+CD206+ cell numbers were calculated on the basis of total cell recovery. Compared to bleomycin-treated WT mice (n = 5), CD206+ cells are increased in bleomycin-treated YKL-40 Tg mice (n = 6). P = 0.036, unpaired Student’s t test. (F to H) When intrapulmonary macrophages were depleted using inhaled clodronate in bleomycin-treated YKL-40 Tg+ mice (n = 6), total lung collagen measured by Sircol was reduced compared to controls (n = 6). P = 0.006, unpaired Student’s t test. mRNA levels of α-SMA were decreased. P = 0.026, unpaired Student’s t test. (H) BAL macrophages were also decreased. P = 0.001, unpaired Student’s t test. n = 6 mice per group.
CD206+ mouse lung macrophages stimulate fibroblast proliferation and survival but not myofibroblast transformation in decellularized lung scaffolds
The pathologic findings of IPF include the histologic pattern of usual interstitial pneumonia, an entity characterized by the juxtaposition of histologically normal lung, established fibrosis, and fibroblastic foci that contained proliferative and contractile myofibroblasts (33). To determine whether macrophages in general, and CD206+ macrophages in particular, influence these endpoints, we used the three-dimensional culture system used above to interrogate macrophage-fibroblast interactions in the context of experimentally induced pulmonary fibrosis. Here, decellularized scaffolds prepared from adult mouse lungs were placed in Transwell plates, seeded with a murine fibroblast cell line, and grown in the presence of total macrophages, CD206+ macrophages, CD206− macrophages, or no macrophages obtained from the bleomycin-exposed lung at the 14-day time point. Our results indicate that unfractionated macrophages obtained from the fibrotic mouse lung exert stimulatory effects on fibroblast density in this model and that this effect is largely attributable to the presence of CD206+ macrophages (Fig. 7A and fig. S14). CD206+ cells also enhanced fibroblast proliferation (based on Ki67 immunostaining, Fig. 7B) and survival (based on TUNEL staining, Fig. 7C). There was no induction of α-SMA expression in any of the conditions studied. In addition, CD206+ macrophages obtained from bleomycin-exposed BRP39−/− or YKL-40+Tg+ mice did not differentially regulate fibroblast density when compared to CD206+ macrophages obtained from bleomycin-exposed wild-type mice (Fig. 7D), indicating that any macrophage-mediated effects on fibrosis in these animals are likely due to the magnitude of the infiltrate rather than inherent phenotypic differences or secretion of CHI3L1 by these cells. These data indicate that CD206+ mouse lung macrophages, which are stimulated by CHI3L1, enact stimulatory effects on fibroblast proliferation and survival but may not directly influence the development of the ultimate effector cells in fibrosis, namely, the activated myofibroblasts.
Fig. 7.

(A) CD206+ macrophages obtained from the bleomycin-exposed mouse lung increase fibroblast density in decellularized murine lungs when compared to scaffolds grown with no cells (P < 0.01, one-way ANOVA with Bonferroni post-test), with CD206+/CD206 macrophages (P < 0.05, one-way ANOVA with Bonferroni post-test), or with CD206 macrophages alone (P < 0.0001, one-way ANOVA with Bonferroni post-test). For all studies, n = 3 experiments. (B) CD206+ macrophages obtained from the bleomycin-exposed mouse lung increase Ki67 immunostaining/proliferation in fibroblast-seeded lung scaffolds when compared to no cells (n = 3 experiments per group; P < 0.0001, one-way ANOVA with Bonferroni post-test), the CD206+/CD206− fraction (n = 3 experiments per group; P < 0.01, one-way ANOVA with Bonferroni post-test), and CD206 macrophages (n = 3 experiments per group; P < 0.0001, one-way ANOVA with Bonferroni post-test). (C) CD206 macrophages reduce fibroblast survival based on TUNEL staining compared to cells cultured with CD206+/CD206 macrophages (P < 0.05, one-way ANOVA with Bonferroni post-test) and CD206+ macrophages only (P < 0.01, one-way ANOVA with Bonferroni post-test). For all comparisons, n = 3 experiments. The dotted line represents the unstimulated control value. (D) CD206+ macrophages obtained from either CHI3L1−/− or CHI3L1 Tg+ mice exposed to bleomycin show no significant difference in fibroblast cell density in the scaffold culture system.
CHI3L1 augments fibroblast proliferation and myofibroblast transformation in decellularized human lung scaffolds
CHI3L1 is reported to stimulate the proliferation of fibroblasts in several settings (25). However, the potential for CHI3L1 to contribute to fibroproliferation via myofibroblast transformation has not been described. To explore this issue, we adapted the three-dimensional culture system described above for the study of soluble mediators in human lung biology. Decellularized scaffolds prepared from nonfibrotic human lungs were seeded with the MRC5 human fibroblast cell line and stimulated with rhCHI3L1. Recombinant human interferon-γ (rhIFN-γ) was used as a negative control. Using this approach, we found that, in contrast to unstimulated cells, fibroblasts grown in the presence of rhCHI3L1 demonstrated increased cell density based on hematoxylin and eosin (H&E) staining (Fig. 8A) and increased proliferation based on Ki67 staining (Fig. 8B). Additionally, the addition of rhCHI3L1 was sufficient to modestly induce α-SMA detection in MRC5 cells cultured in this model (Fig. 8C), and a contractile phenotype was suggested by the shrinking and rupture of the matrix slices noted on H&E stains (Fig. 8A). Cells treated with IFN-γ failed to display a similar response in these parameters, indicating that the fibroblast-stimulating effects are relatively specific to CHI3L1 and not to cytokine stimulation in general (Fig. 8, A to C). These data indicate that CHI3L1 directly regulates fibroblast proliferation and to a smaller extent myofibroblast transformation in the decellularized human lung, thereby demonstrating a role for CHI3L1 in the regulation of all three phases of the fibrotic response.
Fig. 8. Effect of rhCHI3L1 on human lung fibroblast phenotypes grown in the decellularized human lung.

(A to C) Compared to unstimulated cells, exposure to rhCHI3L1 increases (A) fibroblast cell density measured by H&E staining (n = 3 per group; P < 0.0001, one-way ANOVA with Bonferroni post-test; scale bar, 200 μm), (B) proliferation as measured by Ki67 staining (red) (n = 3 per group; P < 0.05, one-way ANOVA with Bonferroni post-test; scale bars, 100 μm), and (C) myofibroblast transformation as measured by α-SMA immunostaining (red) (n = 3 per group; P < 0.001, one-way ANOVA with Bonferroni post-test; scale bars, 100 μm). Fluorescence images are counter-stained with 4′,6-diamidino-2-phenylindole (DAPI) (blue). Values are means ± SEM and are representative of three separate iterations. In the graphs, the dotted horizontal line represents the unstimulated control value.
DISCUSSION
IPF has a prevalence of 132,000 to 200,000 people in the United States, affects millions worldwide, and has a 5-year mortality of 70% (36). In keeping with its clinical impact, efforts have been directed at defining the mechanisms of injury and exaggerated fibroproliferative repair that characterize this disorder. Despite these efforts, therapeutic agents that can be manipulated to completely control the development and progression of pulmonary fibrosis remain elusive. Because CHI3L1 has been implicated in the control of tissue injury and fibroproliferative repair, studies were undertaken to define the regulation and roles of CHI3L1 in IPF and murine models of pulmonary fibrosis. Consistent with other reports (27), these studies demonstrated that the levels of circulating CHI3L1 were increased in patients with IPF and that, in ambulatory patients, this increase was highest in the setting of progressive disease. They also demonstrated that in the bleomycin model, the expression of CHI3L1 was decreased during the early injury phase and returned to and exceeded normal during fibroproliferative repair. When inducible YKL-40 Tg mice were used to eliminate this early decrease in CHI3L1, tissue injury was decreased and fibrosis was ameliorated. Conversely, when the expression of CHI3L1 was exaggerated during repair, matrix gene expression, collagen accumulation, α-SMA expression, and fibrosis were augmented. CHI3L1 also interacted with lung ECM to augment fibroblast proliferation and transformation to activated myofibroblasts. These studies demonstrate that CHI3L1 has multiple effects in the lung where it functions to simultaneously decrease injury and augment repair. They allow for the exciting two-part hypothesis that the injury that initiates fibrotic responses is mediated in part by a decrease in CHI3L1, and that the exaggerated levels of CHI3L1 seen in the lungs and circulation of ambulatory IPF patients represent a protective response that functions to control injury but does so at the cost of exaggerated fibroproliferative repair.
AEs constitute the most devastating complication of IPF. These exacerbations are typically acute, clinically significant deteriorations of unidentifiable cause that transform the frequently noted slow and more or less steady disease decline in ambulatory patients to a disease characterized by rapid deterioration and death (37). Studies of tissues from patients with these AE have revealed DAD, the tissue counterpart of acute respiratory distress syndrome (ARDS), superimposed on usual interstitial pneumonia, the tissue equivalent of IPF. They have also demonstrated that IPF-AE is characterized by enhanced epithelial injury, apoptosis, and proliferation (28). However, the cellular events that prevent DAD in most patients with IPF and their alterations in patients with AE have not been fully defined. Previous studies from our laboratory demonstrated that CHI3L1 is an important inhibitor of oxidant-induced epithelial injury and apoptosis (24), and the studies noted above demonstrated that the expression of CHI3L1 is augmented in IPF. Our murine studies demonstrated that the levels of CHI3L1 are decreased immediately after bleomycin administration and that transgenic CHI3L1 inhibits bleomycin-induced acute lung injury. In keeping with these findings, we noted that the levels of CHI3L1 were not increased in patients with IPF-AE and instead were at or below the levels in normal controls. This allows for the interesting hypothesis that the induction of CHI3L1 in ambulatory IPF patients prevents diffuse lung injury and that a failure of this protective response plays an important role in the development of AE, although this idea is largely speculative at this point and will require further study. These studies also raise the intriguing possibility that rCHI3L1 might be used in the treatment of IPF patients experiencing these often fatal AEs.
Apoptosis of type II alveolar epithelial cells is a persistent finding in lungs from IPF patients (38, 39) and murine models of pulmonary fibrosis (40–42), and it is believed to be a primary event that occurs before excess collagen accumulation (43). Studies of murine models from this and other laboratories have demonstrated that blocking epithelial apoptosis with caspase inhibitors (12, 44), angiotensin system antagonists (45), hepatocyte growth factor (46), and cyclin-dependent kinase inhibitors (47) decreases tissue fibrosis. As a result, therapeutic strategies aimed at manipulating the apoptotic response(s) in IPF have been proposed. Our studies add to this body of knowledge by demonstrating that CHI3L1 is also an important inhibitor of apoptosis in pulmonary fibrosis. Because the injury and repair responses that are seen in IPF are geographically and temporally heterogeneous, these observations raise the possibility that CHI3L1 might also be a useful therapeutic that could control tissue injury. Its utility in this setting would be further enhanced if subsequent studies define ways to separate the antiapoptotic and fibroproliferative effects of this CLP, allowing the protective effects of the former to manifest without engendering excess scarring.
Alternatively activated (M2) macrophages are detected with high frequency in lungs of IPF patients (48), and this differentiation is proposed as a fibrotic mechanism in this disorder (9, 48). M2 macrophages express high levels of YM-1, FIZZ-1, and macrophage mannose receptor (CD206) (49, 50), and produce a variety of soluble mediators such as TGF-β, CCL18, and IL-8 (51, 52). Our previous studies demonstrated that CHI3L1 is a direct stimulator of alternative macrophage activation (19). Here, we showed that the enhanced fibrotic response in YKL-40 Tg mice included increased expression of M2 markers such as YM-1, CD206, and FIZZ-1. In addition, CD206+ macrophages were markedly increased in the lungs of YKL-40 Tg mice compared to wild-type mice after bleomycin administration. This result is consistent with our clinical observation that CD206+ monocytes are detected with increased frequency in the blood of IPF patients with a more progressive disease phenotype. The in vitro work presented in this study indicate that the fibroproliferative effects of CD206+ macrophages include stimulation of fibroblast proliferation but do not extend to activation of myofibroblasts, although it is possible that interactions occurring within the native or fibrotic lung might enable macrophage induction of α-SMA expression via interactions with neighboring cells, ECM components, and/or mechanotransductive pathways. They also demonstrate that CHI3L1 expression by macrophages amplifies but does not initiate fibrotic responses, suggesting that the role of macrophages in experimentally induced lung fibrosis follows a similar pattern.
Our work also shows that stimulation with CHI3L1 alone is sufficient to modestly induce α-SMA expression in cultured fibroblasts, suggesting that although there is redundancy in the mechanism(s) through which CHI3L1 influences fibroblast proliferation, its effects on myofibroblast transformation are enacted more directly. These effects could occur via direct receptor-mediated interactions on α-SMA production or via CHI3L1-induced auto-crine secretion of TGF-β1 by fibroblasts. When viewed in combination, this constellation of findings leads to a paradigm in which CHI3L1 functions as a master regulator of repair and remodeling by exerting cytoprotective effects on structural cells to limit the extent of damage, and stimulatory effects on macrophages and fibroblasts to induce healing. Because we have recently shown that CHI3L1 signals through IL-13Rα2 and interacts with several different signaling pathways including AKT phosphorylation, inflammasome activation, and stimulation of TGF-β1 production (53), it is intriguing to speculate that our reported results are mediated via differential regulation of these downstream components of IL-13Rα2 or by an alternate as yet undescribed receptor or pathway. Better understanding of these mechanisms will be essential to designing compartment-specific, CHI3L1-targeted therapies that might reduce epithelial cell damage without bystander effects on inflammatory cells and fibroblasts, or vice versa.
Our work is limited by several caveats. We have not performed the large-scale replication cohort studies necessary to establish CHI3L1 as a predictive biomarker in IPF, although this has been suggested by previous reports (27). We have not determined the precise relationship between CHI3L1 and either AE or CD206 macrophages, although the lack of a strong statistical correlation between CHI3L1 and CD206+ monocytes in the peripheral blood suggests that the relationship between these entities is unlikely to be linear and may be more robust in target organs. We have not determined the precise identity of the cells expressing CD206 in the IPF circulation, although the high expression of CD14 combined with low to absent expression of CD16 suggests that they might be “classical” monocytes, although the functional relevance of monocyte subsets in the context of IPF has not been defined. We have not determined the mechanism(s) through which CHI3L1 exerts its effects on structural cell death and fibroproliferation, and we have not determined the mechanism(s) though which CHI3L1-stimulated macrophages regulate fibroblast responses either in the intact organ or in the decellularized mammalian lung. We have also not investigated the mechanism of CHI3L1’s stimulation of αSMA expression in cultured fibroblasts though the low level induction of this protein in our model suggests that other factors are more dominant mediators of myofibroblast transformation in the context of pulmonary fibrosis. Last, the relevance of the three-dimensional decellularized lung cultures both to previous in vitro studies (which have largely relied on two-dimensional systems) and to lung biology remains to be determined.
Despite these shortcomings, however, our studies convincingly demonstrate that CHI3L1 exerts complex and seemingly opposed effects in pulmonary fibrosis, with the ability to decrease/inhibit injury and, as a result, decrease fibrosis on one hand, and the ability to augment fibroproliferative repair and tissue scarring on the other. They also demonstrate that CHI3L1 is induced in ambulatory patients with IPF, where it is associated with disease activity and likely represents a protective response that functions to decrease injury and augment healing. Better delineation of the mechanisms involved may advance our understanding of IPF to allow compartment-specific therapies for this otherwise incurable disease.
MATERIALS AND METHODS
Study design
The overall goal of this study aimed to understand the putative role of CHI3L1 in human IPF and in experimentally induced lung fibrosis in mice. Three different experimental approaches were used to achieve this aim. In terms of human studies, a prospective cohort of subjects was assembled to test the hypothesis that elevated circulating CHI3L1 was associated with disease progression (defined by the composite outcome of lung transplantation or death). The sample size of at least 23 subjects per group was calculated using pilot data for a projected increase in circulating CHI3L1 of at least 20 ng/ml between stable and progressive subjects, assuming 80% power and a 95% confidence level (α = 0.05) and including four additional subjects per group to control for dropouts and withdrawal of consent. Secondary hypotheses generated after the initiation of the study were related to the role of CHI3L1 in IPF-AE as well as the relationship to CD206 expression on monocyte-derived cells. The human studies were only performed once with no confirmatory cohort. For the mouse studies, CHI3L1 transgenic and null mutant mice were created and characterized in the context of bleomycin-induced fibrosis to allow for mechanistic studies of the temporally heterogeneous and compartment-specific role that CHI3L1 might play in lung fibrosis. Research staff involved with animal husbandry (such as animal facility personnel) and data acquisition from samples that had been obtained from the animal studies (such as laboratory personnel) were blinded as to the experimental group of the animal or sample. For the three-dimensional culture studies, individuals performing the analysis of the sections were blinded as to study group. Mouse and culture studies were repeated three times with the exception of the bone marrow transplantation (BMT) studies and several of the cell culture studies (which were requested by reviewers during peer review for publication). Outliers were included in the statistical analysis.
Human subjects
All studies were carried out with approval from the Human Investigations Committee at the Yale University School of Medicine with explicit informed consent. Inclusion criteria included age >18 years who had been diagnosed with IPF based on current criteria (33, 54). Healthy, age-matched controls were recruited from Yale’s Program on Aging and from the greater New Haven community. Exclusion criteria included current or recent use of immunosuppression; chronic infection such as HIV or hepatitis; known pulmonary hypertension; cardiovascular, renal, or neoplastic disease; and inability to provide informed consent. Comprehensive clinical data, including age, sex, race and ethnicity, comorbidities, medications, and physiologic impairment as measured by the percentage predicted forced vital capacity (FVC) and diffusion capacity of carbon monoxide (DLCO), were collected. All of these human studies were performed with appropriate institutional approval and with explicit informed consent.
CHI3L1 ELISA
Detection of human CHI3L1 was performed using a commercially available ELISA kit (Quidel) as previously described (55). CHI3L1 levels in mouse BAL samples were quantified using coating and detection antibodies from MedImmune.
Processing of human cells and flow cytometric analysis
After informed consent and enrollment, 40 ml of heparinized peripheral blood was drawn from study subjects, anonymized, and transported to the laboratory. PBMCs were isolated from the whole blood using Ficoll-Paque–based separation (STEMCELL Technologies) and processed for FACS as we have previously described (56). Antibodies against human CD206, CD14, CD11b, and CD16 and appropriate isotype controls were obtained from BD Pharmingen. Flow cytometry and cell sorting were performed using BD FACSCalibur. Data were analyzed using FlowJo software version 8.7.3 (Tree Star Inc.). For all analyses, isotype control staining was subtracted from true antibody staining to determine the percentage of positive cells.
Knockout and transgenic mice
CHI3L1/BRP-39 knockout mice (BRP-39−/−) and CC10-driven, lung-specific, inducible CHI3L1/YKL-40 Tg mice were previously generated in our laboratory (21). YKL-40 Tg mice or their wild-type littermate controls (transgene-negative) were given doxycycline (1 g/liter) in their drinking water for up to 2 weeks. All mice were congenic on a C57BL/6 background and were genotyped as previously described (19). Animal experiments were approved by the Yale School of Medicine Institutional Animal Care and Use Committee in accordance with federal guidelines.
Bleomycin administration
Sex-matched, 8-week-old wild-type, BRP-39−/−, and YKL-40 Tg mice (five or more mice per group) were exposed to a single bleomycin injection (1.25 U/kg; Teva Parenteral Medicines) via intratracheal administration. Mice were sacrificed and evaluated at days 3, 7, 10, 14, and 21.
mRNA analysis of mouse and human samples
Total cellular RNA from human PBMCs, or wild-type and YKL-40 Tg mice lungs, was obtained using TRIzol reagent (Invitrogen) according to the manufacturer’s instructions. mRNA was measured using real-time RT-PCR as described previously (19, 24). The primer sequences for IRF-4, IRF-5, ECM genes, TGF-β1, and macrophage activation markers were obtained from PrimerBank (http://pga.mgh.harvard.edu/primerbank/) or the same as previously used (19, 57–59).
Histologic analysis
Formalin-fixed, paraffin-embedded sections of IPF and nonfibrotic control lungs were obtained from the Department of Pathology at Yale University School of Medicine. Immunohistochemical analysis of IPF lungs was performed as previously described (56). Antibodies against human CHI3L1 and CD206 were obtained from MedImmune and BD Pharmingen. Mouse lungs were removed en bloc, inflated to 25-cm pressure with phosphate-buffered saline (PBS) containing 0.5% low–melting point agarose gel, fixed, embedded in paraffin, sectioned, and stained. H&E and Mallory’s trichrome stains were performed in the Research Histology Laboratory of the Department of Pathology at the Yale University School of Medicine. BAL and lung inflammation was assessed as described previously (19).
Immunofluorescence staining
To localize the expression of CHI3L1, in human lungs, three-color immunofluorescence staining was undertaken on sections using antibodies against human pro-SPC, EMR-1, and CHI3L1 and on cytospins using antibodies against CHI3L1, CCL-18, and iNOS. For mouse studies, paraffin-embedded lungs from wild-type mice were sectioned. Monoclonal anti-BRP-39 (R&D Systems) and antibodies against CC10 (Santa Cruz Biotechnology), SPC (Santa Cruz Biotechnology), and F4/80 (Abcam) were used in these evaluations.
Quantification of lung collagen
Animals were anesthetized, median sternotomy was performed, and right heart perfusion was completed with calcium- and magnesium-free PBS. The heart and lungs were then removed. The right lung was frozen in liquid nitrogen and stored at −80°C until used. Collagen content was determined by quantifying total soluble collagen using the Sircol Collagen Assay kit (Biocolor, Accurate Chemical and Scientific Corp.) according to the manufacturer’s instructions.
TUNEL analysis
End labeling of exposed 3′-OH ends of DNA fragments in paraffin-embedded tissue was undertaken with the TUNEL In Situ Cell Death Detection Kit, AP (Roche Diagnostics). After staining, 8 to 10 random pictures were obtained from each lung, and a minimum of 300 cells were visually evaluated in each section. The labeled cells were expressed as a percentage of total nuclei.
Bone marrow transplantation
Bone marrow transplantation was performed as previously described (59). Female recipient mice were exposed to 1000 cGy of total body irradiation, and 6 hours after dose completion, the recipient mice received 2 million whole bone marrow cells from donor mice via tail vein injection. After BMT, mice were maintained on sulfatrim and given autoclaved food and water for 3 weeks. All animals were treated in accordance with the Institutional Animal Care and Use Committee guidelines. Five mice were included in each group, and the experiment was performed twice for a total of 10 mice per group at each time point.
Flow cytometric analysis for mouse macrophages
Mouse lungs were digested for flow cytometry, total viable cells were quantified using trypan blue staining, and flow cytometric analysis for macrophages was performed according to our previously published method (31). Percentages of live cells coexpressing CD45 F4/80, CD11C, and CD206 were multiplied by total viable cell count of mouse digested lung to determine the absolute number of CD45+F4/80+CD11C+CD206+ cells per either the right lower lobe or entire left lung. Flow cytometry and cell sorting were performed using BD FACSCalibur. Data were analyzed using FlowJo software version 7.5 (Tree Star). For all analyses, isotype control staining was subtracted from true antibody staining to determine the percentage of positive cells.
Clodronate depletion of intrapulmonary macrophages
Macrophages in the lung were depleted in wild-type and YKL-40 Tg mice with liposomal clodronate 5 days after bleomycin administration every 3 days. It was encapsulated in liposomes as described earlier (13).
Preparation of decellularized lungs
Decellularized murine lung scaffolds were prepared according to a modification of previously reported methods (34, 35, 60, 61). In brief, deeply anesthetized mice were subjected to BAL, and lungs were removed en bloc, sliced into 2-mm sections, and placed in zwitterionic CHAPS-based decellularization solution overnight. After this procedure, lungs were extensively rinsed in PBS containing 1% penicillin-streptomycin. Decellularization was confirmed using fluorometric DNA analysis and DAPI staining as previously described (34, 61). Assessment of ECM components was performed using Western blot and immunostaining as we have extensively described (34, 35, 60, 61). For the human scaffolds, freshly resected human lung explants that had been deemed unsuitable for lung transplantation were decellularized and processed as previously described (35).
Three-dimensional culture of fibroblasts in decellularized lungs
Immediately before culture, slices were rinsed with PBS for 5 min, placed in a Transwell insert (Corning Life Sciences), extended with forceps, and incubated at 37°C × 10 min to allow adherence. Scaffolds were seeded with 1 million A9 murine fibroblasts as we have previously described (61), and the bottom of the Transwell plate was incubated with 1 million macrophages of interest (CD206hi, CD206lo, or CD206hi and CD206lo) in serum-containing RPMI medium. The medium was changed every 48 hours, and scaffolds were harvested, fixed in formalin, and paraffin-embedded after 7 days. For the human fibroblast studies, 1 million MRC5 cells were seeded onto decellularized lungs and either grown in serum-supplemented medium containing (for the coculture studies) monocytes from subjects with CD206 above or below the median and harvested after 14 days or grown in serum-supplemented medium containing (for the stimulation studies) rhCHI3L1 (200 ng/ml), rhIFN-γ (50 ng/ml), or no additives and harvested after 72 hours. Harvested scaffolds were either processed for RNA extraction or fixed in formalin and paraffin-embedded for future H&E staining and/or immunofluorescence.
Statistics
Human data are presented as dot plots with means ± SEM unless stated otherwise. Data distribution was determined with the D’Agostino and Pearson omnibus test. Normally distributed data were compared using Student’s t test or ANOVA with Bonferroni post-test as appropriate. Non-normally distributed data in two groups were compared using the non-parametric two-tailed Mann-Whitney test. Logistic regression modeling was used to calculate an unadjusted receiver operating characteristic (ROC) curve as well as an ROC curve adjusted for age, gender, % FVC predicted at baseline, and corrected DLCO, and the C-statistic was calculated for various split points in the continuous variable CHI3L1/YKL-40 to determine the optimal split point. GraphPad Prism 5.0 (GraphPad Software) and SPSS 13.0 (SPSS Inc.) were used for all of these analyses. Mouse data are expressed as means ± SEM. As appropriate, groups were compared by ANOVA; follow-up comparisons between groups were conducted using Student’s t test. A P value of ≤0.05 was considered to be significant.
Supplementary Material
Acknowledgments
Funding: HL109033, 5U01HL112702, American Thoracic Society, Pulmonary Fibrosis Foundation, Yale Translational Lung Research Program (E.L.H.), N01-HHS N272201100019C (R.R.M. and A.C.S.), American Thoracic Society/Hermansky-Pudlak Syndrome Network (Y.Z.), HL093017, U01HL108638 (J.A.E.), and U01HL109118 (E.S.W.).
Footnotes
Author contributions: Y.Z., H.P., and H.S. participated in study design, data acquisition, data analysis, and manuscipt preparation. C.T., Y.G., X.P., X.C., A.M., C.-M.L., R.J.H., R.R.M., A.C.S., M.W.M., and E.S.W. participated in data acquisition and analysis. B.H., M.D.S., and M.G. participated in data analysis. C.G.L., J.A.E., and E.L.H. participated in study design, data analysis, and manuscript preparation.
Competing interests: J.A.E. has submitted a patent on CHI3L1. M.G. has an advisory relationship with InterMune. E.L.H. has consulted for Sanofi, Boehringer, and Pfizer in the area of pulmonary fibrosis. The other authors declare no competing interests.
SUPPLEMENTARY MATERIALS www.sciencetranslationalmedicine.org/cgi/content/full/6/240/240ra76/DC1
REFERENCES AND NOTES
- 1.Wagner GR. Asbestosis and silicosis. Lancet. 1997;349:1311–1315. doi: 10.1016/S0140-6736(96)07336-9. [DOI] [PubMed] [Google Scholar]
- 2.Vanhee D, Gosset P, Wallaert B, Voisin C, Tonnel AB. Mechanisms of fibrosis in coal workers’ pneumoconiosis. Increased production of platelet-derived growth factor, insulin-like growth factor type I, and transforming growth factor beta and relationship to disease severity. Am. J. Respir. Crit. Care Med. 1994;150:1049–1055. doi: 10.1164/ajrccm.150.4.7921435. [DOI] [PubMed] [Google Scholar]
- 3.Abid SH, Malhotra V, Perry MC. Radiation-induced and chemotherapy-induced pulmonary injury. Curr. Opin. Oncol. 2001;13:242–248. doi: 10.1097/00001622-200107000-00006. [DOI] [PubMed] [Google Scholar]
- 4.Thannickal VJ, Toews GB, White ES, Lynch JP, III, Martinez FJ. Mechanisms of pulmonary fibrosis. Annu. Rev. Med. 2004;55:395–417. doi: 10.1146/annurev.med.55.091902.103810. [DOI] [PubMed] [Google Scholar]
- 5.Steen VD, Owens GR, Fino GJ, Rodnan GP, Medsger TA., Jr. Pulmonary involvement in systemic sclerosis (scleroderma) Arthritis Rheum. 1985;28:759–767. doi: 10.1002/art.1780280706. [DOI] [PubMed] [Google Scholar]
- 6.Majumdar S, Li D, Ansari T, Pantelidis P, Black CM, Gizycki M, du Bois RM, Jeffery PK. Different cytokine profiles in cryptogenic fibrosing alveolitis and fibrosing alveolitis associated with systemic sclerosis: A quantitative study of open lung biopsies. Eur. Respir. J. 1999;14:251–257. doi: 10.1034/j.1399-3003.1999.14b03.x. [DOI] [PubMed] [Google Scholar]
- 7.Lewis MJ, Lewis EH, III, Amos JA, Tsongalis GJ. Cystic fibrosis. Am. J. Clin. Pathol. 2003;120:S3–S13. doi: 10.1309/DUUVFR11W595V7FE. [DOI] [PubMed] [Google Scholar]
- 8.Gay SE, Kazerooni EA, Toews GB, Lynch JP, III, Gross BH, Cascade PN, Spizarny DL, Flint A, Schork MA, Whyte RI, Popovich J, Hyzy R, Martinez FJ. Idiopathic pulmonary fibrosis: Predicting response to therapy and survival. Am. J. Respir. Crit. Care Med. 1998;157:1063–1072. doi: 10.1164/ajrccm.157.4.9703022. [DOI] [PubMed] [Google Scholar]
- 9.Reilkoff RA, Bucala R, Herzog EL. Fibrocytes: Emerging effector cells in chronic inflammation. Nat. Rev. Immunol. 2011;11:427–435. doi: 10.1038/nri2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Panos RJ, Mortenson RL, Niccoli SA, King TE., Jr. Clinical deterioration in patients with idiopathic pulmonary fibrosis: Causes and assessment. Am. J. Med. 1990;88:396–404. doi: 10.1016/0002-9343(90)90495-y. [DOI] [PubMed] [Google Scholar]
- 11.Sime PJ, O’Reilly KM. Fibrosis of the lung and other tissues: New concepts in pathogenesis and treatment. Clin. Immunol. 2001;99:308–319. doi: 10.1006/clim.2001.5008. [DOI] [PubMed] [Google Scholar]
- 12.Lee CG, Cho SJ, Kang MJ, Chapoval SP, Lee PJ, Noble PW, Yehualaeshet T, Lu B, Flavell RA, Milbrandt J, Homer RJ, Elias JA. Early growth response gene 1–mediated apoptosis is essential for transforming growth factor β1–induced pulmonary fibrosis. J. Exp. Med. 2004;200:377–389. doi: 10.1084/jem.20040104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray LA, Chen Q, Kramer MS, Hesson DP, Argentieri RL, Peng X, Gulati M, Homer RJ, Russell T, van Rooijen N, Elias JA, Hogaboam CM, Herzog EL. TGF-beta driven lung fibrosis is macrophage dependent and blocked by serum amyloid P. Int. J. Biochem. Cell Biol. 2011;43:154–162. doi: 10.1016/j.biocel.2010.10.013. [DOI] [PubMed] [Google Scholar]
- 14.Gibbons MA, MacKinnon AC, Ramachandran P, Dhaliwal K, Duffin R, Phythian-Adams AT, van Rooijen N, Haslett C, Howie SE, Simpson AJ, Hirani N, Gauldie J, Iredale JP, Sethi T, Forbes SJ. Ly6Chi monocytes direct alternatively activated profibrotic macrophage regulation of lung fibrosis. Am. J. Respir. Crit. Care Med. 2011;184:569–581. doi: 10.1164/rccm.201010-1719OC. [DOI] [PubMed] [Google Scholar]
- 15.Kuwano K, Kunitake R, Kawasaki M, Nomoto Y, Hagimoto N, Nakanishi Y, Hara N. P21Waf1/Cip1/Sdi1 and p53 expression in association with DNA strand breaks in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 1996;154:477–483. doi: 10.1164/ajrccm.154.2.8756825. [DOI] [PubMed] [Google Scholar]
- 16.Kuwano K, Hagimoto N, Kawasaki M, Yatomi T, Nakamura N, Nagata S, Suda T, Kunitake R, Maeyama T, Miyazaki H, Hara N. Essential roles of the Fas-Fas ligand pathway in the development of pulmonary fibrosis. J. Clin. Invest. 1999;104:13–19. doi: 10.1172/JCI5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kuwano K, Miyazaki H, Hagimoto N, Kawasaki M, Fujita M, Kunitake R, Kaneko Y, Hara N. The involvement of Fas–Fas ligand pathway in fibrosing lung diseases. Am. J. Respir. Cell Mol. Biol. 1999;20:53–60. doi: 10.1165/ajrcmb.20.1.2941. [DOI] [PubMed] [Google Scholar]
- 18.Prasse A, Probst C, Bargagli E, Zissel G, Toews GB, Flaherty KR, Olschewski M, Rottoli P, Müller-Quernheim J. Serum CC-chemokine ligand 18 concentration predicts outcome in idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009;179:717–723. doi: 10.1164/rccm.200808-1201OC. [DOI] [PubMed] [Google Scholar]
- 19.Lee CG, Hartl D, Lee GR, Koller B, Matsuura H, Da Silva CA, Sohn MH, Cohn L, Homer RJ, Kozhich AA, Humbles A, Kearley J, Coyle A, Chupp G, Reed J, Flavell RA, Elias JA. Role of breast regression protein 39 (BRP-39)/chitinase 3-like-1 in Th2 and IL-13–induced tissue responses and apoptosis. J. Exp. Med. 2009;206:1149–1166. doi: 10.1084/jem.20081271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bussink AP, Speijer D, Aerts JM, Boot RG. Evolution of mammalian chitinase(-like) members of family 18 glycosyl hydrolases. Genetics. 2007;177:959–970. doi: 10.1534/genetics.107.075846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Funkhouser JD, Aronson NN., Jr. Chitinase family GH18: Evolutionary insights from the genomic history of a diverse protein family. BMC Evol. Biol. 2007;7:96. doi: 10.1186/1471-2148-7-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kzhyshkowska J, Gratchev A, Goerdt S. Human chitinases and chitinase-like proteins as indicators for inflammation and cancer. Biomark Insights. 2007;2:128–146. [PMC free article] [PubMed] [Google Scholar]
- 23.Matsuura H, Hartl D, Kang MJ, Dela Cruz CS, Koller B, Chupp GL, Homer RJ, Zhou Y, Cho WK, Elias JA, Lee CG. Role of breast regression protein–39 in the pathogenesis of cigarette smoke–induced inflammation and emphysema. Am. J. Respir. Cell Mol. Biol. 2011;44:777–786. doi: 10.1165/rcmb.2010-0081OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sohn MH, Kang MJ, Matsuura H, Bhandari V, Chen NY, Lee CG, Elias JA. The chitinase-like proteins breast regression protein-39 and YKL-40 regulate hyperoxia-induced acute lung injury. Am. J. Respir. Crit. Care Med. 2010;182:918–928. doi: 10.1164/rccm.200912-1793OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee CG, Da Silva CA, Dela Cruz CS, Ahangari F, Ma B, Kang MJ, He CH, Takyar S, Elias JA. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011;73:479–501. doi: 10.1146/annurev-physiol-012110-142250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Recklies AD, White C, Ling H. The chitinase 3-like protein human cartilage glycoprotein 39 (HC-gp39) stimulates proliferation of human connective-tissue cells and activates both extracellular signal-regulated kinase- and protein kinase B-mediated signalling pathways. Biochem. J. 2002;365:119–126. doi: 10.1042/BJ20020075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korthagen NM, van Moorsel CH, Barlo NP, Ruven HJ, Kruit A, Heron M, van den Bosch JM, Grutters JC. Serum and BALF YKL-40 levels are predictors of survival in idiopathic pulmonary fibrosis. Respir. Med. 2010;105:106–113. doi: 10.1016/j.rmed.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 28.Konishi K, Gibson KF, Lindell KO, Richards TJ, Zhang Y, Dhir R, Bisceglia M, Gilbert S, Yousem SA, Song JW, Kim DS, Kaminski N. Gene expression profiles of acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2009;180:167–175. doi: 10.1164/rccm.200810-1596OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dela Cruz CS, Liu W, He CH, Jacoby A, Gornitzky A, Ma B, Flavell R, Lee CG, Elias JA. Chitinase 3-like-1 promotes Streptococcus pneumoniae killing and augments host tolerance to lung antibacterial responses. Cell Host Microbe. 2012;12:34–46. doi: 10.1016/j.chom.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Collard HR, Moore BB, Flaherty KR, Brown KK, Kaner RJ, King TE, Jr., Lasky JA, Loyd JE, Noth I, Olman MA, Raghu G, Roman J, Ryu JH, Zisman DA, Hunninghake GW, Colby TV, Egan JJ, Hansell DM, Johkoh T, Kaminski N, Kim DS, Kondoh Y, Lynch DA, Müller-Quernheim J, Myers JL, Nicholson AG, Selman M, Toews GB, Wells AU, Martinez FJ. Acute exacerbations of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2007;176:636–643. doi: 10.1164/rccm.200703-463PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Peng X, Mathai SK, Murray LA, Russell T, Reilkoff R, Chen Q, Gulati M, Elias JA, Bucala R, Gan Y, Herzog EL. Local apoptosis promotes collagen production by monocyte-derived cells in transforming growth factor β1-induced lung fibrosis. Fibrogenesis Tissue Repair. 2011;4:12. doi: 10.1186/1755-1536-4-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 33.Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, Colby TV, Cordier JF, Flaherty KR, Lasky JA, Lynch DA, Ryu JH, Swigris JJ, Wells AU, Ancochea J, Bouros D, Carvalho C, Costabel U, Ebina M, Hansell DM, Johkoh T, Kim DS, King TE, Jr., Kondoh Y, Myers J, Müller NL, Nicholson AG, Richeldi L, Selman M, Dudden RF, Griss BS, Protzko SL, Schünemann HJ, ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: Idiopathic pulmonary fibrosis: Evidence-based guidelines for diagnosis and management. Am. J. Respir. Crit. Care Med. 2011;183:788–824. doi: 10.1164/rccm.2009-040GL. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petersen TH, Calle EA, Zhao L, Lee EJ, Gui L, Raredon MB, Gavrilov K, Yi T, Zhuang ZW, Breuer C, Herzog E, Niklason LE. Tissue-engineered lungs for in vivo implantation. Science. 2010;329:538–541. doi: 10.1126/science.1189345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Booth AJ, Hadley R, Cornett AM, Dreffs AA, Matthes SA, Tsui JL, Weiss K, Horowitz JC, Fiore VF, Barker TH, Moore BB, Martinez FJ, Niklason LE, White ES. Acellular normal and fibrotic human lung matrices as a culture system for in vitro investigation. Am. J. Respir. Crit. Care Med. 2012;186:866–876. doi: 10.1164/rccm.201204-0754OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raghu G, Weycker D, Edelsberg J, Bradford WZ, Oster G. Incidence and prevalence of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2006;174:810–816. doi: 10.1164/rccm.200602-163OC. [DOI] [PubMed] [Google Scholar]
- 37.Papiris SA, Manali ED, Kolilekas L, Kagouridis K, Triantafillidou C, Tsangaris I, Roussos C. Clinical review: Idiopathic pulmonary fibrosis acute exacerbations—Unravelling Ariadne’s thread. Crit. Care. 2010;14:246. doi: 10.1186/cc9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Plataki M, Koutsopoulos AV, Darivianaki K, Delides G, Siafakas NM, Bouros D. Expression of apoptotic and antiapoptotic markers in epithelial cells in idiopathic pulmonary fibrosis. Chest. 2005;127:266–274. doi: 10.1378/chest.127.1.266. [DOI] [PubMed] [Google Scholar]
- 39.Uhal BD, Joshi I, Hughes WF, Ramos C, Pardo A, Selman M. Alveolar epithelial cell death adjacent to underlying myofibroblasts in advanced fibrotic human lung. Am. J. Physiol. 1998;275:L1192–L1199. doi: 10.1152/ajplung.1998.275.6.L1192. [DOI] [PubMed] [Google Scholar]
- 40.Li X, Rayford H, Uhal BD. Essential roles for angiotensin receptor AT1a in bleomycin-induced apoptosis and lung fibrosis in mice. Am. J. Pathol. 2003;163:2523–2530. doi: 10.1016/S0002-9440(10)63607-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hagimoto N, Kuwano K, Nomoto Y, Kunitake R, Hara N. Apoptosis and expression of Fas/Fas ligand mRNA in bleomycin-induced pulmonary fibrosis in mice. Am. J. Respir. Cell Mol. Biol. 1997;16:91–101. doi: 10.1165/ajrcmb.16.1.8998084. [DOI] [PubMed] [Google Scholar]
- 42.Nomoto Y, Kuwano K, Hagimoto N, Kunitake R, Kawasaki M, Hara N. Apoptosis and Fas/Fas ligand mRNA expression in acute immune complex alveolitis in mice. Eur. Respir. J. 1997;10:2351–2359. doi: 10.1183/09031936.97.10102351. [DOI] [PubMed] [Google Scholar]
- 43.Barbas-Filho JV, Ferreira MA, Sesso A, Kairalla RA, Carvalho CR, Capelozzi VL. Evidence of type II pneumocyte apoptosis in the pathogenesis of idiopathic pulmonary fibrosis (IFP)/usual interstitial pneumonia (UIP) J. Clin. Pathol. 2001;54:132–138. doi: 10.1136/jcp.54.2.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang R, Ibarra-Sunga O, Verlinski L, Pick R, Uhal BD. Abrogation of bleomycin-induced epithelial apoptosis and lung fibrosis by captopril or by a caspase inhibitor. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000;279:L143–L151. doi: 10.1152/ajplung.2000.279.1.L143. [DOI] [PubMed] [Google Scholar]
- 45.Uhal BD, Wang R, Laukka J, Zhuang J, Soledad-Conrad V, Filippatos G. Inhibition of amiodarone-induced lung fibrosis but not alveolitis by angiotensin system antagonists. Pharmacol. Toxicol. 2003;92:81–87. doi: 10.1034/j.1600-0773.2003.920204.x. [DOI] [PubMed] [Google Scholar]
- 46.Okada M, Sugita K, Inukai T, Goi K, Kagami K, Kawasaki K, Nakazawa S. Hepatocyte growth factor protects small airway epithelial cells from apoptosis induced by tumor necrosis factor-α or oxidative stress. Pediatr. Res. 2004;56:336–344. doi: 10.1203/01.PDR.0000134255.58638.59. [DOI] [PubMed] [Google Scholar]
- 47.Inoshima I, Kuwano K, Hamada N, Yoshimi M, Maeyama T, Hagimoto N, Nakanishi Y, Hara N. Induction of CDK inhibitor p21 gene as a new therapeutic strategy against pulmonary fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2004;286:L727–L733. doi: 10.1152/ajplung.00209.2003. [DOI] [PubMed] [Google Scholar]
- 48.Pechkovsky DV, Prasse A, Kollert F, Engel KM, Dentler J, Luttmann W, Friedrich K, Müller-Quernheim J, Zissel G. Alternatively activated alveolar macrophages in pulmonary fibrosis—Mediator production and intracellular signal transduction. Clin. Immunol. 2010;137:89–101. doi: 10.1016/j.clim.2010.06.017. [DOI] [PubMed] [Google Scholar]
- 49.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: New molecules and patterns of gene expression. J. Immunol. 2006;177:7303–7311. doi: 10.4049/jimmunol.177.10.7303. [DOI] [PubMed] [Google Scholar]
- 50.Raes G, De Baetselier P, Noël W, Beschin A, Brombacher F, Hassanzadeh Gh G. Differential expression of FIZZ1 and Ym1 in alternatively versus classically activated macrophages. J. Leukoc. Biol. 2002;71:597–602. [PubMed] [Google Scholar]
- 51.Bargagli E, Prasse A, Olivieri C, Muller-Quernheim J, Rottoli P. Macrophage-derived bio-markers of idiopathic pulmonary fibrosis. Pulm. Med. 2011;2011:717130. doi: 10.1155/2011/717130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raes G, Beschin A, Ghassabeh GH, De Baetselier P. Alternatively activated macrophages in protozoan infections. Curr. Opin. Immunol. 2007;19:454–459. doi: 10.1016/j.coi.2007.05.007. [DOI] [PubMed] [Google Scholar]
- 53.He C, Lee CG, Cruz C. S. Dela, Lee CM, Zhou Y, Ahangari F, Ma B, Herzog EL, Rosenberg SA, Li Y, Nour AM, Parikh CR, Schmidt I, Modis Y, Cantley L, Elias JA. Chitinase 3-like 1 regulates cellular and tissue responses via IL-13 receptor α2. Cell Rep. 2013;4:830–841. doi: 10.1016/j.celrep.2013.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.American Thoracic Society; European Respiratory Society, American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am. J. Respir. Crit. Care Med. 2002;165:277–304. doi: 10.1164/ajrccm.165.2.ats01. [DOI] [PubMed] [Google Scholar]
- 55.Chupp GL, Lee CG, Jarjour N, Shim YM, Holm CT, He S, Dziura JD, Reed J, Coyle AJ, Kiener P, Cullen M, Grandsaigne M, Dombret MC, Aubier M, Pretolani M, Elias JA. A chitinase-like protein in the lung and circulation of patients with severe asthma. N. Engl. J. Med. 2007;357:2016–2027. doi: 10.1056/NEJMoa073600. [DOI] [PubMed] [Google Scholar]
- 56.Reilkoff RA, Peng H, Murray LA, Peng X, Russell T, Montgomery R, Feghali-Bostwick C, Shaw A, Homer RJ, Gulati M, Mathur A, Elias JA, Herzog EL. Semaphorin 7a+ regulatory T cells are associated with progressive idiopathic pulmonary fibrosis and are implicated in transforming growth factor-β1–induced pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2013;187:180–188. doi: 10.1164/rccm.201206-1109OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhou Y, Lee JY, Lee CM, Cho WK, Kang MJ, Koff JL, Yoon PO, Chae J, Park HO, Elias JA, Lee CG. Amphiregulin, an epidermal growth factor receptor ligand, plays an essential role in the pathogenesis of transforming growth factor-β-induced pulmonary fibrosis. J. Biol. Chem. 2012;287:41991–42000. doi: 10.1074/jbc.M112.356824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kang HR, Lee CG, Homer RJ, Elias JA. Semaphorin 7A plays a critical role in TGF-β1–induced pulmonary fibrosis. J. Exp. Med. 2007;204:1083–1093. doi: 10.1084/jem.20061273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gan Y, Reilkoff R, Peng X, Russell T, Chen Q, Mathai SK, Homer R, Gulati M, Siner J, Elias J, Bucala R, Herzog E. Role of semaphorin 7a signaling in transforming growth factor β1–induced lung fibrosis and scleroderma-related interstitial lung disease. Arthritis Rheum. 2011;63:2484–2494. doi: 10.1002/art.30386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Petersen TH, Calle EA, Colehour MB, Niklason LE. Matrix composition and mechanics of decellularized lung scaffolds. Cells Tissues Organs. 2012;195:222–231. doi: 10.1159/000324896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sun H, Calle EA, Chen X, Mathur A, Mendez J, Zhao L, Niklason LE, Peng X, Peng H, Herzog EL. Fibroblast engraftment in the decellularized mouse lung occurs via a β1-integrin-dependent, FAK-dependent pathway that is mediated by ERK and opposed by AKT. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014;306:L463–L475. doi: 10.1152/ajplung.00100.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.