

Abstract
Leber hereditary optic neuropathy (LHON) is the most common primary mitochondrial DNA (mtDNA) disorder in the general population. It is an important cause of severe, usually irreversible, visual loss among young adults with a peak age of onset in the second and third decades of life. Management is currently mostly supportive but recent developments in LHON research are pointing the way towards more effective treatments for this blinding mitochondrial disorder.
Introduction
Leber hereditary optic neuropathy (LHON, OMIM 535000) is named after Theodore Leber (1840-1917), a German Ophthalmologist who in 1871 first described the defining clinical features of this disorder. LHON is the most common primary mitochondrial DNA (mtDNA) disorder in the general population, with a minimum prevalence of 1 in 31,000 in the North of England.1,2 Three mtDNA point mutations, which all involve complex I subunits of the mitochondrial respiratory chain, account for the vast majority of LHON cases (Table 1).3,4 Other rarer mtDNA mutations have been confirmed as definitely causing the LHON phenotype, having been reported in more than one pedigree and showing clear segregation with affected disease status. For those putative LHON variants identified in singleton cases or in single families only, additional evidence is required before pathogenicity can be irrefutably ascribed. The m.11778G>A mutation is found in over 70% of all LHON patients, but as a result of a founder event, nearly 90% of all affected carriers of French Canadian descent harbour the m.14484T>C mutation.3,4 In most laboratories worldwide, the diagnostic protocol for LHON involves a targeted molecular approach, specifically looking for the three “primary” mtDNA mutations i.e. m.3460G>A, m.11778G>A, and m.14484T>C. As full mitochondrial genome sequencing still remains a time consuming and expensive assay, it is only indicated if the initial LHON screen is negative and there is a strong clinical suspicion.
Table 1. Mitochondrial DNA variants associated with LHON.
| Mitochondrial Gene | Nucleotide Change | |
|---|---|---|
| Common variants (~ 90%) | MTND1 | m.3460G>A* |
| MTND4 | m.11778G>A* | |
| MTND6 | m.14484T>C* | |
| Rare variants (~ 10%) | MTND1 | m.3376G>A, m.3635G>A*, m.3697G>A, m.3700G>A, m.3733G>A*, m.4025C>T, m.4160T>C, m.4171C>A* |
| MTND2 | m.4640C>A, m.5244G>A | |
| MTND3 | m.10237T>C | |
| MTND4 | m.11696G>A, m.11253T>C | |
| MTND4L | m.10663T>C* | |
| MTND5 | m.12811T>C, m.12848C>T, m.13637A>G, m.13730G>A |
|
| MTND6 | m.14325T>C, m.14568C>T, m.14459G>A*, m.14729G>A, m.14482C>A*, m.14482C>G*, m.14495A>G*, m.14498C>T, m.14568C>T*, m.14596A>T |
|
| MTATP6 | m.9101T>C | |
| MTCO3 | m.9804G>A | |
| MTCYB | m.14831G>A |
These mtDNA variants are definitely pathogenic. They have been identified in ≥ 2 independent LHON pedigrees, showing segregation with affected disease status. The remaining putative LHON mutations have been found in singleton cases or in a single family, and additional evidence is required before pathogenicity can be irrefutably ascribed.3,4
Clinical features of LHON
Telangiectatic vessels around the optic disc and fluctuating oedema of the retinal nerve fibre layer (RNFL) can sometimes be observed in asymptomatic LHON carriers. These oedematous areas have been quantified with optical coherence tomography imaging and RNFL thickening was found to be more prominent in the temporal quadrant, in keeping with the greater vulnerability of the papillomacular retinal ganglion cell (RGC) axons. With more sophisticated psychophysical testing, other subclinical abnormalities of optic nerve function have also been demonstrated, such as reduced contrast sensitivity and depressed colour perception along the red-green (protan) axis.3,4
LHON classically presents with acute or subacute painless loss of central vision. The initial visual loss is severe with most patients achieving best corrected visual acuities of 6/60 or worse.3,4 There is an associated dense central scotoma and a marked reduction in colour perception. Despite this global reduction in optic nerve function, the pupillary light reflexes are relatively preserved, and this distinctive feature has been ascribed to a special class of melanopsin-containing RGCs that are less vulnerable to the downstream consequences of the mtDNA LHON mutations.5 Bilateral optic nerve involvement at first presentation occurs in ~ 25% of acute LHON cases. If sequential, the second eye is invariably affected within one year of disease onset, unilateral optic neuropathy being exceptionally rare in LHON.3,4
In the acute phase, optic disc hyperaemia, peripapillary telangiectatic vessels, vascular tortuosity, and retinal nerve fibre layer oedema can be observed, the latter being due to RGC axonal stasis (Figure 1).3,4 In the pre-molecular era, these fundal abnormalities were particularly informative, allowing a presumptive diagnosis of LHON to be made, especially when supported by a maternal family history of early-onset visual loss. In 20-40% of acute cases, the optic discs look entirely normal and these patients are often incorrectly labelled as having functional visual loss.3,4 Pallor of the neuroretinal rim develops within six weeks of disease onset and it is initially more apparent temporally due to the accelerated loss of RGC axons within the papillomacular bundle.
Figure 1. Fundal appearance in acute and chronic LHON stages.
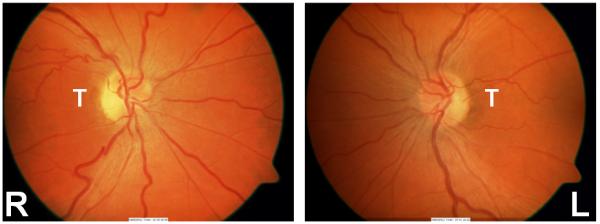
This 47-year-old m.11778G>A male carrier experienced visual loss in his right eye first, followed by his left eye eight months later. These fundal pictures were taken one month after the left eye had become involved. Best corrected visual acuities at that point were counting fingers in the right eye and 6/36 in the left eye. There is vascular tortuosity in both eyes and temporal pallor of the right optic disc (chronic stage) due to the earlier and more pronounced loss of RGC axons within the papillomacular bundle, which sub-serves the central 10° of the visual field. There is mild hyperaemia of the left optic disc, in keeping with the recent onset of visual symptoms in that eye (acute stage). (L = left eye; R = right eye; T = temporal quadrant corresponding to the papillomacular bundle)
Extra-ocular features such as cardiac conduction defects, peripheral neuropathy, dystonia, and myopathy are thought to more common among LHON carriers compared with the general population.3,4 There is also a well-established association between the three primary LHON mutations and a demyelinating illness, which interestingly is clinically and radiologically indistinguishable from multiple sclerosis (Harding’s disease).6 Other rarer pathogenic mtDNA variants have been linked with more atypical “LHON plus” syndromes where the optic neuropathy is complicated by prominent spastic dystonia, ataxia, juvenile-onset encephalopathy, and psychiatric disturbances.3,4
Visual prognosis
The visual prognosis in LHON is poor and most patients remain legally blind. The likelihood of visual recovery is greatest with the m.14484T>C mutation, least with the m.11778G>A mutation, and intermediate with the m.3460G>A mutation.3,4 Although delayed visual recovery has been reported, maximal improvement in visual function usually occurs within the first year, if it occurs at all. The appearance of small islands of vision within the patient’s visual field (fenestrations) can greatly help scanning vision, especially if the central scotoma becomes concurrently less dense.
Genetic counselling
The mitochondrial genome is maternally inherited and thousands of mtDNA molecules are present in metabolically-active cells. As a result of this high-copy number genome, two possible situations can arise, known as homoplasmy and heteroplasmy. Among LHON families, 85-90% of carriers are homoplasmic for the mtDNA mutation, i.e. 100% mutant, whereas the remainder are heteroplasmic harbouring a mixture of both wild-type and mutant mtDNA species.2 The risk of disease conversion is low if the mutational load is below the threshold level of 60%.7 Male LHON carriers can be reassured that their children will not inherit their mitochondrial genetic defect. On the other hand, female LHON carriers will transmit the mutation to all their offspring. For the minority of mothers with heteroplasmic LHON mutations, it is difficult to accurately predict the mutational level that will be transmitted since rapid generational shifts in mitochondrial allele frequencies can occur due to the “mitochondrial bottleneck” operating in the female germline.8 Based on published figures, some indication of recurrence risks can be provided to maternal relatives of a LHON proband (Table 2).
Table 2. Risk of visual loss for relatives of LHON probands.
| Risk of visual loss |
||
|---|---|---|
| m.11778G>A | m.14484T>C | |
| Siblings | ||
| Brother | 25% | 28% |
| Sister | 8% | 5% |
|
| ||
| Sister’s children | ||
| Nephew | 41% | 30% |
| Niece | 7% | 3% |
|
| ||
| Maternal first cousins | ||
| Male | 30% | 19% |
| Female | 7% | 4% |
Although it is not possible to predict whether or when a LHON carrier will be affected, epidemiological studies have identified major risk factors for visual loss, namely age, sex and environmental exposure.2-4 Over 90% of LHON carriers who will experience visual failure will do so before the age of 50 years. In addition, LHON is characterised by a marked sex bias, male carriers having a ~ 50% lifetime risk of developing the optic neuropathy compared with only ~ 10% for female carriers. Unaffected LHON carriers should be strongly advised not to smoke and to minimise their alcohol intake, not only as a general health measure, but because smoking, and to a lesser extent excessive alcohol intake, have been associated with increased risks of disease conversion.3,4 In one large study comparing 196 affected and 206 unaffected carriers from 125 LHON families, light and heavy smokers were twice and three times more likely to lose vision compared to non-smokers, respectively.9
Treatment strategies
Mitochondria provide the bulk of the cell’s adenosine triphosphate (ATP) requirements through the tightly-regulated control of electron flux along the mitochondrial respiratory chain. All three primary LHON mutations; m.3460G>A, m.11778G>A, and m.14484T>C disrupt key polypeptide subunits of complex I, resulting in a significant bioenergetic deficit and raised levels of reactive oxygen species (ROS).10 An intriguing aspect of the pathophysiology of LHON still remains – why are RGCs selectively vulnerable to disturbed mitochondrial function? Several hypotheses have been proposed based on the distinct anatomical, physiological, and cytoskeletal arrangements present within the optic nerve.10 Notwithstanding these unresolved issues, the final pathological outcome in LHON is apoptotic RGC loss and the aims of treatment for this disorder are threefold: (i) to prevent initial visual loss among LHON carriers, (ii) to protect the unaffected fellow eye in patients with unilateral optic neuropathy, and (iii) to preserve visual function in already compromised optic nerves.
Neuroprotection
Various treatment “cocktails” have been used to mitigate the deleterious impact of mitochondrial dysfunction on RGC survival.3,4 Co-enzyme Q10 (CoQ10) is a quinone analogue and based on limited evidence, it is frequently prescribed to patients with mitochondrial disease. Idebenone is a related compound, a shorter-chain synthetic benzoquinone analogue, which is thought to have a better bioavailability profile compared with CoQ10.11 Idebenone is able to bypass complex I inhibition and by shuttling electrons directly from the cytosol to complex III, ATP production is optimised with a decrease in toxic ROS levels.11 This dual mode of action is an attractive therapeutic combination and anecdotally, some patients with LHON have experienced significant visual recovery following treatment with idebenone.3,4 In collaboration with clinical partners in the UK, Germany and Canada, we therefore conducted a multicentre double-blind randomised controlled trial (RCT) to investigate the safety, tolerability, and efficacy of high-dose idebenone in LHON. RHODOS (Rescue of Hereditary Optic Disease Outpatient Study) successfully enrolled 85 patients harbouring the three most common mtDNA LHON mutations: m.3460G>A (n = 11), m.11778G>A (n = 57), and m.14484T>C (n = 17).12 These patients were randomised in a 2:1 ratio to receive either high-dose idebenone (900mg) or placebo over a 24-week treatment period. A major finding of the RHODOS trial is that patients with LHON were more likely to benefit from idebenone if they were treated relatively early in the course of the disease (Figure 2). The efficacy of idebenone has also been evaluated retrospectively in a cohort of affected LHON carriers, all of whom were older than 10 years of age and within one year of disease onset.13 Follow-up clinical data were collected for at least five years and visual outcome was compared between patients who received idebenone (n = 44) against those who were left untreated (n = 59). An increased frequency of visual recovery was observed in the idebenone group and subgroup analysis indicated that LHON carriers harbouring the m.11778G>A mutation were the best responders. The early initiation of idebenone therapy in the acute phase of LHON was the most significant prognostic factor for visual improvement. No adverse drug reactions were reported in both studies and idebenone is currently under review by the European Medicines Agency for regulatory approval.
Figure 2. Change in best visual acuity for patients with discordant visual acuities at baseline.
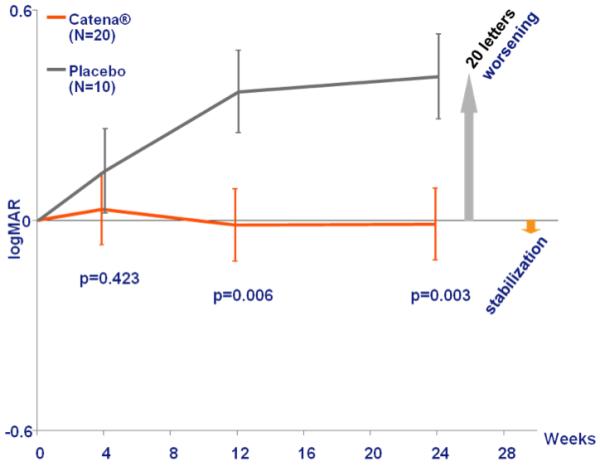
Catena is the formulation of idebenone provided by Santhera Pharmaceuticals (Liestal, Switzerland). Patients randomised to the treatment arm received a total dose of idebenone of 900mg per day, with 300mg taken three times a day during meals. Of the 85 patients enrolled into the RHODOS trial, 30 had discordant visual acuities at baseline, which was defined as a difference of more than 0.2 LogMAR between the two affected eyes.12 This degree of visual disparity is rare in late-stage chronic LHON where patients usually have fairly symmetrical visual loss between the two affected eyes. Patients with discordant visual acuities at baseline were therefore at highest risk of further deterioration in the least affected eye, accounting for the greater treatment effect observed in this particular subgroup. LogMAR = logarithm of the minimum angle of resolution.
Other neuroprotective agents are currently being investigated as potential treatment options in LHON. In an open-label study of four patients with acute LHON treated within 90 days of disease conversion, the antioxidant α-tocotrienol-quinone (EPI-743), a vitamin E derivative, has shown early promise and an RCT is underway to study this compound further (http://www.aosonline.org/annualmeeting/am_program.pdf, Accessed 8th of August 2011). Although their usefulness has yet to be established clinically, a marked amelioration in mitochondrial oxidative function was observed in cellular LHON models following supplementation with oestrogen-based compounds.14 This protective effect provides a plausible explanation for the reduced prevalence of visual loss among female LHON carriers and the therapeutic potential of these oestrogen-based compounds deserve further investigation.
Gene therapy
Gene therapy for primary mtDNA disorders is challenging. The mitochondrial inner membrane is relatively impermeable and a highly-efficient vector is needed to transfect a sufficient number of mitochondria per cell in order to rescue the disease phenotype. To circumvent these technical difficulties, a possible solution is the so-called allotopic approach where the gene of interest is transfected into the nuclear genome, and the encoded protein is engineered with a specific targeting sequence that facilitates its uptake into the mitochondrial compartment.10 RGC loss was dramatically reduced, in both in vitro and in vivo experimental LHON models, by transfecting them with an adenovirus vector containing the human SOD2 gene.15 In these conditions of heightened oxidative stress, the over-expression of the antioxidant enzyme superoxide dismutase is thought to promote RGC survival by exerting an anti-apoptotic influence. Another attractive and more direct approach is to replace the dysfunctional subunit encoded by the mtDNA LHON mutation. Proof of this concept has recently been demonstrated in a rat model harbouring a defective ND4 gene with the m.11778A>G mutation.16 Visual loss was reversed by transfecting RGCs with the wild-type version of the ND4 gene, the level of transgene expression being sufficient to rescue RGCs and maintain normal physiological responses.
Looking into the future
RHODOS is the first RCT for a primary mitochondrial disorder and the results obtained with high-dose idebenone are encouraging. A number of novel neuroprotective agents are currently in the pipeline for other mitochondrial cytopathies and these could also prove beneficial for patients with LHON. Gene therapy is a valid complementary approach to pharmacological intervention but long-term safety data is essential before human clinical trials can be advocated. After two decades of sustained research, we are now at the start of an exciting translational phase not only for LHON, but for other mitochondrially-determined optic neuropathies, which as a group represent an important cause of chronic visual morbidity in the population.
Acknowledgements
PYWM is a Medical Research Council (MRC, UK) Clinician Scientist. PFC is a Wellcome Trust Senior Fellow in Clinical Science and also receives funding from Parkinson’s UK, the MRC Translational Muscle Centre, and the UK NIHR Biomedical Research Centre in Ageing and Age-related Disease. We are grateful to the Editorial Board of Advances in Clinical Neuroscience and Rehabilitation for granting copyright permission for this article.
Footnotes
Conflict of interest
None
References
- 1.Schaefer AM, McFarland R, Blakely EL, He L, Whittaker RG, Taylor RW, et al. Prevalence of mitochondrial DNA disease in adults. Annals of Neurology. 2008;63:35–9. doi: 10.1002/ana.21217. [DOI] [PubMed] [Google Scholar]
- 2.Man PY, Griffiths PG, Brown DT, Howell N, Turnbull DM, Chinnery PF. The epidemiology of Leber hereditary optic neuropathy in the North East of England. American Journal of Human Genetics. 2003;72:333–9. doi: 10.1086/346066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yu-Wai-Man P, Griffiths PG, Hudson G, Chinnery PF. Inherited mitochondrial optic neuropathies. Journal of Medical Genetics. 2009;46:145–58. doi: 10.1136/jmg.2007.054270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraser JA, Biousse V, Newman NJ. The Neuro-ophthalmology of mitochondrial disease. Survey of Ophthalmology. 2010;55(4):299–334. doi: 10.1016/j.survophthal.2009.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.La Morgia C, Ross-Cisneros FN, Sadun AA, Hannibal J, Munarini A, Mantovani V, et al. Melanopsin retinal ganglion cells are resistant to neurodegeneration in mitochondrial optic neuropathies. Brain. 2010;133:2426–38. doi: 10.1093/brain/awq155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harding AE, Sweeney MG, Miller DH, Mumford CJ, Kellar-Wood H, Menard D, et al. Occurrence of a multiple sclerosis-like illness in women who have a Leber’s hereditary optic neuropathy mitochondrial DNA mutation. Brain. 1992;115:979–89. doi: 10.1093/brain/115.4.979. [DOI] [PubMed] [Google Scholar]
- 7.Chinnery PF, Andrews RM, Turnbull DM, Howell NN. Leber hereditary optic neuropathy: Does heteroplasmy influence the inheritance and expression of the G11778A mitochondrial DNA mutation? American Journal of Medical Genetics. 2001;98:235–43. doi: 10.1002/1096-8628(20010122)98:3<235::aid-ajmg1086>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- 8.Cree LM, Samuels DC, Chinnery PF. The inheritance of pathogenic mitochondrial DNA mutations. Biochimica et Biophysica Acta-Molecular Basis of Disease. 2009;1792:1097–1102. doi: 10.1016/j.bbadis.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirkman MA, Yu-Wai-Man P, Korsten A, Leonhardt M, Dimitriadis K, De Coo IF, et al. Gene-environment interactions in Leber hereditary optic neuropathy. Brain. 2009;132:2317–26. doi: 10.1093/brain/awp158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies - Disease mechanisms and therapeutic strategies. Progress in Retinal and Eye Research. 2011;30:81–114. doi: 10.1016/j.preteyeres.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haefeli RH, Erb M, Gemperli AC, Robay D, Courdier Fruh I, Anklin C, et al. NQO1-dependent redox cycling of idebenone: effects on cellular redox potential and energy levels. PLoS One. 2011;6:e17963. doi: 10.1371/journal.pone.0017963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klopstock K, Yu Wai Man P, Dimitriadis K, Rouleau J, Heck S, Bailie M, et al. A randomized placebo-controlled trial of idebenone in Leber’s hereditary optic neuropathy. Brain. 2011;134:2677–86. doi: 10.1093/brain/awr170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carelli V, La Morgia C, Valentino ML, Rizzo G, Carbonelli M, De Negri AM, et al. Idebenone Treatment In Leber’s Hereditary Optic Neuropathy. Brain. 2011;134:e188. doi: 10.1093/brain/awr180. [DOI] [PubMed] [Google Scholar]
- 14.Giordano C, Montopoli M, Perli E, Orlandi M, Fantin M, Ross-Cisneros FN, et al. Oestrogens ameliorate mitochondrial dysfunction in Leber’s hereditary optic neuropathy. Brain. 2011;134:220–34. doi: 10.1093/brain/awq276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qi XP, Sun L, Hauswirth WW, Lewin AS, Guy J. Use of mitochondrial antioxidant defenses for rescue of cells with a Leber hereditary optic neuropathy-causing mutation. Archives of Ophthalmology. 2007;125:268–72. doi: 10.1001/archopht.125.2.268. [DOI] [PubMed] [Google Scholar]
- 16.Ellouze S, Augustin S, Bouaita A, Bonnet C, Simonutti M, Forster V, et al. Optimized allotopic expression of the human mitochondrial ND4 prevents blindness in a rat model of mitochondrial dysfunction. American Journal of Human Genetics. 2008;83:373–87. doi: 10.1016/j.ajhg.2008.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]