

Abstract
Inflammation increases the abundance of inducible nitric oxide synthase (iNOS), leading to enhanced production of nitric oxide (NO), which can modify proteins by S-nitrosylation. Enhanced NO production increases the activities of the transcription factors p53 and nuclear factor κB (NF-κB) in several models of disease-associated inflammation. S-Nitrosylation inhibits the activity of the protein deacetylase SIRT1. SIRT1 limits apoptosis and inflammation by deacetylating p53 and p65 (also known as RelA), a subunit of NF-κB. We showed in multiple cultured mammalian cell lines that NO donors or inflammatory stimuli induced S-nitrosylation of SIRT1 within CXXC motifs, which inhibited SIRT1 by disrupting its ability to bind zinc. Inhibition of SIRT1 reduced deacetylation and promoted activation of p53 and p65, leading to apoptosis and increased expression of proinflammatory genes. In rodent models of systemic inflammation, Parkinson’s disease, or aging-related muscular atrophy, S-nitrosylation of SIRT1 correlated with increased acetylation of p53 and p65 and activation of p53 and NF-κB target genes, suggesting that S-nitrosylation of SIRT1 may represent a proinflammatory switch common to many diseases and aging.
INTRODUCTION
Nitric oxide (NO) exerts physiological effects in most cell types and tissues. NO is produced by three isoforms of NO synthase (NOS) that are widely distributed: inducible NOS (iNOS), neuronal NOS (nNOS), and endothelial NOS (eNOS). iNOS- and nNOS-derived NO are frequently implicated in the pathogenesis of aging-related disorders, including type 2 diabetes (1, 2), neurodegeneration (3–5), atherosclerosis (6), and muscle atrophy (7, 8). These divergent pathologies commonly involve nuclear factor κB (NF-κB)–mediated inflammation and p53-mediated apoptosis (9, 10). The effects of NO on inflammation and apoptosis are mediated primarily through S-nitrosylation (the covalent attachment of NO to cysteine thiols) of proteins, including NF-κB– and p53-related proteins (11–15). Thus, increased S-nitrosylation may contribute to cellular injury (termed nitrosative stress) (16) in many inflammatory conditions. Furthermore, co-morbidities are frequently observed among aging-related disorders, suggesting that these disorders could have common mechanisms, perhaps involving nitrosative stress.
Sir2 family proteins (known as sirtuins) are nicotinamide adenine di-nucleotide (NAD+)–dependent histone deacetylases (HDACs) that are conserved from bacteria to humans. Human SIRT1 is the closest homolog of the yeast gene SIR2 (17). In addition to lysine deacetylation, some sirtuins, including SIRT1, exhibit mono–adenosine diphosphate (ADP)–ribosyltransferase activity (17, 18), although the biological role of this activity is not fully understood (18). Deletion of SIR2 decreases the life span of yeast (19). In mammals, SIRT1 plays a crucial role in regulating multiple cellular processes, including apoptosis, cellular senescence, and inflammation (20–24). Dysregulation of SIRT1 activity is implicated in a number of mouse models of aging-related disorders, including type 2 diabetes (25), Alzheimer’s disease (26), and muscle wasting (27). Activation of SIRT1 ameliorates symptoms in these models (25–29), whereas inhibition of SIRT1 exacerbates obesity-induced insulin resistance and diabetes in mice (30–31).
SIRT1 deacetylates several transcriptional factors including p53 (22, 23), the p65 (also known as RelA) subunit of nuclear factor-κB (NF-κB) (20, 24), and peroxisome proliferator–activated receptor-γ coactivator-1α (PGC-1α) (32). The prototypic form of NF-κB is a dimer consisting of p65 and p50 or p52 subunits. Acetylation of p65, which contains a transcriptional activation domain and a DNA binding domain, increases transcriptional activity of NF-κB. Deacetylation by SIRT1 inhibits p53 and NF-κB activities, suppressing apoptosis and inflammation (20, 22, 23). p53 and p65 are also deacetylated and thereby inhibited by class I and class II HDACs (33, 34).
The catalytic domain of Sir2 family HDACs contains two adjoining, phylogenetically conserved Cys-X-X-Cys (CXXC) motifs (Fig. 1A). S-Nitrosylation of the CXXC motif of SIRT1 by NO generated by nNOS inactivates SIRT1, increasing the abundance of acetylated PGC-1α (35), which is deacetylated exclusively by SIRT1 (32). Thus, S-nitrosylation of SIRT1 may decrease its ability to deacetylate additional substrates, including p53 and p65.
Fig. 1. S-Nitrosylation of the CXXC motif reversibly inactivates SIRT1.

(A) Amino acid sequence of the region containing the CXXC motifs in SIRT1 homologs. (B) Graph of the deacetylase activity of FLAG-wild-type (WT) or mutated SIRT1 (M1, M2, or M3) immunopurified from COS-7 cells. CRL, control plasmid. Western blot shows the relative abundance of these proteins in transfected cells. P < 0.01 CRL, M1, M2, or M3 compared to WT, Kruskal-Wallis test. Data are means ± SEM of three independent experiments. (C and D) Western blots for S-nitrosylated SIRT1 (SNO-SIRT1) or SIRT1 in lysates from COS-7 cells transfected FLAG-tagged WT SIRT1 or SIRT1M3 and exposed to the NO donor SNAP (300 μM) and carmustine (80 μM) (C) or recombinant SIRT1 incubated with SNAP or DTT (D). Data are representative of three independent experiments. (E and H) Graphs of the deacetylase activity of immunopurified FLAG-SIRT1 from COS-7 cells exposed to GSNO (0.6 mM) (E) or recombinant SIRT1 exposed to SNAP (0.6 mM) (H). Immunopurified FLAG-SIRT1 or recombinant proteins were incubated with the reducing agent DTT (5 mM), the Zn2+ chelator TPEN (200 μM), or the Ca2+ chelator EGTA (0.6 mM). Western blot in (E) shows the relative S-nitrosylation of SIRT1 used in this assay. *P < 0.05, one-way analysis of variance (ANOVA) with the Newman-Keuls post hoc test compared to control (E); P < 0.02 between samples, Kruskal-Wallis test (H). Data are means ± SEM of three independent experiments. (F and G) Autoradiographs of 65Zn2+ bound to immunopurified FLAG-SIRT1, but not FLAG-SIRT1M3 (F) or recombinant SIRT1 (G). In (G), 65Zn2+ binding is shown for the same membrane before and after incubation. Coomassie brilliant blue (CBB) shows total protein. Data are representative of two independent experiments.
The effects of NO on the activities of p53 and NF-κB have been studied primarily under conditions of nitrosative stress. Nitrosative stress activates p53 through an unknown mechanism, leading to apoptosis (36, 37). In cultured cells, iNOS-induced S-nitrosylation of p65 and upstream signaling molecules, in particular, inhibitory κB kinase (IKK), decreases NF-κB activity (12–14). iNOS is an NF-κB target gene (38), suggesting that inhibition of NF-κB by iNOS may be a negative feedback mechanism. Conversely, in most rodent models of human diseases that involve increased production of NO, including ischemia-reperfusion injury, genetic or pharmacological inhibition of iNOS decreases NF-κB activity (39–42). Thus, NO can increase or decrease NF-κB activity, presumably dependent on cellular context and target genes.
Here, we demonstrated in cultured cells that NO increased the acetylation of p53 and p65 and activated p53- and NF-κB–dependent transcription by suppressing SIRT1-mediated deacetylation as a result of S-nitrosylation within the CXXC motifs of SIRT1. S-Nitrosylation inactivated SIRT1 by disrupting Zn2+ binding. S-Nitrosylation of SIRT1 was associated with increased acetylation of p53 and p65 in three rodent models of human diseases—endotoxemia, Parkinson’s disease, and sarcopenia—each of which involves NO, p53, and NF-κB (3–5, 7, 8, 10, 43–53). Thus, S-nitrosylation of SIRT1 may represent a common mechanism for the regulation of inflammation and apoptosis in multiple inflammatory and degenerative disorders.
RESULTS
S-Nitrosylation of CXXC motifs reversibly inactivates SIRT1
S-Nitrosylation of the CXXC motif of SIRT1 is associated with increased acetylation of PGC-1α (35). To test directly whether S-nitrosylation of the CXXC motif abolishes the catalytic activity of SIRT1, we evaluated the NAD+-dependent deacetylase activity of SIRT1 in vitro with synthetic acetylated peptides. We also assessed the in vitro ADP-ribosyltransferase activity of SIRT1 with recombinant histone H3. We generated mutants of FLAG-tagged SIRT1 in which cysteines in the first (SIRT1M1), second (SIRT1M2), or both first and second (SIRT1M3) CXXC motifs were replaced by serines (Fig. 1, A and B). We expressed FLAG-tagged wild-type or mutated SIRT1 in COS-7 cells, performed immunopurification for the FLAG tag, and tested the deacetylase and ADP-ribosyltransferase activities of SIRT in vitro in the presence of excess NAD+. We found that substituting one or both CXXC motifs with SXXS eliminated the ability of SIRT1 to deacetylate synthetic peptides (Fig. 1B). In addition, FLAG-SIRT1, but not FLAG-SIRT1M3, was able to ribosylate recombinant histone H3 in vitro (fig. S1A). FLAG-SIRT1G261A, which is deficient in ribosylase activity but has deacetylase activity (17), did not ribosylate recombinant histone H3 (fig. S1A).
Oxidative stress often accompanies nitrosative stress (54–56), and oxidative stress can enhance NO-mediated S-nitrosylation (57, 58). Human and mouse SIRT1 each contain 19 cysteine residues. To determine whether or not cysteine residues other than those in the CXXC motifs are S-nitrosylated, we exposed COS-7 cells with the NO donor S-nitroso-N-acetylpenicillamine (SNAP) in the presence of carmustine (also known as BCNU). Carmustine inhibits glutathione reductase (59) and enhances S-nitrosylation (57, 58), and thus was used to facilitate S-nitrosylation of SIRT1. For the detection of S-nitrosylated cysteines, we used the “biotin-switch” method in the presence of ascorbate. This method promotes the conversion of S-nitrosothiols (SNO) into biotinylated thiols (57, 60). Consistent with a previous report (35), we found that wild-type SIRT1, but not SIRT1M3, was S-nitrosylated in COS-7 cells exposed to SNAP and carmustine (Fig. 1C), confirming that the CXXC motifs are the major S-nitrosylation site in SIRT1. We found that incubating recombinant SIRT1 in vitro with SNAP without carmustine induced S-nitrosylation and reduced the ability of SIRT1 to deacetylate synthetic peptides (Fig. 1, D and H). Incubating recombinant SIRT1 with dithiothreitol (DTT), which reduces both nitrosylated and oxidized thiols (SOX) (61, 62), reversed SNAP-induced S-nitrosylation and inactivation of SIRT1 (Fig. 1, D and H and fig. S1B).
The CXXC motif in some proteins is a target of SOX, such as the formation of disulfide bonds and sulfenic acid (63). NO donors can cause S-oxidation reactions in addition to S-nitrosylation (64). Ascorbate decomposes SNO but not SOX (57, 60), whereas DTT reduces both SNO and SOX (61, 62). The biotin-switch method using ascorbate detects S-nitrosylated proteins by converting SNO, but not SOX, into biotinylated thiols (57, 60). In contrast, when DTT is used in lieu of ascorbate, both SNO and SOX are detected as biotinylated thiols (62, 65). Thus, we can differentially detect SNO alone using ascorbate, and total of SNO and SOX using DTT in the biotin-switch method (fig. S2A). We found that exposure of COS-7 cells to SNAP in the presence of carmustine increased both SNO-FLAG-SIRT1 alone and the total of SNO-FLAG-SIRT1 and SOX-FLAG-SIRT1 to a similar extent (fig. S2B). In contrast, SNAP exposure in the presence of carmustine did not increase SNO-FLAG-SIRT1M3 or the total of SNO-FLAG-SIRT1M3 and SOX-FLAG-SIRT1M3 (fig. S2B). To further study whether SNAP and carmustine increase SOX-FLAG-SIRT1, we decomposed SNO-FLAG-SIRT1 by preincubation of the cell lysates with ascorbate and then detected SNO-FLAG SIRT1 by the biotin-switch method using DTT (fig. S2C). When the cell lysates were preincubated with ascorbate, biotinylated FLAG-SIRT1 was not detected, indicating that there was little, if any, SOX-FLAG-SIRT1 induced by exposure to SNAP and carmustine. Like ascorbate, HgCl2 also specifically decomposes SNO (66). To further corroborate the specificity of the biotin-switch method for detecting S-nitrosylation, we preincubated the cell lysates with HgCl2 before the biotin-switch method using ascorbate and found that the preincubation with HgCl2 abolished the detection of biotinylated FLAG-SIRT1 from COS-7 cells exposed to SNAP and carmustine (fig. S3A). In contrast, when we omitted ascorbate in the biotin-switch procedure, biotinylated FLAG-SIRT1 was not detected, indicating ascorbate-dependent detection of SNO-SIRT1 (fig. S3B). Thus, S-nitrosylation of the CXXC motif is likely to be the major modification of SIRT1 in response to NO.
To study the biological effects of NO donor, such as apoptotic changes, we exposed the cells to GSNO, a major intracellular SNO donor, without adding carmustine. SNAP and GSNO have different half-lives and cell permeabilities (67). We found that FLAG-SIRT1 from cells grown in the presence of GSNO was S-nitrosylated and had reduced ability to deacetylate synthetic peptides in vitro compared to that isolated from cells grown in control media (Fig. 1E). Moreover, using DTT to reduce FLAG-SIRT1 isolated from GSNO-exposed COS-7 cells reversed S-nitrosylation and restored deacetylase activity (Fig. 1E).
Two adjoining CXXC motifs serve as a Zn2+ finger in many proteins, including steroid nuclear receptors (68). Whereas Zn2+ bound to wild-type recombinant SIRT1 or immunopurified FLAG-SIRT1 (Fig. 1F and fig. S4A), Zn2+ did not bind to immunopurified FLAG-SIRT1M3 (Fig. 1F). Furthermore, adding SNAP to Zn2+-bound recombinant SIRT1 reduced Zn2+ binding (Fig. 1G). The Zn2+ chelator N,N,N′,N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), but not the Ca2+ chelator EGTA, prevented the ability of DTT to restore the deacetylase activity of recombinant SIRT1 exposed to SNAP (Fig. 1H and fig. S4B) or GNSO (fig. S4C). Likewise, TPEN prevented the ability of DTT to restore the ability of S-nitrosylated recombinant SIRT1 to deacetylate recombinant histone H3 (fig. S4D). In the absence of NO donors, TPEN was less effective than SNAP at suppressing the deacetylase activity of recombinant SIRT1 (fig. S4E), indicating that unlike the NO donors, TPEN alone cannot fully inactivate SIRT1. This suggests that Zn2+ binding to SIRT1 may be relatively stable even in the presence of TPEN unless S-nitrosylation of the CXXC motif releases Zn2+. Thus, S-nitrosylation of the CXXC motif disrupts Zn2+ binding and thereby inactivates SIRT1.
S-Nitrosylation of SIRT1 increases the acetylation and activation of p53, leading to cell death
We and others have shown that inhibition of SIRT1 reduces cell viability in the absence or presence of DNA-damaging agents (22, 23, 69). NO causes p53-dependent apoptotic cell death (36, 37), and acetylation of p53 increases its transcriptional activity (70, 71). Expression of SIRT1M3 in COS-7 cells decreased viability and enhanced the ability of etoposide (fig. S5A) or bleomycin (fig. S5B) to reduce viability, suggesting that SIRT1M3 exerts a dominant negative effect on endogenous SIRT1. Expression of SIRT1M3 or catalytically inactive SIRT1H355Y (22, 23) in COS-7 cells increased the abundance of acetylated p53 in the presence or absence of bleomycin (Fig. 2A). Overexpressing wild-type SIRT1 in COS-7 cells decreased the basal amount of acetylated p53, and this effect was reversed by exposing cells to GSNO (fig. S6A).
Fig. 2. SIRT1 S-nitrosylation mediates NO-induced p53 acetylation, leading to cell death.

(A) Western blot of lysates from COS-7 cells transfected with WT or mutated SIRT1 and treated with bleomycin (10 μg/ml) or vehicle (dimethyl sulfoxide). CRL, control plasmid. **P < 0.01 compared to control plasmid; ††P < 0.01 compared to WT. One-way ANOVA with Tukey’s post hoc test. Data are means ± SEM of three biological replicates from one of three independent experiments. (B) Western blots of lysates from H1299 cells expressing exogenous wild-type exposed to GSNO (0.5 mM), reduced (GSH; 0.5 mM) or oxidized glutathione (GSSG; 0.5 mM), or 8-bromo cGMP (Br-cGMP; 0.5 mM). *P < 0.05, **P < 0.01 compared to control; †P < 0.05, ††P < 0.01 compared to GSH; ‡P < 0.05, ‡‡P < 0.01 compared to GSSG; §§P < 0.01 compared to Br-cGMP. One-way ANOVA with Tukey’s post hoc test. Data are means ± SEM from three independent experiments. (C to F) Western blot of lysates from COS-7 cells transfected with the indicated siRNAs and transfected with WT SIRT1, SIRT1M3, or control plasmids, or exposed to GSNO (1 mM) with or without TSA (1 μM). *P < 0.05 compared to control siRNA + control plasmid, ††P < 0.01 compared to SIRT1 siRNA + WT SIRT1 (C); *P < 0.05 compared to WT SIRT1 without GSNO (D); **P < 0.01 compared to control, ††P < 0.01 compared to TSA alone, ‡‡P < 0.01 compared to GSNO alone (E); **P < 0.01 compared to control siRNA alone, ††P < 0.01 compared to control siRNA + TSA (F). One-way ANOVA with Tukey’s post hoc test. Data are means ± SEM of three biological replicates from one of two independent experiments. (G and H) Western blot of C2C12 myotubes exposed to LPS (10μg/ml)andcytokines[TNF-α (10ng/ml)andIFN-γ (200ng/ml)]orHepa1c1c7 cells exposed to LPS (10 μg/ml) and cytokines [TNF-α (5 ng/ml) and IFN-γ (50 ng/ml)] with or without the iNOS inhibitor L-NIL (200 μM) or iNOS knockdown. Blots are representative of three independent experiments.
We also examined the NO-induced changes in the acetylation of p53 and apoptosis in the non–small-cell lung carcinoma cell line H1299, which does not express endogenous p53. Exposing H1299 cells transfected with a plasmid encoding p53 (Fig. 2B and fig. S6B) to GSNO increased the abundance of acetylated p53, similar to exposing COS-7 cells to SNAP (fig. S6C). Exposing H1299 cells expressing exogenous wild-type p53 to GSNO increased the abundance of cleaved poly(ADP-ribose) polymerase 1 (PARP1) (Fig. 2B and figs. S6D and S7A) and caspase-3 (fig. S7A) and reduced the number of cells (fig. S7B), consistent with the activation of apoptosis. Acetylation of Lys382 in p53 increases the ability of p53 to activate transcription (70, 71), and this residue is the major site targeted by SIRT1 (22, 23). Thus, we tested whether acetylation of Lys382 was required for the ability of NO to reduce cell viability. We transfected H1299 cells with plasmids encoding wild-type p53 or K382A p53 (72). Exposing cells to GSNO increased the acetylation of wild-type, but not K382A, p53 (fig. S7C). Furthermore, GSNO or SNAP decreased the viability of H1299 cells expressing wild-type, but not K382A, p53 (fig. S7C).
NO signals through cyclic guanosine monophosphate (cGMP)– or S-nitrosylation–dependent mechanisms (73). Exposing H1299 cells expressing p53 to a cell-permeable cGMP analog (8-bromo-cGMP) did not increase the abundance of acetylated p53 (Fig. 2B) or decrease cell viability (fig. S8A). Moreover, exposing COS-7 cells to an inhibitor of soluble guanylyl cyclase, 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (74), did not decrease the ability of GSNO to increase the abundance of acetylated p53 or cleaved PARP1 (fig. S8B) or decrease the viability (fig. S8A) of p53-expressing H1299 cells, suggesting that NO-induced acetylation of p53 is independent of cGMP. In contrast to GSNO, neither reduced (GSH) nor oxidized glutathione (GSSG) increased p53 acetylation or PARP1 cleavage (Fig. 2B and fig. S8A), supporting the specificity of the NO-induced effects of GSNO.
To investigate the role of the CXXC motif of SIRT1 in the inhibition of deacetylation of p53, we expressed wild-type SIRT1 or SIRT1M3 in COS-7 cells with small interfering RNA (siRNA)–mediated knockdown of endogenous SIRT1. Knockdown of SIRT1 increased the abundance of acetylated p53 in unstimulated cells, and reconstitution with wild-type SIRT1, but not with SIRT1M3, decreased the abundance of acetylated p53 in these cells (Fig. 2C). GSNO increased the abundance of acetylated p53 in cells with SIRT1 knockdown that expressed exogenous wild-type SIRT1, but not SIRT1M3 (Fig. 2D). These results suggest that GSNO is likely to increase p53 acetylation by S-nitrosylation of the CXXC motif in SIRT1.
In addition to SIRT1, a class III HDAC, class I and class II HDACs can deacetylate p53 (33, 34). Thus, we tested whether trichostatin A (TSA), a specific inhibitor of class I and class II HDACs (75), increased the acetylation of p53 in cells exposed to GSNO or with knockdown of SIRT1. Exposing COS-7 cells to GSNO and TSA increased the abundance of acetylated p53 to a greater extent than did GSNO or TSA alone (Fig. 2E). If the inhibition of SIRT1 does not play a major role in the effect of GSNO on the abundance of acetylated p53 and if the inhibition of class I and class II HDACs has an important role, then GSNO could increase the abundance of p53 acetylation in SIRT1-deficient cells. However, we observed that SIRT1 knockdown and exposure to GSNO increased the abundance of acetylated p53 to a comparable extent in COS-7 cells (Fig. 2F), and that GSNO failed to further increase the abundance of acetylated p53 in COS-7 cells with SIRT1 knockdown (Fig. 2F). These results suggest that SIRT1 is required for the GSNO-induced increase in the abundance of p53 acetylation in COS-7 cells. However, TSA increased the abundance of acetylated p53 in COS-7 cells with knockdown of SIRT1 (Fig. 2F), suggesting that inhibition of SIRT1 is the major contributor to the increased abundance of p53 by GSNO in COS-7 cells, but both SIRT1 and other HDACs influence the acetylation of p53.
Inflammatory disease is associated with activation of iNOS and p53-dependent cell death (54, 76, 77). Therefore, we investigated the role of iNOS in SIRT1-dependent deacetylation of p53. Overexpressing iNOS in COS-7 cells increased the abundance of S-nitrosylated SIRT1 and acetylated p53, and these effects could be reversed by an inhibitor of iNOS (L-NIL) (78) (fig. S9A). Moreover, stimulating C2C12 myotubes, which activate iNOS expression in response to inflammatory stimuli (79), with a mixture of proinflammatory molecules, including lipopolysaccharide (LPS), tumor necrosis factor–α (TNF-α), and interferon-γ (IFN-γ), increased the abundance of S-nitrosylated SIRT1 and acetylated p53 and the ability of p53 to bind to target DNA, and these effects could be reversed by L-NIL (Fig. 2G and fig. S9B). Likewise, a second iNOS inhibitor (1400W) (80) prevented the ability of the mixture of LPS and cytokines to increase the acetylation of p53 in C2C12 myotubes (fig. S9C).
We also tested the role of iNOS in mouse hepatoma Hepa1c1c7 cells, which induce iNOS expression in response to inflammatory stimuli (81). In Hepa1c1c7 cells transfected with siRNAs targeting iNOS, the mixture of LPS and cytokines did not increase the release of nitrite into the media (fig. S9D) or the abundance of S-nitrosylated SIRT1 or acetylated p53 compared to Hepa1c1c7 cells transfected with control siRNAs (Fig. 2H and fig. S9, E and F). Thus, activation of inflammatory signaling causes iNOS-dependent S-nitrosylation of SIRT1 that inhibits the deacetylation of p53.
S-Nitrosylation of SIRT1 increases the acetylation and activation of p65
In addition to p53, SIRT1 deacetylates p65, thereby attenuating the activity of NF-κB (20, 24). NO and iNOS can either increase or decrease NF-κB activity depending on the cellular context or pathophysiological condition (12–14, 39–42). Similar to p53, the abundance of acetylated p65 was increased in COS-7 cells grown in the presence of GSNO or SNAP compared to cells grown in control media (Fig. 3, A and B). p65 is deacetylated by class I and class II HDACs in addition to SIRT1 (34), and exposing COS-7 cells to TSA increased the abundance of acetylated p65 in the presence or absence of GSNO (Fig. 3B). Moreover, exposing C2C12 myotubes to LPS and cytokines increased the abundance of S-nitrosylated SIRT1 (Fig. 3C) and acetylated p65 (Fig. 3D and fig. S10), the ability of p65 to bind to target DNA (Fig. 3E), and the expression of NF-κB target genes (Fig. 3F), and exposure to L-NIL inhibited these effects. We confirmed the increased ability of LPS and cytokines to induce iNOS-derived NO generation by measuring nitrite accumulation in the culture media of C2C12 myotubes (Fig. 3G). COS-7 cells with knockdown of SIRT1 had increased abundance of acetylated p65 that was further increased by exposure to TSA, but not GSNO (Fig. 3H). Thus, S-nitrosylation of SIRT1 inhibits the deacetylation of p65.
Fig. 3. S-Nitrosylation–mediated inactivation of SIRT1 leads to increased acetylation of p65.

(A and B) Western blots of lysates from COS-7 cells exposed to SNAP (300 μM) (A) or GSNO (1 mM) and TSA (1 μM) (B). (C to G) C2C12 myotubes exposed to LPS (10 μg/ml) and cytokines [TNF-α (10 ng/ml) and IFN-γ (200 ng/ml)] with or without L-NIL (200 μM). (C and D) Western blot. (E) p65 DNA binding activity. (F) mRNA abundance of NF-κB target genes. (G) Nitrite in the culture media. *P < 0.05, **P < 0.01, ***P < 0.001 compared to control; ††P < 0.01, †††P < 0.001 compared to cytokine + L-NIL. One-way ANOVA with Tukey’s post hoc test. (H) Western blot of lysates from COS-7 cells transfected with the indicated siRNAs and exposed to GSNO (1 mM) with or without TSA (1 μM). **P < 0.01 compared to control, †P < 0.05 compared to control siRNA + TSA; ‡‡P < 0.01 compared to SIRT1 siRNA + GSNO. One-way ANOVA with Tukey’s post hoc test. Data are means ± SEM of three biological replicates from one of two (B and H) or three (A and C to G) independent experiments.
iNOS deficiency prevents endotoxin-induced S-nitrosylation of SIRT1 and acetylation of p53 and p65
Endotoxemia is implicated in multiple age-related diseases, including type 2 diabetes, Parkinson’s disease, atherosclerosis, and muscle wasting (82–85). Furthermore, the prevalence of systemic inflammatory response syndrome (SIRS) appears to be greater in the elderly population (86), and SIRS can be modeled in mice by injection of endotoxins, such as LPS (87). To investigate the relevance of NO in SIRS, we examined the effects of LPS injection in iNOS knockout mice (88). LPS injection increased the abundance of iNOS and cleaved caspase-3 in the liver of wild-type mice, but not iNOS knockout mice (Fig. 4, A to C). Moreover, LPS increased the S-nitrosylation of SIRT1, the abundance of acetylated p53 and p65, the binding of p53 and p65 to target DNA, and the expression of p53 and NF-κB target genes in the liver of wild-type mice, but not iNOS knockout mice (Fig. 4, D to F, and fig. S11).
Fig. 4. iNOS deficiency prevents endotoxin-induced SIRT1 S-nitrosylation and acetylation of p53 and p65 in mouse liver.

(A to F) Western blot and quantification of lysates from livers of WT mice or iNOS knockout (iNOS−/−) mice injected with LPS [27.5 mg/kg, intraperitoneal (ip)]. *P < 0.05, **P < 0.01, ***P < 0.001 compared to WT without LPS; †P < 0.05, †††P < 0.001 compared to iNOS−/− with LPS. One-way ANOVA with Tukey’s post hoc test. n = 8 mice per group.
S-Nitrosylation of SIRT1 parallels acetylation of p53 and p65 in a mouse model of Parkinson’s disease
NO, p53, and NF-κB play important roles in neurodegenerative disorders, including Parkinson’s disease (4, 5, 10, 46, 49, 89). In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)–induced mouse model of Parkinson’s disease, the initial inflammatory and apoptotic responses cause chronic inflammation and Parkinsonian-like symptoms (90). Inhibition of either nNOS or iNOS prevents MPTP-induced symptoms (4, 5, 49). However, nNOS activation in neurons precedes activation of iNOS, which occurs mainly in microglia and astrocytes in this model (49). We injected mice daily with the nNOS inhibitor 7-nitroindazole (7-NI) for 3 days after injection of MPTP. We analyzed protein changes in the nigrastriatum at 3 days and behavioral symptoms at 7 days after MPTP injections. Consistent with previous reports (4, 5), we found that injection of 7-NI prevented MPTP-induced motor symptoms (bradykinesia) (Fig. 5A) and the reduction of tyrosine hydroxylase (TH) in the nigrastriatum (Fig. 5B). Neither MPTP nor the combination of MPTP and 7-NI affected the abundance of nNOS, p53, or p65 (fig. S12, A to C). However, MPTP increased the abundance of S-nitrosylated SIRT1 and acetylated p53 and p65, and these effects were reversed by injection of 7-NI (Fig. 5, C to F, and fig. S12, Band C). Moreover, MPTP increased the ability of p53 and p65 to bind target DNA (fig. S12D) and promote the expression of p53 and NF-κB target genes in an nNOS activity–dependent manner (fig. S12, E to H). Previous studies have shown that nNOS is not detectable in TH-positive dopaminergic neurons in the substantia nigra pars compacta (91), but nNOS is present in neurons in the adjacent substantia nigra pars reticulata (92) and in the striatum (93). The striatum contains dopamine nerve terminals, a proposed primary target of MPTP in Parkinson’s disease (5). NO acts as both an intracellular signaling molecule and an intercellular messenger (94, 95). Thus, our data suggest that nNOS in the striatum or the substantia nigra pars reticulata may contribute to S-nitrosylation of SIRT1 in the substantia nigra pars compacta of MPTP-injected mice.
Fig. 5. S-Nitrosylation of SIRT1 parallels acetylation of p53 and p65 in a mouse model of Parkinson’s disease.

(A to F) Mice were injected with MPTP (80 mg/kg, ip) and the nNOS inhibitor 7-NI (50 mg/kg, ip) and monitored 7 days after MPTP injection for bradykinesia (prolonged immobility time during the tail suspension test) (A), or the nigra-striatum was dissected 3 days after MPTP injection and analyzed by Western blot for the indicated proteins (B to F). *P < 0.05, **P < 0.05 compared to control; †P < 0.05, ††P < 0.01 compared to MPTP + 7-NI. One-way ANOVA with Tukey’s post hoc test. n = 8 mice per group.
Increased abundance of iNOS is associated with S-nitrosylation of SIRT1 and acetylation and activation of p53 and p65 in skeletal muscle of aged rats
Aging-associated inflammation and induction of iNOS are involved in the development of sarcopenia, an age-related muscle wasting disorder (7, 8). We found that skeletal muscle of aged rats had increased numbers of TUNEL-positive nuclei, which are indicative of apoptosis, compared to young rats (Fig. 6A). Moreover, the mRNA abundance of Fas and FasL (fig. S13A), which encode proteins that induce of apoptosis (96) and are targets of p53 and NF-κB, as well as Atrogin-1 and Murf1 (fig. S13B), which encode muscle-specific ubiquitin ligases involved in muscle wasting and are targets of NF-κB (44, 97), were increased in skeletal muscle of aged rats compared to young rats. The skeletal muscle of aged rats had increased abundance of iNOS, S-nitrosylated SIRT1, and acetylated p53 and p65 (Fig. 6B and fig. S13, C and D). The abundance of p53 (fig. S13C), but not p65 (fig. S13D), was greater in skeletal muscle of aged rats compared to young counterparts. The skeletal muscle of aged rats had increased binding of p53 and p65 to target DNA (fig. S13E).
Fig. 6. Increased abundance of iNOS is associated with S-nitrosylation of SIRT1 and acetylation and activation of p53 and p65 in skeletal muscle of aged rats.
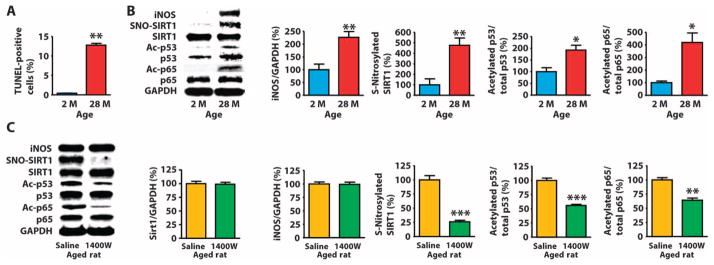
(A and B) Terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling (TUNEL) (A) or Western blot of lysates (B) of skeletal muscle from rats at the indicated ages. *P < 0.05, **P < 0.01, 28 months (28 M) compared to 2 months (2 M). Unpaired two-tailed Student’s t test. n = 8 mice per group. (C) Western blot of skeletal muscle of 23-month-old rats injected with saline or the iNOS inhibitor 1400W (10 mg/kg, ip) for 10 days. **P < 0.01, ***P < 0.001 compared to saline. Unpaired two-tailed Student’s t test. n = 7 mice per group.
We gave aged rats daily injections of saline or 1400W for 10 days and found that inhibition of iNOS decreased the S-nitrosylation of SIRT1, the acetylation and DNA binding of p53 and p65, and the expression of p53 and NF-κB target genes in skeletal muscle (Fig. 6C and fig. S14), suggesting that iNOS may contribute to acetylation and activation of p53 and p65 in skeletal muscle of aged rats, at least in part, by increasing S-nitrosylation of SIRT1.
DISCUSSION
Nitrosative stress involving dysregulated protein S-nitrosylation is implicated in many age-related inflammatory disorders. SIRT1, p53, and NF-κB play pivotal roles in inflammation and apoptosis in conditions characterized by nitrosative stress. Our findings demonstrate that S-nitrosylation of the CXXC motif in SIRT1 is likely a critical factor in an inflammatory cascade, comprising activation of iNOS or nNOS, S-nitrosylation of SIRT1, and activation of p65 (NF-κB) and p53. Thus, aberrant S-nitrosylation of SIRT1 may result in pathologic activation of p53 and NF-κB to induce apoptosis and inflammation in multiple aging and degenerative disorders (Fig. 7).
Fig. 7. The proposed SIRT1 nitrosative stress–sensitive switch.

SIRT1 decreases apoptosis and inflammation by deacetylating proteins in the p53 and NF-κB pathways. SIRT1 deacetylates p53 and the p65 subunit of NF-κB when the CXXC zinc finger motif is bound to zinc (Zn2+) (solid green). NO derived from iNOS or nNOS S-nitrosylates the cysteine thiol groups (SNO) in the CXXC motif, disrupting zinc binding, which renders SIRT1 inactive. Acetylation of p53 and p65 increases apoptosis and inflammation (dashed green). S-Nitrosylation and resultant inactivation of SIRT1 are reversible. Ac, acetyl group; SH, sulfhydryl group.
The NO-induced increase in the acetylation of p53 and p65 was SIRT1-dependent in cultured cells, supporting the notion that CXXC motifs in SIRT1 are a major molecular target of nitrosative stress. However, class I and class II HDACs also deacetylate p53 and p65, and our data do not exclude the possibility that these HDACs may also contribute to NO-mediated activation of p53 and p65 in vivo.
Comorbidity of inflammation-associated disorders is a hallmark of aging. For example, individuals with Parkinson’s disease often have muscle atrophy and metabolic endotoxemia (83). Furthermore, the incidence of nitrosative stress increases with age (98–100). Thus, common molecular mechanisms such as S-nitrosylation of SIRT1 may underlie the pathogenesis of age-related disorders.
Our findings help clarify the molecular mechanisms by which NO causes or enhances apoptosis and inflammation. p53 plays an important role in NO-induced apoptosis in various cell types, including hepatocytes and skeletal muscle cells (36, 101). On the basis of studies using exogenous NO donors, it has been shown that S-nitrosylation can inhibit the binding of p53 to E3 ubiquitin ligases including MDM2 and HDM2, and thereby prevent degradation of p53 (15). Our data demonstrate that endogenous S-nitrosylation of SIRT1 induced by inflammatory stimuli correlates with p53 activation in multiple pathophysiological conditions, suggesting that inhibition of deacetylation of p53 may serve as a critical mechanism by which NO induces apoptosis. NF-κB can have anti- or proapoptotic functions, and SIRT1-mediated deacetylation of p65 promotes either cell survival (20) or apoptosis (24). In rodent models of endotoxemia, Parkinson’s disease, and sarcopenia, activation of NF-κB was associated with apoptotic changes, consistent with reports indicating that NF-κB activation plays a key role in the pathogenesis of these conditions (7, 44, 51, 89). NO and iNOS can either increase or decrease NF-κB activity (11–14, 39–42). Most studies show that inhibition of iNOS decreases NF-κB activity in rodents (39–42). However, S-nitrosylation of NF-κB inhibits NF-κB–mediated gene transcription in human lung carcinoma epithelial A549 cells and mouse macrophage Raw 264.7 cells (13, 14), suggesting that the effects of NO on NF-κB activity vary depending on cell type, the cellular context, and possibly the various promoters of NF-κB target genes. Because NF-κB drives transcription of the gene encoding iNOS (38), S-nitrosylation of NF-κB was proposed as a negative feedback mechanism to regulate the abundance of iNOS. Inhibition of NF-κB signaling by NO is well characterized and may involve S-nitrosylation of p65, p50, or IKK (12–14). In contrast, much less is known about the manner in which NO enhances NF-κB activity. One study has suggested that NO may directly activate NF-κB (102). Our finding that NO can reversibly activate NF-κB through inhibitory S-nitrosylation of SIRT1, resulting in enhanced acetylation of p65, identified a positive feedback loop composed of NF-κB, iNOS, SIRT1, and p65 that enhances and sustains the inflammatory response (Fig. 7).
Concerted activation of p53, NF-κB, and iNOS or nNOS plays a crucial role in the pathogenesis of inflammatory and degenerative diseases. Pharmacological inhibition or genetic disruption of iNOS, p53, or NF-κB prevents LPS-induced liver damage (48, 51, 52) and skeletal muscle wasting (7, 8, 44, 45, 53). Inhibition or genetic disruption of iNOS or nNOS, p53, or NF-κB rescues MPTP-induced loss of dopaminergic neurons (4, 5, 10, 49, 89). Apoptosis and inflammation are involved in the development of these conditions. Cell death, including apoptosis, contributes to both local and systemic inflammatory responses through the release of damage-associated molecular patterns (DAMPs) (103). Thus, p53 activation is a likely component of progressive, chronic inflammation. In addition to p53 and p65, acetylation of PGC-1α, a substrate of SIRT1, reduces its function (32), leading to mitochondrial dysfunction and subsequent oxidative and nitrosative stress. Therefore, the increased acetylation of PGC-1α, which decreases its ability to activate transcription and occurs in conditions that promote S-nitrosylation of SIRT1 (35), may also contribute to NO-induced inflammation (Fig. 7). Thus, SIRT1 may be a hub for inflammatory and cell death signaling pathways in aging-related disorders. S-Nitrosylation of SIRT1, in particular, is increased in multiple inflammatory and aging models, where it may function as a proinflammatory switch and may represent a molecular signature of cellular pathophysiology, including cellular senescence, apoptosis, and inflammation.
MATERIALS AND METHODS
Animals
Male C57BL/6 and iNOS knockout mice on C57BL/6 background at 7 weeks of age were purchased from The Jackson Laboratory. Male F344 rats at 2 months and 23 to 28 months of age were purchased from Taconic Farms and the National Institute on Aging, respectively. The study was approved by the Institutional Animal Care Committees of the Massachusetts General Hospital and University of Texas. In the mouse model of endotoxemia, the mice received a bolus injection of LPS (27.5 mg/kg, ip) or phosphate-buffered saline (PBS), the liver was collected at 7 hours after the LPS injection under anesthesia with pentobarbital sodium (50 mg/kg, ip), and then the animals were euthanized with an overdose of pentobarbital (200 mg/kg, ip). To induce Parkinsonian syndrome, the mice were injected four times with MPTP (20 mg/kg, ip) at 2-hour intervals (104). The mice were injected with 7-NI (50 mg/kg, ip) or vehicle, once daily for 3 days starting at 30 min before the first injection of MPTP. At 3 days after the first injection of MPTP, the brain was excised under anesthesia, and the nigrastriatum was dissected for biochemical analyses. The tail suspension test (104) was performed at 7 days after MPTP injection. To study age-related alterations in skeletal muscle, gastrocnemius muscle was excised under anesthesia from rats at 2 and 28 months of age. To study the role of iNOS in these age-related alterations, the rats at 23 months of age were injected daily with 1400W (10 mg/kg, ip) or saline for 10 days.
Plasmids
Mouse SIRT1 complementary DNA (cDNA) (Upstate, #21-198) was subcloned into pCDNA3.1-N3-FLAG plasmid, provided by A. Yamakawa. cDNAs for SIRT1 mutants (M1: C363S and C366S; M2: C387S and C390S; M3: C363S, C366S, C387S, and C390S) were generated by site-directed mutagenesis using a commercial kit (QuikChange Multi Site-Directed Mutagenesis Kit, Stratagene) and primers (M1: 5′-CCTTTGCAACAGCATCTTCCCTGATTAGTAAGTACAAAGTTGATTGTG-3′; M2: 5′-GGTAGTTCCTCGGTCCCCTAGGTCCCCAGCTGATGAGC-3′; and M3: M1 and M2 primers). SIRT1 mutants, SIRT1H355Y and SIRT1G261A, were also generated using the following primers: H355Y, 5′-CAAAGGATCCTTCAGTGTTATGGTTCCTTTGCAACAGC-3′; G261A, 5′-GTTTCTGTCTCCTGTGCGATTCCTGACTTCAGATC-3′. pCDNA3.1-iNOS (105) was a gift from B. C. Kone. pFC-p53 was purchased from Stratagene. p53 K382A (72) was a gift from S.-C. Lee. pCDNA3.1 (Invitrogen) was used as the negative control plasmid in all experiments. All plasmids were verified by sequencing of the mutation sites.
Reagents
SNAP, GSNO, L-NIL, and 1400W were purchased from Cayman Chemical. Nicotinamide, TSA, antibody recognizing FLAG M2, methyl methanethiosulfonate (MMTS), N-ethylmaleimide (NEM), LPS, ascorbate, dithiothreitol (DTT), streptavidin immobilized on agarose CL-4B, MPTP, and 7-NI were purchased from Sigma. TNF-α was purchased from Roche. IFN-γ was purchased from R&D Systems. Maleimide-PEG2-biotin and N-(6-(biotinamido)hexyl)-3′-(2′-pyridyldithio)-propionamide (biotin-HPDP) were purchased from Pierce. The antibodies used in this study were acetylated p53 (Lys379; #2570), p53 (#9282), p65 (#4764), histone H3 (#9715), acetylated histone H3 (Lys9; #9649) (Cell Signaling Technology); acetylated p65 NF-κB (Lys310; Abcam, ab19870); p53 (Santa Cruz Biotechnology, sc-6243); and SIRT1 (#07-131) and iNOS (#06-573) (Millipore). The siRNAs used in this study were iNOS (#M-42006-01), SIRT1 (5′-TGAAGTGCCTCAGATATTA-3′), and a negative control siRNA (5′-AACACTTGTCACTACTTTCTC-3′) (Dharmacon) (69).
Cell culture
H1299, COS-7, C2C12, and Hepa1c1c7 cells were purchased from the American Type Culture Collection and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin at 37°C and 5% CO2. C2C12 myoblasts were differentiated into myotubes by culturing in DMEM containing 2% horse serum for 7 days. Plasmids were transfected using Lipofectamine 2000 (Invitrogen), and siRNAs were transfected using Lipofectamine RNAiMAX (Invitrogen). Cells were exposed to SNAP (300 μM) for 1 to 2 hours or GSNO (1 mM), TSA (3 μM), GSH (1 mM), GSSG (1 mM), ODQ (3 μM), or Br-cGMP (500 μM) for 10 to 24 hours. These reagents were added 24 hours after plasmid transfection or 48 hours after siRNA transfection. For experiments involving plasmids and siRNAs, plasmids were transfected 48 hours after siRNA transfection, and small molecules were added 24 hours later. For experiments with SNAP, the culture medium was changed to serum-free medium; 90 min later, the cells were incubated with 80 μM carmustine for 30 min and then exposed to SNAP (300 μM) for 2 hours unless otherwise indicated in the figure legends. Hepa1c1c7 cells were stimulated with LPS (10 μg/ml), TNF-α (10 ng/ml), and IFN-γ (200 ng/ml) for 24 hours in the presence or absence of L-NIL (200 μM). Differentiated C2C12 myotubes were stimulated with LPS (10 μg/ml), TNF-α (5 ng/ml), and IFN-γ (50 ng/ml) for 72 hours in the presence or absence of L-NIL (200 μM) or 1400W (50 μM), and the culture medium was replaced with fresh medium containing cytokines and LPS every 24 hours.
Measurement of nitrite concentration
Nitrite accumulation in the culture media was measured by using Griess reagent according to the manufacturer’s instructions (Sigma).
Detection of S-nitrosylated protein
The biotin-switch assay was performed as previously described (58, 106). Briefly, liver, nigrastriatum, and muscle were washed with PBS, minced, and homogenized in homogenization buffer A [PBS-HCl (pH 3.5), 150 mM NaCl, 1 mM EDTA, 1 mM diethylenetriamine pentaacetic acid (DTPA), 7.5% SDS, 2% CHAPS, 0.1 mM neocuproine, 80 μM carmustine, 1 mM phenylmethylsulfonyl fluoride (PMSF), protease inhibitor cocktail (Sigma)] as previously described (106). Cultured cells were lysed in lysis buffer B [PBS-HCl (pH 3.5), 150 mM NaCl, 1 mM EDTA, 1 mM DTPA, 2.5% SDS, 0.5% NP-40, 0.1 mM neocuproine, 80 μM carmustine, 1 mM PMSF, protease inhibitor cocktail] (106). After centrifugation to remove insoluble material, tissue or cell supernatants were mixed with 2 volumes of blocking buffer [PBS-HCl (pH 3.5), 150 mM NaCl, 1 mM EDTA, 1 mM DTPA, 0.1 mM neocuproine, 30 mM MMTS] (with or without 2.5% SDS for cell lysates and tissue homogenates, respectively) and incubated at 50°C for 20 min with vortex every 2 min. After precipitation using −20°C acetone, pellets were resuspended in modified HENS buffer [25 mM Hepes (pH 7.7), 1% SDS, 1 mM EDTA, 1 mM DTPA, and 0.1 mM neocuproine], neutralized with HEN buffer containing 0.5% Triton X-100 and 1 mM DTPA, and biotinylated with pyridyldithiol (HPDP)–biotin (0.2 mM) (Pierce) in the presence of sodium ascorbate (4 mM). After precipitation with −20°C acetone, streptavidin-agarose beads (Sigma) were added, and the samples were incubated for 1 hour at room temperature. The beads were washed with HEN buffer containing 0.5% NP-40, and proteins were eluted by incubation with elution buffer [20 mM Hepes (pH 7.7), 1 mM EDTA, 100 mM NaCl, 200 mM DTT] for 30 min and separated by SDS–polyacrylamide gel electrophoresis (SDS-PAGE) followed by immunoblotting with a SIRT1 antibody. In fig. S3A, the cell lysates were incubated with or with-out HgCl2 (100 μM) before addition of MMTS and the biotin switch method.
To evaluate the role of CXXC motifs in S-nitrosylation of SIRT1 and assess S-nitrosylation and oxidative thiol modifications of SIRT1, COS-7 cells were transfected with FLAG-tagged wild-type SIRT1 or SIRT1M3. After 2 hours of serum starvation, the cells were exposed to SNAP (1 mM) and carmustine (80 μM) for 2 hours. The cells were lysed in lysis buffer A, clarified by centrifugation, mixed with 1 volume of blocking buffer [PBS-HCl (pH 3.5), 150 mM NaCl, 1 mM EDTA, 1 mM DTPA, 2.5% SDS, 0.1 mM neocuproine, and 20 mM NEM], and incubated at 50°C for 20 min with vortex every 2 min. Lysates were biotinylated with NEM-PEG2-biotin (0.2 mM) (Pierce) in the presence of ascorbate sodium (4 mM) for the detection of SNO or with DTT (4 mM) for the detection of both SNO and SOX (fig. S2). After precipitation with −20°C acetone, antibody recognizing FLAG M2 (Sigma) and protein G and protein A agarose suspension (#IP05, Calbiochem) were added, and the samples were incubated at room temperature for 1 hour or at 4°C overnight. The immunopurified complexes were washed five times with wash buffer [50 mM Hepes (pH 7.7), 150 mM NaCl, 0.1% NP-40, 1 mM EDTA] and separated by SDS-PAGE. Biotinylated SIRT1 was detected with horseradish peroxidase (HRP)–conjugated streptavidin (Pierce).
Immunoblotting
Immunoblotting was performed as described previously (106). Briefly, tissue samples were powdered under liquid nitrogen and homogenized in ice-cold homogenization buffer A [50 mM Hepes (pH 8.0), 150 mM NaCl, 2 mM EDTA, 7.5% lithium dodecyl sulfate, 2% CHAPS, 10% glycerol, 10 mM sodium fluoride, 2 mM sodium vanadate, 1 mM PMSF, 10 mM sodium pyrophosphate, protease inhibitor cocktail, 5 μM TSA, and 10 mM nicotinamide]. Cultured cells were lysed in lysis buffer B [50 mM Hepes (pH 8.0), 150 mM NaCl, 2 mM EDTA, 2.5% lithium dodecyl sulfate, 2% CHAPS, 10% glycerol, 10 mM sodium fluoride, 2 mM sodium vanadate, 1 mM PMSF, 10 mM sodium pyrophosphate, protease inhibitor cocktail, 5 μM TSA, and 10 mM nicotinamide]. TSA and nicotinamide were added to prevent deacetylation during the experimental procedure. After incubation at 8°C for 10 min, the samples were centrifuged at 13,000g for 10 min at 8°C. For the detection of acetylated and total p53 and p65 from rodent tissue samples, to completely solubilize proteins including those in the nuclear and chromatin fractions, pulverized samples were homogenized in ice-cold hypotonic buffer [20 mM Hepes (pH 7.5), 10 mM MgCl2, 10 mM KCl, 10 mM sodium fluoride, 1 mM PMSF, 2 mM EDTA, 1 mM DTT, 20% glycerol, protease inhibitor cocktail, 5 μM TSA, and 10 mM nicotinamide] for 15 min on ice, and the samples were centrifuged at 13,000g for 10 min at 4°C. Then, the precipitates were homogenized in lysis buffer A. Aliquots of the supernatant containing equal amounts of protein, determined by detergent-compatible protein assay kit (Bio-Rad), were boiled for 5 min in Laemmli sample buffer, separated by SDS-PAGE, and transferred onto nitrocellulose membranes (Bio-Rad). The membranes were blocked in 2% blocking reagent (GE Healthcare) for 1 hour at room temperature, incubated with primary antibodies for 2 hours at room temperature or overnight at 4°C, and incubated with secondary antibodies [antibody recognizing rabbit-immunoglobulin G (IgG) or mouse-IgG] conjugated to HRP for 1 hour at 4°C. Enhanced chemiluminescence reagent (ECL Advance, GE Healthcare) was used to detect HRP activity. Bands of interest were scanned by ScanMaker (Microtek) and quantified by National Institutes of Health (NIH) Image 1.62 software (NTIS).
SIRT1 deacetylase activity assay
For the immunopurification of FLAG-tagged wild-type or mutated SIRT1, equal amounts of cell lysates in buffer A [50 mM tris-HCl (pH 7.6), 150 mM NaCl, 0.1% NP-40, 2 mM sodium vanadate, 10 mM sodium fluoride, 1 mM PMSF, protease inhibitor cocktail, 1 mM DTT] were incubated with antibody recognizing FLAG M2 (Sigma) for 2 hours at 4°C. Protein A/G PLUS-Agarose beads (#sc-2003, Santa Cruz Biotechnology) were added for 1 hour. Immunopurified protein complexes were washed three times with buffer A and washed once with buffer B [25 mM tris-HCl (pH 8.0), 100 mM NaCl, 10% glycerol]. FLAG-tagged SIRT1 was eluted by incubating with buffer B containing FLAG peptide (Sigma; 20 μg/ml).
Recombinant human SIRT1 protein (Biomol) or immunopurified wild-type or mutated SIRT1 were assayed for in vitro deacetylase activity using a commercial kit (Sirt1 Fluorescent Activity Assay/Drug Discovery Kit, Biomol) according to the manufacturer’s instructions. Fluorescence was measured using a fluorometric reader (Victor3V, PerkinElmer) with excitation at 360 nm and emission detection at 460 nm. Immunopurified SIRT1 or recombinant SIRT1 (1 U) was incubated with 50 μM deacetylase substrate in the presence of NAD+ (250 μM) at 37°C for 45 min. For the assay of immunopurified SIRT1, 2.5 mM TSA was added to inhibit endogenous class I and class II HDACs. SIRT1 deacetylase activity was evaluated as a nicotinamide-inhibitable activity by subtracting the fluorescence intensities measured in the presence of nicotinamide (2 mM), a SIRT1 inhibitor (107), from those measured in the absence of nicotinamide according to the instructions of the manufacturer (Biomol). Recombinant SIRT1 was pre-incubated with SNAP (1 mM) for 30 min at 37°C and then incubated for an additional 30 min with or without DTT (5 mM) in the presence and absence of TPEN (200 μM). In a separate experiment, recombinant SIRT1 was pre-incubated with or without SNAP (0.6 mM) or GSNO (0.6 mM) at 37°C for 30 min and then incubated with or without DTT (4 mM) in the presence and absence of different concentrations of TPEN (2, 20, 100, or 200 μM) or EGTA (0.6 mM) for 30 min. The deacetylase activity of SIRT1 was also evaluated in vitro by deacetylation of recombinant histone H3 (Sigma) performed in SIRT1 activity assay buffer (Biomol) containing recombinant SIRT1 (1 U), histone H3 (0.5 μg), and NAD+ (250 μM) for 30 min at 37° C. The reaction was stopped by adding SDS sample buffer and boiling for 5 min. After separation by SDS-PAGE, immunoblot analysis was performed using antibodies for acetylated histone H3 and histone H3.
In vitro ADP-ribosylation assay
The in vitro ADP-ribosylation assay was performed as previously described (108) with minor modifications, using FLAG-tagged wild-type and mutated SIRT1 and immunopurified from COS-7 cells as described above. The reaction was carried out in SIRT1 activity assay buffer (Biomol) containing immunopurified SIRT1, 32P-labeled NAD+ (10 μCi, Amersham Biosciences), unlabeled NAD+ (10 μM), and histone H3 (3 μg) for 1 hour at 37°C in the presence of a PARP inhibitor, PJ34 (3 μM, EMD Biosciences), and an inhibitor for arginine mono-ADP-ribosyltransferases, m-iodobenzylguanidine (0.5 mM, MIBG, Sigma). The reaction was terminated by adding SDS sample buffer and boiling for 5 min. After separation by SDS-PAGE, incorporation of 32P-labeled NAD+ was evaluated by autoradiography.
Isolation of total RNA and quantitative reverse transcription polymerase chain reaction
Total RNA was isolated with an RNeasy Mini Kit (Qiagen). The first-strand cDNA was synthesized from 1 μg of total RNA using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). Real-time reverse transcription polymerase chain reaction analyses were performed using 10 ng of cDNA and TaqMan probes (Applied Biosystems) for BCL2-associated X protein (Bax: Mm00432051_m1, Rn02532082_g1), TNF receptor superfamily member 6 (Fas: Mm01204974_m1, Rn00685720_m1), Fas ligand (Fasl: Mm00438864_m1, Rn00563754_m1), toll-like receptor 4 (TLR4: Mm00445273_m1, Rn00569848_m1), Atrogin-1 (Mm00499523_m1, Rn00591730_m1), muscle-specific RING finger protein 1 (Murf1: Mm01185221_m1, Rn00590197_m1), and 18S ribosomal RNA (Hs99999901_s1), conducted with Mastercycler ep realplex (Eppendorf). The abundances of Bax, Fas, FasL, TLR4, Atrogin-1, and Murf1 mRNAs were normalized to 18S ribosomal RNA.
Measurement of DNA binding of p53 and p65
DNA binding activities of p53 and p65 were measured by TransAM p53 or p65 NF-κB Transcription Factor Assay Kit (Active Motif).
Evaluation of apoptotic nuclei in skeletal muscle
Apoptotic nuclei were detected in gastrocnemius muscle of rats by fluorescence TUNEL staining using the ApopTag Fluorescein In Situ Apoptosis Detection Kit (Promega). The numbers of TUNEL-positive nuclei were normalized to those of DAPI (4′,6-diamidino-2-phenylindole)–positive nuclei.
Zn2+ binding assay
FLAG-tagged mouse wild-type SIRT1 or mutant SIRT1M3 proteins were purified from transfected COS-7 cells as described above. FLAG-SIRT1, FLAG-SIRT1M3, recombinant human wild-type SIRT1 (Biomol), and recombinant Akt1 (Millipore) were denatured in SDS sample buffer containing 8% β-mercaptoethanol and separated by SDS-PAGE with running buffer containing 5 μM TPEN. After electrophoretic transfer (5 μM TPEN in transfer buffer) to nitrocellulose membranes (Bio-Rad), the membranes were incubated in tris-HCl buffer (10 mM tris-HCl, pH 7.7) containing DTT (20 mM) for 1 hour at room temperature. The membranes were rinsed with tris-KCl buffer [10 mM tris-HCl (pH 7.5), 100 mM KCl], incubated with tris-KCl buffer containing 65ZnCl2 (5 μCi/10 ml) (National Isotope Development Center) for 1 hour at room temperature, washed twice with tris-KCl buffer containing 0.05% Tween 20 for 1 hour, and exposed on BioMax films (Kodak) with enhancer plates at −80°C overnight. For the experiment with NO donors, immediately after autoradiography, the same membranes were incubated with tris-KCl buffer containing SNAP (100 μM) for 1 hour and then exposed to new films overnight at −80°C.
Supplementary Material
Fig. S1. Mutation of the CXXC motif in SIRT1 abolishes its ADP-ribosyltransferase activity, and SNAP decreases SIRT1 deacetylase activity.
Fig. S2. S-Nitrosylation is a major thiol modification of SIRT1 in cultured cells exposed to SNAP and carmustine.
Fig. S3. The specific detection of SIRT1 S-nitrosylation is confirmed by the effect of pre-incubation with HgCl2 and by ascorbate-dependent biotinylation.
Fig. S4. TPEN blocks reactivation of NO donor–exposed recombinant SIRT1 by DTT.
Fig. S5. SIRT1M3 decreases cell viability.
Fig. S6. NO donors increase the abundance of acetylated p53.
Fig. S7. GSNO and SNAP decrease viable cell numbers in a p53-dependent manner.
Fig. S8. GSNO induces p53 acetylation and decreases viable cell numbers independent of cGMP.
Fig. S9. Inhibition of iNOS prevents the ability of inflammatory molecules or iNOS overexpression to induce S-nitrosylation of SIRT1 and acetylation of p53.
Fig. S10. Inhibition of iNOS prevents the ability of inflammatory molecules to induce acetylation of p65.
Fig. S11. iNOS deficiency prevents the ability of LPS to induce acetylation and activation of p53 and p65 in mouse liver.
Fig. S12. Inhibition of nNOS prevents the ability of MPTP to induce acetylation and activation of p53 and p65 in the nigrastriatum of MPTP-injected mice.
Fig. S13. Acetylation and activation of p53 and p65 are increased in skeletal muscle of aged rats compared to young rats.
Fig. S14. Inhibition of iNOS decreases acetylation and activation of p53 and p65 in skeletal muscle of aged rats.
Acknowledgments
We are grateful to B. C. Kone at University of Texas Medical School, S.-C. Lee at National Taiwan University, and A. Yamakawa at Dana-Farber Cancer Institute for providing cDNAs for iNOS and acetylation-resistant p53 mutant and pcDNA3 3x FLAG, respectively. We thank J. Avruch at Massachusetts General Hospital, Harvard Medical School, for helpful discussion, D. T. Hess at Case Western Reserve University for critical reading and helpful comments, and K. Shinozaki and M. Saito for technical assistance.
Funding: This work was supported by grants from the NIH (R01-DK-058127), American Diabetes Association (7-08-RA-77), and Shriners Hospitals for Children (71000, 85800) to M.K.; NIH (R01-GM-099921, 5P01-HL-075443-08) and Defense Advanced Research Project Agency (N66001-13-C-4054) to J.S.; NIH (R01-AG-039732) to H.S.; JSPS KAKENHI (25860231) and Takeda Scientific Foundation to S.S.; Atherosclerosis Research Foundation to K.S.; MEXT KAKENHI (24108007) to N.S.; and MEXT KAKENHI (21592304) to K.C.
Footnotes
Author contributions: S.S. and K.C. designed the experiments, performed the experiments, analyzed the data, and contributed to the writing of the manuscript; M.S., N.S., M.Y., T.T., H.N., and A.I. designed and performed the experiments and analyzed the data; F.I. contributed to the study design, data interpretation, and writing of the manuscript; Y.Y., H.I., Y.O., and K.S. contributed to the study design and writing of the manuscript; M.E.S. contributed to the study design and performed the experiments; H.S. designed and performed the experiments and contributed to the writing of the manuscript; J.S.S. contributed to the study design and wrote the manuscript; M.K. designed the study and experiments, analyzed the data, and wrote the manuscript.
Competing interests: All authors declare that they have no competing interests. J. Stamler has licensed technology to Novartis within the field of S-nitrosylation that does not have a direct relationship to this work.
Data and materials availability: A material transfer agreement is required for expression constructs and cDNAs of wild-type and mutants of SIRT1. Expression constructs of iNOS and p53K382A should be requested to B. C. Kone at University of Texas Medical School and S.-C. Lee at National Taiwan University, respectively. All other reagents are commercially available.
REFERENCES AND NOTES
- 1.Perreault M, Marette A. Targeted disruption of inducible nitric oxide synthase protects against obesity-linked insulin resistance in muscle. Nat Med. 2001;7:1138–1143. doi: 10.1038/nm1001-1138. [DOI] [PubMed] [Google Scholar]
- 2.Sugita H, Fujimoto M, Yasukawa T, Shimizu N, Sugita M, Yasuhara S, Martyn JA, Kaneki M. Inducible nitric-oxide synthase and NO donor induce insulin receptor substrate-1 degradation in skeletal muscle cells. J Biol Chem. 2005;280:14203–14211. doi: 10.1074/jbc.M411226200. [DOI] [PubMed] [Google Scholar]
- 3.Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA, Kaneki M. S-nitrosylation of Drp1 mediates beta-amyloid-related mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hantraye P, Brouillet E, Ferrante R, Palfi S, Dolan R, Matthews RT, Beal MF. Inhibition of neuronal nitric oxide synthase prevents MPTP-induced parkinsonism in baboons. Nat Med. 1996;2:1017–1021. doi: 10.1038/nm0996-1017. [DOI] [PubMed] [Google Scholar]
- 5.Przedborski S, Jackson-Lewis V, Yokoyama R, Shibata T, Dawson VL, Dawson TM. Role of neuronal nitric oxide in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurotoxicity. Proc Natl Acad Sci USA. 1996;93:4565–4571. doi: 10.1073/pnas.93.10.4565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuhlencordt PJ, Chen J, Han F, Astern J, Huang PL. Genetic deficiency of inducible nitric oxide synthase reduces atherosclerosis and lowers plasma lipid peroxides in apolipoprotein E-knockout mice. Circulation. 2001;103:3099–3104. doi: 10.1161/01.cir.103.25.3099. [DOI] [PubMed] [Google Scholar]
- 7.Di Marco S, Mazroui R, Dallaire P, Chittur S, Tenenbaum SA, Radzioch D, Marette A, Gallouzi IE. NF-κB-mediated MyoD decay during muscle wasting requires nitric oxide synthase mRNA stabilization, HuR protein, and nitric oxide release. Mol Cell Biol. 2005;25:6533–6545. doi: 10.1128/MCB.25.15.6533-6545.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hall DT, Ma JF, Marco SD, Gallouzi IE. Inducible nitric oxide synthase (iNOS) in muscle wasting syndrome, sarcopenia, and cachexia. Aging. 2011;3:702–715. doi: 10.18632/aging.100358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cai D, Yuan M, Frantz DF, Melendez PA, Hansen L, Lee J, Shoelson SE. Local and systemic insulin resistance resulting from hepatic activation of IKK-β and NF-κB. Nat Med. 2005;11:183–190. doi: 10.1038/nm1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP. p53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann Neurol. 2002;52:597–606. doi: 10.1002/ana.10350. [DOI] [PubMed] [Google Scholar]
- 11.Connelly L, Palacios-Callender M, Ameixa C, Moncada S, Hobbs AJ. Biphasic regulation of NF-κB activity underlies the pro- and anti-inflammatory actions of nitric oxide. J Immunol. 2001;166:3873–3881. doi: 10.4049/jimmunol.166.6.3873. [DOI] [PubMed] [Google Scholar]
- 12.Katsuyama K, Shichiri M, Marumo F, Hirata Y. NO inhibits cytokine-induced iNOS expression and NF-κB activation by interfering with phosphorylation and degradation of IκB-α. Arterioscler Thromb Vasc Biol. 1998;18:1796–1802. doi: 10.1161/01.atv.18.11.1796. [DOI] [PubMed] [Google Scholar]
- 13.Kelleher ZT, Matsumoto A, Stamler JS, Marshall HE. NOS2 regulation of NF-κB by S-nitrosylation of p65. J Biol Chem. 2007;282:30667–30672. doi: 10.1074/jbc.M705929200. [DOI] [PubMed] [Google Scholar]
- 14.Marshall HE, Stamler JS. Inhibition of NF-κB by S-nitrosylation. Biochemistry. 2001;40:1688–1693. doi: 10.1021/bi002239y. [DOI] [PubMed] [Google Scholar]
- 15.Schonhoff CM, Daou MC, Jones SN, Schiffer CA, Ross AH. Nitric oxide-mediated inhibition of Hdm2–p53 binding. Biochemistry. 2002;41:13570–13574. doi: 10.1021/bi026262q. [DOI] [PubMed] [Google Scholar]
- 16.Shahani N, Sawa A. Nitric oxide signaling and nitrosative stress in neurons: Role for S-nitrosylation. Antioxid Redox Signal. 2011;14:1493–1504. doi: 10.1089/ars.2010.3580. [DOI] [PubMed] [Google Scholar]
- 17.Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795–800. doi: 10.1038/35001622. [DOI] [PubMed] [Google Scholar]
- 18.Moazed D. Enzymatic activities of Sir2 and chromatin silencing. Curr Opin Cell Biol. 2001;13:232–238. doi: 10.1016/s0955-0674(00)00202-7. [DOI] [PubMed] [Google Scholar]
- 19.Delaney JR, Sutphin GL, Dulken B, Sim S, Kim JR, Robison B, Schleit J, Murakami CJ, Carr D, An EH, Choi E, Chou A, Fletcher M, Jelic M, Liu B, Lockshon D, Moller RM, Pak DN, Peng Q, Peng ZJ, Pham KM, Sage M, Solanky A, Steffen KK, Tsuchiya M, Tsuchiyama S, Johnson S, Raabe C, Suh Y, Zhou Z, Liu X, Kennedy BK, Kaeberlein M. Sir2 deletion prevents lifespan extension in 32 long-lived mutants. Aging Cell. 2011;10:1089–1091. doi: 10.1111/j.1474-9726.2011.00742.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen J, Zhou Y, Mueller-Steiner S, Chen LF, Kwon H, Yi S, Mucke L, Gan L. SIRT1 protects against microglia-dependent amyloid-β toxicity through inhibiting NF-κB signaling. J Biol Chem. 2005;280:40364–40374. doi: 10.1074/jbc.M509329200. [DOI] [PubMed] [Google Scholar]
- 21.Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG, Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002;21:2383–2396. doi: 10.1093/emboj/21.10.2383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L, Gu W. Negative control of p53 by Sir2α promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 23.Vaziri H, Dessain SK, Eaton ENg, Imai SI, Frye RA, Pandita TK, Guarente L, Weinberg RA. SIRT1hSIR2 functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 24.Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, Mayo MW. Modulation of NF-κB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004;23:2369–2380. doi: 10.1038/sj.emboj.7600244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gillum MP, Kotas ME, Erion DM, Kursawe R, Chatterjee P, Nead KT, Muise ES, Hsiao JJ, Frederick DW, Yonemitsu S, Banks AS, Qiang L, Bhanot S, Olefsky JM, Sears DD, Caprio S, Shulman GI. SirT1 regulates adipose tissue inflammation. Diabetes. 2011;60:3235–3245. doi: 10.2337/db11-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Donmez G, Wang D, Cohen DE, Guarente L. SIRT1 suppresses β-amyloid production by activating the α-secretase gene ADAM10. Cell. 2010;142:320–332. doi: 10.1016/j.cell.2010.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 27.Lee D, Goldberg AL. SIRT1 protein, by blocking the activities of transcription factors FoxO1 and FoxO3, inhibits muscle atrophy and promotes muscle growth. J Biol Chem. 2013;288:30515–30526. doi: 10.1074/jbc.M113.489716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Y, Xu S, Giles A, Nakamura K, Lee JW, Hou X, Donmez G, Li J, Luo Z, Walsh K, Guarente L, Zang M. Hepatic overexpression of SIRT1 in mice attenuates endoplasmic reticulum stress and insulin resistance in the liver. FASEB J. 2011;25:1664–1679. doi: 10.1096/fj.10-173492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banks AS, Kon N, Knight C, Matsumoto M, Gutiérrez-Juárez R, Rossetti L, Gu W, Accili D. SirT1 gain of function increases energy efficiency and prevents diabetes in mice. Cell Metab. 2008;8:333–341. doi: 10.1016/j.cmet.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Luu L, Dai FF, Prentice KJ, Huang X, Hardy AB, Hansen JB, Liu Y, Joseph JW, Wheeler MB. The loss of Sirt1 in mouse pancreatic beta cells impairs insulin secretion by disrupting glucose sensing. Diabetologia. 2013;56:2010–2020. doi: 10.1007/s00125-013-2946-5. [DOI] [PubMed] [Google Scholar]
- 31.Wang RH, Kim HS, Xiao C, Xu X, Gavrilova O, Deng CX. Hepatic Sirt1 deficiency in mice impairs mTorc2/Akt signaling and results in hyperglycemia, oxidative damage, and insulin resistance. J Clin Invest. 2011;121:4477–4490. doi: 10.1172/JCI46243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 33.Ito A, Kawaguchi Y, Lai CH, Kovacs JJ, Higashimoto Y, Appella E, Yao TP. MDM2-HDAC1-mediated deacetylation of p53 is required for its degradation. EMBO J. 2002;21:6236–6245. doi: 10.1093/emboj/cdf616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Y, Smith PW, Jones DR. Breast cancer metastasis suppressor 1 functions as a corepressor by enhancing histone deacetylase 1-mediated deacetylation of RelA/p65 and promoting apoptosis. Mol Cell Biol. 2006;26:8683–8696. doi: 10.1128/MCB.00940-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kornberg MD, Sen N, Hara MR, Juluri KR, Nguyen JV, Snowman AM, Law L, Hester LD, Snyder SH. GAPDH mediates nitrosylation of nuclear proteins. Nat Cell Biol. 2010;12:1094–1100. doi: 10.1038/ncb2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee MH, Jang MH, Kim EK, Han SW, Cho SY, Kim CJ. Nitric oxide induces apoptosis in mouse C2C12 myoblast cells. J Pharmacol Sci. 2005;97:369–376. doi: 10.1254/jphs.fpj04017x. [DOI] [PubMed] [Google Scholar]
- 37.Messmer UK, Brüne B. Nitric oxide-induced apoptosis: p53-dependent and p53-independent signalling pathways. Biochem J. 1996;319:299–305. doi: 10.1042/bj3190299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xie QW, Kashiwabara Y, Nathan C. Role of transcription factor NF-κB/Rel in induction of nitric oxide synthase. J Biol Chem. 1994;269:4705–4708. [PubMed] [Google Scholar]
- 39.Cuzzocrea S, Chatterjee PK, Mazzon E, Dugo L, De Sarro A, Van de Loo FA, Caputi AP, Thiemermann C. Role of induced nitric oxide in the initiation of the inflammatory response after postischemic injury. Shock. 2002;18:169–176. doi: 10.1097/00024382-200208000-00014. [DOI] [PubMed] [Google Scholar]
- 40.Hierholzer C, Kalff JC, Billiar TR, Bauer AJ, Tweardy DJ, Harbrecht BG. Induced nitric oxide promotes intestinal inflammation following hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2004;286:G225–G233. doi: 10.1152/ajpgi.00447.2002. [DOI] [PubMed] [Google Scholar]
- 41.Katada K, Bihari A, Badhwar A, Yoshida N, Yoshikawa T, Potter RF, Cepinskas G. Hindlimb ischemia/reperfusion-induced remote injury to the small intestine: Role of inducible nitric-oxide synthase-derived nitric oxide. J Pharmacol Exp Ther. 2009;329:919–927. doi: 10.1124/jpet.108.148460. [DOI] [PubMed] [Google Scholar]
- 42.Skidgel RA, Gao XP, Brovkovych V, Rahman A, Jho D, Predescu S, Standiford TJ, Malik AB. Nitric oxide stimulates macrophage inflammatory protein-2 expression in sepsis. J Immunol. 2002;169:2093–2101. doi: 10.4049/jimmunol.169.4.2093. [DOI] [PubMed] [Google Scholar]
- 43.Bar-Shai M, Carmeli E, Reznick AZ. The role of NF-κB in protein breakdown in immobilization, aging, and exercise: From basic processes to promotion of health. Ann N Y Acad Sci. 2005;1057:431–447. doi: 10.1196/annals.1356.034. [DOI] [PubMed] [Google Scholar]
- 44.Cai D, Frantz JD, Tawa NE, Jr, Melendez PA, Oh BC, Lidov HG, Hasselgren PO, Frontera WR, Lee J, Glass DJ, Shoelson SE. IKKβ/NF-κB activation causes severe muscle wasting in mice. Cell. 2004;119:285–298. doi: 10.1016/j.cell.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 45.Chung L, Ng YC. Age-related alterations in expression of apoptosis regulatory proteins and heat shock proteins in rat skeletal muscle. Biochim Biophys Acta. 2006;1762:103–109. doi: 10.1016/j.bbadis.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 46.da Costa CA, Sunyach C, Giaime E, West A, Corti O, Brice A, Safe S, Abou-Sleiman PM, Wood NW, Takahashi H, Goldberg MS, Shen J, Checler F. Transcriptional repression of p53 by parkin and impairment by mutations associated with autosomal recessive juvenile Parkinson’s disease. Nat Cell Biol. 2009;11:1370–1375. doi: 10.1038/ncb1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dajani R, Sanlioglu S, Zhang Y, Li Q, Monick MM, Lazartigues E, Eggleston T, Davisson RL, Hunninghake GW, Engelhardt JF. Pleiotropic functions of TNF-α determine distinct IKKβ-dependent hepatocellular fates in response to LPS. Am J Physiol Gastrointest Liver Physiol. 2007;292:G242–G252. doi: 10.1152/ajpgi.00043.2006. [DOI] [PubMed] [Google Scholar]
- 48.Guler R, Olleros ML, Vesin D, Parapanov R, Vesin C, Kantengwa S, Rubbia-Brandt L, Mensi N, Angelillo-Scherrer A, Martinez-Soria E, Tacchini-Cottier F, Garcia I. Inhibition of inducible nitric oxide synthase protects against liver injury induced by mycobacterial infection and endotoxins. J Hepatol. 2004;41:773–781. doi: 10.1016/j.jhep.2004.07.031. [DOI] [PubMed] [Google Scholar]
- 49.Liberatore GT, Jackson-Lewis V, Vukosavic S, Mandir AS, Vila M, McAuliffe WG, Dawson VL, Dawson TM, Przedborski S. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat Med. 1999;5:1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 50.Liu TH, Robinson EK, Helmer KS, West SD, Castaneda AA, Chang L, Mercer DW. Does upregulation of inducible nitric oxide synthase play a role in hepatic injury? Shock. 2002;18:549–554. doi: 10.1097/00024382-200212000-00011. [DOI] [PubMed] [Google Scholar]
- 51.Ogushi I, Iimuro Y, Seki E, Son G, Hirano T, Hada T, Tsutsui H, Nakanishi K, Morishita R, Kaneda Y, Fujimoto J. Nuclear factor κB decoy oligodeoxynucleotides prevent endotoxin-induced fatal liver failure in a murine model. Hepatology. 2003;38:335–344. doi: 10.1053/jhep.2003.50298. [DOI] [PubMed] [Google Scholar]
- 52.Schäfer T, Scheuer C, Roemer K, Menger MD, Vollmar B. Inhibition of p53 protects liver tissue against endotoxin-induced apoptotic and necrotic cell death. FASEB J. 2003;17:660–667. doi: 10.1096/fj.02-0774com. [DOI] [PubMed] [Google Scholar]
- 53.Schwarzkopf M, Coletti D, Sassoon D, Marazzi G. Muscle cachexia is regulated by a p53–PW1/Peg3-dependent pathway. Genes Dev. 2006;20:3440–3452. doi: 10.1101/gad.412606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kaneki M, Shimizu N, Yamada D, Chang K. Nitrosative stress and pathogenesis of insulin resistance. Antioxid Redox Signal. 2007;9:319–329. doi: 10.1089/ars.2006.1464. [DOI] [PubMed] [Google Scholar]
- 55.Kuo PC, Abe KY, Schroeder RA. Oxidative stress increases hepatocyte iNOS gene transcription and promoter activity. Biochem Biophys Res Commun. 1997;234:289–292. doi: 10.1006/bbrc.1997.6562. [DOI] [PubMed] [Google Scholar]
- 56.Nakamura T, Lipton SA. Molecular mechanisms of nitrosative stress-mediated protein misfolding in neurodegenerative diseases. Cell Mol Life Sci. 2007;64:1609–1620. doi: 10.1007/s00018-007-6525-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forrester MT, Foster MW, Stamler JS. Assessment and application of the biotin switch technique for examining protein S-nitrosylation under conditions of pharmacologically induced oxidative stress. J Biol Chem. 2007;282:13977–13983. doi: 10.1074/jbc.M609684200. [DOI] [PubMed] [Google Scholar]
- 58.Yasukawa T, Tokunaga E, Ota H, Sugita H, Martyn JA, Kaneki M. S-nitrosylation-dependent inactivation of Akt/protein kinase B in insulin resistance. J Biol Chem. 2005;280:7511–7518. doi: 10.1074/jbc.M411871200. [DOI] [PubMed] [Google Scholar]
- 59.Eklöw L, Moldéus P, Orrenius S. Oxidation of glutathione during hydroperoxide metabolism. A study using isolated hepatocytes and the glutathione reductase inhibitor 1,3-bis(2-chloroethyl)-1-nitrosourea. Eur J Biochem. 1984;138:459–463. doi: 10.1111/j.1432-1033.1984.tb07938.x. [DOI] [PubMed] [Google Scholar]
- 60.Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: A physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001;3:193–197. doi: 10.1038/35055104. [DOI] [PubMed] [Google Scholar]
- 61.Al-Ani B, Hewett PW, Ahmed S, Cudmore M, Fujisawa T, Ahmad S, Ahmed A. The release of nitric oxide from S-nitrosothiols promotes angiogenesis. PLOS One. 2006;1:e25. doi: 10.1371/journal.pone.0000025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ratnayake S, Dias IH, Lattman E, Griffiths HR. Stabilising cysteinyl thiol oxidation and nitrosation for proteomic analysis. J Proteomics. 2013;92:160–170. doi: 10.1016/j.jprot.2013.06.019. [DOI] [PubMed] [Google Scholar]
- 63.Conway ME, Yennawar N, Wallin R, Poole LB, Hutson SM. Identification of a peroxide-sensitive redox switch at the CXXC motif in the human mitochondrial branched chain aminotransferase. Biochemistry. 2002;41:9070–9078. doi: 10.1021/bi020200i. [DOI] [PubMed] [Google Scholar]
- 64.Mohr S, Hallak H, de Boitte A, Lapetina EG, Brüne B. Nitric oxide-induced S-glutathionylation and inactivation of glyceraldehyde-3-phosphate dehydrogenase. J Biol Chem. 1999;274:9427–9430. doi: 10.1074/jbc.274.14.9427. [DOI] [PubMed] [Google Scholar]
- 65.Rogers LK, Leinweber BL, Smith CV. Detection of reversible protein thiol modifications in tissues. Anal Biochem. 2006;358:171–184. doi: 10.1016/j.ab.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 66.Kostka P, Park JK. Fluorometric detection of S-nitrosothiols. Methods Enzymol. 1999;301:227–235. doi: 10.1016/s0076-6879(99)01086-1. [DOI] [PubMed] [Google Scholar]
- 67.Al-Sa’doni HH, Khan IY, Poston L, Fisher I, Ferro A. A novel family of S-nitrosothiols: Chemical synthesis and biological actions. Nitric Oxide. 2000;4:550–560. doi: 10.1006/niox.2000.0315. [DOI] [PubMed] [Google Scholar]
- 68.Freedman LP, Luisi BF, Korszun ZR, Basavappa R, Sigler PB, Yamamoto KR. The function and structure of the metal coordination sites within the glucocorticoid receptor DNA binding domain. Nature. 1988;334:543–546. doi: 10.1038/334543a0. [DOI] [PubMed] [Google Scholar]
- 69.Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y, Kaneki M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras–MAPK signaling in human cancer cells. Oncogene. 2006;25:176–185. doi: 10.1038/sj.onc.1209049. [DOI] [PubMed] [Google Scholar]
- 70.Gu W, Roeder RG. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell. 1997;90:595–606. doi: 10.1016/s0092-8674(00)80521-8. [DOI] [PubMed] [Google Scholar]
- 71.Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, Pelicci PG. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature. 2000;406:207–210. doi: 10.1038/35018127. [DOI] [PubMed] [Google Scholar]
- 72.Wang YH, Tsay YG, Tan BC, Lo WY, Lee S-C. Identification and characterization of a novel p300-mediated p53 acetylation site, lysine 305. J Biol Chem. 2003;278:25568–25576. doi: 10.1074/jbc.M212574200. [DOI] [PubMed] [Google Scholar]
- 73.Lima B, Forrester MT, Hess DT, Stamler JS. S-nitrosylation in cardiovascular signaling. Circ Res. 2010;106:633–646. doi: 10.1161/CIRCRESAHA.109.207381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schrammel A, Behrends S, Schmidt K, Koesling D, Mayer B. Characterization of 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one as a heme-site inhibitor of nitric oxide-sensitive guanylyl cyclase. Mol Pharmacol. 1996;50:1–5. [PubMed] [Google Scholar]
- 75.Khan N, Jeffers M, Kumar S, Hackett C, Boldog F, Khramtsov N, Qian X, Mills E, Berghs SC, Carey N, Finn PW, Collins LS, Tumber A, Ritchie JW, Jensen PB, Lichenstein HS, Sehested M. Determination of the class and isoform selectivity of small-molecule histone deacetylase inhibitors. Biochem J. 2008;409:581–589. doi: 10.1042/BJ20070779. [DOI] [PubMed] [Google Scholar]
- 76.Ak P, Levine AJ. p53 and NF-κB: Different strategies for responding to stress lead to a functional antagonism. FASEB J. 2010;24:3643–3652. doi: 10.1096/fj.10-160549. [DOI] [PubMed] [Google Scholar]
- 77.Sunico CR, Nakamura T, Rockenstein E, Mante M, Adame A, Chan SF, Newmeyer TF, Masliah E, Nakanishi N, Lipton SA. S-Nitrosylation of parkin as a novel regulator of p53-mediated neuronal cell death in sporadic Parkinson’s disease. Mol Neurodegen. 2013;8:29. doi: 10.1186/1750-1326-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moore WM, Webber RK, Jerome GM, Tjoeng FS, Misko TP, Currie MG. L-N6-(1-Iminoethyl)lysine: A selective inhibitor of inducible nitric oxide synthase. J Med Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- 79.Williams G, Brown T, Becker L, Prager M, Giroir BP. Cytokine-induced expression of nitric oxide synthase in C2C12 skeletal muscle myocytes. Am J Physiol. 1994;267:R1020–R1025. doi: 10.1152/ajpregu.1994.267.4.R1020. [DOI] [PubMed] [Google Scholar]
- 80.Boer R, Ulrich WR, Klein T, Mirau B, Haas S, Baur I. The inhibitory potency and selectivity of arginine substrate site nitric-oxide synthase inhibitors is solely determined by their affinity toward the different isoenzymes. Mol Pharmacol. 2000;58:1026–1034. [PubMed] [Google Scholar]
- 81.Gharavi N, El-Kadi AO. Role of nitric oxide in downregulation of cytochrome P450 1a1 and NADPH: Quinone oxidoreductase 1 by tumor necrosis factor-α and lipopolysaccharide. J Pharm Sci. 2007;96:2795–2807. doi: 10.1002/jps.20910. [DOI] [PubMed] [Google Scholar]
- 82.Doyle A, Zhang G, Abdel Fattah EA, Eissa NT, Li YP. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011;25:99–110. doi: 10.1096/fj.10-164152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Forsyth CB, Shannon KM, Kordower JH, Voigt RM, Shaikh M, Jaglin JA, Estes JD, Dodiya HB, Keshavarzian A. Increased intestinal permeability correlates with sigmoid mucosa α-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. PLOS One. 2011;6:e28032. doi: 10.1371/journal.pone.0028032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Trøseid M, Nestvold TK, Rudi K, Thoresen H, Nielsen EW, Lappegård KT. Plasma lipopolysaccharide is closely associated with glycemic control and abdominal obesity: Evidence from bariatric surgery. Diabetes Care. 2013;36:3627–3632. doi: 10.2337/dc13-0451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wiesner P, Choi SH, Almazan F, Benner C, Huang W, Diehl CJ, Gonen A, Butler S, Witztum JL, Glass CK, Miller YI. Low doses of lipopolysaccharide and minimally oxidized low-density lipoprotein cooperatively activate macrophages via nuclear factor κB and activator protein-1: Possible mechanism for acceleration of atherosclerosis by subclinical endotoxemia. Circ Res. 2010;107:56–65. doi: 10.1161/CIRCRESAHA.110.218420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Starr ME, Saito H. Sepsis in old age: Review of human and animal studies. Aging Dis. 2014;5:126–136. doi: 10.14336/AD.2014.0500126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Takaoka Y, Goto S, Nakano T, Tseng HP, Yang SM, Kawamoto S, Ono K, Chen CL. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) prevents lipopolysaccharide (LPS)-induced, sepsis-related severe acute lung injury in mice. Sci Rep. 2014;4:5204. doi: 10.1038/srep05204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.MacMicking JD, Nathan C, Hom G, Chartrain N, Fletcher DS, Trumbauer M, Stevens K, Xie QW, Sokol K, Hutchinson N, Chen H, Mudget JS. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell. 1995;81:641–650. doi: 10.1016/0092-8674(95)90085-3. [DOI] [PubMed] [Google Scholar]
- 89.Ghosh A, Roy A, Liu X, Kordower JH, Mufson EJ, Hartley DM, Ghosh S, Mosley RL, Gendelman HE, Pahan K. Selective inhibition of NF-κB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104:18754–18759. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Novikova L, Garris BL, Garris DR, Lau YS. Early signs of neuronal apoptosis in the substantia nigra pars compacta of the progressive neurodegenerative mouse 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid model of Parkinson’s disease. Neuroscience. 2006;140:67–76. doi: 10.1016/j.neuroscience.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 91.Mitkovski M, Padovan-Neto FE, Raisman-Vozari R, Ginestet L, da-Silva CA, Del-Bel EA. Investigations into potential extrasynaptic communication between the dopaminergic and nitrergic systems. Front Physiol. 2012;3:372. doi: 10.3389/fphys.2012.00372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gonzá-Hernández T, Rodríguez M. Compartmental organization and chemical profile of dopaminergic and GABAergic neurons in the substantia nigra of the rat. J Comp Neurol. 2000;421:107–135. doi: 10.1002/(sici)1096-9861(20000522)421:1<107::aid-cne7>3.3.co;2-6. [DOI] [PubMed] [Google Scholar]
- 93.Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7:615–624. doi: 10.1016/0896-6273(91)90374-9. [DOI] [PubMed] [Google Scholar]
- 94.Gow AJ, Chen Q, Hess DT, Day BJ, Ischiropoulos H, Stamler JS. Basal and stimulated protein S-nitrosylation in multiple cell types and tissues. J Biol Chem. 2002;277:9637–9640. doi: 10.1074/jbc.C100746200. [DOI] [PubMed] [Google Scholar]
- 95.Murad F. The nitric oxide-cyclic GMP signal transduction system for intracellular and intercellular communication. Recent Prog Horm Res. 1994;49:239–248. doi: 10.1016/b978-0-12-571149-4.50016-7. [DOI] [PubMed] [Google Scholar]
- 96.Suda T, Takahashi T, Golstein P, Nagata S. Molecular cloning and expression of the Fas ligand, a novel member of the tumor necrosis factor family. Cell. 1993;75:1169–1178. doi: 10.1016/0092-8674(93)90326-l. [DOI] [PubMed] [Google Scholar]
- 97.Dodd SL, Hain B, Senf SM, Judge AR. Hsp27 inhibits IKKβ-induced NF-κB activity and skeletal muscle atrophy. FASEB J. 2009;23:3415–3423. doi: 10.1096/fj.08-124602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McCann SM, Mastronardi C, de Laurentiis A, Rettori V. The nitric oxide theory of aging revisited. Ann N Y Acad Sci. 2005;1057:64–84. doi: 10.1196/annals.1356.064. [DOI] [PubMed] [Google Scholar]
- 99.Nakamura T, Tu S, Akhtar MW, Sunico CR, Okamoto S, Lipton SA. Aberrant protein S-nitrosylation in neurodegenerative diseases. Neuron. 2013;78:596–614. doi: 10.1016/j.neuron.2013.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Santhanam L, Lim HK, Lim HK, Miriel V, Brown T, Patel M, Balanson S, Ryoo S, Anderson M, Irani K, Khanday F, Di Costanzo L, Nyhan D, Hare JM, Christianson DW, Rivers R, Shoukas A, Berkowitz DE. Inducible NO synthase–dependent S-nitrosylation and activation of arginase1 contribute to age-related endothelial dysfunction. Circ Res. 2007;101:692–702. doi: 10.1161/CIRCRESAHA.107.157727. [DOI] [PubMed] [Google Scholar]
- 101.Ronco MT, de Alvarez ML, Monti JA, Carrillo MC, Pisani GB, Lugano MC, Carnovale CE. Role of nitric oxide increase on induced programmed cell death during early stages of rat liver regeneration. Biochim Biophys Acta. 2004;1690:70–76. doi: 10.1016/j.bbadis.2004.05.004. [DOI] [PubMed] [Google Scholar]
- 102.Yakovlev VA, Barani IJ, Rabender CS, Black SM, Leach JK, Graves PR, Kellogg GE, Mikkelsen RB. Tyrosine nitration of IκBα: A novel mechanism for NF-κB activation. Biochemistry. 2007;46:11671–11683. doi: 10.1021/bi701107z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zheng Y, Gardner SE, Clarke MC. Cell death, damage-associated molecular patterns, and sterile inflammation in cardiovascular disease. Arterioscler Thromb Vasc Biol. 2011;31:2781–2786. doi: 10.1161/ATVBAHA.111.224907. [DOI] [PubMed] [Google Scholar]
- 104.Yamada M, Kida K, Amutuhaire W, Ichinose F, Kaneki M. Gene disruption of caspase-3 prevents MPTP-induced Parkinson’s disease in mice. Biochem Biophys Res Commun. 2010;402:312–318. doi: 10.1016/j.bbrc.2010.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kuncewicz T, Balakrishnan P, Snuggs MB, Kone BC. Specific association of nitric oxide synthase-2 with Rac isoforms in activated murine macrophages. Am J Physiol Renal Physiol. 2001;281:F326–F336. doi: 10.1152/ajprenal.2001.281.2.F326. [DOI] [PubMed] [Google Scholar]
- 106.Shinozaki S, Choi CS, Shimizu N, Yamada M, Kim M, Zhang T, Shiota G, Dong HH, Kim YB, Kaneki M. Liver-specific inducible nitric-oxide synthase expression is sufficient to cause hepatic insulin resistance and mild hyperglycemia in mice. J Biol Chem. 2011;286:34959–34975. doi: 10.1074/jbc.M110.187666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- 108.Tanny JC, Moazed D. Coupling of histone deacetylation to NAD breakdown by the yeast silencing protein Sir2: Evidence for acetyl transfer from substrate to an NAD breakdown product. Proc Natl Acad Sci USA. 2001;98:415–420. doi: 10.1073/pnas.031563798. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Mutation of the CXXC motif in SIRT1 abolishes its ADP-ribosyltransferase activity, and SNAP decreases SIRT1 deacetylase activity.
Fig. S2. S-Nitrosylation is a major thiol modification of SIRT1 in cultured cells exposed to SNAP and carmustine.
Fig. S3. The specific detection of SIRT1 S-nitrosylation is confirmed by the effect of pre-incubation with HgCl2 and by ascorbate-dependent biotinylation.
Fig. S4. TPEN blocks reactivation of NO donor–exposed recombinant SIRT1 by DTT.
Fig. S5. SIRT1M3 decreases cell viability.
Fig. S6. NO donors increase the abundance of acetylated p53.
Fig. S7. GSNO and SNAP decrease viable cell numbers in a p53-dependent manner.
Fig. S8. GSNO induces p53 acetylation and decreases viable cell numbers independent of cGMP.
Fig. S9. Inhibition of iNOS prevents the ability of inflammatory molecules or iNOS overexpression to induce S-nitrosylation of SIRT1 and acetylation of p53.
Fig. S10. Inhibition of iNOS prevents the ability of inflammatory molecules to induce acetylation of p65.
Fig. S11. iNOS deficiency prevents the ability of LPS to induce acetylation and activation of p53 and p65 in mouse liver.
Fig. S12. Inhibition of nNOS prevents the ability of MPTP to induce acetylation and activation of p53 and p65 in the nigrastriatum of MPTP-injected mice.
Fig. S13. Acetylation and activation of p53 and p65 are increased in skeletal muscle of aged rats compared to young rats.
Fig. S14. Inhibition of iNOS decreases acetylation and activation of p53 and p65 in skeletal muscle of aged rats.