

Abstract
Background
Rapid diagnostic tests (RDTs) detecting histidine-rich protein 2 (PfHRP2) antigen are used to identify individuals with Plasmodium falciparum infection even in low transmission settings seeking to achieve elimination. However, these RDTs lack sensitivity to detect low-density infections, produce false negatives for P. falciparum strains lacking pfhrp2 gene and do not detect species other than P. falciparum.
Methods
Results of a PfHRP2-based RDT and Plasmodium nested PCR were compared in a region of declining malaria transmission in southern Zambia using samples from community-based, cross-sectional surveys from 2008 to 2012. Participants were tested with a PfHRP2-based RDT and a finger prick blood sample was spotted onto filter paper for PCR analysis and used to prepare blood smears for microscopy. Species-specific, real-time, quantitative PCR (q-PCR) was performed on samples that tested positive either by microscopy, RDT or nested PCR.
Results
Of 3,292 total participants enrolled, 12 (0.4%) tested positive by microscopy and 42 (1.3%) by RDT. Of 3,213 (98%) samples tested by nested PCR, 57 (1.8%) were positive, resulting in 87 participants positive by at least one of the three tests. Of these, 61 tested positive for P. falciparum by q-PCR with copy numbers ≤ 2 x 103 copies/μL, 5 were positive for both P. falciparum and Plasmodium malariae and 2 were positive for P. malariae alone. RDT detected 32 (53%) of P. falciparum positives, failing to detect three of the dual infections with P. malariae. Among 2,975 participants enrolled during a low transmission period between 2009 and 2012, sensitivity of the PfHRP2-based RDT compared to nested PCR was only 17%, with specificity of >99%. The pfhrp gene was detected in 80% of P. falciparum positives; however, comparison of copy number between RDT negative and RDT positive samples suggested that RDT negatives resulted from low parasitaemia and not pfhrp2 gene deletion.
Conclusions
Low-density P. falciparum infections not identified by currently used PfHRP2-based RDTs and the inability to detect non-falciparum malaria will hinder progress to further reduce malaria in low transmission settings of Zambia. More sensitive and specific diagnostic tests will likely be necessary to identify parasite reservoirs and achieve malaria elimination.
Electronic supplementary material
The online version of this article (doi:10.1186/s12936-015-0544-3) contains supplementary material, which is available to authorized users.
Keywords: PfHRP2-RDT, Nested PCR, Quantitative PCR, Malaria, Elimination, Zambia
Background
A substantial reduction in the burden of malaria has been achieved in several countries of sub-Saharan Africa [1]. Sustaining this level of malaria control and progressing towards elimination will depend on sensitive and specific diagnostic tools to identify persistent reservoirs of infection. Although microscopy remains the diagnostic gold standard, malaria rapid diagnostic tests (RDTs) are widely used throughout malaria endemic areas, particularly in rural settings where access to health facilities with trained microscopists is limited. As transmission declines, detection of infected individuals is critical to achieving and sustaining control. Evaluation of RDT performance in regions of low transmission is necessary.
Zambia has recorded a significant decrease in parasite prevalence as documented by malaria indicator surveys, with parasite prevalence among children younger than five years of age decreasing from 21.8% in 2006 to 14.9% in 2012 [2]. Southern Province, Zambia reported a parasite prevalence of less than 10% and is considered a potential area for malaria elimination. RDTs based on detecting histidine-rich protein 2 (PfHRP2), an antigen produced only by Plasmodium falciparum, have been used in Zambia since their national introduction in 2009 [3]. However, RDTs lack the sensitivity to detect low-density infections compared to more sensitive molecular methods such as polymerase chain reaction (PCR) [4-6]. Furthermore, P. falciparum parasites with a deletion of the pfhrp2 gene can cause patent bloodstream infection but false negative RDT results [7-11]. Identification of other human malaria parasite species is not possible with PfHRP2-based RDTs and non-falciparum malaria may become more apparent as transmission decreases. To assess the validity of PfHRP2-based RDTs in a hypoendemic area moving towards elimination, results of a PfHRP2-based RDT were compared to those of a Plasmodium nested PCR. Using real-time, quantitative PCR (q-PCR), identification of parasite species, quantification of parasitaemia and potential pfhrp gene deletions were assessed. This is the first study to examine the presence of non-P.falciparum species and pfhrp gene deletion in southern Zambia and the findings have implications for RDT-based strategies to achieve malaria elimination in this region.
Methods
The study consisted of community-based, cross-sectional surveys conducted in the catchment area of Macha Hospital located in Choma District, Southern Province, Zambia between 2008 and 2012. Macha Hospital is located approximately 70 km from the nearest town of Choma on a plateau at an altitude of approximately 1,100 metres above sea level and in a habitat characterised as Miombo woodland. There is a single rainy season from approximately November through April, followed by a cool, dry season from April to August and a hot, dry season from August to November. The catchment area is populated by traditional villagers living in small scattered homesteads. Anopheles arabiensis is the primary vector responsible for malaria transmission [12], which peaks during the rainy season. The Southern Province of Zambia historically had hyperendemic P. falciparum transmission but the parasite prevalence and number of hospitalizations for malaria declined dramatically over the past decade [13].
Cross-sectional surveys were conducted every other month beginning February 2008. Household and participant selection was as described previously [14], but in brief, households were randomly selected from satellite images and all household residents were eligible to participate. Informed consent was obtained from participating adults and from parents or guardians of children younger than 16 years. Questionnaires were administered to collect information on age, sex and history of recent malaria and treatment. Finger prick blood samples were used to prepare thick and thin blood smears for microscopy, RDT (ICT Diagnostics, Cape Town, South Africa) and spotted onto filter paper (Whatman 903™ Protein Saver card) as dried blood spots (DBS). The ICT Diagnostics RDT detects PfHRP2 antigen. Blood smear and DBS samples were transported to a molecular laboratory at Macha Research Trust, located within the study area, where both microscopy and Plasmodium nested PCR analysis were performed. Storage of DBS was at −20°C, with each filter paper card individually sealed in a plastic bag containing desiccant prior to nucleic acid extraction and PCR analysis. DBS collected from February to September 2008 were initially stored at room temperature (within individually sealed plastic bags with desiccant) but subsequently stored at −20°C.
Microscopic examination was performed at the Macha Research Trust laboratory within approximately three days of sample collection. Thin blood smears were air-dried, fixed with methanol and Giemsa-stained whereas thick blood smears were air dried and then Giemsa-stained without fixing with methanol. Parasitological diagnosis was made independently by two microscopists, with discrepancies resolved by a third reader. Parasite count was recorded per 1,000 white blood cells.
All laboratory assays except q-PCR were performed at the Macha Research Trust laboratory in Zambia. A Chelex© extraction method was used to recover parasite DNA from dried blood spots [15] within approximately one year of sample collection for DBS collected from 2010 to 2012 and approximately three to five years for DBS collected in 2008 and 2009. Positive and negative control samples spotted as dried blood on filter paper were included in each extraction experiment. Positive controls consisted of parasitized blood from laboratory cultures at 1,000 parasites/μL. Negative controls consisted of blood from individuals with no travel history to malaria endemic areas. The dried blood spots were placed in 1.5 mL microcentrifuge tubes, 1 mL of 0.1% weight by volume saponin in 1x phosphate buffered saline (PBS) was added and the mixture was incubated for 10 minutes at room temperature. The tubes were centrifuged for two minutes at 14,000 rpm, the supernatant discarded and 1 mL of 1 × PBS was added. The tubes were again centrifuged for 2 minutes at 14,000 rpm, the supernatant discarded and 150 μL of 2% weight by volume Chelex© solution and 50 μl of DNase free water were added and the tubes boiled for 8 minutes. The tubes were then centrifuged for one minute at 14,000 rpm and approximately 150 μL of DNA was stored at −20°C.
A Plasmodium nested PCR assay was performed within approximately one month following the DNA extraction for detection of asexual stage parasite DNA using two sets of primers targeting a segment of the mitochondrial cytochrome b gene (cytb) conserved in the four major human Plasmodium parasites [16]. In the primary PCR step, 6 μL of DNA extract was pipetted into 0.2 mL tubes containing a 19 μL reaction mix made up of DNase free water and final concentrations of dNTPs, 10× PCR buffer, magnesium chloride, forward and reverse primers and DNA Taq polymerase as 0.2 mΜ, 1×, 3 mΜ, 1 μΜ and 2 units in 25 μL reaction mix, respectively. In the nested PCR step, 3 μL of the primary PCR product was added to 0.2 mL PCR tubes containing 22 μL of reaction mix containing DNase free water and final concentrations of dNTPs, 10× buffer, magnesium chloride, forward and reverse primers and Taq DNA polymerase as 0.2 mΜ, 1×, 2.5 mΜ, 1 μΜ and 2 units in 25 μL reaction mix, respectively. No-template controls were included in each experiment and reactions were run in a Techne™ TC-412 thermo cycler (See Additional file 1). Amplified product was detected by electrophoresis on 1% agarose gel and viewed under UV light as an 815 base pair DNA band.
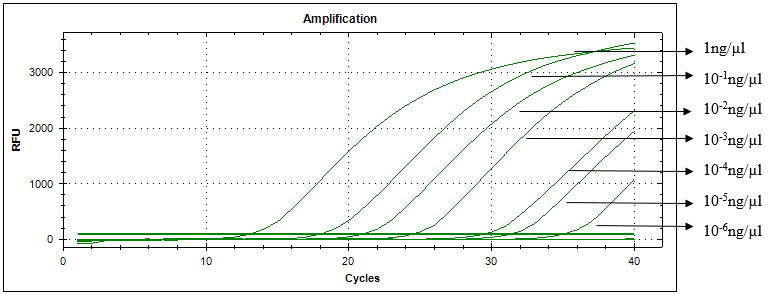
DNA extracts from individuals positive by either microscopy, RDT or nested PCR were shipped with ice packs to the United States and stored at −20°C prior to further analyses by q-PCR. P. falciparum, P. malariae and pfhrp gene detection by q-PCR using specific primer sets was performed at the Johns Hopkins Bloomberg School of Public Health between July and October 2013. Amplification was demonstrated to require ten-fold dilution with nuclease free water to minimize inhibition of the q-PCR reaction due to substrate in the crude Chelex DNA extract. Primers for species-specific cytb were used to detect P. falciparum and P. malariae. The primers used for the detection of the pfhrp gene were designed to amplify the highly conserved secretary leader of pfhrp as previously reported [17]. The primer was designed to confirm the presence of pfhrp2 or pfhrp3 in a single reaction, with expected product sizes of 278 base pairs and 259 base pairs for pfhrp2 and pfhrp3, respectively (See Additional file 1). Five μL of reaction mix containing 5 μL of 2x iQ™ SYBR® Green supermix (Bio-Rad) and 400 nΜ primer were added in duplicate to a 384-well plate along with 5 μL of ten-fold diluted DNA extract. Standards were generated by serial dilution of genomic DNA. Ten-fold dilutions of laboratory-cultured 3D7 genomic DNA were used as standards for P. falciparum and pfhrp gene detection (See Additional file 2), whereas P. malariae genomic DNA from a single infected individual was used as a standard for detection of P. malariae. The loaded plates were centrifuged for 5 minutes at 2,000 rpm and reactions were run in a Bio-Rad CFX384™ real time thermo cycler. Baseline thresholds were uniform for all experiments of the same assay and cycle count values falling within the range set by the standards were used to determine positive results for each q-PCR assay. Absolute quantification was used to determine gene copy numbers for the P. falciparum and pfhrp q-PCR assays based on the standard curve generated from the 3D7 dilutions (See Additional file 3).
Results
A total of 3,292 study participants were enrolled from 2008 through 2012 (See Additional file 4). Of these 3,292 participants, 12 (0.4%) were positive by microscopy and 42 (1.3%) were positive by PfHRP2-based RDT. Nested PCR was performed on 3,213 (98%) samples and 57 (1.8%) positive individuals were identified. In 2008, nested PCR was performed on 276 (87%) of the 317 samples collected; however, the prevalence of nested PCR positivity (3.6%) was lower than RDT positivity (10%) presumably as a consequence of long storage time. Hence, results from 2008 were not included in the comparisons between RDT and nested PCR as potential nested PCR positive but RDT and microscopy negative samples may have been missed. Consequently, 2,975 samples were used for comparison of RDT and nested PCR for the study period 2009 to 2012.
Parasite prevalence by microscopy, RDT and nested PCR from 2009 to 2012
Of the 2,975 participants enrolled in the cross sectional surveys during the four-year period of low transmission from 2009 through 2012, only four individuals were positive for malaria by microscopy (0.13%) and ten by RDT (0.34%). Of the 2,937 (99%) samples tested by nested PCR, 47 were positive (1.6%). The parasite prevalence by RDT was 0.74% in 2009, 0.23% in 2010, 0.4% in 2011 and 0% in 2012, whereas the parasite prevalence by nested PCR was 2.7% in 2009, 1.8% in 2010, 1.5% in 2011 and 0.44% in 2012 (Figure 1). The sensitivity of the PfHRP2-based RDT compared to the nested PCR was only 17% (Table 1). In this study setting in which all household residents were tested regardless of symptoms, in contrast to a clinical setting where the prevalence of malaria would be higher, the positive predictive value of the RDT compared to nested PCR was 80%, with a negative predictive value of 99%.
Figure 1.
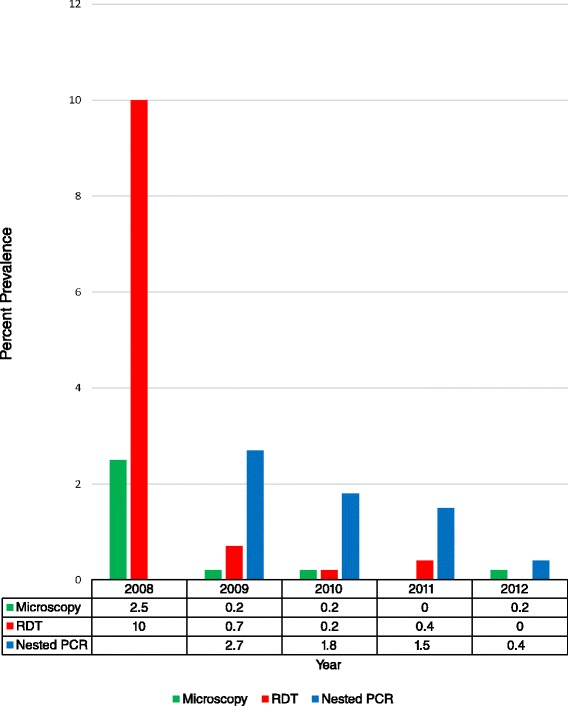
Parasite prevalence as measured by microscopy, Pf HRP2-RDT and Plasmodium nested PCR. Prevalence by microscopy and ICT Diagnostics RDT per year on a total of 3, 292 participants and by nested PCR on 3,213 (98%) participants enrolled between 2008 and 2012. *PCR data for 2008 excluded.
Table 1.
Comparison of Pf HRP2-based RDT results with microscopy and nested PCR from 2009 – 2012
| Microscopy positive | Microscopy negative | Total | PCR positive | PCR negative | Total | |
|---|---|---|---|---|---|---|
| RDT positive | 2 | 8 | 10 | 8 | 2 | 10 |
| RDT negative | 2 | 3280 | 3282 | 39 | 2888 | 2927 |
| Total | 4 | 3288 | 3292 | 47 | 2890 | 2937* |
*Of the 2,975 enrolled during this period, 2,937 (99%) were tested by both RDT and nested PCR.
Sensitivity of PfHRP2-based RDT compared to microscopy = 0.50.
Specificity of PfHRP2-based RDT compared to microscopy = > 0.99.
Positive predictive value of PfHRP2-based RDT compared to microscopy = 20%.
Negative predictive value of PfHRP2-based RDT compared to microscopy = > 99%.
Sensitivity of PfHRP2-based RDT compared to PCR = 0.17.
Specificity of PfHRP2-based RDT compared to PCR = > 0.99.
Positive predictive value of PfHRP2-based RDT compared to PCR = 80%.
Negative predictive value of PfHRP2-based RDT compared to PCR = 99%.
Identification of Plasmodium species
The q-PCR for P. falciparum and P. malariae was performed on 49 samples collected from 2009 through 2012 that were positive for malaria by RDT, microscopy or nested PCR as well as an additional 38 samples collected during 2008 and positive by at least one of the three tests, yielding a total of 87 samples on which q-PCR was performed.
Of these 87 samples, 56 (64%) were positive by q-PCR for P. falciparum alone, five were positive for both P. falciparum and P. malariae and two were positive for P. malariae alone (Table 2). Thus, 61 (70%) were positive for P. falciparum and seven for P. malariae (0.2% of all participants). The q-PCR was negative for 24 individuals who tested positive by either microscopy, RDT or nested PCR. Of these 24, one was positive by microscopy only, eight were positive by RDT only, thirteen were positive by nested PCR only, and two were positive by both RDT and nested PCR.
Table 2.
Plasmodium species identification by q-PCR
| P. falciparum | P. malariae | P. falciparum and P. malariae | |
|---|---|---|---|
| Total positive by q-PCR* | 56 | 2 | 5 |
| Also positive by microscopy | 10 | 0 | 1 |
| Also positive by RDT | 30 | 0 | 2 |
| Also positive by nested PCR | 37 | 2 | 3 |
*Of the 87 tested samples that were positive by RDT, microscopy or PCR, 24 were negative by q-PCR.
Of the 42 individuals positive by RDT from 2008 through 2012, 32 were positive for P. falciparum by q-PCR. Eight of the 10 individuals positive by RDT but negative by q-PCR were also negative by microscopy and nested PCR, suggesting these individuals had persistent PfHRP2 antigenaemia in the absence of visible parasites or parasite DNA. Only one of these eight individuals reported prior treatment, but with an herbal medicine and not artemisinin combination therapy. The remaining two samples were positive by nested PCR, which suggested either false positive results for both RDT and nested PCR or false negative q-PCR. Of the 12 individuals positive by microscopy, only one was negative by q-PCR, suggestive of either false positive microscopy or false negative q-PCR.
Quantification of P. falciparum parasitaemia
The P. falciparum genome size is 23 mega bases [18] and on the assumption that the average weight of one mole of a base pair is 650 g, 1 ng of parasite genomic DNA corresponds to 40,000 copies of the target gene. The limit of detection for P. falciparum q-PCR was determined to be 0.04 copies/μL. The cytb gene copy numbers quantified by q-PCR for the 61 samples positive for P. falciparum ranged from 1 × 10−1 copies/μL to 2 × 103 copies/μL. The mean copy number for individuals positive by both RDT and P. falciparum cytb q-PCR was 97 copies/μL (range 0.05 to 2178 copies/μL, SD = 381) in contrast to a mean copy number of 8 copies/μL (range 0.1 to 96 copies/μL, SD = 18) for individuals who were RDT negative but P. falciparum q-PCR positive (Table 3).
Table 3.
Pfcytb and Pfhrp copy numbers in individuals positive for P. falciparum by q-PCR
| P. falciparum q-PCR positive | Pfhrp q-PCR result (N) | Pfcytb mean copy number/μL | Pfhrp mean copy number/μL | |
|---|---|---|---|---|
| RDT positive | 32 | positive (26) | 97 | 1827 |
| negative (6) | NA | |||
| RDT negative | 29 | positive (23) | 8 | 237 |
| negative (6) | NA |
NA = not amplified.
Detection of pfhrp gene in P. falciparum infections
The limit of detection for the pfhrp gene by q-PCR was 4 copies/μL. The pfhrp gene was present in 49 (80%) of the 61 samples confirmed to have P. falciparum DNA by q-PCR. Of the 32 samples positive by both RDT and q-PCR, the pfhrp gene was detectable in 26 (81%). Of the 29 RDT-negative but q-PCR positive samples for P. falciparum, the pfhrp gene was detected in 23 (79%). The mean copy number of Pfcytb q-PCR was approximately 13-fold lower among RDT negative samples compared to RDT positive samples. Similarly, the mean pfhrp copy number was approximately 7-fold lower among RDT negative samples compared to RDT positive samples (Table 3).
Discussion
In a region of declining malaria transmission in southern Zambia, a PfHRP2-based RDT had low sensitivity in detecting P. falciparum infections and failed to detect a small number of mixed infections with P. malariae. Failure to detect the pfhrp gene in approximately 20% of infections with P. falciparum was likely a consequence of low parasite density rather than deletion of the pfhrp gene. Although the absolute prevalence of malaria was low, PCR detected four to nine times the number of infections than the PfHRP2-based RDT.
Field evaluations of PfHRP2-based RDTs have reported high sensitivities in medium to high malaria transmission settings [19,20] where parasite densities commonly exceed 200 parasite/μL. However, recent studies conducted in pre-elimination settings in sub-Saharan Africa have shown a reduction in sensitivity of RDTs for malaria detection. The sensitivity of RDT compared to PCR in Zanzibar was 76.5% [21], higher than observed in Zambia. In regions of declining transmission, this may reflect low-level parasitaemia in individuals with clinical immunity acquired when transmission was higher. Incorporation of more sensitive diagnostics may be necessary in such settings to eliminate the residual infectious reservoir, consistent with recent recommendations by the WHO Malaria Policy Advisory Committee on the role of nucleic acid-based malaria diagnosis in low transmission settings [22]. If transmission remains low for an extended period, clinical immunity may be lost, resulting in more frequent symptomatic infections with higher levels of parasitaemia and increased sensitivity of RDTs.
Non-falciparum Plasmodium species have previously been described in Zambia although they constitute a small proportion of infections. Infection with Plasmodium ovale was described in the 1960’s [23] and infection with P. malariae was estimated to comprise 2-4% of malaria infections in Zambia. In 2012, the prevalence of infection with P. malariae was 2.1% in Nchelenge District in northern Zambia [24].
Although P. falciparum not expressing PfHRP2 has been reported from some regions [25], the pfhrp2 gene was present in the majority of the circulating P. falciparum parasites in southern Zambia. This is the first report of prevalence of pfhrp2 in circulating P. falciparum parasites in Zambia and continued monitoring for the occurrence of pfhrp gene deletions will be necessary as PfHRP2-based RDTs continue to be used.
Conclusions
Current efforts to achieve malaria elimination in southern Zambia using reactive case detection based on RDTs may be insufficiently sensitive to interrupt transmission. Alternative strategies, such as focal drug administration or the use of more sensitive and specific diagnostic tests, may be necessary to eliminate the parasite reservoir.
Acknowledgements
NL gratefully acknowledges Fogarty Training Grant (7D43TW001587-08) awarded to Dr. Nirbhay Kumar which funded her work in the laboratory of Dr. David Sullivan at The Johns Hopkins Bloomberg School of Public Health. This work was supported by the Johns Hopkins Malaria Research Institute, the Bloomberg Family Foundation and the Division of Microbiology and Infectious Diseases, National Institutes of Allergies and Infectious Diseases, National Institutes of Health as part of the International Centers of Excellence for Malaria Research (U19 AI089680). Thanks are also due to the field team and laboratory staff at Macha Research Trust and most importantly to participants from the Macha community who provided the data for this study.
Additional files
Primer sequences and reaction conditions for PCR assays. Amplification of asexual-stage Plasmodium parasite used primers targeting a region of the mitochondrial cytochrome b (cytb) gene conserved in the four major human malaria parasites. Identification of species and detection of pfhrp gene were based on species-specific cytb primers and primers amplifying a highly conserved secretary leader of the pfhrp gene respectively.
{kind=link}
Amplification curve for Plasmodium falciparum q-PCR. Q-PCR amplification curve for P. falciparum detection was generated by Bio-Rad CFX-384 Thermocycler using ten-fold dilutions of 3D7 genomic DNA from a 1 nanogram stock.
{kind=link}
Standard curve for absolute quantification of Plasmodium falciparum parasitaemia. Quantification was based on 1 nanogram of P. falciparum genomic DNA corresponding to 40,000 target gene copy number.
Number of samples tested by Microscopy, RDT, nested PCR and the q-PCR per study year.
Footnotes
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
NL carried out the molecular analysis, analysed data and drafted the manuscript. TK and DS analysed data and reviewed the manuscript. HH coordinated field sample collection. SM and PET supervised field and laboratory data collection. CS obtained Fogarty Training Grant funding and reviewed the manuscript. WJM set up the project, participated in study design and drafted the manuscript. All authors read and approved the manuscript.
Contributor Information
Natasha M Laban, Email: natasha.laban@macharesearch.org.
Tamaki Kobayashi, Email: tkobaya2@jhu.edu.
Harry Hamapumbu, Email: harry.hamapumbu@macharesearch.org.
David Sullivan, Email: dsulliv7@jhmi.edu.
Sungano Mharakurwa, Email: sungano.mharakurwa@macharesearch.org.
Philip E Thuma, Email: phil.thuma@macharesearch.org.
Clive J Shiff, Email: cshiff1@jhu.edu.
William J Moss, Email: wmoss1@jhu.edu.
References
- 1.O’Meara WP, Mangeni JN, Steketee R, Greenwood B. Changes in the burden of malaria in sub-Saharan Africa. Lancet Infect Dis. 2010;10:545–55. doi: 10.1016/S1473-3099(10)70096-7. [DOI] [PubMed] [Google Scholar]
- 2.Government of the Republic of Zambia MoH . Zambia National Malaria Indicator Survey 2012. 2013. [Google Scholar]
- 3.Yukich JO, Bennett A, Albertini A, Incardona S, Moonga H, Chisha Z, et al. Reductions in artemisinin-based combination therapy consumption after the nationwide scale up of routine malaria rapid diagnostic testing in Zambia. Am J Trop Med Hyg. 2012;87:437–46. doi: 10.4269/ajtmh.2012.12-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harris I, Sharrock WW, Bain LM, Gray KA, Bobogare A, Boaz L, et al. A large proportion of asymptomatic Plasmodium infections with low and sub-microscopic parasite densities in the low transmission setting of Temotu Province, Solomon Islands: challenges for malaria diagnostics in an elimination setting. Malar J. 2010;9:254. doi: 10.1186/1475-2875-9-254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golassa L, Enweji N, Erko B, Aseffa A, Swedberg G. Detection of a substantial number of sub-microscopic Plasmodium falciparum infections by polymerase chain reaction: a potential threat to malaria control and diagnosis in Ethiopia. Malar J. 2013;12:352. doi: 10.1186/1475-2875-12-352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McMorrow ML, Aidoo M, Kachur SP. Malaria rapid diagnostic tests in elimination settings–can they find the last parasite? Clin Microbiol Infect. 2011;17:1624–31. doi: 10.1111/j.1469-0691.2011.03639.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Koita OA, Doumbo OK, Ouattara A, Tall LK, Konare A, Diakite M, et al. False-negative rapid diagnostic tests for malaria and deletion of the histidine-rich repeat region of the hrp2 gene. Am J Trop Med Hyg. 2012;86:194–8. doi: 10.4269/ajtmh.2012.10-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kumar N, Pande V, Bhatt RM, Shah NK, Mishra N, Srivastava B, et al. Genetic deletion of HRP2 and HRP3 in Indian Plasmodium falciparum population and false negative malaria rapid diagnostic test. Acta Trop. 2013;125:119–21. doi: 10.1016/j.actatropica.2012.09.015. [DOI] [PubMed] [Google Scholar]
- 9.Maltha J, Gamboa D, Bendezu J, Sanchez L, Cnops L, Gillet P, et al. Rapid diagnostic tests for malaria diagnosis in the Peruvian Amazon: impact of pfhrp2 gene deletions and cross-reactions. PLoS One. 2012;7:e43094. doi: 10.1371/journal.pone.0043094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gamboa D, Ho MF, Bendezu J, Torres K, Chiodini PL, Barnwell JW, et al. A large proportion of P. falciparum isolates in the Amazon region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS One. 2010;5:e8091. doi: 10.1371/journal.pone.0008091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Houze S, Hubert V, Le Pessec G, Le Bras J, Clain J. Combined deletions of pfhrp2 and pfhrp3 genes result in Plasmodium falciparum malaria false-negative rapid diagnostic test. J Clin Microbiol. 2011;49:2694–6. doi: 10.1128/JCM.00281-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kent RJ, Thuma PE, Mharakurwa S, Norris DE. Seasonality, blood feeding behavior, and transmission of Plasmodium falciparum by Anopheles arabiensis after an extended drought in southern Zambia. Am J Trop Med Hyg. 2007;76:267–74. [PMC free article] [PubMed] [Google Scholar]
- 13.Moss WJ, Norris DE, Mharakurwa S, Scott A, Mulenga M, Mason PR, et al. Challenges and prospects for malaria elimination in the Southern Africa region. Acta Trop. 2012;121:207–11. doi: 10.1016/j.actatropica.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sutcliffe CG, Kobayashi T, Hamapumbu H, Shields T, Kamanga A, Mharakurwa S, et al. Changing individual-level risk factors for malaria with declining transmission in southern Zambia: a cross-sectional study. Malar J. 2011;10:324. doi: 10.1186/1475-2875-10-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kain KC, Lanar DE. Determination of genetic variation within Plasmodium falciparum by using enzymatically amplified DNA from filter paper disks impregnated with whole blood. J Clin Microbiol. 1991;29:1171–4. doi: 10.1128/jcm.29.6.1171-1174.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steenkeste N, Incardona S, Chy S, Duval L, Ekala MT, Lim P, et al. Towards high-throughput molecular detection of Plasmodium: new approaches and molecular markers. Malar J. 2009;8:86. doi: 10.1186/1475-2875-8-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan DJ, Jr, Ayala YM, Goldberg DE. An unexpected 5’ untranslated intron in the P. falciparum genes for histidine-rich proteins II and III. Mol Biochem Parasitol. 1996;83:247–51. doi: 10.1016/S0166-6851(96)02768-5. [DOI] [PubMed] [Google Scholar]
- 18.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moonasar D, Goga AE, Kruger PS, La Cock C, Maharaj R, Frean J, et al. Field evaluation of a malaria rapid diagnostic test (ICT Pf) S Afr Med J. 2009;99:810–3. [PubMed] [Google Scholar]
- 20.Hopkins H, Bebell L, Kambale W, Dokomajilar C, Rosenthal PJ, Dorsey G. Rapid diagnostic tests for malaria at sites of varying transmission intensity in Uganda. J Infect Dis. 2008;197:510–8. doi: 10.1086/526502. [DOI] [PubMed] [Google Scholar]
- 21.Shakely D, Elfving K, Aydin-Schmidt B, Msellem MI, Morris U, Omar R, et al. The usefulness of rapid diagnostic tests in the new context of low malaria transmission in Zanzibar. PLoS One. 2013;8:e72912. doi: 10.1371/journal.pone.0072912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.WHO . Recommendation for malaria diagnostics in low transmission settings. Geneva: World Health Organization; 2014. [Google Scholar]
- 23.Wolfe HL. Plasmodium ovale in Zambia. Bull World Health Organ. 1968;39:947–8. [PMC free article] [PubMed] [Google Scholar]
- 24.Nambozi M, Malunga P, Mulenga M, Van Geertruyden JP, D’Alessandro U. Defining the malaria burden in Nchelenge District, northern Zambia using the World Health Organization malaria indicators survey. Malar J. 2014;13:220. doi: 10.1186/1475-2875-13-220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheng Q, Gatton ML, Barnwell J, Chiodini P, McCarthy J, Bell D, et al. Plasmodium falciparum parasites lacking histidine-rich protein 2 and 3: a review and recommendations for accurate reporting. Malar J. 2014;13:283. doi: 10.1186/1475-2875-13-283. [DOI] [PMC free article] [PubMed] [Google Scholar]