

Abstract
Background
We recently expressed a potent and noncytotoxic short hairpin (sh)RNA directed against chemokine (c-c motif) receptor 5 (CCR5) using lentiviral mediated transduction of CD34+ hematopoietic progenitor cells (HPCs) and demonstrated the stable reduction of CCR5 expression in T-lymphocytes.
Methods
In the present study, we further assessed the activity of the shRNA through HPC transduction and differentiation into macrophages derived from fetal liver CD34+ (FL-CD34+) HPCs. Transduced lentiviral vector encoding the human CCR5 shRNA was stably maintained in FL-CD34+ cells and in the terminally differentiated macrophages using macrophage colony-stimulating factor, granulocyte macrophage colony-stimulating factor, interleukin-3 and stem cell factor.
Results
Quantitative real-time polymerase chain reaction for CCR5 mRNA indicated over 90% reduction of CCR5 mRNA levels in CCR5 shRNA-transduced population. The cells with knockdown of CCR5 expression acquired resistance to R5 tropic HIV-1 NFN-SX strain. We also developed a novel approach utilizing a mCherry-CCR5 chimeric reporter to assess the effectiveness of CCR5 target down-regulation in macrophages directly. Both the shRNA and the reporter were maintained throughout HPC differentiation to macrophages without apparent cytotoxicity.
Conclusions
The present study demonstrates a novel method to simply and directly assess the function of small interfering RNA and the effective inhibition of HIV-1 infection by a potential potent shRNA to CCR5 delivered into macrophages derived from HPCs.
Keywords: shRNA, CCRS, HIV-1, lentiviral vector, macrophages, hematopoietic progenitor cells
Introduction
HIV-1 has become one of the major public health threats ever since the 1980s. Although the drug cocktails used in highly active antiretroviral therapy (HAART) have markedly suppressed the progression to AIDS in HIV-infected individuals [1–3], drug-related toxicity has always been a primary concern [4–6]. In addition, the lifelong treatment of HAART has also led investigators to seek an alternative or adjuvant therapeutic method that requires less frequent intake of medicine.
RNA interference (RNAi) is a powerful method for suppressing gene expression, with a tremendous potential for therapeutic applications. Small interfering (si)RNAs recognize cognate mRNAs and induce sequence specific RNA degradation through a highly conserved cellular mechanism [7]. Hematopoietic progenitor cells (HPCs) are progenitor cells of all types of blood lineages; therefore, RNAi technique combined with HPC transplant can provide a stable modulation of gene expression and potentially treat HIV-1. Several vector systems have been developed to express short-hairpin RNAs (shRNAs) to produce siRNAs that target HIV-1 viral proteins and/or cellular components [8–12]. Because of the genetic diversity of HIV-1 virus resulting from high mutation rate during viral replication, designing siRNAs to target cellular components confers more stability and effectiveness [13,14]. Recently, chemokine receptor 5 (CCR5) emerged as an ideal cellular target for anti-HIV-1 therapy by siRNA because of its utilization by most strains of HIV-1 as a co-receptor to enter target cells such as T-lymphocytes and macrophages [15,16]. It is apparently dispensable for normal immune functions. Most importantly, individuals homozygous for a 32-bp deletion in this gene are resistant to HIV-1 infection [17–20].
Gene therapy utilizing siRNA has shown marked efficacy of down-regulating gene expression in vivo [21,22]; however, we and others find that the majority of siRNAs and shRNAs exhibit cytotoxicity in long-term expression [23–26]. We reported a cytotoxicity (two-fold decrease in transduced peripheral blood mononuclear cells over 2 weeks of culture) in primary human lymphocytes transduced with shRNAs directed to CCR5 as well as shRNAs directed to irrelevant targets such as luciferase and LacZ [27]. The cytotoxic effects were alleviated when shRNAs were expressed from the weaker H1 promoter, although the potency was also reduced [27]. We intensively screened a random library of shRNA directed to human CCR5 (huCCR5) sequences expressed using the H1 promoter within a lentiviral vector. We identified one most potent and noncytotoxic shRNA that stably down-regulates CCR5 among the shRNAs characterized to date [28,29]. We tested the function and safety of an analogous shRNA sequence that targets rhesus macaque CCR5 by lentiviral vector-mediated transduction of cytokine mobilized peripheral blood rhesus CD34+ cells followed by autologous transplant into myeloablated rhesus macaques [28]. The shRNA-transduced lymphocytes are less susceptible to simian immunodeficiency virus infection ex vivo and a long-term expression (up to 14 months) of this siRNA in rhesus macaques was observed. Importantly, no apparent toxicity was observed despite expression of siRNA during hematopoietic cell differentiation over the period of the study.
In addition to T lymphocytes, macrophages comprise another primary target cell for HIV-1 [30]. They are among the cells to be first infected by HIV-1, and have been proposed to form a reservoir of HIV-1 in infected individuals. Because CCR5 is also an essential co-receptor for HIV-1 targeting to macrophages [31], genetic modification of HPCs by siRNA directed to CCR5 would render the progeny macrophages resistant to HIV-1 infection.
In the present study, we tested the efficacy and safety of our unique CCR5 shRNA delivered by HPC transduction and inhibition of macrophages to HIV-1 infection. We also tested a novel reporter system that assesses the siRNA effect directly and easily. Our studies demonstrate a potent and noncytotoxic shRNA therapeutic approach in HPCs for the treatment of HIV-1 infection.
Materials and methods
Antibodies
The antibodies used for flow cytometry in the present study were: PE-CD14 (clone MφP9; BD Biosciences, San Jose, CA, USA); PE-CD68 (clone Y1/82 A; BioLegend, San Diego, CA, USA); PECY7-CD11b (clone ICRF44; BioLegend); Alexa647-CD206 (clone 15-2; BioLegend); PE-CD4 (clone RPA-T4; eBioscience, San Diego, CA, USA); PE-CXCR4 (clone 12G5; BioLegend); and PECY5-CCR5 (clone 2d7; BD Biosciences). As a control, the cells were also stained with respective isotype control. Isotype control staining was used to determine background levels.
Cell culture and differentiation into macrophage
293T and huCCR5-293T cells [29] were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, 100 μg/ml streptomycin and 2 mM glutamine. Human primary monocytes were isolated by adherence from leukopacks after Ficoll-Paque PLUS (GE Healthcare, Milwaukee, WI, USA) purification [32]. Monocytes were then cultured in RPMI medium containing 10% FBS and 10 ng/ml of macrophage colony-stimulating factor (MCSF) for 7 days to differentiate into macrophages. FL-CD34+ cells were prepared from human fetus ranging in age from 16–24 weeks as previously described [33]. Briefly, fragments of fetal liver tissues were washed in phosphate-buffered saline (PBS), and a single-cell suspension was obtained by digestion in collagenase (507 U/ml), hyaluronidase (2400 U/ml) and DNase (300 Kunitz units; Gibco, Grand Island, NY, USA) for 2 h at 37 °C. Erythrocytes were removed from the cell suspension by centrifugation over Ficoll-Hypaque (Sigma, St Louis, MO, USA). CD34+ cells were isolated using CD34 progenitor cell isolation kit (Miltenyi BioTec, Auburn, CA, USA). Isolated FL-CD34+ cells were then stained with anti-CD34 antibody and analysed by FACS to determine cell purity. For differentiation of macrophage from FL-CD34+ cells, cells were cultured in RPMI containing 20% FBS and 20 ng/ml of interleukin (IL)-3, 50 ng/ml of stem cell factor (SCF) and 20 ng/ml of MCSF for 9 days. The medium was then switched to RPMI containing 10% human AB serum and 5 ng/ml of granulocyte macrophage colony-stimulating factor (GM-CSF) and further cultured for 5 days.
Vector construction
The lentiviral vector encoding CCR5 shRNA 1005 (sh1005) (5′-GAGCAAGCTCAGTTTACACC-3′) under the human H1 RNA polymerase III promoter was described previously [28]. CCR5 shRNA containing three mismatched nucleotides was used as a control (mutant CCR5 shRNA; 5′-GAGCAAGCTCTCGTTACACC-3′). The firefly luciferase shRNA served as an irrelevant control (Luc shRNA; 5′-AATCGATATTGTTACAACA-3′). The lentiviral vector expressing chimeric construct of mCherry and full-length CCR5 was prepared by polymerase chain reaction (PCR) amplification for CCR5 using the primers: 5′-GC GACGTACGTTCGAATGGATTATCAAGTGTCAAGTCC-3′ and 5′-TGCGCGATATCGCTAGCTCACAAGCCCACAGAT ATTTCC-3′. The lentiviral vector expressing chimeric construct of mCherry and CCR5 target sequence, which contains the 20 nucleotide predicted shRNA target sequence (5′-GAGCAAGCTCAGTTTACACC-3′), was also prepared by PCR amplification using the primers: 5′-G ACCATGATTACGCCAAGCTTGCATGCCTGCAGGTCG-3′ and 5′-CGTGCGCTAGCGATCGGGTGTAAACTGAGCTTGC TCGCGGAATTCTAGA GTCGCGG-3′. The amplified PCR fragments were cloned into the FG12 lentiviral vector [28].
Virus production and titration
We generated lentiviral vector stocks using an HIV-1 based reporter virus, packaging plasmid pCMV R8.2ΔVpr and the VSV-G envelope protein-coding plasmid by calcium phosphate-mediated transient transfection as described previously [34]. After 48 and 72 h, lentiviral vector particles were harvested and concentrated by ultracentrifugation and resuspended in a 100-fold lower volume of Hanks' balanced salt solutions and stored at −80 °C. The viral titer was measured by anti-p24 Gag enzyme-linked immunosorbent assay (ELISA) and the infectious titer was determined in 293T cells by infecting with vector in the presence of 8 μg/ml of polybrene. Reporter gene expression was monitored by flow cytometry. Data were collected on a Cytomics FC500 (Beckman Coulter, Fullerton, CA, USA) and analysed using FCS express (De Novo Software, Los Angeles, CA, USA). CCR5-tropic NFN-SX virus was also produced by calcium phosphate-mediated transient transfection with the infectious proviral plasmid in 293T cells. The virus supernatants were filtered with 0.22-μm filters and stored at −80 °C without concentration. The viral titer was measured by anti-p24 Gag ELISA and the multiplicity of infection (MOI) was determined by serial dilution of virus supernatant on human peripheral blood monocyte-derived macrophages.
Virus transduction
Macrophages derived from FL-CD34+ cells were challenged with NFN-SX strain at an MOI of 0.01. As a control, cells were also challenged with VSV-G pseudotyped NFN-SX strain deleted envelope (VSV-G pseudo/NFN-SXΔEnv). Culture supernatants were collected at 7-day intervals for 21 days and assayed for p24 antigen by ELISA. For lentiviral vector transduction, FL-CD34+ cells were incubated with respective vectors (MOI of 5) overnight on a retronectin-coated plate in accordance with the manufacturer's instructions (Takara Bio Inc., Otsu, Japan). Transduced cells were sorted by flow cytometry for enhanced green fluorescent protein (EGFP) expression 48 h post-transduction. The sorting purity of EGFP + population was over 90% as determined by flow cytometric analysis of the sorted cells.
Phagocytosis
Phagocytosis assay with Texas Red-conjugated Zymosan bioparticles was performed in accordance with the manufacturer's instructions (Molecular Probes, Eugene, OR, USA). Briefly, 1 × 105 macrophage cells were incubated with opsonized Zymosan bioparticles at a MOI = 50 for 90 min. The cells were then washed with cold 1× PBS three times to remove extra bioparticles. Pictures were taken using an Olympus IX50 inverted camera at ×200 magnification (Olympus, Center Valley, PA, USA). The cells were then incubated with 0.25% trypsin for 8 min, washed with medium containing serum and fixed in 1% paraformaldehyde for flow cytometry.
Colony formation assay
FL-CD34+ cells (2 × 103) transduced with either sh1005 or Luc shRNA were plated in 1 ml of methylcellulose medium supplemented with 10 ng/ml of IL-3, 50 ng/ml of SCF, 10 ng/ml of GM-CSF and 3 units/ml of Erythropoietin (Stem Cell Technology, Vancouver, Canada). Cells were cultured at 37 °C with 5% CO2 for 14 days and then colonies (>30 cells) were scored under fluorescence microscope (Olympus IX50). Pictures of colonies were taken with an Olympus camera DP10.
Cotransfection
Lentiviral vector encoding either mCherry-CCR5 target (1 μg) or mCherry-CCR5 (1 μg) was cotransfected with either 3 μg of CCR5 shRNA (1005) or CCR5 mutant shRNA encoding pBluescript onto 293T cells (1 × 105) in a 12-well plate. FuGENE (Roche Diagnostics, Basel, Switzerland) was used for cotransfection in accordance with the manufacturer's instructions. Forty-eight hours post-transfection, cells were analysed for mCherry expression by flow cytometry and by a fluorescence microscope (Leica DM IRB; Leica Microsystems, Wetzlar, Germany).
Real-time PCR
All real-time PCR quantifications were performed using the Bio-Rad iQ5 system (Bio-Rad, Hercules, CA, USA) in parallel with a set of known quantitative standards. For quantification of CCR5 mRNA, total RNA was extracted from approximately 1 × 106 cells with RNeasy (Qiagen, Valencia, CA, USA) and used for quantitative real-time reverse transcriptase (RT)-PCR. RNA standards for CCR5 mRNA and β-actin mRNA quantification were made by serial dilution of in vitro transcribed human CCR5 RNA and β-actin RNA using T7 RNA polymerase (MEGAscript T7; Ambion, Austin, TX, USA). The iScript one-step RT-PCR kit for probes (Bio-Rad) was used with 50 ng of total RNA for amplification of CCR5 and β-actin as a control. The primers used were: CCR5: forward 5′-GTCCCCTTCTGGGCTCACTAT-3′; reverse, 5′-CCCTGTCAAGAGTTGACACATTGTA-3′; probe: FAM-5′-TCCAAAGTCCCACTGGGCGGCAG-3′-BHQ1. β-actin: forward 5′-CGAGCGCGGCTACAGCTT-3′; reverse, 5′-CCTTA ATGTCACGCACGATT-3′; probe, probe: HEX-5′-ACC ACCACGGCCGAGCGG-3′-BHQ2. All primers and probe were synthesized by Biosearch Technologies Inc. (Novato, CA, USA). All RT-PCR reactions were carried out as follows: reverse transcription at 50 °C for 10 min, inactivation of reverse transcriptase at 95 °C for 5 min, and subsequently 45 cycles in two phases consisting of 95 °C for 15 s, and 58 °C for 30 s. CCR5 mRNA was normalized using the endogenous β-actin mRNA as a reference.
Results
CCR5 shRNA 1005 specifically reduces the cell surface expression of CCR5 in CCR5-293T cells
The CCR5 shRNA 1005 (sh1005) published previously was constructed to express the potent CCR5 shRNA transcripts driven by the H1 PoI III promoter within a FG12 lentiviral vector cassette (Figure 1A). As controls, we generated a mutant CCR5 shRNA (muCCR5 shRNA) that is three nucleotides different from the sh1005 and a shRNA against firefly luciferase (Luc shRNA) (Figure 1A). As previously reported, the FG12 vector also expresses EGFP under the human UbiC internal promoter for tracking transduced cells [9]. To examine the specificity of sh1005 in targeting, we first tested the suppression of CCR5 expression on the cell surface by sh1005 in 293T cells expressing human CCR5 (CCR5-293T). We transduced the CCR5-293T cells with lentiviral vectors expressing either sh1005 or muCCR5 shRNA. Cells transduced by sh1005 showed an almost complete reduction of CCR5 expression, whereas no reduction of CCR5 expression was observed in the muCCR5 shRNA-transduced cells (Figure 1B). These results indicated a highly specific and potent inhibition of CCR5 expression by sh1005 in CCR5-293T cells.
Figure 1.
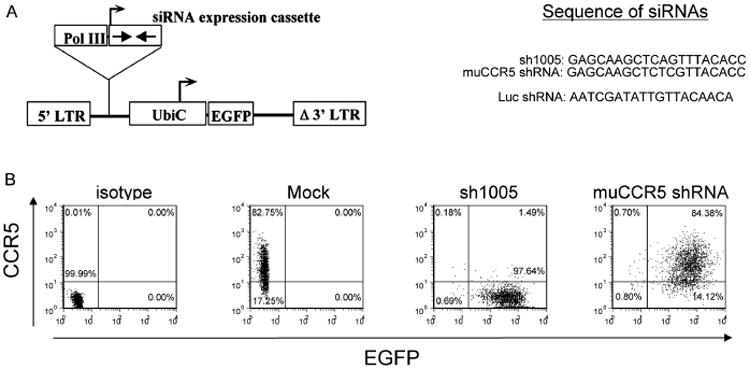
CCR5 shRNA can specifically reduce the cell surface expression of CCR5 in CCR5-293T cells. (A) Schematic diagram of the siRNA-expressing lentiviral vector. The shRNA is expressed under the control of a human H1-RNA Pol III promoter (Pol III). The vector also contains a human ubiquitin C promoter (UbiC) driving the EGFP marker gene for tracking transduced cells. (B) CCR5-293T cells (1 × 105) were transduced with lentiviral vectors encoding CCR5 shRNA 1005 (sh1005) or the mutant CCR5 shRNA (muCCR5 shRNA). The cells were harvested 2 days after virus transduction and analysed by flow cytometry with PE-Cy5 conjugated anti-human CCR5 or isotype control antibody staining (isotype). The transduced (EGFP+) and nontransduced (EGFP−) cells were gated based upon their EGFP expression. The number in each quadrant represents the percentage in each population
sh1005 transduction does not affect hematopoietic differentiation of FL-CD34+ cells
The ultimate goal of the present study was to test suppression of CCR5 expression in macrophages derived from FL-CD34+ cells. We first examined whether the shRNA transduction affects hematopoietic differentiation of the FL-CD34+ cells. To examine whether FL-CD34+ cells behave similarly to peripheral blood mononuclear cells (PBMCs) in macrophage differentiation, macrophages derived from PBMCs or FL-CD34+ cells were stained for cell surface markers. The results obtained demonstrated that macrophages derived from both PBMCs and FL-CD34+ cells exhibited similar expression of cell surface markers, CD14, CD68, CD11b and CD206 (Figure 2). We also stained the cells for HIV receptors, CD4, CXCR4 and CCR5 and observed similar levels of cell surface expression on both macrophages. Having confirmed that macrophages derived from FL-CD34+ cells share the same cell surface marker expression as those derived from PBMCs, we further examined whether the transduction of either sh1005 or muCCR5 shRNA affects macrophage differentiation in FL-CD34+ cells. As indicated by the CD14, CD11b and CD206 surface marker staining, no apparent adverse effects on macrophage differentiation were observed in shRNA-transduced cells (Figure 3A). In addition, macrophages transduced with either sh1005 or muCCR5 shRNA were functionally competent for phagocytic activity similar to that showed by the mock cells (Figure 3B). Furthermore, we confirmed that the surface expression of CCR5 was clearly reduced in cells transduced with sh1005 compared to cells transduced with muCCR5 shRNA (Figure 3C). These results indicate that the expression of sh1005 in FL-CD34+ cells does not affect the differentiation into macrophages nor their phagocytic activities.
Figure 2.
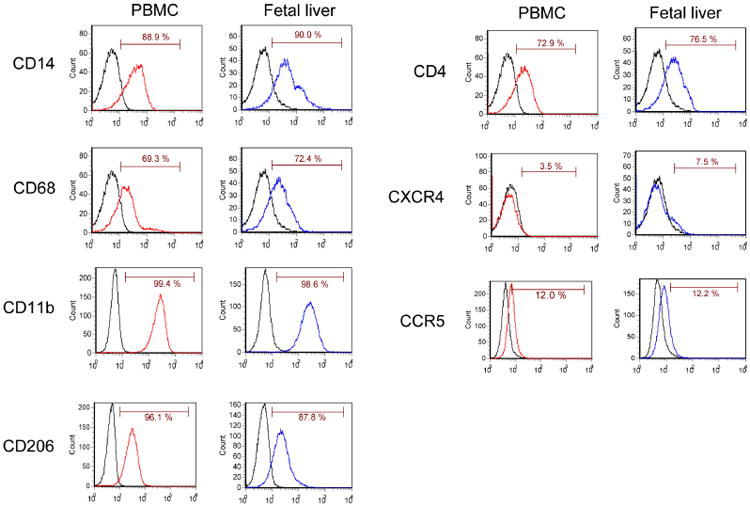
Phenotypic FACS analysis of macrophages derived from PBMC and FL-CD34+ cells. FL-CD34+ cells (2 × 105) were cultured in the presence of 20 ng/ml of IL-3, 50 ng/ml of SCF and 20 ng/ml of MCSF for 9 days and then with 5 ng/ml of GM-CSF for 5 days to differentiate to macrophages. Monocytes (1 × 106) derived from PBMCs were cultured in the presence of 10 ng/ml of MCSF for 7 days to differentiate to macrophages. The attached cells were gently scraped off the bottom of the plate and 2 × 105 cells were stained with antibodies to CD14, CD68, CD11b and CD206 (macrophage markers), and CD4, CXCR4 and CCR5 (receptors for HIV-1). The expression of these surface markers was analysed by flow cytometry. The results are shown as a histogram. The number in each panel represents the percentage of positive population
Figure 3.

shRNA transduction does not grossly affect differentiation to macrophages derived from FL-CD34+ cells. (A) FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors encoding sh1005 or muCCR5 shRNA as a control. Mock are cells without shRNA transduction. The cells were treated with 20 ng/ml of IL-3, 50 ng/ml of SCF and 20 ng/ml of MCSF for 9 days and then with 5 ng/ml of GM-CSF for 5 days to differentiate to the macrophages. The attached cells were gently scraped off the bottom of the plate and 1 × 105 cells were analysed for CD14, CD11b and CD206 expression by flow cytometry. (B) 1 × 105 macrophage cells were incubated with an MOI of 50 Texas Red-conjugated Zymosan particles for 90 min. Cells were then washed with cold PBS for three times and were analysed by fluorescence microscopy and flow cytometry. The flow cytometry results are exhibited as EGFP versus Zymosan bioparticle (Texas Red) dot plots. (C) Surface CCR5 staining of macrophages derived from sh1005 and muCCR5 shRNA-transduced cells.−, isotype control; +, CCR5 staining
To further ascertain the effect of shRNA transduction on hematopoietic differentiation of the FL-CD34+ cells, we next performed colony-forming assays. Mock-, sh1005-or Luc shRNA-transduced FL-CD34+ cells were cultured in cytokine supplemented methylcellulose medium for 14 days and colony-forming units of granulocyte and macrophage, granulocyte, erythroid, macrophage and megakaryocyte, and erythoid were scored. As shown in Table 1, the total number of colonies as well as the percentage of different kinds of colonies in each group of mock, sh1005 or Luc shRNA-transduced FL-CD34+ cells are not apparently different, further indicating that hematopoietic differentiation is not grossly affected by shRNA transduction. The above studies of both cell surface markers and colony-forming assays indicated that the H1 promoter-driven sh1005 expression posed no adverse effect on FL-CD34+ cell differentiation and no apparent cytotoxicities on the cells.
Table 1. shRNA transduction of FL-CD34+ cells does not grossly affect the colony formation in the methylcellulose assay.
| Total | GM | GEMM | Erythroid | EGFP + | %EGFP | |
|---|---|---|---|---|---|---|
| Mock | 104 | 66 | 36 | 2 | 0 | 0 |
| Luc shRNA | 104 | 53 | 48 | 3 | 29 | 28 |
| sh1005 | 83 | 46 | 35 | 2 | 35 | 42 |
FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors expressing sh1005 or Luc shRNA. Viable cells were quantified, plated (2 × 103 cells/ml), and cultured in humidified chambers for hematopoietic colony-formation using standard methylcellulose medium containing 10 ng/ml of IL-3, 50 ng/ml of SCF, 10 ng/ml of GM-CSF and 3 units/ml of erythropoietin. Cultures were scored at day 14 for granulocyte-macrophage colony (GM), granulocyte, erythroid, macrophage and megakaryocyte (GEMM), and erythroid colony (Erythroid) under the microscope.
sh1005 reduces expression of mCherry-CCR5 chimera in 293T cells and macrophages derived from FL-CD34+ cells
The staining of CCR5 on the cell surface of macrophages is a measure of CCR5 down-regulation by shRNA; however, low concentrations of CCR5 on the cell surface (Figure 2) and potential disruption of CCR5 expression by technical manipulation renders quantitative analysis difficult in macrophages. Therefore, we developed an approach to simply assess CCR5 target down-regulation in macrophages directly. We constructed a fluorescent reporter fused with CCR5 sequences to allow us to assess CCR5 down-regulation quantitatively. The reporter plasmid expresses the mCherry gene fused to CCR5 coding sequences downstream. The chimeric mRNA is driven by ubiquitin C promoter within the FG12 lentiviral vector cassette. Because we demonstrated that CCR5 is down-regulated by sh1005, we hypothesized that the mCherry-CCR5 chimeric mRNA will be degraded in sh1005-transduced cells, thus resulting in inhibition of mCherry expression.
We first tested the feasibility of this reporter system in 293T cells. 293T cells were cotransfected with lentiviral vectors expressing sh1005 or muCCR5 shRNA and either mCherry-CCR5 chimera or mCherry encoding lentiviral vector (Figure 4). With mCherry-CCR5 target plus sh1005, the expression of mCherry diminished. muCCR5 shRNA did not affect mCherry expression in mCherry-CCR5 target transuded cells. Transduction of mCherry alone was not affected by either sh1005 or muCCR5 shRNA.
Figure 4.

Specific knockdown of mCherry-CCR5 chimera expression by sh1005 in 293T cells. 293T cells (1 × 105) were cotransfected with lentiviral vectors expressing sh1005 or the muCCR5 shRNA and either mCherry-CCR5 chimera or mCherry encoding lentiviral vector by FuGENE. The cells were then cultured for 2 days and the mCherry expression was analysed by fluorescence microscopy (A) and flow cytometry (B). The results are exhibited as side scatter versus mCherry dot plots. The quadrant lines were defined by mock 293T cells (data not shown) and the percentage numbers are indicated
We further modified the reporter to include only the CCR5 20 nucleotide shRNA target sequence (5′-GAGCAAGCTCAGTTTACACC-3′) downstream from the mCherry gene, providing even greater assessment of target specificity. We first tested the effectiveness of the mCherry-CCR5 (20 nucleotides) target (CCR5 target) in 293T cells. 293T cells were cotransfected with lentiviral vectors expressing sh1005 or muCCR5 shRNA plus either the CCR5 target or mCherry encoding lentiviral vector (Figure 5). With the CCR5 target plus sh1005, the expression of mCherry diminished. muCCR5 shRNA did not affect mCherry expression in CCR5 target-transduced cells.
Figure 5.
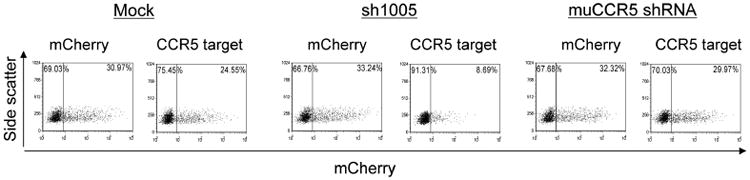
Specific knockdown of mCherry-CCR5 target expression by sh1005 in 293T cells. 293T cells (1 × 105) were cotransfected with lentiviral vectors expressing sh1005 or muCCR5 shRNA and either mCherry-CCR5 target or mCherry encoding lentiviral vector by FuGENE. The cells were then cultured for 2 days and the mCherry expression was analysed by flow cytometry. The results are exhibited as side scatter versus mCherry dot plots. The quadrant lines were defined by mock 293T cells (data not shown) and the percentage numbers are indicated
We next assessed the effectiveness of this reporter in macrophages derived from FL-CD34+ cells. We transduced the CCR5 target into either sh1005 or Luc shRNA-transduced FL-CD34+ cells. The doubly-transduced FL-CD34 + cells were cultured in cytokines for 14 days to allow differentiation to macrophages. mCherry expression in FL-CD34+ cells 3 days post-transduction were monitored by flow cytometry and decreased mCherry expression was only observed in sh1005-transduced cells (Figure 6). mCherry expression in macrophages were monitored by fluorescence microscopy and by flow cytometry. Fluorescence microscopy showed a significant reduction of mCherry expression in CCR5 shRNA-transduced cells compared to the Luc shRNA-transduced cells (Figure 7). Flow cytometric analysis showed similar results to the microscopic data, with an approximately three-fold reduction of mCherry expression in sh1005-transduced cells compared to the Luc shRNA-transduced cells (Figure 7). No obvious toxicities were observed in FL-CD34+ cells and throughout the differentiation to macrophages. These data demonstrate that the mCherry-CCR5 target was an effective and efficient reporter system, which can be used directly to monitor the effect of shRNA vector.
Figure 6.

Specific knockdown of mCherry-CCR5 target expression by sh1005 in FL-CD34+ cells. FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors expressing sh1005 or Luc shRNA. The cells were harvested 3 days after virus transduction, sorted by the EGFP expression, and further transduced with the lentiviral vector encoding mCherry-CCR5 target sequence at an MOI of 0.5. The mCherry expression was monitored by flow cytometry 3 days after the transduction. The results are exhibited as mCherry dot plots versus EGFP dot plots. The number in each quadrant represents the percentage in each population
Figure 7.
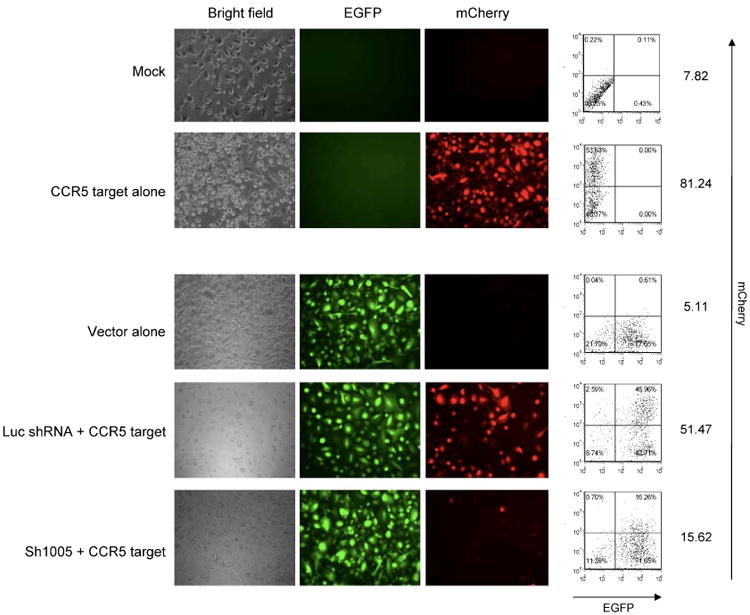
Specific knockdown of mCherry-CCR5 target expression by sh1005 in macrophages derived from FL-CD34+ cells. FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors expressing sh1005 or Luc shRNA. The cells were harvested 3 days after virus transduction, sorted by the EGFP expression, and further transduced with the lentiviral vector encoding mCherry-CCR5 target sequence at an MOI of 0.5. FL-CD34+ cells were then differentiated to macrophages with 20 ng/ml of IL-3, 50 ng/ml of SCF and 20 ng/ml of MCSF for 9 days and then with 5 ng/ml of GM-CSF for 5 days. The effect of shRNA was monitored by the expressions of mCherry by florescence microscopy and by flow cytometry. The results are exhibited as mCherry dot plots versus EGFP dot plots. The number in each quadrant represents the percentage in each population. The numbers on right side of each panel show the mean fluorescent intensity of the mCherry expression
Knockdown of endogenous CCR5 expression by sh1005 in macrophages derived from FL-CD34+ cells
To confirm the effect of sh1005 on the expression of endogenous CCR5, we measured the amount of endogenous CCR5 mRNA by real-time RT-PCR (Figure 8) in macrophages derived from FL-CD34+ cells. In mock or Luc shRNA-transduced macrophages derived from FL-CD34+ cells, there was no difference in the level of endogenous CCR5 mRNA expression. By contrast, in the sh1005-transduced cells, the level of endogenous CCR5 mRNA expression was decreased to over 90% compared to those in mock cells or Luc shRNA-transduced cells. These results indicated that sh1005 can potently knockdown endogenous CCR5 mRNA expression.
Figure 8.
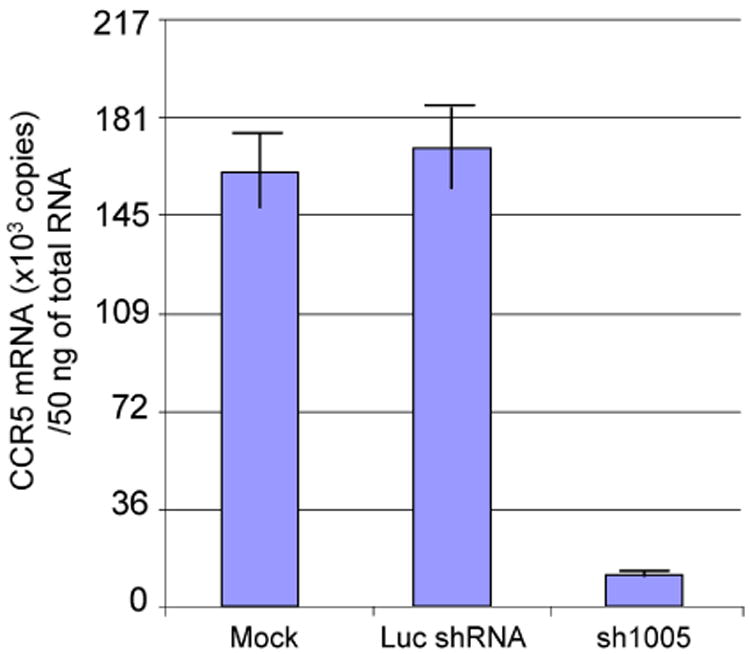
sh1005 can effectively reduce the levels of endogenous CCR5 mRNA in macrophages derived from FL-CD34+ cells. FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors expressing sh1005 or Luc shRNA. The cells were harvested 3 days after virus transduction and sorted by the EGFP expression. The cells were then differentiated to macrophages with 20 ng/ml of IL-3, 50 ng/ml of SCF and 20 ng/ml of MCSF for 9 days and then with 5 ng/ml of GM-CSF for 5 days. Total RNA were isolated from macrophages using the Qiagen RNeasy extraction kit and treated with DNase I. Quantification of mRNA was performed using IQ5 with iScript one-step RT-PCR kit. RNA standards for CCR5 mRNA quantification were made by serial dilution of in vitro transcribed human CCR5 RNA using T7 RNA polymerase
Knockdown of CCR5 expression on macrophages derived from FL-CD34+ cells rendered resistance to R5 tropic HIV-1 NFN-SX infection
We evaluated the anti-HIV activity of sh1005 in macrophages derived from FL-CD34+ cells using R5-tropic HIV-1 (NFN-SX). VSV-G pseudotyped NFN-SXΔEnv was used as a positive control for virus infection. Viral p24 antigen levels were determined by the ELISA every 7 days post-infection to quantify the levels of HIV-1 production. No virus infection served as a control for p24 background measurement. The p24 levels in the cells transduced with sh1005 were similar to mock levels (Figure 9), indicating that sh1005 strongly impaired the infectivity of NFN-SX in the macrophages derived from FL-CD34+ cells. By contrast, nontransduced (−) or muCCR5-transduced cells exhibited identical virus production as measured by capsid p24 levels (Figure 9). VSV-G pseudotyped NFN-SXΔEnv showed similar kinetics for p24 production in nontransduced, sh1005, or muCCR5 shRNA-transduced cells, demonstrating that the sh1005 expression did not affect virus production itself. Together, these data indicated that sh1005 was effective in inhibiting HIV-1 infection in FL-CD34+ cell-derived macrophages.
Figure 9.

Stable knockdown of CCR5 expression on macrophages derived from FL-CD34+ cells rendered resistance to R5 tropic HIV-1 NFN-SX infection. FL-CD34+ cells (2 × 105) were transduced with lentiviral vectors expressing sh1005 or muCCR5 shRNA. The cells were harvested 3 days after virus transduction, sorted by the EGFP expression. FL-CD34+ cells (1 × 105) stably expressed sh1005 or the muCCR5 shRNA were treated with 20 ng/ml of IL-3, 50 ng/ml of SCF and 20 ng/ml of MCSF for 9 days and then with 5 ng/ml of GM-CSF for 5 days to differentiate to macrophages. The cells were then infected with either wild-type NFN-SX or VSV-G pseudotyped NFN-SXΔEnv, and cell free culture supernatant (1.0 ml) was harvested every 7 days post-infection for 21 days. Virus replication was monitored by measuring p24 antigen in culture supernatants. Mock, cells not infected by NFN-SX; −, nontransduced cells
Discussion
HAART is currently the only effective clinical treatment for HIV-1 infection [1–3]. Modification of human genes in HPCs to forms that are resistant to HIV-1 infection might provide an alternative or adjuvant therapy to avoid the serious drug toxicities associated with frequent antiretroviral intake [11,12]. RNAi offers an attractive alternative to other gene therapeutic reagents as a result of its small size, ease of manipulation, and ease of combining with other reagents, which is an essential requirement for HIV-1 disease. Transplantation of HPCs with anti-HIV infection RNAi elements would result in reconstitution of a hematopoietic system that should be protected from the infection of HIV-1. Because HPC transplantation is for a lifetime, the challenge is to identify shRNAs with sufficient potency to down-regulate the gene in question over sustained lengths of time, but without toxicity to the cells bearing them.
In the present study, we investigated the safety and efficacy of a CCR5 shRNA (sh1005), which was previously shown to have potency as well as noncytotoxicity in nonhuman primates [28,29], with respect to inhibiting HIV-1 infection in macrophages derived from FL-CD34+ cells. We confirmed that sh1005 is noncytotoxic and does not affect the differentiation of FL-CD34+ cells into macrophages by cell surface marker staining, phagocytosis assay and colony-forming assays. When we tested the function of the sh1005 in FL-CD34+ cell-derived macrophages, over 90% of the endogenous CCR5 was down-regulated by real-time RT-PCR analysis. In addition, sh1005 almost completely inhibited HIV-1 infection in FL-CD34+ cell-derived macrophages, although it did not affect transduction by VSV-G pseudotyped NFN-SXΔEnv. The potency of sh1005 in inhibiting HIV-1 infection supports its potential future application in the clinic.
A different shRNA to CCR5 was tested previously in a similar HPC system [12]. Although that study reported no obvious cytotoxicity using the strong U6 promoter, the present study showed that selection of a shRNA that maintains potency with a weaker H1 promoter is possible. The use of a weaker promoter should be safer in clinical applications where the shRNA is expressed long-term, such as when delivered by stem cell transplant.
In addition to the characterization of safety and potency of sh1005 in macrophages, we developed a novel approach to facilitate the examination of CCR5 knockdown on macrophages without surface staining by expressing the mCherry gene fused to CCR5 coding sequences downstream. Using this mCherry-CCR5 chimera, we easily observed shRNA-mediated down-regulation in macrophage cells transduced with sh1005. This method is technically easy and sensitive when CCR5 expression is low on the cell surface. It can be used in various studies to directly assess the effect of siRNA.
HIV-1 kills and the body replenishes new T-cells and monocytes/macrophages each day [35–37]. One concept underlying successful anti-HIV therapy is reconstitution with gene-modified hematopoietic progenitor and stem cells that provide a continual and lifelong source of protected T-cells and monocyte/macrophages. Proof concept for a stem cell therapy was provided by a recent case study in which an HIV-1 positive individual with concurrent acute myeloid lymphoma was treated by transplant of allogeneic stem cells specifically chosen from a CCR5Δ32 homozygous donor [38]. The CCR5Δ32 donor cells apparently completely and rapidly replaced the recipient cells within 61 days and the patient has remained undetectable for HIV-1 for more than 200 days in the absence of antiretroviral therapy.
Overall, the present study demonstrates a proof concept of preventing HIV-1 infection by the genomic modification of HPCs. However, monotherapies for HIV-1 disease might fail as a result of the development of mutations in HIV-1 that confer resistance to the therapy. In the case of CCR5, variants of HIV-1, termed X4, exist in nature that utilize an alternative co-receptor, CXCR4 [39–41]. The role of these X4 viruses in HIV-1 pathogenesis remains unclear [42–45] but must be taken into consideration. Effective therapeutic application of siRNAs for HIV-1 disease will eventually require combinations of multiple reagents directed against HIV-1. Multiple gene therapeutic reagents can easily be incorporated into a single vector. HPC therapy for HIV-1 disease provides a model for the treatment of other stem cell-based diseases in which genetic modification will be beneficial.
Supplementary Material
Acknowledgments
M.L. and M.K. designed and performed the studies, analysed data, and wrote the manuscript. We thank R. Lee for editorial assistance with the manuscript. We thank the Gene and Cellular Therapy Core Laboratory of UCLA for providing fetal liver for CD34+ cell purification. We thank the Virology Core Facility of UCLA for p24 measurement. We thank the Flow Cytometry Core Facility of UCLA for cell sorting. This work was supported by US National Institutes of Health grant AI055281, California Institute for Regenerative Medicine grant RS1-00172 and Center for AIDS Research grant AI028697. There are no ethical issues regarding this study.
Footnotes
K.N.C. contributed to the mCherry-CCR5 reporter experiments. N.P. contributed to the colony assay of FL-CD34+ cells. A.D.S. contributed to the construction of shRNA and mCherry lentiviral vectors. I.S.Y.C. supervised the study.
The authors declare that there are no competing financial interests.
References
- 1.Gulick RM, Mellors JW, Havlir D, et al. Simultaneous versus sequential initiation of therapy with indinavir, zidovudine, and lamivudine for HIV-1 infection: 100-week follow-up. JAMA. 1998;280:35–41. doi: 10.1001/jama.280.1.35. [DOI] [PubMed] [Google Scholar]
- 2.Gulick RM, Mellors JW, Havlir D, et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. New Engl J Med. 1997;337:734–739. doi: 10.1056/NEJM199709113371102. [DOI] [PubMed] [Google Scholar]
- 3.Hammer SM, Squires KE, Hughes MD, et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. New Engl J Med. 1997;337:725–733. doi: 10.1056/NEJM199709113371101. [DOI] [PubMed] [Google Scholar]
- 4.Brau N, Leaf HL, Wieczorek RL, et al. Severe hepatitis in three AIDS patients treated with indinavir. Lancet. 1997;349:924–925. doi: 10.1016/S0140-6736(05)62700-6. [DOI] [PubMed] [Google Scholar]
- 5.d'Arminio Monforte A, Lepri AC, Rezza G, et al. Insights into the reasons for discontinuation of the first highly active antiretroviral therapy (HAART) regimen in a cohort of antiretroviral naive patients. ICONA Study Group. Italian Cohort of Antiretroviral-Naive Patients. AIDS. 2000;14:499–507. doi: 10.1097/00002030-200003310-00005. [DOI] [PubMed] [Google Scholar]
- 6.Nunez M, Lana R, Mendoza JL, et al. Risk factors for severe hepatic injury after introduction of highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2001;27:426–431. doi: 10.1097/00126334-200108150-00002. [DOI] [PubMed] [Google Scholar]
- 7.Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–811. doi: 10.1038/35888. [DOI] [PubMed] [Google Scholar]
- 8.Li MJ, Bauer G, Michienzi A, et al. Inhibition of HIV-1 infection by lentiviral vectors expressing Pol III-promoted anti-HIV RNAs. Mol Ther. 2003;8:196–206. doi: 10.1016/s1525-0016(03)00165-5. [DOI] [PubMed] [Google Scholar]
- 9.Qin XF, An DS, Chen IS, et al. Inhibiting HIV-1 infection in human T cells by lentiviral-mediated delivery of small interfering RNA against CCR5. Proc Natl Acad Sci USA. 2003;100:183–188. doi: 10.1073/pnas.232688199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Novina CD, Murray MF, Dykxhoorn DM, et al. siRNA-directed inhibition of HIV-1 infection. Nat Med. 2002;8:681–686. doi: 10.1038/nm725. [DOI] [PubMed] [Google Scholar]
- 11.Li MJ, Kim J, Li S, et al. Long-term inhibition of HIV-1 infection in primary hematopoietic cells by lentiviral vector delivery of a triple combination of anti-HIV shRNA, anti-CCR5 ribozyme, and a nucleolar-localizing TAR decoy. Mol Ther. 2005;12:900–909. doi: 10.1016/j.ymthe.2005.07.524. [DOI] [PubMed] [Google Scholar]
- 12.Anderson J, Akkina R. Complete knockdown of CCR5 by lentiviral vector-expressed siRNAs and protection of transgenic macrophages against HIV-1 infection. Gene Ther. 2007;14:1287–1297. doi: 10.1038/sj.gt.3302958. [DOI] [PubMed] [Google Scholar]
- 13.Das AT, Brummelkamp TR, Westerhout EM, et al. Human immunodeficiency virus type 1 escapes from RNA interference-mediated inhibition. J Virol. 2004;78:2601–2605. doi: 10.1128/JVI.78.5.2601-2605.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Westerhout EM, ter Brake O, Berkhout B. The virion-associated incoming HIV-1 RNA genome is not targeted by RNA interference. Retrovirology. 2006;3:57. doi: 10.1186/1742-4690-3-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choe H, Farzan M, Sun Y, et al. The beta-chemokine receptors CCR3 and CCR5 facilitate infection by primary HIV-1 isolates. Cell. 1996;85:1135–1148. doi: 10.1016/s0092-8674(00)81313-6. [DOI] [PubMed] [Google Scholar]
- 16.Rana S, Besson G, Cook DG, et al. Role of CCR5 in infection of primary macrophages and lymphocytes by macrophage-tropic strains of human immunodeficiency virus: resistance to patient-derived and prototype isolates resulting from the delta ccr5 mutation. J Virol. 1997;71:3219–3227. doi: 10.1128/jvi.71.4.3219-3227.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dean M, Carrington M, Winkler C, et al. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Hemophilia Growth and Development Study, Multicenter AIDS Cohort Study, Multicenter Hemophilia Cohort Study, San Francisco City Cohort, ALIVE Study. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 18.Huang Y, Paxton WA, Wolinsky SM, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2:1240–1243. doi: 10.1038/nm1196-1240. [DOI] [PubMed] [Google Scholar]
- 19.Liu R, Paxton WA, Choe S, et al. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply exposed individuals to HIV-1 infection. Cell. 1996;86:367–377. doi: 10.1016/s0092-8674(00)80110-5. [DOI] [PubMed] [Google Scholar]
- 20.Samson M, Libert F, Doranz BJ, et al. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature. 1996;382:722–725. doi: 10.1038/382722a0. [DOI] [PubMed] [Google Scholar]
- 21.Xia H, Mao Q, Paulson HL, et al. siRNA-mediated gene silencing in vitro and in vivo. Nat Biotechnol. 2002;20:1006–1010. doi: 10.1038/nbt739. [DOI] [PubMed] [Google Scholar]
- 22.Banerjea A, Li MJ, Bauer G, et al. Inhibition of HIV-1 by lentiviral vector-transduced siRNAs in T lymphocytes differentiated in SCID-hu mice and CD34+ progenitor cell-derived macrophages. Mol Ther. 2003;8:62–71. doi: 10.1016/s1525-0016(03)00140-0. [DOI] [PubMed] [Google Scholar]
- 23.Fish RJ, Kruithof EK. Short-term cytotoxic effects and long-term instability of RNAi delivered using lentiviral vectors. BMC Molecular Biol. 2004;5:9. doi: 10.1186/1471-2199-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Grimm D, Streetz KL, Jopling CL, et al. Fatality in mice due to oversaturation of cellular microRNA/short hairpin RNA pathways. Nature. 2006;441:537–541. doi: 10.1038/nature04791. [DOI] [PubMed] [Google Scholar]
- 25.Jackson AL, Burchard J, Schelter J, et al. Widespread siRNA ‘off-target’ transcript silencing mediated by seed region sequence complementarity. RNA. 2006;12:1179–1187. doi: 10.1261/rna.25706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Snove O, Jr, Rossi JJ. Toxicity in mice expressing short hairpin RNAs gives new insight into RNAi. Genome Biol. 2006;7:231. doi: 10.1186/gb-2006-7-8-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.An DS, Qin FX, Auyeung VC, et al. Optimization and functional effects of stable short hairpin RNA expression in primary human lymphocytes via lentiviral vectors. Mol Ther. 2006;14:494–504. doi: 10.1016/j.ymthe.2006.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.An DS, Donahue RE, Kamata M, et al. Stable reduction of CCR5 by RNAi through hematopoietic stem cell transplant in non-human primates. Proc Natl Acad Sci USA. 2007;104:13110–13115. doi: 10.1073/pnas.0705474104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shimizu S, Kamata M, Kittipongdaja P, et al. Characterization of a potent non-cytotoxic shRNA directed to the HIV-1 co-receptor CCR5. Genet Vaccines Ther. 2009;7:8. doi: 10.1186/1479-0556-7-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zaitseva M, Blauvelt A, Lee S, et al. Expression and function of CCR5 and CXCR4 on human Langerhans cells and macrophages: implications for HIV primary infection. Nat Med. 1997;3:1369–1375. doi: 10.1038/nm1297-1369. [DOI] [PubMed] [Google Scholar]
- 31.Wu L, Paxton WA, Kassam N, et al. CCR5 levels and expression pattern correlate with infectability by macrophage-tropic HIV-1, in vitro. J Exp Med. 1997;185:1681–1691. doi: 10.1084/jem.185.9.1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bennett S, Breit SN. Variables in the isolation and culture of human monocytes that are of particular relevance to studies of HIV. J Leukoc Biol. 1994;56:236–240. doi: 10.1002/jlb.56.3.236. [DOI] [PubMed] [Google Scholar]
- 33.Akkina RK, Rosenblatt JD, Campbell AG, et al. Modeling human lymphoid precursor cell gene therapy in the SCID-hu mouse. Blood. 1994;84:1393–1398. [PubMed] [Google Scholar]
- 34.Kamata M, Nagaoka Y, Chen IS. Reassessing the role of APOBEC3G in human immunodeficiency virus type 1 infection of quiescent CD4+ T-cells. PLoS Pathog. 2009;5:e1000342. doi: 10.1371/journal.ppat.1000342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wei X, Ghosh SK, Taylor ME, et al. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–122. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 36.Ho DD, Neumann AU, Perelson AS, et al. Rapid turnover of plasma virions and CD4 lymphocytes in HIV-1 infection. Nature. 1995;373:123–126. doi: 10.1038/373123a0. [DOI] [PubMed] [Google Scholar]
- 37.Coffin JM. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 38.Hutter G, Nowak D, Mossner M, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. New Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- 39.Doranz BJ, Rucker J, Yi Y, et al. A dual-tropic primary HIV-1 isolate that uses fusin and the beta-chemokine receptors CKR-5, CKR-3, and CKR-2b as fusion cofactors. Cell. 1996;85:1149–1158. doi: 10.1016/s0092-8674(00)81314-8. [DOI] [PubMed] [Google Scholar]
- 40.Endres MJ, Clapham PR, Marsh M, et al. CD4-independent infection by HIV-2 is mediated by fusin/CXCR4. Cell. 1996;87:745–756. doi: 10.1016/s0092-8674(00)81393-8. [DOI] [PubMed] [Google Scholar]
- 41.Feng Y, Broder CC, Kennedy PE, et al. HIV-1 entry cofactor: functional cDNA cloning of a seven-transmembrane, G protein-coupled receptor. Science. 1996;272:872–877. doi: 10.1126/science.272.5263.872. [DOI] [PubMed] [Google Scholar]
- 42.Langford SE, Ananworanich J, Cooper DA. Predictors of disease progression in HIV infection: a review. AIDS Res Ther. 2007;4:11. doi: 10.1186/1742-6405-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ray N, Doms RW. HIV-1 coreceptors and their inhibitors. Curr Top Microbiol Immunol. 2006;303:97–120. doi: 10.1007/978-3-540-33397-5_5. [DOI] [PubMed] [Google Scholar]
- 44.Regoes RR, Bonhoeffer S. The HIV coreceptor switch: a population dynamical perspective. Trends Microbiol. 2005;13:269–277. doi: 10.1016/j.tim.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 45.Tsibris AM, Kuritzkes DR. Chemokine antagonists as therapeutics: focus on HIV-1. Annu Rev Med. 2007;58:445–459. doi: 10.1146/annurev.med.58.080105.102908. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.