

Abstract
Mitochondria have a well-recognized role in the production of ATP and the intermediates needed for macromolecule biosynthesis, such as nucleotides. Mitochondria also participate in the activation of signaling pathways. Overall, accumulating evidence now suggests that mitochondrial bioenergetics, biosynthesis and signaling are required for tumorigenesis. Thus, emerging studies have begun to demonstrate that mitochondrial metabolism is potentially a fruitful arena for cancer therapy. In this Perspective, we highlight recent developments in targeting mitochondrial metabolism for the treatment of cancer.
Historically, mitochondrial metabolism has been erroneously viewed as inconsequential for the metabolic demands of rapidly proliferating cells1,2. Furthermore, the mitochondria have been ascribed to produce copious amounts of reactive oxygen species (ROS) that promote DNA damage and genetic instability3. This view mainly stems from two long-standing observations. The first observation, made in the 1920s, states that cancer cells take up glucose and produce large quantities of lactate in the presence of ample oxygen4. This is referred to as aerobic glycolysis or the Warburg effect. This observation led to the hypothesis that mitochondria are damaged in tumors; thus, cancer cells primarily use glycolysis as the major metabolic pathway for proliferation2. A second observation is that tumors frequently produce high levels of mitochondrial ROS to invoke genetic instability and ultimately tumorigenesis5. Consequently, mitochondrial dysfunction was designated as a metabolic hallmark of cancer cells.
The notion that mitochondria in tumor cells are dysfunctional was challenged in the 1950s6 by the demonstration that, much like cells from normal tissues such as the liver, tumor cells effectively oxidized the fatty acid palmitate7. Moreover, enzymes of the tricarboxylic acid (TCA) cycle had similar activities in the mitochondria of tumors as in the mitochondria of normal tissues8, and the high rate of glycolysis is a hallmark of both normal and neoplastic cells9. Importantly, these observations and models have largely been validated since the 1950s. Recent studies provided genetic evidence that mitochondrial metabolism is essential for tumorigenesis10–12. Accordingly, in this Perspective, we highlight emerging data that point to mitochondrial metabolism as a target for cancer therapy.
Biochemical functions of the mitochondria
In classical biochemistry textbooks, the major tasks of the mitochondria are the production of ATP and the metabolites necessary to fulfill the bioenergetic and biosynthetic demands of the cell. Mitochondria use multiple carbon fuels to produce ATP and metabolites, including pyruvate, which is generated from glycolysis; amino acids such as glutamine; and fatty acids. These carbon fuels feed into the TCA cycle in the mitochondrial matrix to generate the reducing equivalents NADH and FADH2, which deliver their electrons to the electron transport chain (ETC). The transfer of electrons through the ETC is coupled to the pumping of hydrogen ions from the matrix to the intermembrane space by complexes I, III and IV. Protein pumping generates the proton motive force, composed of a small chemical component (DpH) and the large electrical component membrane potential (Dψ), which Complex V (the ATP synthase) uses to generate ATP from ADP and Pi (i.e., oxidative phosphorylation). In addition to generating NADH and FADH2, the TCA cycle generates intermediates that can funnel into multiple biosynthetic metabolic pathways to produce glucose, amino acids, lipids, heme and nucleotides. Thus, the mitochondria operate as a central hub of both catabolic (the breakdown of large macromolecules to produce energy) and anabolic (the production of large macromolecules from small metabolic intermediates using energy) metabolism (Fig. 1).
Figure 1. Mitochondria function as bioenergetic, biosynthetic and signaling organelles.

Cancer cells catabolize pyruvate and glutamine through the TCA cycle, resulting in the generation of reducing equivalents such as NADH that donate electrons to the ETC. The ETC generates a proton gradient that is used for production of ATP, i.e., oxidative phosphorylation (blue). TCA cycle intermediates can also be directed into biosynthetic pathways (green) that allow for the production of macromolecules (lipids, amino acids and nucleotides). Finally, mitochondrial production of ROS and metabolites act as signaling molecules to alter protein function. Mitochondrial one-carbon metabolism produces NADPH to prevent accumulation of ROS in the mitochondrial matrix. NADPH maintains antioxidant activity of glutathione peroxidase (GPX) and thioredoxin reductases (TrxRs). TA, aminotransferase; VDAC, voltage-dependent anion channel. TRX, thioredoxin; GSH, glutathione; SOD2, superoxide dismutase 2, mitochondrial; 5,10-CH2-THF, 5,10-methylene-tetrahydrofolate.
Mitochondria function as signaling organelles
Mitochondria have to alter their bioenergetic and biosynthetic functions to meet the metabolic demands of the cell. Furthermore, to ensure that the mitochondria have the ability to meet the metabolic demands of the cell, the mitochondria have to continuously communicate their fitness to the rest of the cell. We postulate that when cells receive cues to begin proliferation, a metabolically demanding process, an internal mitochondrial metabolic checkpoint is assessed before robustly activating transcriptional programs needed for cell proliferation. There are two modes of communication between the mitochondria and the rest of the cell: anterograde and retrograde signaling. Anterograde signaling denotes signal transduction from the cytosol to the mitochondria, which occurs by the rapid sequestration of calcium into the mitochondrial matrix in the presence of high cytosolic calcium. This increase in mitochondrial calcium activates multiple TCA cycle enzymes to stimulate mitochondrial oxidative metabolism. Signal transduction from the mitochondria to the cytosol is referred to as retrograde signaling (Fig. 1). Initially, the discovery that cytochrome c is released to initiate apoptosis in mammals sparked the idea of retrograde signaling. Today, there are multiple mechanisms of retrograde signaling, including the release of metabolites and ROS. Metabolites such as citrate may be released into the cytosol and cleaved by ATP citrate lyase to yield acetyl-CoA and oxaloacetate13. Acetyl-CoA levels can regulate protein acetylation, a reversible post-translational modification that alters protein activity14. ROS (in the nanomolar range) such as hydrogen peroxide (H2O2) released into the cytosol from the mitochondria can reversibly oxidize certain thiols within proteins to modify protein function15. It is important to note the emergence of several modes of mitochondrial signaling, including the expression of phosphatases16 and kinases17 in the mitochondrial matrix as well as the presence of signaling platforms on the mitochondrial outer membrane18,19. Thus, the concept of mitochondria as signaling organelles is burgeoning20.
Mitochondrial metabolism is required for tumor growth
Currently, the consensus in the cancer metabolism field is that tumor cells robustly engage in both glycolysis and mitochondrial metabolism to provide the necessary building blocks for macromolecule (nucleotides, lipids and amino acids) synthesis as well as ATP and NADPH, which are essential for cell proliferation21. The high rate of glycolysis in tumor cells is a consequence of deregulating signaling pathways such as the PI3K pathway and activation of oncogenes such as MYC and KRAS, which allows for the generation of glycolytic intermediates that can funnel into multiple subsidiary biosynthetic pathways necessary for cell proliferation, such as the pentose phosphate pathway for NADPH and nucleotide production22. Consequently, many tumors are addicted to glucose. Oncogenic activation also increases mitochondrial metabolism to generate ATP and TCA cycle intermediates used as precursors for macromolecule synthesis23. For example, the TCA cycle intermediate citrate is exported to the cytosol for lipid synthesis (acetyl-CoA) and nucleotide synthesis (oxaloacetate)24. It is essential for cells to replenish TCA cycle intermediates that are siphoned for the biosynthesis of macromolecules. One mechanism by which cells restore their TCA cycle intermediates is through the stepwise oxidation of glutamine to generate the TCA cycle intermediate α-ketoglutarate, which is further metabolized through the TCA cycle to produce oxaloacetate25. Subsequently, oxaloacetate can combine with acetyl-CoA to generate another molecule of citrate for macromolecule synthesis. Thus, many cancer cells exhibit addiction to glutamine. As a consequence of oxidative metabolism, which occurs in cancer cells, copious amounts of ROS are produced by the mitochondrial ETC. The high levels of mitochondrial ROS activate signaling pathways proximal to the mitochondria to promote cancer cell proliferation and tumorigenesis26. However, if the ROS are allowed to accumulate, cells will undergo death27. Therefore, cancer cells generate an abundance of NADPH in the mitochondria and the cytosol to sustain high antioxidant activity and prevent the buildup of potentially detrimental ROS28,29. Thus, both glucose-dependent metabolic pathways and mitochondrial metabolism are essential for tumor cell proliferation.
Although the majority of cancer cells display functional mitochondria, there are small subsets of cancer cells that display mutations in TCA cycle proteins or in subunits of various mitochondrial ETC complexes30,31. Notably, loss of function in genes encoding the TCA cycle enzymes succinate dehydrogenase and fumarate hydratase result in accumulation of succinate and fumarate, respectively30. These cancer cells that are dependent on glycolysis for ATP production should have impaired mitochondrial metabolism, preventing the generation of TCA cycle intermediates and ROS. However, recent studies demonstrated that tumor cells with impaired TCA cycle or ETC function cannot generate ATP but are still able to generate the TCA cycle intermediate citrate by ‘reductive carboxylation’, a process where glutamine generates α-ketoglutarate, which takes a reverse path to produce citrate32–34. In these TCA cycle or ETC mutant cells, the increased rate of glycolysis exclusively provides the ATP necessary for cellular proliferation and survival. In addition, glycolysis also provides metabolic intermediates for macromolecule synthesis. The TCA cycle or ETC mutant cancer cells also generate ROS for cell proliferation10,35. Studies have consistently demonstrated that diminishing or ablating TCA cycle or ETC proteins that enhance tumorigenesis and metastasis requires an increase in the rate of mitochondrial ROS production36–40. Taken together, these observations indicate that some tumors can rely exclusively on glycolysis to meet the bioenergetics needs of the cancer cell, but mitochondrial function is still required to meet the biosynthetic demands and ROS generation required for cell proliferation41.
Targeting bioenergetics for cancer therapy
The majority of ATP in tumor cells is produced by the mitochondria42,43. Nevertheless, as noted in the previous section, upregulating glycolysis to produce ATP can compensate for the lack of ATP production by the mitochondria. Thus, targeting mitochondrial ATP production for cancer therapy might not be an effective strategy. However, we postulate that there are three reasons to be optimistic that targeting mitochondrial ATP production will emerge as a viable therapeutic strategy against cancer. First, the centers of many solid tumors are poorly perfused and exist in nutrient-poor environments with limited glucose and oxygen44. These tumors typically are not highly proliferative but continue to survive45. The ETC is able to function optimally at oxygen levels as low as 0.5%46. Therefore, poorly perfused tumors have limited glucose availability but just enough oxygen to generate mitochondrial ATP. Consequently, a drug blocking mitochondrial ATP production would induce cell death in poorly perfused tumors. Second, there are subsets of tumors that show a heavy dependence on oxidative phosphorylation for ATP47–49. These tumor cells are likely to be sensitive to drugs that limit mitochondrial ATP production owing to a failure of glycolytic compensation. Third, inhibiting mitochondrial ATP production would synergize with therapies that diminish glycolysis, such as the clinically used inhibitors of the PI3K signaling pathway50. The key consideration in targeting mitochondrial ATP production is that normal cells use mitochondrial ATP production for survival; thus, the therapeutic index may be limited. The only exception would be if cancer cells selectively uptake the inhibitors of mitochondrial ATP production compared to normal cells. On the basis of recent evidence, we suggest that the widely used antidiabetic drug metformin may be a viable anticancer agent that targets mitochondrial ATP production without invoking toxicity in normal tissues.
In diabetic patients, the therapeutic effect of metformin is a result of decreasing hepatic gluconeogenesis to diminish circulating insulin levels51. Epidemiological studies have suggested that patients taking metformin to control their blood glucose levels are less likely to develop cancer52. In addition, metformin increases the survival rate of patients that have already developed cancer53. Furthermore, multiple laboratory-based studies have also provided evidence that metformin may serve as an anticancer agent54–56. Owing to the strong retrospective clinical evidence and laboratory-based experiments, there are more than 100 ongoing clinical trials assessing metformin’s anticancer effects in combination with current standard treatments57.
There are two mechanisms to explain the antitumor effects of metformin that are not necessarily mutually exclusive58. First, metformin decreases blood glucose and, consequently, circulating insulin levels. Insulin is a known mitogen for many cancer cells. Insulin and insulin growth factors (IGFs) stimulate the protumorigenic PIK3 signaling pathway in tumor cells that are positive for insulin and/or insulin growth factor receptor59. It is important to note that not all cancers are insulin sensitive, and therefore reduction of insulin levels would be irrelevant. A second possible mechanism by which metformin acts as an anticancer agent is through inhibition of mitochondrial ETC complex I to diminish tumor growth (Fig. 2). In 2000, two studies demonstrated that metformin in vitro inhibits mitochondrial complex I60,61. Recent work has provided a mechanistic understanding of how metformin and related biguanides inhibit complex I62. Yet, it was only recently shown in experimental models of cancer that the in vivo antitumorigenic effects of metformin are also dependent on inhibiting mitochondrial complex I63. Specifically, metformin inhibits mitochondrial ATP production and induces cell death when glycolytic ATP diminishes because of limited glucose availability. In the presence of ample glucose, metformin decreases proliferation through mechanisms that are not fully understood. Like other complex I inhibitors in prostate cancer cells, metformin does not limit the ability to generate TCA cycle intermediates owing to induction of glutamine-dependent reductive carboxylation64. However, in breast cancer cells, metformin diminishes TCA cycle intermediate production through complex I inhibition65. A leading mechanism to explain metformin’s antiproliferative effect is that metformin decreases mitochondrial ATP production, resulting in AMPK activation and diminished mTOR activity, a necessity for the anabolism of proliferating cells66. It is likely that this mechanism cannot solely explain the antiproliferative effect of metformin as many cancers that are unable to activate AMPK are still susceptible to the drug67.
Figure 2. Targeting mitochondrial bioenergetic capacity.

Mitochondrial ATP generation is necessary for tumorigenesis, and few molecules have demonstrated success in preclinical models of cancer. Specifically, (1) the biguanides metformin and phenformin inhibit mitochondrial complex I; (2) VLX600 inhibits the ETC at multiple sites; (3) tigecycline inhibits the mitochondrial ribosomal machinery and consequently the translation of ETC subunits; (4) gamitrinib is an inhibitor of mitochondrial chaperone proteins, such as TRAP-1 and HSP-90. Loss of these chaperones decreases ETC complex stability and subsequently reduces electron transport function. These therapies all share the same ultimate outcome with a decrease in mitochondrial ETC function to reduce mitochondrial bioenergetic capacity. mtDNA, mitochondrial DNA.
An important concern is whether metformin, as an anticancer agent, has a favorable therapeutic index. Metformin requires organic cation transporters (OCTs) that are present in a few tissues, such as liver and kidney68. Thus, the current dosing of metformin for diabetes has a remarkable safety profile. Tumor cells can also express OCTs to allow the uptake of metformin. However, not all tumor cells express OCTs; thus, metformin would not accumulate in these tumors and therefore not inhibit mitochondrial complex I. The uptake of the positively charged metformin at normal pH into the mitochondrial matrix to inhibit complex I requires a robust mitochondrial membrane potential63. Going forward, clinical trials using metformin as an anticancer agent should assess the expression levels of OCTs and mitochondrial genes in the tumors to identify those likely to be susceptible to metformin, that is, those with highest expression. The combination therapy of metformin with PI3K inhibitors that reduce glucose uptake and glycolysis will also be more efficacious than treatment with metformin alone. Lastly, it is not clear whether the antidiabetic dosing of metformin used in current clinical trials will allow for accumulation to levels necessary to inhibit mitochondrial complex I in tumors. It might be possible to escalate the dosing of metformin to higher levels than those currently used for diabetes without causing toxicity.
An alternative to metformin is the drug phenformin, another biguanide that inhibits mitochondrial complex I. Phenformin is less polar and more lipid soluble and exhibits a higher affinity for mitochondrial membranes than metformin. Phenformin is readily transported into tumor cells compared to metformin; thus, phenformin displays greater antineoplastic activity69. Recently, it has been demonstrated that phenformin also inhibits mitochondrial complex I to exert its antitumor effects in experimental models of cancer70. Additionally, cells impaired in glucose utilization were most sensitive to phenformin70. An important drawback of phenformin clinically is that phenformin increases the incidence of lactic acidosis, and thus it has been withdrawn for use in humans in most parts of the world. By contrast, metformin has a lower risk of inducing lactic acidosis and an excellent safety profile. Nevertheless, multiple experimental studies using phenformin have provided insight into which cancers phenformin may be most effective at treating. Specifically, studies have demonstrated that phenformin is effective in an experimental model of non–small-cell lung carcinoma (NSCLC) driven by oncogenic Kras and loss of LKB1 but not in NSCLC driven by oncogenic Kras and loss of p53 (ref. 71). Phenformin also increases the therapeutic benefit of BRAF inhibitors in melanoma driven by BRAFV600E mutations72. BRAF inhibitors increased mitochondrial respiration in BRAFV600E melanomas, making them susceptible to phenformin72. On the basis of these studies, it is worth exploring phenformin as a possible cancer therapy as lactic acidosis can be monitored.
Besides the biguanide family, other compounds and pathways have recently been identified in preclinical models as possible therapeutic approaches that ultimately seem to impair mitochondrial bioenergetic capacity (Fig. 2). Although these compounds have not undergone clinical trials for safety and therapeutic efficacy, they highlight the progress in targeting mitochondrial ATP production in cancer cells as a therapeutic strategy. A recent study identified the compound VLX600, an ETC inhibitor that markedly reduced colon cancer tumor growth73. VLX600 displayed an antitumor effect in conditions where glucose availability was limiting, such as in the center of the tumor or in tumor cells where the rate of proliferation produces such a high bioenergetic demand that it cannot be met through a compensatory induction of glycolysis.
Emerging data suggest that, in addition to the effect of direct ETC inhibitors, diminishing mitochondrial protein translation and stability may provide another opportunity to impede mitochondrial bioenergetic capacity. The US Food and Drug Administration–approved antibiotic tigecycline inhibits mitochondrial protein translation and displays robust antitumorigenic properties in numerous experimental models of leukemia74. There are 13 subunits of the ETC that are encoded by mitochondrial DNA, and disrupting translation of these proteins leads to major defects in mitochondrial ETC function. Intriguingly, leukemic cells have been shown to perform higher rates of fatty acid oxidation, and therefore they may be more dependent on mitochondrial ATP production for survival. Protein stability of ETC proteins is maintained in part by mitochondria-localized chaperones such as HSP90 and TRAP1, which are abundant in tumor cells. Gamitrinib, a small-molecule inhibitor of HSP90 and TRAP-1 ATPase activity, was engineered to selectively accumulate in the mitochondria, diminish mitochondrial ATP production and display antitumorigenic properties in experimental models of cancer75. Collectively, these data suggest that, although historically ignored in tumorigenesis, mitochondrial energy production is required for many cancer types, and there is strong evidence to suggest that its inhibition may provide a valuable clinical target.
Targeting mitochondrial biosynthetic function
As noted above, TCA cycle intermediates are used for the biosynthesis of macromolecules. Glutamine is a major carbon source to replenish the TCA cycle intermediates and to sustain their utilization for biosynthesis in many tumor cells76. Glutamine is the most abundant amino acid in plasma. MYC- and KRAS-driven cancer cells are addicted to glutamine10,77–79. Glutamine is converted into glutamate, a precursor of glutathione, by glutaminases (GLSs). Next, glutamate is converted into α-ketoglutarate either by glutamate dehydrogenase (GLUD) or by aminotransferases. Glutamine catabolism to α-ketoglutarate fulfills multiple functions, including (i) generation of the TCA cycle reducing equivalents NADH and FADH2, which are used by the ETC to generate ATP; (ii) replenishment of TCA cycle intermediates, as these intermediates are siphoned off to support macromolecule biosynthesis; (iii) production of NADPH by malic enzyme; and (iv) utilization of α-ketoglutarate as a substrate for dioxygenases such as prolyl hydroxylases, histone demethylases and 5-methylcytosine hydroxylases (Fig. 3). Thus, targeting glutamine catabolism is an appealing idea. Indeed, two specific GLS inhibitors, compound 968 (ref. 80) and bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide81, diminish glutamine catabolism and delay tumor growth in experimental models of cancer. Targeting glutamate conversion to α-ketoglutarate by aminotransferases with nonspecific inhibitors also diminishes tumor growth82,83. These studies give credence to the development of drugs that target glutamine catabolism. It is important to note that not all tumors display addiction to glutamine catabolism. However, using [13C]glutamine infusion in patients to determine whether glutamine is being catabolized will help predict which tumors will respond to therapies targeting glutamine metabolism76.
Figure 3. Targeting mitochondrial biosynthetic production.

Mitochondrial TCA cycle intermediates are siphoned off for utilization as precursors for macromolecule production. Specifically, citrate is transported into the cytosol, where it is converted into acetyl-CoA (a precursor for fatty acid synthesis) and oxaloacetate. Thus, the TCA cycle intermediates have to be replenished for the cycle to continue functioning. Many cancer cells use glutamine, the most abundant amino acid in plasma, to replenish the TCA cycle. Glutaminase converts glutamine to glutamate, which is a precursor for glutathione synthesis. Next, glutamate is converted into the TCA cycle intermediate α-ketoglutarate by GLUD or aminotransferases (TA). The α-ketoglutarate generated from glutamine replenishes the TCA cycle and is also used as a substrate for dioxygenase-dependent reactions. Inhibition of GLS (1) or TA/GLUD (2) have shown efficacy in preclinical models of cancer. (3) Short-term inhibition of autophagy has also shown therapeutic efficacy in preclinical models of cancer. Autophagy generates metabolic substrates for mitochondrial metabolism, particularly glutamine.
Another approach to diminish the replenishment of TCA cycle intermediates is targeting autophagy in established tumors. Autophagy sustains mitochondrial metabolism in tumor cells by providing substrates such as glutamine to replenish TCA cycle intermediates, which are used for macromolecule biosynthesis12. Autophagy inhibition in mouse models of BRAFV600-driven tumors impairs mitochondrial metabolism and increases survival of BRAFV600 tumor–bearing mice84. Similar to the synthetic lethality observed with phenformin and BRAFV600 inhibitors in melanoma, the combination of BRAFV600 inhibitors with autophagy inhibitors is also likely to be successful. Chloroquine, a US Food and Drug Administration–approved small-molecule inhibitor of autophagy in ongoing clinical trials for cancer, has been demonstrated to impair mitochondrial metabolism and diminish tumor growth. One cautionary note about autophagy inhibitors is that they should be used acutely as long-term use induces toxicity. On the basis of these studies, we suggest that acute administration of chloroquine in combination with phenformin might be effective, as this would target both TCA cycle flux and ETC function.
Targeting mitochondrial redox capacity
Dietary antioxidants have been widely used as anticancer agents. Yet, they have consistently failed to reduce tumor burden in prospective human clinical trials85. Furthermore, antioxidants raised the incidence of lung cancer in smokers and accelerated tumor burden in mouse models of NSCLC86,87. One reason for the failure of dietary antioxidants is that they do not dampen localized mitochondrially generated ROS.
Mitochondria are proximal to the signaling pathways that are responsive to ROS. Thus, mitochondrially targeted antioxidants effectively reduce cell proliferation in vitro and tumorigenesis in vivo10,40,88,89. Mitochondrially targeted antioxidants have undergone clinical trials in patients suffering from Parkinson’s disease and thus could undergo clinical trials as anticancer agents90. A caveat to this approach is that mitochondrial ROS are used by normal cells including immune and stem cells to function optimally15, and thus the therapeutic index of mitochondrial-targeted antioxidants is not clear.
Cancer cells increase their antioxidant capacity to counterbalance the increased production of ROS by cancer cells91. This permits cancer cells to generate high levels of H2O2 to activate proximal signaling pathways that promote proliferation without allowing buildup of ROS to levels that would otherwise induce cancer cell death or senescence. NADPH is required to maintain multiple antioxidant defense systems. The cytosol has multiple sources of NADPH generation, including the pentose-phosphate pathway, malic enzyme 1, IDH1 and one-carbon metabolism28. One-carbon metabolism in the cytosol and mitochondria generates folate intermediates required for nucleotide synthesis as well as NADPH92. NADPH generation in the mitochondria occurs from one-carbon metabolism29, and one-carbon metabolism in the mitochondria is initiated by serine catabolism by SHMT2. A recent study demonstrated that SHMT2 is necessary for redox balance in Myc-transformed cells under hypoxia, a metabolic feature common to many tumors93. Thus, diminishing SHTM2 levels decreased tumor growth93. RNA profiling of 1,454 metabolic enzymes across 1,981 tumors covering 19 cancer types identified MTHFD2, an enzyme in the mitochondrial one-carbon metabolism, as consistently being the differentially expressed enzyme between cancer and normal cells94. Loss of MTHFD2 increases ROS levels and sensitizes cancer cells to oxidant-induced cell death94. MTHFD2 is not highly expressed in normal adult proliferating cells; thus, MTHFD2 may represent a viable therapeutic target (Fig. 4). Furthermore, targeting mitochondrial one-carbon metabolic enzymes in conjugation with other therapies known to increase ROS including superoxide dismutase 1 (ref. 95), glutathione synthesis96 and glutaminase97 inhibition might be effective. It is interesting to note that there has been a shift in ongoing studies from using dietary antioxidants to examining whether inhibition of antioxidants is an effective cancer therapy95,96,98.
Figure 4. Targeting mitochondrial redox signaling and balance.

Mitochondrial ETC generates superoxide (O2•−) that can be converted into hydrogen peroxide (H2O2) by SOD2 or traverse through voltage-dependent anion channels (VDACs) into the cytosol. Subsequently, SOD1 converts superoxide into H2O2 to activate redox-dependent signaling. Mitochondrial ROS are necessary for cancer cell proliferation. Thus, mitochondrial-targeted antioxidants are effective in decreasing cancer cell proliferation. Mitochondria prevent accumulation of ROS to levels that would be detrimental to mitochondrial metabolism by increasing antioxidant capacity. NADPH is necessary to maintain the antioxidant function of glutathione peroxidase (GPXs) and thioredoxin reductase (TrxR). Mitochondria maintain redox balance by using SHMT2 and MTHFD2, enzymes in one-carbon metabolism, indicating that this pathway may be a therapeutic target.
Conclusion
The pioneering work, which started in the early 1990s, demonstrated that mitochondria-localized antiapoptotic proteins could be successful targets for cancer therapy99. However, only in the past few years has mitochondrial metabolism emerged as a target for cancer therapy, owing to the reemergence of mitochondria as a central metabolic organelle required for tumorigenesis. There remain three major challenges in translating preclinical effective drugs that target mitochondrial metabolism to the clinical trials in patients. First, for any given drug that targets mitochondrial metabolism, the toxicity to normal cells needs to be established. Second, any drug has to traverse not only the cell membrane but also the two mitochondrial membranes. The conjunction of a lipophilic cation to small molecules increases the accumulation in the mitochondrial matrix to 1,000-fold greater in concentration than outside the cell100. The uptake of lipophilic cations into the mitochondria occurs because of the large membrane potential generated by the ETC across the mitochondrial inner membrane (Fig. 5). Commonly used chemotherapeutic agents such as cisplatin can be targeted to the mitochondrial matrix by conjugation to lipophilic cations to target mitochondrial DNA101. The third challenge is to understand more about the basic biology of mitochondrial metabolism in regulating tumorigenesis. This will allow for the design of combination therapies using mitochondrial metabolic inhibitors with other anticancer agents. For example, diminishing oncogenic signaling results in initial pancreatic tumor shrinkage but also in frequent relapse owing to the fraction of surviving cells that are highly dependent on mitochondrial bioenergetics and sensitive to inhibitors targeting oxidative phosphorylation102. It is an exciting time to begin thinking about rationally targeting mitochondrial metabolism.
Figure 5. Targeting mitochondrial metabolism.
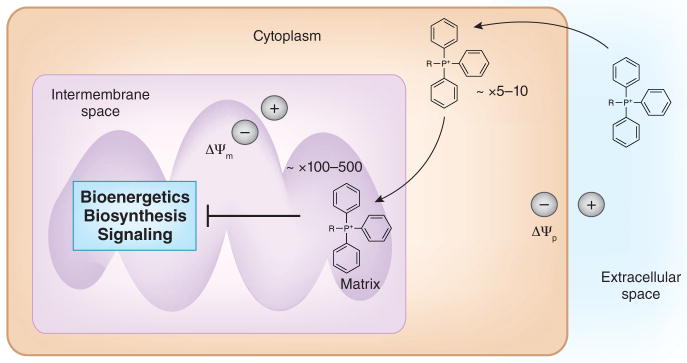
Small molecules conjugated to lipophilic cations such as tetraphenylphosphonium (TPP) result in their accumulation in the mitochondrial matrix. Lipophilic conjugated small molecules will first traverse across the plasma membrane and accumulate in the cytosol. This process is driven by the plasma membrane potential (Δψp). Subsequently, the mitochondrial membrane potential (Δψm) will drive the accumulation of these molecules into the mitochondria by several hundredfold.
Acknowledgments
This work is supported US National Institutes of Health grants R01CA123067 to N.S.C. and 5T32HL076139-10 to S.E.W. We apologize to all investigators whose work could not be cited due to reference limitations.
Footnotes
Competing financial interests
The authors declare competing financial interests: details accompany the online version of the paper.
References
- 1.Bonnet S, et al. A mitochondria-K channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 2.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- 3.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nat Rev Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 5.Cross CE, et al. Oxygen radicals and human disease. Ann Intern Med. 1987;107:526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 6.Weinhouse S. On respiratory impairment in cancer cells. Science. 1956;124:267–269. doi: 10.1126/science.124.3215.267. [DOI] [PubMed] [Google Scholar]
- 7.Weinhouse S, Millington RH, Wenner CE. Metabolism of neoplastic tissue. I The oxidation of carbohydrate and fatty acids in transplanted tumors. Cancer Res. 1951;11:845–850. [PubMed] [Google Scholar]
- 8.Wenner CE, Spirtes MA, Weinhouse S. Metabolism of neoplastic tissue. II A survey of enzymes of the citric acid cycle in transplanted tumors. Cancer Res. 1952;12:44–49. [PubMed] [Google Scholar]
- 9.Weinhouse S. Studies on the fate of isotopically labeled metabolites in the oxidative metabolism of tumors. Cancer Res. 1951;11:585–591. [PubMed] [Google Scholar]
- 10.Weinberg F, et al. Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci USA. 2010;107:8788–8793. doi: 10.1073/pnas.1003428107. This report provides genetic and pharmacologic evidence that mitochondrial metabolism and ROS are required for tumor growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fogal V, et al. Mitochondrial p32 protein is a critical regulator of tumor metabolism via maintenance of oxidative phosphorylation. Mol Cell Biol. 2010;30:1303–1318. doi: 10.1128/MCB.01101-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Guo JY, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes Dev. 2011;25:460–470. doi: 10.1101/gad.2016311. This report provides evidence that autophagy sustains mitochondrial metabolism that is essential for tumor growth. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wellen KE, et al. ATP-citrate lyase links cellular metabolism to histone acetylation. Science. 2009;324:1076–1080. doi: 10.1126/science.1164097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kaelin WG, Jr, McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012;48:158–167. doi: 10.1016/j.molcel.2012.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pagliarini DJ, et al. Involvement of a mitochondrial phosphatase in the regulation of ATP production and insulin secretion in pancreatic β cells. Mol Cell. 2005;19:197–207. doi: 10.1016/j.molcel.2005.06.008. [DOI] [PubMed] [Google Scholar]
- 17.Acin-Perez R, Gatti DL, Bai Y, Manfredi G. Protein phosphorylation and prevention of cytochrome oxidase inhibition by ATP: coupled mechanisms of energy metabolism regulation. Cell Metab. 2011;13:712–719. doi: 10.1016/j.cmet.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Huang LJ, et al. NH2-terminal targeting motifs direct dual specificity A-kinase-anchoring protein 1 (D-AKAP1) to either mitochondria or endoplasmic reticulum. J Cell Biol. 1999;145:951–959. doi: 10.1083/jcb.145.5.951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-κB and IRF 3. Cell. 2005;122:669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 20.Chandel NS. Mitochondria as signaling organelles. BMC Biol. 2014;12:34. doi: 10.1186/1741-7007-12-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ward PS, Thompson CB. Metabolic reprogramming: a cancer hallmark even Warburg did not anticipate. Cancer Cell. 2012;21:297–308. doi: 10.1016/j.ccr.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441–464. doi: 10.1146/annurev-cellbio-092910-154237. [DOI] [PubMed] [Google Scholar]
- 23.Dang CV. Links between metabolism and cancer. Genes Dev. 2012;26:877–890. doi: 10.1101/gad.189365.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hatzivassiliou G, et al. ATP citrate lyase inhibition can suppress tumor cell growth. Cancer Cell. 2005;8:311–321. doi: 10.1016/j.ccr.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 25.DeBerardinis RJ, Cheng T. Q’s next: the diverse functions of glutamine in metabolism, cell biology and cancer. Oncogene. 2010;29:313–324. doi: 10.1038/onc.2009.358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schieber M, Chandel NS. ROS function in redox signaling and oxidative stress. Curr Biol. 2014;24:R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov. 2013;12:931–947. doi: 10.1038/nrd4002. A superb review on targeting ROS for cancer therapy. [DOI] [PubMed] [Google Scholar]
- 28.Fan J, et al. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298–302. doi: 10.1038/nature13236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lewis CA, et al. Tracing compartmentalized NADPH metabolism in the cytosol and mitochondria of mammalian cells. Mol Cell. 2014;55:253–263. doi: 10.1016/j.molcel.2014.05.008. This paper reports the importance of mitochondrial one-carbon metabolism in generating mitochondrial NADPH. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang M, Soga T, Pollard PJ. Oncometabolites: linking altered metabolism with cancer. J Clin Invest. 2013;123:3652–3658. doi: 10.1172/JCI67228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wallace DC. A mitochondrial paradigm of metabolic and degenerative diseases, aging, and cancer: a dawn for evolutionary medicine. Annu Rev Genet. 2005;39:359–407. doi: 10.1146/annurev.genet.39.110304.095751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mullen AR, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature. 2012;481:385–388. doi: 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wise DR, et al. Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc Natl Acad Sci USA. 2011;108:19611–19616. doi: 10.1073/pnas.1117773108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Metallo CM, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature. 2012;481:380–384. doi: 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sullivan LB, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell. 2013;51:236–248. doi: 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guzy RD, Sharma B, Bell E, Chandel N, Schumacker P. Loss of the SdhB, but not the SdhA, subunit of complex II triggers reactive oxygen species–dependent hypoxia-inducible factor activation and tumorigenesis. Mol Cell Biol. 2008;28:718–731. doi: 10.1128/MCB.01338-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo DK, et al. Mitochondrial genome instability and ROS enhance intestinal tumorigenesis in APC(Min/+) mice. Am J Pathol. 2012;180:24–31. doi: 10.1016/j.ajpath.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petros JA, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci USA. 2005;102:719–724. doi: 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ishikawa K, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science. 2008;320:661–664. doi: 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 40.Porporato PE, et al. A mitochondrial switch promotes tumor metastasis. Cell Rep. 2014;8:754–766. doi: 10.1016/j.celrep.2014.06.043. [DOI] [PubMed] [Google Scholar]
- 41.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zu XL, Guppy M. Cancer metabolism: facts, fantasy, and fiction. Biochem Biophys Res Commun. 2004;313:459–465. doi: 10.1016/j.bbrc.2003.11.136. A report that highlights that most cancer cells generate the majority of their ATP from mitochondria. [DOI] [PubMed] [Google Scholar]
- 43.Fan J, et al. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol Syst Biol. 2013;9:712. doi: 10.1038/msb.2013.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jain RK, Munn LL, Fukumura D. Dissecting tumour pathophysiology using intravital microscopy. Nat Rev Cancer. 2002;2:266–276. doi: 10.1038/nrc778. [DOI] [PubMed] [Google Scholar]
- 45.Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891–899. doi: 10.1038/nrc1478. [DOI] [PubMed] [Google Scholar]
- 46.Rumsey WL, Schlosser C, Nuutinen EM, Robiolio M, Wilson DF. Cellular energetics and the oxygen dependence of respiration in cardiac myocytes isolated from adult rat. J Biol Chem. 1990;265:15392–15402. [PubMed] [Google Scholar]
- 47.Caro P, et al. Metabolic signatures uncover distinct targets in molecular subsets of diffuse large B cell lymphoma. Cancer Cell. 2012;22:547–560. doi: 10.1016/j.ccr.2012.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Vazquez F, et al. PGC1a expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. 2013;23:287–301. doi: 10.1016/j.ccr.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Haq R, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1a and MITF. Cancer Cell. 2013;23:302–315. doi: 10.1016/j.ccr.2013.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelman JA, et al. Effective use of PI3K and MEK inhibitors to treat mutant Kras G12D and PIK3CA H1047R murine lung cancers. Nat Med. 2008;14:1351–1356. doi: 10.1038/nm.1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bailey CJ, Turner RC. Metformin. N Engl J Med. 1996;334:574–579. doi: 10.1056/NEJM199602293340906. [DOI] [PubMed] [Google Scholar]
- 52.Evans JM, Donnelly LA, Emslie-Smith AM, Alessi DR, Morris AD. Metformin and reduced risk of cancer in diabetic patients. Br Med J. 2005;330:1304–1305. doi: 10.1136/bmj.38415.708634.F7. A seminal study that was the first to report a potential association of metformin use with reduced cancer incidence. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dowling RJ, Niraula S, Stambolic V, Goodwin PJ. Metformin in cancer: translational challenges. J Mol Endocrinol. 2012;48:R31–R43. doi: 10.1530/JME-12-0007. [DOI] [PubMed] [Google Scholar]
- 54.Buzzai M, et al. Systemic treatment with the antidiabetic drug metformin selectively impairs p53-deficient tumor cell growth. Cancer Res. 2007;67:6745–6752. doi: 10.1158/0008-5472.CAN-06-4447. [DOI] [PubMed] [Google Scholar]
- 55.Memmott RM, et al. Metformin prevents tobacco carcinogen–induced lung tumorigenesis. Cancer Prev Res (Phila) 2010;3:1066–1076. doi: 10.1158/1940-6207.CAPR-10-0055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tomimoto A, et al. Metformin suppresses intestinal polyp growth in ApcMin/+ mice. Cancer Sci. 2008;99:2136–2141. doi: 10.1111/j.1349-7006.2008.00933.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollak M. Overcoming drug development bottlenecks with repurposing: repurposing biguanides to target energy metabolism for cancer treatment. Nat Med. 2014;20:591–593. doi: 10.1038/nm.3596. [DOI] [PubMed] [Google Scholar]
- 58.Birsoy K, Sabatini DM, Possemato R. Untuning the tumor metabolic machine: targeting cancer metabolism: a bedside lesson. Nat Med. 2012;18:1022–1023. doi: 10.1038/nm.2870. [DOI] [PubMed] [Google Scholar]
- 59.Pollak M. The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159–169. doi: 10.1038/nrc3215. [DOI] [PubMed] [Google Scholar]
- 60.El-Mir MY, et al. Dimethylbiguanide inhibits cell respiration via an indirect effect targeted on the respiratory chain complex I. J Biol Chem. 2000;275:223–228. doi: 10.1074/jbc.275.1.223. [DOI] [PubMed] [Google Scholar]
- 61.Owen MR, Doran E, Halestrap AP. Evidence that metformin exerts its anti-diabetic effects through inhibition of complex 1 of the mitochondrial respiratory chain. Biochem J. 2000;348:607–614. [PMC free article] [PubMed] [Google Scholar]
- 62.Bridges HR, Jones AJ, Pollak MN, Hirst J. Effects of metformin and other biguanides on oxidative phosphorylation in mitochondria. Biochem J. 2014;462:475–487. doi: 10.1042/BJ20140620. A detailed study of how metformin and related biguanides inhibit mitochondrial oxidative phosphorylation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wheaton WW, et al. Metformin inhibits mitochondrial complex I of cancer cells to reduce tumorigenesis. eLife. 2014;3:e02242. doi: 10.7554/eLife.02242. A report that indicates that metformin’s anti-cancer effects in vivo are due to complex I inhibition. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Fendt SM, et al. Metformin decreases glucose oxidation and increases the dependency of prostate cancer cells on reductive glutamine metabolism. Cancer Res. 2013;73:4429–4438. doi: 10.1158/0008-5472.CAN-13-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Janzer A, et al. Metformin and phenformin deplete tricarboxylic acid cycle and glycolytic intermediates during cell transformation and NTPs in cancer stem cells. Proc Natl Acad Sci USA. 2014;111:10574–10579. doi: 10.1073/pnas.1409844111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hardie DG. AMPK: a target for drugs and natural products with effects on both diabetes and cancer. Diabetes. 2013;62:2164–2172. doi: 10.2337/db13-0368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Storozhuk Y, et al. Metformin inhibits growth and enhances radiation response of non-small cell lung cancer (NSCLC) through ATM and AMPK. Br J Cancer. 2013;108:2021–2032. doi: 10.1038/bjc.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Emami Riedmaier A, Fisel P, Nies AT, Schaeffeler E, Schwab M. Metformin and cancer: from the old medicine cabinet to pharmacological pitfalls and prospects. Trends Pharmacol Sci. 2013;34:126–135. doi: 10.1016/j.tips.2012.11.005. [DOI] [PubMed] [Google Scholar]
- 69.Appleyard MV, et al. Phenformin as prophylaxis and therapy in breast cancer xenografts. Br J Cancer. 2012;106:1117–1122. doi: 10.1038/bjc.2012.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Birsoy K, et al. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature. 2014;508:108–112. doi: 10.1038/nature13110. A study demonstrating that sensitivity to biguanides increases in cancer cells that harbor mutations in complex I genes or have impaired glucose utilization. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shackelford DB, et al. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013;23:143–158. doi: 10.1016/j.ccr.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yuan P, et al. Phenformin enhances the therapeutic benefit of BRAFV600E inhibition in melanoma. Proc Natl Acad Sci USA. 2013;110:18226–18231. doi: 10.1073/pnas.1317577110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang X, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nat Commun. 2014;5:3295. doi: 10.1038/ncomms4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Škrtić M, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer Cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chae YC, et al. Control of tumor bioenergetics and survival stress signaling by mitochondrial HSP90s. Cancer Cell. 2012;22:331–344. doi: 10.1016/j.ccr.2012.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hensley CT, Wasti AT, DeBerardinis RJ. Glutamine and cancer: cell biology, physiology, and clinical opportunities. J Clin Invest. 2013;123:3678–3684. doi: 10.1172/JCI69600. An excellent review of glutamine metabolism and its potential as a therapeutic target. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wise DR, et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci USA. 2008;105:18782–18787. doi: 10.1073/pnas.0810199105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gao P, et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762–765. doi: 10.1038/nature07823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gaglio D, et al. Oncogenic K-Ras decouples glucose and glutamine metabolism to support cancer cell growth. Mol Syst Biol. 2011;7:523. doi: 10.1038/msb.2011.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang JB, et al. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell. 2010;18:207–219. doi: 10.1016/j.ccr.2010.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Le A, et al. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 2012;15:110–121. doi: 10.1016/j.cmet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Thornburg JM, et al. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008;10:R84. doi: 10.1186/bcr2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Qing G, et al. ATF4 Regulates MYC-mediated neuroblastoma cell death upon glutamine deprivation. Cancer Cell. 2012;22:631–644. doi: 10.1016/j.ccr.2012.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Strohecker AM, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E-driven lung tumors. Cancer Discov. 2013;3:1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Bjelakovic G, Gluud C. Surviving antioxidant supplements. J Natl Cancer Inst. 2007;99:742–743. doi: 10.1093/jnci/djk211. [DOI] [PubMed] [Google Scholar]
- 86.Sayin VI, et al. Antioxidants accelerate lung cancer progression in mice. Sci Transl Med. 2014;6:221ra15. doi: 10.1126/scitranslmed.3007653. [DOI] [PubMed] [Google Scholar]
- 87.The α-Tocopherol β-Carotene Cancer Prevention Study Group. The effect of vitamin-E and β-carotene on the incidence of lung-cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 88.Cheng G, et al. Mitochondria-targeted drugs synergize with 2-deoxyglucose to trigger breast cancer cell death. Cancer Res. 2012;72:2634–2644. doi: 10.1158/0008-5472.CAN-11-3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nazarewicz RR, et al. Does scavenging of mitochondrial superoxide attenuate cancer prosurvival signaling pathways? Antioxid Redox Signal. 2013;19:344–349. doi: 10.1089/ars.2013.5185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Snow BJ, et al. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov Disord. 2010;25:1670–1674. doi: 10.1002/mds.23148. [DOI] [PubMed] [Google Scholar]
- 91.DeNicola GM, et al. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106–109. doi: 10.1038/nature10189. The paper identifies mitochondrial one-carbon metabolic enzyme MTHFD2 as a potential cancer therapeutic target across multiple tumors. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Locasale JW. Serine, glycine and one-carbon units: cancer metabolism in full circle. Nat Rev Cancer. 2013;13:572–583. doi: 10.1038/nrc3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ye J, et al. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov. 2014 doi: 10.1158/2159-8290.CD-14-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Nilsson R, et al. Metabolic enzyme expression highlights a key role for MTHFD2 and the mitochondrial folate pathway in cancer. Nat Commun. 2014;5:3128. doi: 10.1038/ncomms4128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Glasauer A, Sena LA, Diebold LP, Mazar AP, Chandel NS. Targeting SOD1 reduces experimental non-small-cell lung cancer. J Clin Invest. 2014;124:117–128. doi: 10.1172/JCI71714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Trachootham D, et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell. 2006;10:241–252. doi: 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 97.Martin-Rufián M, et al. Both GLS silencing and GLS2 overexpression synergize with oxidative stress against proliferation of glioma cells. J Mol Med (Berl) 2014;92:277–290. doi: 10.1007/s00109-013-1105-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Raj L, et al. Selective killing of cancer cells by a small molecule targeting the stress response to ROS. Nature. 2011;475:231–234. doi: 10.1038/nature10167. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Fesik SW. Promoting apoptosis as a strategy for cancer drug discovery. Nat Rev Cancer. 2005;5:876–885. doi: 10.1038/nrc1736. [DOI] [PubMed] [Google Scholar]
- 100.Murphy MP, Smith RA. Targeting antioxidants to mitochondria by conjugation to lipophilic cations. Annu Rev Pharmacol Toxicol. 2007;47:629–656. doi: 10.1146/annurev.pharmtox.47.120505.105110. An excellent review that details mechanisms by which small molecules preferentially can be delivered into mitochondrial matrix. [DOI] [PubMed] [Google Scholar]
- 101.Marrache S, Pathak RK, Dhar S. Detouring of cisplatin to access mitochondrial genome for overcoming resistance. Proc Natl Acad Sci USA. 2014;111:10444–10449. doi: 10.1073/pnas.1405244111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Viale A, et al. Oncogene ablation-resistant pancreatic cancer cells depend on mitochondrial function. Nature. 2014;514:628–632. doi: 10.1038/nature13611. [DOI] [PMC free article] [PubMed] [Google Scholar]