

Summary
Initially identified as mammalian homologs to yeast Ste20 kinases, the Mst1/2 kinases have been widely investigated subsequent to their rediscovery as key components of the Hippo tumor suppressor pathway in flies. To date, our understanding of Mst substrates and downstream signaling outstrips our knowledge of how these enzymes are controlled by upstream signals. While much remains to be discovered regarding the mechanisms of Mst regulation, it is clear that Mst1 kinase activity is governed at least in part by its state of dimerization, including self-association and also heterodimerization with a variety of other signaling partners. Here, we review the basic architecture of Mst signaling and function and discuss recent advances in our understanding of how these important kinases are regulated.
Keywords: Serine/threonine protein kinases, signal transduction, tumor suppressor, dimerization
Hippo pathway architecture in Drosophila and mammalian cells
The serine/threonine-specific protein kinases Mammalian Sterile-Twenty-like (Mst)1/2 are the defining components of the Hippo signaling pathway. This pathway controls organ size and tissue homeostasis by regulating apoptosis and cell proliferation [1, 2]. Mst1/2 were initially discovered in mammalian cells as members of the Ste20 family [3, 4] and, shortly thereafter, biochemically isolated as kinases activated by extreme stress [5]. Subsequently, an ortholog of these kinases, as well as other core components of what came to be known as the Hippo pathway, were discovered in Drosophila melanogaster by genetic screens designed to identify genes that regulate organ size. Following these pioneering studies in flies, conditional gene-deletion studies in mice confirmed a conserved role for Mst1/2 as a regulator of organ size and as a potential tumor suppressor [6–9]. Since that time, a great deal of attention has been paid towards defining the elements and regulation of the Hippo pathway in both flies and in mammals. However, it is becoming increasingly clear that while most of the Hippo pathway machinery is highly conserved in multicellular organisms, the organization and functions of this pathway differ substantially in various model systems. In this review, we focus on the regulation of mammalian Mst1/2, in particular with respect to how this process differs from what has been learned in Drosophila.
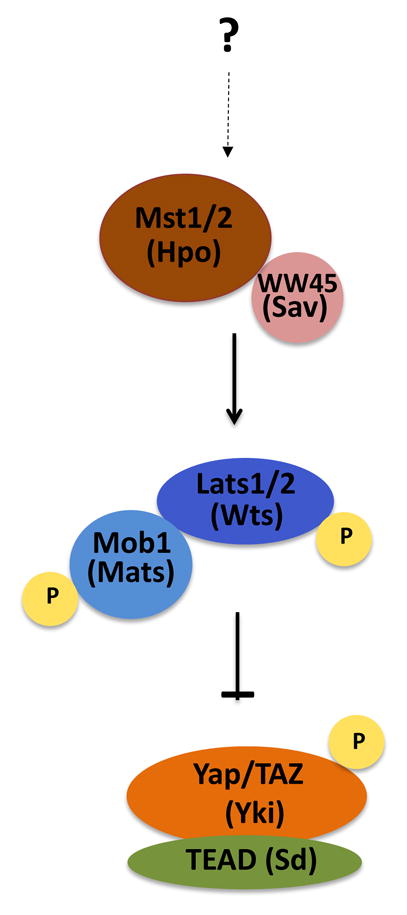
Four proteins - Hpo, Sav, Wts and Mats - constitute the core components of the Hippo pathway in Drosophila, homologous to mammalian Mst1/2, WW45, Lats1/2, and Mob1 respectively (Box 1). In mammals, Mst1/2, in conjunction with WW45, phosphorylates Mob1 and Lats1/2, leading to their activation [10, 11]. Activated Lats1/2 phosphorylates and inactivates a transcriptional co-activator Yes-associated protein (Yap) and/or its partner Taz by promoting its cytoplasmic sequestration [12, 13]. Yap is an oncogene that enhances transcription of genes involved in cell proliferation by partnering with TEAD family of transcription factors; inactivation of Yap by the Hippo pathway kinase cascade suppresses cell proliferation and promotes apoptosis [1, 13, 14].
Box 1. Mst-less Hippo signaling: a cautionary note regarding nomenclature.
Strictly defined, the core Hippo pathway comprises the four signaling proteins Hpo/Mst, its partner Sav/WW45, its substrate Wts/Lats, and the Wts/Lats binding partner Mob/Mats. However, in recent years the term has also been more loosely applied to any signaling cascade that results in inactivation of the transcriptional co-activator Yki/Yap, whether or not Hpo/Mst is involved. As it seems both formally incorrect and misleading to refer to the Hippo pathway absent involvement of the protein for which the pathway is named, we recommend restricting the use of this term as originally defined. It would be more appropriate to use a more general term, such as the Yap pathway, to describe pathways that regulate Yap phosphorylation independently of Mst.
While the core kinase cascade of the Hippo pathway leading from the protein kinase Hpo/Mst to the transcriptional coactivator Yap/Yki is well-established and highly conserved between insects and mammalian organisms, the upstream regulation of this pathway appears to be organized differently in different model organisms. Genetic experiments in Drosophila have uncovered several upstream regulators of the Hippo pathway, including the apical membrane proteins Merlin (Mer), Expanded (Ex), and Kibra (Fig. 1A). However, it is important to note that in mammalian cells, direct links between Mst1/2 and these membrane proteins have not been established, and recent evidence strongly suggests that the mammalian Hippo pathway deviates significantly from the fly model. Indeed, in some cases it is unclear if the central kinase for which the pathway is named is a necessary component of the signaling module in mammalian cells (Box 1). For example, genetic experiments in mice have linked Merlin to the Hippo pathway components Lats and Yap, but not to Mst1/2 [15]. Rather than acting upstream of Mst1/2, it has been suggested that Merlin instead acts in parallel to Mst1/2 to activate Lats [16]. According to this model, Merlin directly binds to Lats and recruits it to the plasma membrane, where it is subsequently phosphorylated by active Mst1/2 (Fig. 1B). While these findings help clarify the heretofore obscure biochemical connection between Merlin and Mst, questions remain regarding their physiological relevance. One such question is why the overgrowth phenotype of Mer mutants in Drosophila is so much milder that that of Hpo, Sav, or Wts. The most straightforward interpretation of the data is that the role of Mer in Hippo pathway activation is relatively minor compared to those of the core components, but this is a question that will certainly bear further examination given the important role of Merlin in human cancer.
Fig. 1. Upstream links to the Hippo pathway in flies and mammals.

(A) In Drosophila, Ex, Mer and Kibra upon activation by Fat, activate Hpo-Sav complex. Activated Hpo-Sav complex phosphorylates and activates Wts-Mts complex. Activated Wt-Mts phosphorylates Yki, leading to its cytoplasmic retention and inactivation. In the absence of Hpo signaling, Yki binds transcription factor Sd and promotes cell proliferation and inhibition of apoptosis. (B) In mammals, Merlin interaction with Lats leads to recruitment of Lats to the plasma membrane. At the plasma membrane Lats is phosphorylated and activated by Mst. Activated Lats phosphorylates Yap leading to its inactivation and cytoplasmic retention. Pointed arrowheads indicate activation and blunted ends indicate inhibition. Solid lines indicate direct interaction while dashed lines indicate unknown mechanism.
Interestingly, disruption of the MST1 gene in humans [17] or in mice [6] is not associated with tissue overgrowth, but rather with low numbers of naïve T-cells, mediated by signaling failure from an Mst1 complex that is normally activated downstream of T-cell receptor ligation or chemokine stimulation [18]. This Mst-regulated signaling pathway, which affects T cell polarization and integrin LFA-1 clustering, contains components that are not typical of the core Hippo signaling pathway as initially defined in flies, and also is independent of Yap/Taz [6]. Here, the small GTPase Rap1 serves as an upstream activator, coupling to Mst1 via the Rap1-binding protein Rassf5b (a.k.a. NORE1b or RAPL), and the downstream signal output is clustering of the integrin Lymphocyte Function Associated antigen (LFA)-1. It has been proposed that the regulation of LFA-1 clustering is mediated by Mst1-catalyzed phosphorylation of the exchange factor DENND1C, which then activates the vesicle transport GTPase Rab13, thus facilitating the delivery of the LFA-1 to the leading edge of lymphocytes [19]. Mst1 also affects lymphocyte motility, a function that is thought to be mediated by Mst-catalyzed phosphorylation of Mob1, which then activates the exchange factor Dock8, and its target small GTPases, Rac and Rho [20] (Fig. 2). Thus, in this setting, Mst is regulated and functions in a signaling cascade that is, in almost all respects, not congruent with the Hippo pathway as commonly understood. Beyond species-specific variations, these differences may reflect a general separation of Mst regulation and usage in epithelial versus lymphoid cells.
Fig. 2. Hippo-pathway independent Mst signaling.
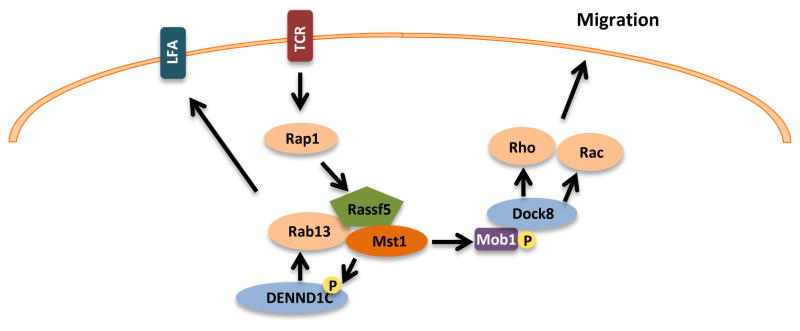
In lymphocytes, Mst1/2 is activated by chemokines and T-cell receptor (TCR) ligation. These signals are propagated by activation of the small GTPase Rap1, which binds to the adaptor protein Rassf5. Rassf5 contains a SARAH domain, which mediates interaction with Mst1/2. Mst has been shown to phosphorylate the guanine-nucleotide exchange factor (GEF) DENND1C, leading to activation of the small GTPase Rab13, and subsequent clustering of the integrin LFA-1. Activated Mst1 also phosphorylates the Rac/Rho GEF Dock8, promoting cell migration.
As emerging genetic data from mammalian systems show that Mst plays key roles in organ development and in immune function, and that these kinases participate in signaling pathways that fundamentally differ from that of the Drosophila Hippo pathway, it is worth reviewing the biochemical mechanisms governing Mst activation, with an emphasis on recent advances in this area.
Mst1/2: domain structure and dimerization
Mst1 and Mst2 are highly homologous by sequence and are identically organized, with an N-terminal kinase domain and a C-terminal regulatory region (Fig. 3). The C-terminal regulatory region can further be divided into two functional domains, an autoinhibitory domain, and a coiled-coil dimerization domain known as a SARAH domain [3]. Mst1/2 contains two caspase-cleavage sites between the kinase and the autoinhibitory domain [21]. Caspase-mediated cleavage of Mst1/2 at these sites removes the autoinhibitory and SARAH domains and activates Mst1/2 during apoptosis [22]. When transiently overexpressed, Mst1/2 itself induces apoptosis, leading to further Mst activation via caspase-mediated cleavage [4, 22]. Interestingly, the active, cleaved form of Mst1/2 is found at substantial levels in normal mouse liver (but not in the spleen), in the absence of apoptosis [7]. How these tissue-specific, cleaved forms are generated, and why they fail to trigger cell death, is currently unknown, but it has been speculated that these truncated, activated forms of Mst1/2 play an important role in hepatocytes in maintaining a differentiated, non-proliferative state [7].
Fig. 3. Schematic representation of the domain structure of Mst1/2.
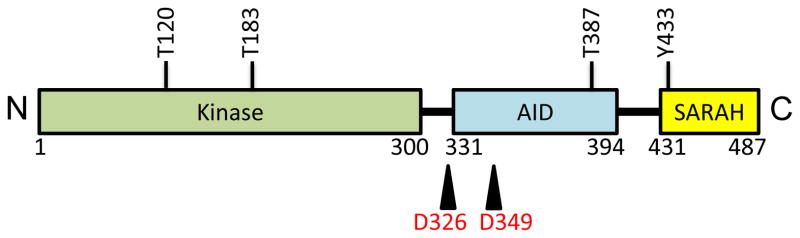
Full-length Mst1/2 is composed of N-terminal kinase domain (green), a C-terminal SARAH domain (yellow), and an auto-inhibitory domain between the kinase and the SARAH domain (blue). Mst1 is regulated by phosphorylation at T120, T183, T387 and Y433, and caspase cleavage at D326 and D349 (equivalent phosphorylation and caspase cleavage sites are present in Mst2).
Mst1 and Mst2 have long been shown to form homodimers, though the functional significance of these homodimers are still a matter of considerable debate. Homodimerization was first suspected because immunoprecipitates from epitope-tagged, exogenous Mst1-transfected cells contained a band that co-migrated with endogenous Mst1 [4]. Later, immunoprecipitation and gel filtration experiments demonstrated that endogenous Mst1 and Mst2 could self-associate [3, 23]. Further studies showed that the extreme C-terminal domain of Mst1 and Mst2 (later termed the SARAH domain) is required for homodimerization of Mst1 and Mst2 [3, 23, 24]. Even though SARAH domain is the major region of interaction in Mst1/2 homodimer, a recent study showed that kinase domain of Mst1 and Mst2 could also interact to form weak dimer in solution, with a dissociation constant of 36 μM [23]. Using heteronuclear NMR spectroscopy, Hwang et al. showed that the SARAH domain of each monomer comprises two helices; a short N-terminal helix (residues 433–437) h1 and a long C-terminal helix (residues 441–480) h2. In addition, they showed that the SARAH domain from the two monomers interact in a head-to-tail manner to form an antiparallel helix dimer, in which helix h1 of one monomer is folded toward helix h1′ of the other monomer. The homodimer interface is mainly stabilized by hydrophobic interactions, but in addition to these interactions, hydrogen bonds and electrostatic interactions between the helices also stabilize the dimer interface [25].
Unlike mammalian Mst1/2, the N-terminal kinase domains of Drosophila Hpo interact with each other and this interaction has been shown to be critical for Hpo autophosphorylation and trans-kinase activity. The SARAH-domain mediated dimerization was not shown to have a major impact on the kinase activity of Hpo in flies. These observations suggest that dimerization of the kinase domain is more important for the activation of Drosophila Hpo than for mammalian Msts [26].
In addition to forming Mst1-Mst1 and Mst2-Mst2 homodimers, these kinases are known to form heterodimers with other SARAH domain containing proteins such as the Ras-association domain family (RASSF) proteins. The RASSF proteins are tumor suppressor proteins implicated in a diverse range of biological functions such as apoptosis, autophagy, cell cycle, DNA repair and microtubule dynamics [27–37]. The RASSF family of proteins consists of ten members (RASSF1-10), all of which contain a conserved Ras association (RA) domain [38]. Of these ten members, only RASSF1-6 contains a SARAH (Salvador-RASSF-Hippo) domain and, not coincidentally, only these six members have the ability to bind Mst1/2 [38]. Mst1 binding to all six SARAH domain containing RASSF family members has been shown by a yeast-two hybrid screen, though not all of these interactions have been corroborated with endogenous proteins in mammalian cells [38, 39]. Among the six RASSF family member proteins, the interaction between Rassf5 and Mst1/2 has been extensively studied. Two recent studies compared the crystal structure of the Mst1/2-RASSF5 heterodimer to that of the Mst1/2 homodimer. Similar to Mst1 homodimers, Mst1-RASSF5 heterodimers form an antiparallel helix; however, there were several key structural differences between the Mst1-RASSF5 heterodimer and the Mst1 homodimers. First, the short N-terminal helix h1 present in the Mst1 homodimer was missing in the Mst2-RASSF5 heterodimer. Second, the h2 helix of Mst1 SARAH domain was longer in the Mst2-RASSF5 complex than in the Mst1 homodimer. Third, a sharp distortion of the α-helix of Mst1 monomer was observed in the Mst1-RASSF5 heterodimer at P453 and P472. Interestingly, this study also showed that Rassf5-Mst1 heterodimers are more stable than the Mst1 homodimer, as there are more extensive polar and nonpolar interactions in the heterodimer [40]. As discussed in the next section, the higher affinity for heterodimer formation may have implications regarding Mst activation, and also raise questions about the abundance of endogenous Mst/Mst homodimers vs. heterodimeric complexes with other SARAH-domain containing proteins.
Regulation of Mst1/2 activity
Mst1 and Mst2 can be activated by various apoptotic and stress stimuli, such as staurosporine, UV radiation, hydrogen peroxide, TNF-α, retinoic acid, okadaic acid, and several anti-cancer drugs [22, 41–45]. Although several mechanisms such as phosphorylation, SARAH-mediated dimerization, and interaction with other proteins have been proposed to regulate Mst1/2 activity, much remains to be learned regarding the mechanisms of Mst1/2 activation. In the following sections we will discuss some important mechanisms of regulation of Mst1/2.
Phosphorylation
In response to apoptotic or stress stimuli, Mst1/2 is autophosphorylated at multiple sites in the activation loop, which results in the activation of Mst1/2 [46, 47]. Among these autophosphorylation sites, phosphorylation at T183 and T180 is essential for Mst1 and Mst2 activation, respectively [48]. These sites can be also be transphosphorylated by the Ste20-family kinase TAO kinase-3, resulting in Mst activation [49]. Besides autophosphorylation, Mst1/2 are also regulated by transphosphorylation by protein kinases such as Akt and c-Abl. Akt and Mst are thought to operate in opposition, as the former promotes cell survival and the latter apoptosis. Akt binds to the C-terminus of Mst1 and phosphorylates Mst1 at T120 and T387 residues. The phosphorylation of Mst1 by Akt prevents caspase-mediated cleavage of Mst1 and hence prevents activation of Mst1 and its downstream target FOXO3 [50, 51]. Recently it was shown that the mTOR signaling pathway also regulates Mst1 phosphorylation at T120 and that this phosphorylation results in loss of Mst1 function in prostrate cancer cells [52]. Interestingly, the protein phosphatase M family members, PHLPP1/2 (pleckstrin homology (PH) domain leucine-rich repeat protein phosphatase), have been shown to reverse the phosphorylation of Mst1 at T387, the Akt target phosphorylation site. PHLPP1/2 interacts and dephosphorylates Mst1 on the inhibitory T387 site, thereby promoting Mst1 activation and Mst1-induced apoptosis [53]. PHLPP1/2-induced dephosphorylation of Mst1 at T387 has also been shown to be required for cellular contact inhibition [54].
Akt also phosphorylates Mst2 at T117 and T384 residues in response to mitogens and oncogenic Ras [55, 56].. Akt-induced Mst2 phosphorylation inhibits Mst2 kinase activity in several ways: by preventing Mst2 association with its activator, RASSF1A, by promoting Mst2 interaction with its inhibitor, Raf-1, and by preventing caspase-mediated cleavage of Mst2 [55, 56]. Akt-induced Mst1/2 phosphorylation and inactivation could be an important mechanism by which Akt promotes cell survival.
c-Abl-mediated tyrosine phosphorylation has also been shown to regulate Mst1 and Mst2 activity. Two groups recently demonstrated that protein kinase c-Abl phosphorylates Mst1 at Y433 and Mst2 at Y81 in response to oxidative stress, leading to Mst1 and Mst2 activation and induction of neuronal cell death [57, 58]. However, these studies relied primarily on overexpression approaches, and it is not clear if c-Abl is an important regulator of Mst1/2 under physiological conditions.
Homodimerization
In COS cells, endogenous Mst1 is found in high molecular complexes from approximately 145 to 443 kDa, with an average of 200 kDa, suggesting that Mst1 exists primarily in multimeric complexes [3]. Exogenously expressed Mst1 readily forms homodimers, and these dimers require the presence of the SARAH domain [3]. Many studies have investigated the importance of Mst1/2 homodimerization in regulating Mst1/2 activity, with contradictory conclusions. Creasy et al. demonstrated that dimerization has no effect on kinase activity of Mst1, as a deletion mutant lacking only the SARAH (but not the autoinhibitory) domain had the same kinase activity as wild-type Mst1 [3]. However, Anand et al. showed that a dimerization-defective Mst1 mutant had ability to bind to and phosphorylate FOXO1, an important downstream target of Mst1 [24]. These results suggest that Mst1 dimerization is required for Mst1 kinase activity, at least towards FOXO1. Another study demonstrated that full-length Mst2, but not the isolated kinase domain of Mst2, undergoes autophosphorylation at T180. As autophosphorylation at T180 is important for Mst2 activation, this study suggested that Mst2 homodimerization is important for regulation of Mst2 activity [23].. In another study, reconstitution of Raf1 in Raf1−/− cells caused Mst2 homodimers to disassemble. Further, Raf1 reconstitution abrogated Mst2 kinase activity, suggesting that Mst2 homodimerization is required for Mst2 activity [59]. While these cell-based studies suggest that Mst dimerization and activity are related, none are definitive, as it is difficult to separate dimerization per se from other binding events (e.g., to FOXO1) or phosphorylation events (e.g., Raf1). In our view, what is needed to resolve this question is a “cleaner” biochemical approach using recombinant enzymes.
Heterodimerization with RASSF family proteins
SARAH-mediated heterodimerization of Mst1/2 with RASSF proteins has been shown to both positively and negatively regulate Mst1/2 activity [48, 60]. For example, as mentioned previously, in T cells, Rassf5 complexes with Mst1, and overexpression of an activated form of the small GTPase Rap1 activates Mst1 [18]. Importantly, gene deletion studies confirmed that this interaction is required for Mst1 activation by T-cell receptor or chemokine stimulation [18]. Supporting this model, one group showed that overexpression of RASSF1A in 293T cells increased the kinase activity of Mst1/2 and promoted Mst1-mediated apoptosis [60]. In contrast, another group reported that overexpression of RASSF1A in cells suppressed autoactivation of Mst1 [48]. It is possible that these apparently contradictory results are related to the different expression levels of RASSF1A used in each study, as RASSF1A was expressed at close to physiological levels in the first study, whereas expression of RASSF1A was much higher in the second.
At least four mechanisms have been proposed for Mst1/2 activation by RASSF1 (Fig. 4). First, RASSF1A has been shown to activate Mst1/2 by preventing protein phosphatase 2A-mediated dephosphorylation of Mst1 at Thr-183 and Mst2 at Thr-180. In addition to preventing dephosphorylation, RASSF1A has also been shown to stabilize the Mst2 protein [61]. Third, RASSF1A binding to Mst2 has been shown to release Mst2 from an inhibitory complex with Raf-1, leading to the activation of Mst2. Finally, RASSF1A has been shown to promote the interaction of Mst2 with its substrate Lats1, leading to the activation of Lats1 and downstream Yap1 [62].
Fig. 4. Mst1/2 regulation by Rassf1.

Mechanistic models of Mst1/2 activation by Rassf1. A. Rassf1 can activate Mst1 and Mst2 is by preventing dephosphorylation by PP2A. B. Rassf1 can activate Mst2 by releasing it from its inhibitory complex with Raf-1. C. Rassf1 can activate Mst2 by preventing its degradation. D. Rassf1 can activate Mst2 by promoting its interaction with its substrate, Lats. Pointed arrowheads indicate activation while blunted ends indicate inhibition.
As with RASSF1, there are contradictory reports regarding Mst1 regulation by RASSF2. Song et al. reported that RASSF2 activates Mst1-mediated histone H2B phosphorylation in vitro, but that it inhibits Mst1-mediated FOXO3 phosphorylation in intact cells [63]. These results suggest that regulation of Mst1 activity by RASSF2 in intact cells may be more complex than a simple association between the two proteins. It is possible that in intact cells, RASSF2 recruits other proteins into the Mst1-RASSF2 complex, resulting in inhibition of Mst1 activity, or affects Mst1 substrate specificity.
The interaction of Mst2 with RASSF5 exemplifies one other aspect of regulation by heterodimerization; namely, temporal considerations. In this case, it has been reported that the effects of RASSF5 binding to Mst2 depend on whether association occurs before or after Mst2 is activated. In this model, if the two proteins interact before Mst2 activation, disruption of the Mst2 homodimer by RASSF5 prevents subsequent Mst2 activation; if after, there is no effect on Mst2 activity. Thus, RASSF5 can act as a context-dependent negative regulator of Mst2, depending on whether it binds to Mst2 before or after Mst2 activation [23] (Fig. 5).
Fig. 5. Regulation of Mst2 by Rassf5.
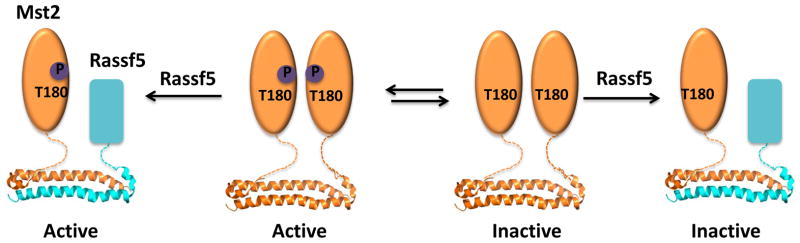
Rassf5 can act as an activator or inhibitor of Mst2, depending on whether it binds Mst2 before or after activation-loop phosphorylation. When Rassf5 binds Mst2, before activation-loop phosphorylation, it blocks Mst2 homodimerization and hence activation, but when Rassf5 binds Mst2 after activation-loop phosphorylation, it promotes Mst2 activation and function.
Regulation by non-SARAH domain protein interactions
Mst2 has been shown to interact with Raf-1, a key component of Ras/Raf/MEK/ERK mitogen-activated protein kinase signaling pathway that plays a crucial role in regulation of cell proliferation, differentiation and cell survival [64–66]. Raf-1 interaction has been shown to negatively regulate Mst2 kinase activity [55]. O’Neill et al. demonstrated that Mst2 was constitutively phosphorylated at activating threonine residues in Raf1−/− cells but not in Raf1+/+ cells. Reconstitution of Raf-1 or a kinase-dead form of Raf-1 in Raf1−/− cells abrogated Mst2 phosphorylation and activation, suggesting that Raf-1 inhibits Mst2 independent of Raf-1 kinase activity. Further examination revealed that Raf-1 inhibits Mst2 activation by two mechanisms: first, by preventing homodimerization of Mst2, and second, by recruiting a phosphatase that removes activating phosphorylations on Mst2 [59, 67]. While Mst2 is negatively regulated by Raf-1, conversely, Mst2 regulates Raf-1 [55, 68]. Mst2 knockdown has been shown to enhance the inhibitory phosphorylation of Raf-1 on Ser259, suggesting that, in response to mitogen stimulation, Mst2 positively regulates Raf-1 activity by suppressing the Ser259 inhibitory phosphorylation on Raf-1 [69].
Antioxidant enzymes such as thioredoxin and peroxiredoxin-1 (Prdx1) have also been shown to bind Mst1 and to regulate Mst1 activation by oxidative stress [79–81]. Similar to the model of Mst2/RASSF5 interaction (Fig. 5), Chae et al. suggested that association of thioredoxin inactivates Mst1 by preventing its homodimerization and autophosphorylation. In this schema, H2O2 alleviates inhibition of Mst1 by disrupting the interaction of Mst1 with thioredoxin [72]. Prdx1 represents a second antioxidant enzyme that interacts with Mst1 [70, 71]. In this case, however, the interaction is promoted, rather than disrupted, by oxidant stress [71]. While one study showed that association of Mst1 with Prdx1 was required for H2O2-induced Mst1 activation and induction of apoptosis [70],, another reported that Mst1 interaction with Prdx1 led to phosphorylation and inactivation of Prdx1. In this setting, inactivation of Prdx1 by Mst1 resulted in the accumulation of H2O2 in cells, which might reinforce Mst1 activation by positive feedback [71].
Concluding Remarks
As central components of a recently discovered tumor suppressor pathway, the Mst kinases have deservedly garnered much attention. Based on pioneering experiments in Drosophila, the central role for Mst1/2 in regulating Yap has been established, but it is now apparent that in mammals Mst1/2 have roles other than Yap activation, and also that Yap can be activated independently of Mst1/2. These findings mean that Mst signaling is more complex than initially supposed. It is clear that much work needs to be done in terms of understanding how Msts are activated, particularly in mammalian systems, and also to determine if the two isoforms of Mst have unique regulation or functions. Other open questions remain regarding the relationship of Mst dimerization and activity and how such dimerization is regulated. Given the multiplicity of relevant genetic model systems now available and recent advances in structural analysis of the Mst kinase and SARAH domains, a more complete picture of the workings of this important kinase should soon come into view.
Highlights.
Mst1/Mst2/Hippo are core, conserved components of a tumor suppressor pathway.
Mst/Hippo signaling has both similarities and key differences in mammals and flies.
Homo- and heterodimerization play important roles in Mst regulation.
Acknowledgments
Financial Support: This work was supported by grants from the NIH (R01 CA148805 and R01 CA098830) to JC, and an appropriation from the state of Pennsylvania to the Fox Chase Cancer Center (P30 CA006927).
We would like to thank Jeffrey Peterson for valuable comments on the manuscript.
Footnotes
Conflict of Interest Statement: The authors declare no conflicts of interest
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Zhao B, et al. The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes & development. 2010;24:862–874. doi: 10.1101/gad.1909210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer cell. 2008;13:188–192. doi: 10.1016/j.ccr.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Creasy CL, et al. The Ste20-like protein kinase, Mst1, dimerizes and contains an inhibitory domain. The Journal of biological chemistry. 1996;271:21049–21053. doi: 10.1074/jbc.271.35.21049. [DOI] [PubMed] [Google Scholar]
- 4.Creasy CL, Chernoff J. Cloning and characterization of a human protein kinase with homology to Ste20. J Biol Chem. 1995;270:21695–21700. doi: 10.1074/jbc.270.37.21695. [DOI] [PubMed] [Google Scholar]
- 5.Taylor LK, et al. Newly identified stress-responsive protein kinases, Krs-1 and Krs-2. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:10099–10104. doi: 10.1073/pnas.93.19.10099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhou D, et al. The Nore1B/Mst1 complex restrains antigen receptor-induced proliferation of naive T cells. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:20321–20326. doi: 10.1073/pnas.0810773105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou D, et al. Mst1 and Mst2 maintain hepatocyte quiescence and suppress hepatocellular carcinoma development through inactivation of the Yap1 oncogene. Cancer cell. 2009;16:425–438. doi: 10.1016/j.ccr.2009.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Song H, et al. Mammalian Mst1 and Mst2 kinases play essential roles in organ size control and tumor suppression. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:1431–1436. doi: 10.1073/pnas.0911409107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oh S, et al. Crucial role for Mst1 and Mst2 kinases in early embryonic development of the mouse. Molecular and cellular biology. 2009;29:6309–6320. doi: 10.1128/MCB.00551-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Praskova M, et al. MOBKL1A/MOBKL1B phosphorylation by MST1 and MST2 inhibits cell proliferation. Curr Biol. 2008;18:311–321. doi: 10.1016/j.cub.2008.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan EH, et al. The Ste20-like kinase Mst2 activates the human large tumor suppressor kinase Lats1. Oncogene. 2005;24:2076–2086. doi: 10.1038/sj.onc.1208445. [DOI] [PubMed] [Google Scholar]
- 12.Hao Y, et al. Tumor suppressor LATS1 is a negative regulator of oncogene YAP. The Journal of biological chemistry. 2008;283:5496–5509. doi: 10.1074/jbc.M709037200. [DOI] [PubMed] [Google Scholar]
- 13.Zhao B, et al. Inactivation of YAP oncoprotein by the Hippo pathway is involved in cell contact inhibition and tissue growth control. Genes & development. 2007;21:2747–2761. doi: 10.1101/gad.1602907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhao B, et al. TEAD mediates YAP-dependent gene induction and growth control. Genes & development. 2008;22:1962–1971. doi: 10.1101/gad.1664408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang N, et al. The Merlin/NF2 tumor suppressor functions through the YAP oncoprotein to regulate tissue homeostasis in mammals. Developmental cell. 2010;19:27–38. doi: 10.1016/j.devcel.2010.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin F, et al. Spatial organization of Hippo signaling at the plasma membrane mediated by the tumor suppressor Merlin/NF2. Cell. 2013;154:1342–1355. doi: 10.1016/j.cell.2013.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nehme NT, et al. MST1 mutations in autosomal recessive primary immunodeficiency characterized by defective naive T-cell survival. Blood. 2012;119:3458–3468. doi: 10.1182/blood-2011-09-378364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Katagiri K, et al. Spatiotemporal regulation of the kinase Mst1 by binding protein RAPL is critical for lymphocyte polarity and adhesion. Nature immunology. 2006;7:919–928. doi: 10.1038/ni1374. [DOI] [PubMed] [Google Scholar]
- 19.Nishikimi A, et al. Rab13 acts downstream of the kinase Mst1 to deliver the integrin LFA-1 to the cell surface for lymphocyte trafficking. Science signaling. 2014;7:ra72. doi: 10.1126/scisignal.2005199. [DOI] [PubMed] [Google Scholar]
- 20.Mou F, et al. The Mst1 and Mst2 kinases control activation of rho family GTPases and thymic egress of mature thymocytes. The Journal of experimental medicine. 2012;209:741–759. doi: 10.1084/jem.20111692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee KK, et al. MST, a physiological caspase substrate, highly sensitizes apoptosis both upstream and downstream of caspase activation. The Journal of biological chemistry. 2001;276:19276–19285. doi: 10.1074/jbc.M005109200. [DOI] [PubMed] [Google Scholar]
- 22.Graves JD, et al. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J. 1998;17:2224–2234. doi: 10.1093/emboj/17.8.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ni L, et al. Structural Basis for Autoactivation of Human Mst2 Kinase and Its Regulation by RASSF5. Structure. 2013 doi: 10.1016/j.str.2013.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Anand R, et al. Biochemical analysis of MST1 kinase: elucidation of a C-terminal regulatory region. Biochemistry. 2008;47:6719–6726. doi: 10.1021/bi800309m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang E, et al. Structural insight into dimeric interaction of the SARAH domains from Mst1 and RASSF family proteins in the apoptosis pathway. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:9236–9241. doi: 10.1073/pnas.0610716104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jin Y, et al. Dimerization and cytoplasmic localization regulate Hippo kinase signaling activity in organ size control. The Journal of biological chemistry. 2012;287:5784–5796. doi: 10.1074/jbc.M111.310334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donninger H, et al. The RASSF1A tumor suppressor. J Cell Sci. 2007;120:3163–3172. doi: 10.1242/jcs.010389. [DOI] [PubMed] [Google Scholar]
- 28.Dammann R, et al. Epigenetic inactivation of a RAS association domain family protein from the lung tumour suppressor locus 3p21.3. Nature genetics. 2000;25:315–319. doi: 10.1038/77083. [DOI] [PubMed] [Google Scholar]
- 29.van der Weyden L, Adams DJ. The Ras-association domain family (RASSF) members and their role in human tumourigenesis. Biochim Biophys Acta. 2007;1776:58–85. doi: 10.1016/j.bbcan.2007.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vos MD, et al. RASSF2 is a novel K-Ras-specific effector and potential tumor suppressor. The Journal of biological chemistry. 2003;278:28045–28051. doi: 10.1074/jbc.M300554200. [DOI] [PubMed] [Google Scholar]
- 31.Eckfeld K, et al. RASSF4/AD037 is a potential ras effector/tumor suppressor of the RASSF family. Cancer research. 2004;64:8688–8693. doi: 10.1158/0008-5472.CAN-04-2065. [DOI] [PubMed] [Google Scholar]
- 32.Allen NP, et al. RASSF6 is a novel member of the RASSF family of tumor suppressors. Oncogene. 2007;26:6203–6211. doi: 10.1038/sj.onc.1210440. [DOI] [PubMed] [Google Scholar]
- 33.Clark GJ, et al. RASSF Family Proteins. Mol Biol Int. 2012;2012:938916. doi: 10.1155/2012/938916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dallol A, et al. RASSF1A interacts with microtubule-associated proteins and modulates microtubule dynamics. Cancer research. 2004;64:4112–4116. doi: 10.1158/0008-5472.CAN-04-0267. [DOI] [PubMed] [Google Scholar]
- 35.Shivakumar L, et al. The RASSF1A tumor suppressor blocks cell cycle progression and inhibits cyclin D1 accumulation. Molecular and cellular biology. 2002;22:4309–4318. doi: 10.1128/MCB.22.12.4309-4318.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vos MD, et al. A role for the RASSF1A tumor suppressor in the regulation of tubulin polymerization and genomic stability. Cancer research. 2004;64:4244–4250. doi: 10.1158/0008-5472.CAN-04-0339. [DOI] [PubMed] [Google Scholar]
- 37.Vos MD, et al. Ras uses the novel tumor suppressor RASSF1 as an effector to mediate apoptosis. The Journal of biological chemistry. 2000;275:35669–35672. doi: 10.1074/jbc.C000463200. [DOI] [PubMed] [Google Scholar]
- 38.Avruch J, et al. Protein kinases of the Hippo pathway: regulation and substrates. Semin Cell Dev Biol. 2012;23:770–784. doi: 10.1016/j.semcdb.2012.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khokhlatchev A, et al. Identification of a novel Ras-regulated proapoptotic pathway. Current biology: CB. 2002;12:253–265. doi: 10.1016/s0960-9822(02)00683-8. [DOI] [PubMed] [Google Scholar]
- 40.Hwang E, et al. Structural basis of the heterodimerization of the MST and RASSF SARAH domains in the Hippo signalling pathway. Acta crystallographica Section D, Biological crystallography. 2014;70:1944–1953. doi: 10.1107/S139900471400947X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu ML, et al. UV irradiation-induced apoptosis leads to activation of a 36-kDa myelin basic protein kinase in HL-60 cells. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:8977–8982. doi: 10.1073/pnas.93.17.8977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lehtinen MK, et al. A conserved MST-FOXO signaling pathway mediates oxidative-stress responses and extends life span. Cell. 2006;125:987–1001. doi: 10.1016/j.cell.2006.03.046. [DOI] [PubMed] [Google Scholar]
- 43.Watabe M, et al. Activation of MST/Krs and c-Jun N-terminal kinases by different signaling pathways during cytotrienin A-induced apoptosis. The Journal of biological chemistry. 2000;275:8766–8771. doi: 10.1074/jbc.275.12.8766. [DOI] [PubMed] [Google Scholar]
- 44.Lee KK, et al. Proteolytic activation of MST/Krs, STE20-related protein kinase, by caspase during apoptosis. Oncogene. 1998;16:3029–3037. doi: 10.1038/sj.onc.1201840. [DOI] [PubMed] [Google Scholar]
- 45.Kakeya H, et al. Caspase-mediated activation of a 36-kDa myelin basic protein kinase during anticancer drug-induced apoptosis. Cancer Res. 1998;58:4888–4894. [PubMed] [Google Scholar]
- 46.Glantschnig H, et al. Mapping of MST1 kinase sites of phosphorylation. Activation and autophosphorylation. The Journal of biological chemistry. 2002;277:42987–42996. doi: 10.1074/jbc.M208538200. [DOI] [PubMed] [Google Scholar]
- 47.Graves JD, et al. Both phosphorylation and caspase-mediated cleavage contribute to regulation of the Ste20-like protein kinase Mst1 during CD95/Fas-induced apoptosis. The Journal of biological chemistry. 2001;276:14909–14915. doi: 10.1074/jbc.M010905200. [DOI] [PubMed] [Google Scholar]
- 48.Praskova M, et al. Regulation of the MST1 kinase by autophosphorylation, by the growth inhibitory proteins, RASSF1 and NORE1, and by Ras. Biochem J. 2004;381:453–462. doi: 10.1042/BJ20040025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Boggiano JC, et al. Tao-1 phosphorylates Hippo/MST kinases to regulate the Hippo-Salvador-Warts tumor suppressor pathway. Developmental cell. 2011;21:888–895. doi: 10.1016/j.devcel.2011.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jang SW, et al. Akt phosphorylates MstI and prevents its proteolytic activation, blocking FOXO3 phosphorylation and nuclear translocation. The Journal of biological chemistry. 2007;282:30836–30844. doi: 10.1074/jbc.M704542200. [DOI] [PubMed] [Google Scholar]
- 51.Yuan Z, et al. Phosphoinositide 3-kinase/Akt inhibits MST1-mediated pro-apoptotic signaling through phosphorylation of threonine 120. The Journal of biological chemistry. 2010;285:3815–3824. doi: 10.1074/jbc.M109.059675. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 52.Collak FK, et al. Threonine-120 phosphorylation regulated by phosphoinositide-3-kinase/Akt and mammalian target of rapamycin pathway signaling limits the antitumor activity of mammalian sterile 20-like kinase 1. The Journal of biological chemistry. 2012;287:23698–23709. doi: 10.1074/jbc.M112.358713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qiao M, et al. Mst1 is an interacting protein that mediates PHLPPs’ induced apoptosis. Mol Cell. 2010;38:512–523. doi: 10.1016/j.molcel.2010.03.017. [DOI] [PubMed] [Google Scholar]
- 54.Jung S, et al. PHLPP1 regulates contact inhibition by dephosphorylating Mst1 at the inhibitory site. Biochemical and biophysical research communications. 2014;443:1263–1269. doi: 10.1016/j.bbrc.2013.12.129. [DOI] [PubMed] [Google Scholar]
- 55.Romano D, et al. Proapoptotic kinase MST2 coordinates signaling crosstalk between RASSF1A, Raf-1, and Akt. Cancer research. 2010;70:1195–1203. doi: 10.1158/0008-5472.CAN-09-3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim D, et al. Regulation of proapoptotic mammalian ste20-like kinase MST2 by the IGF1-Akt pathway. PloS one. 2010;5:e9616. doi: 10.1371/journal.pone.0009616. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 57.Liu W, et al. Regulation of neuronal cell death by c-Abl-Hippo/MST2 signaling pathway. PloS one. 2012;7:e36562. doi: 10.1371/journal.pone.0036562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xiao L, et al. The c-Abl-MST1 signaling pathway mediates oxidative stress-induced neuronal cell death. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:9611–9619. doi: 10.1523/JNEUROSCI.0035-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.O’Neill E, et al. Role of the kinase MST2 in suppression of apoptosis by the proto-oncogene product Raf-1. Science. 2004;306:2267–2270. doi: 10.1126/science.1103233. [DOI] [PubMed] [Google Scholar]
- 60.Oh HJ, et al. Role of the tumor suppressor RASSF1A in Mst1-mediated apoptosis. Cancer Res. 2006;66:2562–2569. doi: 10.1158/0008-5472.CAN-05-2951. [DOI] [PubMed] [Google Scholar]
- 61.Guo C, et al. The tumor suppressor RASSF1A prevents dephosphorylation of the mammalian STE20-like kinases MST1 and MST2. J Biol Chem. 2011;286:6253–6261. doi: 10.1074/jbc.M110.178210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Matallanas D, et al. RASSF1A elicits apoptosis through an MST2 pathway directing proapoptotic transcription by the p73 tumor suppressor protein. Mol Cell. 2007;27:962–975. doi: 10.1016/j.molcel.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song H, et al. Role of the tumor suppressor RASSF2 in regulation of MST1 kinase activity. Biochem Biophys Res Commun. 2010;391:969–973. doi: 10.1016/j.bbrc.2009.11.175. [DOI] [PubMed] [Google Scholar]
- 64.Kolch W. Meaningful relationships: the regulation of the Ras/Raf/MEK/ERK pathway by protein interactions. Biochem J. 2000;351(Pt 2):289–305. [PMC free article] [PubMed] [Google Scholar]
- 65.Avruch J, et al. Ras activation of the Raf kinase: tyrosine kinase recruitment of the MAP kinase cascade. Recent Prog Horm Res. 2001;56:127–155. doi: 10.1210/rp.56.1.127. [DOI] [PubMed] [Google Scholar]
- 66.Baccarini M. An old kinase on a new path: Raf and apoptosis. Cell Death Differ. 2002;9:783–785. doi: 10.1038/sj.cdd.4401070. [DOI] [PubMed] [Google Scholar]
- 67.O’Neill E, Kolch W. Taming the Hippo: Raf-1 controls apoptosis by suppressing MST2/Hippo. Cell Cycle. 2005;4:365–367. doi: 10.4161/cc.4.3.1531. [DOI] [PubMed] [Google Scholar]
- 68.Romano D, et al. Protein interaction switches coordinate Raf-1 and MST2/Hippo signalling. Nature cell biology. 2014;16:673–684. doi: 10.1038/ncb2986. [DOI] [PubMed] [Google Scholar]
- 69.Kilili GK, Kyriakis JM. Mammalian Ste20-like kinase (Mst2) indirectly supports Raf-1/ERK pathway activity via maintenance of protein phosphatase-2A catalytic subunit levels and consequent suppression of inhibitory Raf-1 phosphorylation. The Journal of biological chemistry. 2010;285:15076–15087. doi: 10.1074/jbc.M109.078915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Morinaka A, et al. Oligomeric peroxiredoxin-I is an essential intermediate for p53 to activate MST1 kinase and apoptosis. Oncogene. 2011;30:4208–4218. doi: 10.1038/onc.2011.139. [DOI] [PubMed] [Google Scholar]
- 71.Rawat SJ, et al. The tumor suppressor Mst1 promotes changes in the cellular redox state by phosphorylation and inactivation of peroxiredoxin-1 protein. The Journal of biological chemistry. 2013;288:8762–8771. doi: 10.1074/jbc.M112.414524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chae JS, et al. Thioredoxin-1 functions as a molecular switch regulating the oxidative stress-induced activation of MST1. Free radical biology & medicine. 2012;53:2335–2343. doi: 10.1016/j.freeradbiomed.2012.10.527. [DOI] [PubMed] [Google Scholar]