

Abstract
Glycogen synthase kinase-3 (GSK3) may be the busiest kinase in most cells, with over 100 known substrates to deal with. How does GSK3 maintain control to selectively phosphorylate each substrate, and why was it evolutionarily favorable for GSK3 to assume such a large responsibility? GSK3 must be particularly adaptable for incorporating new substrates into its repertoire, and we discuss the distinct properties of GSK3 that may contribute to its capacity to fulfill its roles in multiple signaling pathways. The mechanisms regulating GSK3 (predominantly post-translational modifications, substrate priming, cellular trafficking, protein complexes) have been reviewed previously, so here we focus on newly identified complexities in these mechanisms, how each of these regulatory mechanism contributes to the ability of GSK3 to select which substrates to phosphorylate, and how these mechanisms may have contributed to its adaptability as new substrates evolved. The current understanding of the mechanisms regulating GSK3 is reviewed, as are emerging topics in the actions of GSK3, particularly its interactions with receptors and receptor-coupled signal transduction events, and differential actions and regulation of the two GSK3 isoforms, GSK3α and GSK3β. Another remarkable characteristic of GSK3 is its involvement in many prevalent disorders, including psychiatric and neurological diseases, inflammatory diseases, cancer, and others. We address the feasibility of targeting GSK3 therapeutically, and provide an update of its involvement in the etiology and treatment of several disorders.
Keywords: Alzheimer’s disease, depression, G protein-coupled receptors, glycogen synthase kinase-3, lithium, neuroinflammation
1. Introduction
Not long ago, glycogen synthase kinase-3 (GSK3) seemed to be a rather curious enzyme in a number of ways, but perhaps not one of especially widespread importance (Grimes and Jope, 2001; Woodgett, 2001). GSK3 was considered of some interest because it has the unconventional characteristics for a kinase of being constitutively active, its substrates usually need to be pre-phosphorylated by another kinase, and it is inhibited, rather than activated, in response to stimulation of the two main signaling pathways known to impinge on GSK3, the insulin and Wnt pathways. These characteristics made GSK3 somewhat idiosyncratic compared with other kinases, but did not engender especially widespread attention. However, two key developments in particular transformed this perspective to the current state of widespread interest. One of these developments was the discovery that GSK3 is a key kinase contributing to abnormal phosphorylation of the microtubule-binding protein tau in the process thought to cause neurofibrillary tangles in Alzheimer’s disease (Hanger et al., 1992; Mandelkow et al., 1992). This finding brought GSK3 to the attention of the many investigators, as well as pharmaceutical companies, who were looking for therapeutic targets for Alzheimer’s disease. The other development was the finding that GSK3 is inhibited by lithium (Klein and Melton, 1996; Stambolic et al., 1996). Lithium is the classical mood stabilizer used in the treatment of bipolar disorder, so this finding raised the prospect that GSK3 may have a central role in bipolar disorder, and it also provided a relatively selective inhibitor of GSK3 that opened the door for discoveries of other targets of GSK3. Thus, during the last 20 years the interest in GSK3 and its known actions swung to the opposite pole, and it now seems like GSK3 touches almost every aspect of cellular signaling and is involved in an unparalleled number of disease processes.
In conjunction with these discoveries and the ensuing enormous growth in recognizing new actions of GSK3, many additional inhibitors of GSK3 were developed as GSK3 began to be seriously considered as a therapeutic target (Eldar-Finkelman and Martinez, 2011). Potential therapeutic applications include many prevalent conditions, such as cancer, cardiovascular diseases, diabetes, inflammatory conditions, neurodegenerative diseases, and psychiatric diseases, as well as other less prevalent conditions. Such an expansive influence on cellular signaling and association with multiple disease processes has stimulated skeptics to inquire how GSK3 can manage all of its tasks in an organized fashion, and how such a pervasive kinase can reasonably be thought of as a feasible therapeutic target. Although these conundrums remain to be definitively answered, much recent progress reviewed here is leading to a more complete understanding of this enigmatic kinase and its potential as a therapeutic target. Moreover, substantial evidence supports the conclusion that inhibition of GSK3 is an important action contributing to the mood stabilizing action of lithium in bipolar disease, demonstrating that inhibition of GSK3 is, in fact, feasible for safe therapeutic interventions.
A single review cannot adequately cover all aspects concerning GSK3, and there are now excellent reviews available that are focused on many of the individual processes and diseases involving GSK3, which are cited below. Therefore, this review is focused on conceptualizations concerning the actions and regulation of GSK3, on misunderstandings and ambiguities about GSK3, and on new directions that are developing in GSK3 research. Many of these topics are best considered in discussions of the mechanisms regulating GSK3 and its associations with several classes of diseases.
2. Regulation of GSK3-mediated substrate phosphorylation
GSK3 refers to two paralogs that are commonly referred to as isoforms, GSK3α and GSK3β. A theoretical analysis found that GSK3β has more predicted substrates than any other kinase (Linding et al., 2007). The mean number of substrates for the 68 kinases examined was 12, the average was 70 substrates, and the predicted number of substrates for GSK3β was over 500 (Linding et al., 2007). While this is almost certainly an overestimation, about 100 proteins phosphorylated by GSK3 have already been reported, although further analyses are required before all can be considered to be bona fide in vivo GSK3 substrates (Sutherland, 2011). Nonetheless, the high number of GSK3 substrates raises the question of how can GSK3 phosphorylate so many proteins in a cell with any discretion? Furthermore, why would one kinase (although there are actually two, GSK3α and GSK3β) have evolved to phosphorylate so many substrates when nature has so many kinases to choose from? Although these questions have not been answered satisfactorily, they suggest that there must be certain characteristics of GSK3 that are particularly useful and versatile which outweigh the functional constraints and complexities required to provide discretion among many substrates. Since GSK3’s activity as a kinase is not particularly different from other kinases, we suggest that the mechanisms regulating GSK3 are particularly adaptable for incorporation into new signaling pathways without perturbing those already existent. Thus, a key to GSK3’s actions may be the multiple regulatory mechanisms available to orchestrate its substrate-specific actions, which are discussed in the following sections of this review. However, this promiscuity also appears to have provided multiple interactions that can be disrupted to result in unbridled actions of GSK3 contributing to multiple types of diseases, which are discussed later in this review. We originally categorized these crucial mechanisms that confer specificity for signaling pathways and for substrates to include the regulatory phosphorylation of GSK3 itself, the regulation of substrate availability, the subcellular localization of GSK3 and its substrates, and the incorporation of GSK3 into protein complexes (Jope and Johnson, 2004), categories that remain the primary mechanisms known to regulate GSK3.
2.1. Regulatory post-translational modifications of GSK3
Inhibitory serine-phosphorylation is the most frequently examined mechanism that regulates the activity of GSK3, although the intricate beauty of this mechanism is often underappreciated. Two key functional domains of GSK3 have been identified (Figure 1), a primed-substrate binding domain that recruits substrates to GSK3, and a kinase domain that phosphorylates the substrate (Frame et al., 2001; ter Haar et al., 2001; Dajani et al., 2003). The former domain provides a binding site for most GSK3 substrates, those that are primed by being pre-phosphorylated. Although GSK3 can phosphorylate a few non-primed substrates at Ser-Pro sites, the most common target for phosphorylation by GSK3 is the pre-phosphorylated sequence, S/T-X-X-X-S/T(P), where GSK3 phosphorylates a serine/threonine four residues N-terminal to a pre-phosphorylated serine/threonine. However, the number of intervening residues between the primed site and the GSK3 target site can be more (Cole et al., 2004) or less (Singh et al., 2012) than four, not surprisingly suggesting that the three-dimensional structure of the substrate influences its interactions with GSK3. The priming phosphorylation allows the substrate to bind the primed-substrate binding domain, placing the target serine/threonine adjacent to the kinase domain of GSK3 to facilitate its phosphorylation (Figure 1). Substrates often contain three or four adjacent S/T-X-X-X-S/T(P) motifs, allowing GSK3 to phosphorylate every fourth residue in a string of sequential sites as it creates its own primed substrate in a stepwise fashion. For example, GSK3 phosphorylates serines 652, 648, 644 and 640 in glycogen synthase, and residues 41, 37, and 33 in β-catenin. In some cases, non-primed substrates may contain an acidic residue four amino acids C-terminal from the GSK3 target site in place of the primed phosphorylated residue, such that the acidic residue can interact with the primed-substrate binding site, but this is not always the case.
Figure 1. Serine9-phosphorylation of GSK3β inhibits its phosphorylation of primed substrates.

(A) Representation of GSK3β based on a recently reported crystal structure (Stamos et al., 2014) showing the adjacent kinase domain and primed-substrate binding domain (based on PDB ID: 4nm0). (B) Phosphorylated serine-9 in the N-terminal tail of GSK3β binds the primed substrate binding domain (based on PDB ID: 4nm3). (C) Primed substrates first associate with the primed substrate binding domain of GSK3β, which places a Ser/Thr four residues N-terminal to the primed phosphorylated Ser/Thr adjacent to the kinase domain of GSK3β to facilitate substrate phosphorylation. (D) Phosphorylated serine-9 in the N-terminal tail of GSK3β inhibits the association of primed substrates with the primed substrate binding domain of GSK3β.
The frequent requirement for a primed substrate is central to two mechanisms that regulate GSK3’s actions: inhibitory serine phosphorylation of GSK3, and control of substrate availability, which is discussed in section 2.2. Phosphorylation of serine-21 in GSK3α or of serine-9 in GSK3β causes the N-terminal tail of GSK3 to act as a pre-phosphorylated substrate, or pseudosubstrate (Frame et al., 2001). This phosphorylated serine tail self-associates in the primed-substrate binding pocket, hindering the binding of primed substrates, and thus diminishing primed-substrate phosphorylation by GSK3 (Figure 1). Multiple signaling pathways feed into this site to increase the serine-phosphorylation of GSK3, which can be mediated by Akt, protein kinase A (PKA), protein kinase C, p70 S6 kinase, and other kinases. Thus, many signaling pathways that activate these kinases can inhibit GSK3 by phosphorylating the inhibitory serines on GSK3. An intriguing exception to this regulatory mechanism is the recent discovery that GSK3 phosphorylates AMP-activated kinase (AMPK) to inhibit its activity (Suzuki et al., 2013). Remarkably, Akt, which usually inhibits GSK3 by serine-9/21 phosphorylation, promotes AMPK phosphorylation by GSK3, showing that in some circumstances Akt and GSK3 cooperatively modulate signaling pathways. However, in general, measuring the serine-9/21 phosphorylation of GSK3 provides a valuable assessment of conditions that regulate the activity of GSK3 via this mechanism, but it is useful to consider four caveats to this conclusion.
Not all substrates of GSK3 are primed, so these non-primed substrates may not require binding to the primed substrate binding domain to be phosphorylated by GSK3, depending on the existence of an acidic residue in place of the primed residue. Thus, the serine-phosphorylation inhibitory mechanism does not necessarily regulate the phosphorylation of non-primed substrates by GSK3. Therefore, if a non-primed substrate is under investigation, examining changes in the serine-phosphorylation of GSK3 should be interpreted cautiously. Thus, it is critical to understand the characteristics of GSK3 substrates when investigating their regulatory interactions with GSK3.
The activity of GSK3 in certain protein complexes is not affected by serine-phosphorylation of GSK3. This has been most well-established, and most often misinterpreted, for Wnt signaling, which is impervious to the serine-phosphorylation status of GSK3 bound to Axin, as discussed in detail in section 2.4.
The serine-phosphorylation status of GSK3 is probably most often undergoing oscillations. These may be rapid, such as in depolarizing-repolarizing neurons, or slower, such as changes associated with varying levels of circulating hormones that regulate GSK3 (discussed in section 3.2) or associated with the circadian rhythm in the suprachiasmatic nucleus and liver of mice, as well as in cultured cells (Iitaka et al., 2005). These fluctuations may influence the basal level of GSK3 phosphorylation and responsiveness to interventions depending on the time scales of the fluctuations and experimental manipulations.
One of the most often over-looked characteristic of the serine-phosphorylation inhibitory mechanism is that it does not cause absolute inhibition of GSK3 activity. In contrast, the phospho-Ser9/21 domain is inhibitory by a mechanism that is competitive with primed substrates (Frame et al., 2001). The Ser-9/21 tail of GSK3 is in a dynamic state of phosphorylation and dephosphorylation, and fluctuates in its binding and release from the primed-substrate binding pocket. Thus, as the concentration of the primed substrate increases, it is able to out-compete the phospho-Ser9/21 domain for binding to GSK3. One ramification of this is that only at low concentrations of the primed substrate is its phosphorylation by GSK3 blocked by phospho-Ser9/21, but as the primed substrate concentration increases it displaces phospho-Ser9/21 and once again can be phosphorylated by GSK3. Thus, inhibitory serine-phosphorylation of GSK3 does not absolutely eliminate its phosphorylation of primed substrates, but this mechanism allows an accumulation of the primed substrate that reaches a new steady-state level at which it reemerges as a substrate of GSK3.
Thus altogether, there are many intricacies associated with the serine-phosphorylation mechanism of regulating GSK3, which can lead to misinterpretations of findings but should instead provide important clues about the regulation of signaling systems.
Phosphorylation of ser21-GSK3α and ser9-GSK3β can be mediated by a large number of different kinases, as noted above. This is important because it allows multiple signaling pathways to impinge on GSK3, and for GSK3 to act as an integrator of signals. It seems likely, but not yet demonstrated in sufficient detail, that the formation of protein complexes has an important role in allowing multiple signals and their particular kinases to regulate GSK3, as discussed in section 2.4. It is also intriguing to note that these are not one-way interactions, but that the activity of GSK3 can also regulate the actions of some GSK3-inhibiting kinases. This bi-directionality has been studied most with the Akt-GSK3 interaction, in which Akt not only inhibits GSK3 but GSK3 can also regulate Akt. For example, GSK3 can regulate Akt in a complex with β-arrestin, which is described in section 3. GSK3α has also been shown to regulate Akt in a manner unique from GSK3β, as GSK3α has been reported to promote the activating phosphorylation of Akt, which may be a cell type specific mechanism for GSK3α to attenuate its own actions (Lu et al., 2011). Additionally, GSK3α can also inhibit the activity of Akt via phosphorylation on a recently discovered regulatory site at threonine312-Akt (Gulen et al., 2012). Also, GSK3 promotes the activity of protein phosphatase-1 (PP1) by phosphorylating to inhibit its inhibitor, called I-2. This activation of PP1 not only increases GSK3 activity, but also may de-phosphorylate upstream kinases, thereby diminishing their induction of phosphoserine-GSK3 (DePaoli-Roach, 1984; Zhang et al., 2003). Thus, there are clearly bi-directional interactions between GSK3 and kinases that phosphorylate GSK3, which are likely driven by specific signaling pathways in a cell type-specific manner, and there are mechanisms by which active GSK3 can perpetuate its own activity.
GSK3 is also phosphorylated at other sites besides serine9/21, but their regulatory outcomes remain unclear. Phosphorylation on Tyr216-GSK3β and Tyr279-GSK3α is required for maximal activity (Hughes et al., 1993; Frame and Cohen, 2001). During translation, GSK3 phosphorylates itself on these residues so that an active kinase is immediately synthesized (Cole et al., 2004b). This is another fascinating characteristic of GSK3, during synthesis it can be a tyrosine kinase, but mature GSK3 is a serine/threonine kinase. Although still unsettled, it appears that phosphotyrosine phosphatases can deactivate GSK3 under certain conditions, because changes in phosphotyrosine-GSK3 levels have been reported in a wide variety of conditions. However, more research is needed to clarify how and when GSK3 is regulated by changes in tyrosine phosphorylation. GSK3β is also phosphorylated on ser389/390 and ser43, but little is known about specific signaling pathways that mediate these modifications and their functional outcomes.
Investigators have long thought that GSK3 must be regulated by additional post-translational mechanisms because it seemed that more control is necessary for it to distinguish among numerous substrates and to be uniquely regulated by specific signaling pathways (Jope and Johnson, 2004). Such modifications are finally beginning to be identified. GSK3 has been reported to be cleaved to activated fragments by calpain (Goñi-Oliver et al., 2007; Goñi-Oliver et al., 2009) and by matrix metalloproteinase-2 (Kandasamy and Schulz, 2009), which may affect its selection of substrates to phosphorylate. A regulatory effect of acetylation on GSK3 activity was recently reported (Monteserin-Garcia et al., 2013). Furthermore, GSK3β was recently found to be inhibited by mono-ADP-ribosylation (Feijs et al., 2013; Rosenthal et al., 2013), and to be regulated by citrullination (Stadler et al., 2013). These and other phosphorylation-independent post-translational mechanisms seem likely to contribute to regulating the multiple actions of GSK3. In the near future we foresee important progress being made in determining which particular pathways utilize each of these modifications, and the identification of additional post-translational modifications that regulate GSK3.
2.2. Substrate pre-phosphorylation and availability
Substrate-recognition by GSK3 is an often underappreciated but crucial mechanism for regulating the actions of GSK3. As noted in the previous section, for GSK3 to phosphorylate the majority of its substrates, two signals must coincide temporally and spatially: induction of the priming phosphorylation of the substrate and GSK3 activation. As noted above, most GSK3 substrates must be primed by phosphorylation on a residue approximately 4 residues C-terminal to the GSK3 target residue, S/T-X-X-X-S/T(P) (Figure 1). This means that GSK3 does not recognize the substrate unless another signaling pathway is activated that primes, by pre-phosphorylating, the substrate. This is an exquisite mechanism for limiting the action of GSK3: signals must be generated to activate a kinase to pre-phosphorylate the substrate, GSK3 must be co-localized with the primed substrate, and the timing of signals that activate GSK3 (e.g., by phosphoserine dephosphorylation) all must be coordinated to determine if and when GSK3 phosphorylates the substrate. This provides an elegant mechanism for preventing spurious actions of GSK3 and to temporally limit the lifetime of primed substrates. For example, the transcription factor cyclic AMP response element binding protein (CREB) is not recognized by GSK3 unless a CREB-activating pathway is turned on that induces phosphorylation of CREB on serine-133, which activates CREB. This creates a primed phosphorylation site for GSK3, and in this way intracellular signaling pathways direct GSK3 to its target. However, since GSK3 is constitutively partially serine-phosphorylated, and thus partially inhibited from recognizing primed substrates, another signal coinciding in time and intracellular location is required to reduce serine-phosphorylation of GSK3 to allow it to recognize phosphoserine133-CREB. In other words, for nuclear CREB, a signal must cause serine-dephosphorylation of GSK3 in the nucleus at a time when CREB is phosphorylated on serine-133. Since phosphorylation of CREB on serine-129 by GSK3 inactivates CREB, we envision this as a mechanism for ensuring that the activation of CREB is transient, an outcome that may be employed in transient learning events that require active CREB for a short time. Thus, this provides an exquisite mechanism for cycling the actions of GSK3 substrates.
2.3. Subcellular localization
GSK3 has traditionally been considered to be largely a cytosolic protein. However, GSK3 is also present within the mitochondria and nucleus, as well as other subcellular compartments, where its levels and/or activation state can be regulated by localized signaling activities. Mitochondrial GSK3 may be especially important in oxidative stress and certain apoptotic conditions (Bijur and Jope, 2003; King et al., 2001). However, little is known about mechanisms regulating the mitochondrial level and activity of GSK3, or the mitochondrial substrates of GSK3, topics that may provide important information about potential pathological roles of mitochondrial GSK3 in diseases that involve mitochondrial dysfunction.
In contrast to its mitochondrial actions, many nuclear effects of GSK3 have been identified, particularly those involving the regulation of gene expression. GSK3 enters the nucleus via an intrinsic nuclear localization sequence in GSK3β (Meares and Jope, 2007), and the nuclear levels of GSK3β rapidly increase in response to some apoptotic stimuli (Bijur and Jope 2001). Conversely, the GSK3-binding protein Frat promotes nuclear export of GSK3 (Franca-Koh et al., 2002). The distinct N-terminal region of GSK3α causes nuclear exclusion that can be counter-activated by calcium signaling (Azoulay-Alfaguter et al., 2011).
Actions of GSK3 affecting nuclear functions, specifically gene expression, include indirect effects via its regulation of many transcription factors, and more direct effects resulting from the regulation of recently identified epigenetic mechanisms. Transcription factors are the largest known class of GSK3 substrates, providing a mechanism by which GSK3 can influence the expression of many genes. Among the many transcription factors regulated by GSK3 are Fos/Jun AP-1, CREB, heat shock factor 1, nuclear factor of activated T cells (NFAT), myc, C/EBP, NF-κB, p53, signal transducer and activator of transcription-3 (STAT3), as well as many others that have been reviewed previously (Grimes and Jope, 2001; Jope and Johnson, 2004; Sutherland, 2011). More recently, evidence has begun to reveal significant modulatory effects of GSK3 on proteins involved in epigenetics. Evidence is particularly strong that GSK3 contributes to the regulation of histone modifications that play a central role in epigenetics. GSK3 can directly phosphorylate histone 1.5 (Happel et al., 2009) and promotes histone 3 phosphorylation, although the mechanism was not identified (Gupta et al., 2014). GSK3 also phosphorylates several histone deacetylases (HDACs). GSK3 phosphorylates HDAC3, which promotes its activity (Bardai and D’Mello, 2011), and HDAC4, a modification that targets HDAC4 for degradation (Cernotta et al., 2011), and indirect evidence suggests that GSK3 may also phosphorylate HDAC6 to regulate its activity (Chen et al., 2010). Conversely, several HDAC inhibitors have been reported to increase the inhibitory serine-phosphorylation of GSK3 (De Sarno et al., 2002). There is also evidence that members of the sirtuin (Sirt) family of deacetylases regulate GSK3. Sirt2 activity is associated with inhibition of GSK3 (Dan et al., 2012; Si et al., 2013), whereas Sirt1 has been reported to either activate (Monteserin-Garcia et al., 2013) or inhibit (Li et al., 2013) GSK3. GSK3 also modulates post-translational modifications of histones by regulating several histone acetyltransferases (HATs). GSK3 directly phosphorylates and activates the HAT Tip60 (Charvet et al., 2011) (Figure 3), which was shown to mediate the previously reported promotion by GSK3 of apoptosis induced by the transcription factor p53 (Watcharasit et al., 2002) and to promote autophagy (Lin et al., 2012). Steroid receptor coactivator-1 was identified as a HAT (Spencer et al., 1997) and both steroid receptor coactivator-1 (Hartmaier et al., 2012) and its paralog steroid receptor coactivator-3 (Wu et al., 2007) are phosphorylated by GSK3, which promotes their degradation. CLOCK is a transcription factor crucial for circadian function that has intrinsic HAT activity (Bellet and Sassone-Corsi, 2010), and GSK3 phosphorylates both CLOCK (Spengler et al., 2009) and its interacting transcription factor BMAL1 (Sahar et al., 2010), which regulates their stabilities and the circadian rhythm. The GSK3 inhibitor lithium regulates expression of T08D10.2, the Caenorhabditis elegans analog of the histone demethylase LSD-1, in a GSK3-dependent manner (McColl et al., 2008). In addition to its effects on histones and their modifying enzymes, GSK3 can also modulate DNA-modifying enzymes. GSK3 phosphorylates and reduces the stability of DNA methyltransferase1 (Dnmt1) (Sun et al., 2007; Lin et al., 2010a; Li et al., 2013), and GSK3 also regulates the stability of Dnmt3a2, although the mechanism remains to be identified (Popkie et al., 2010). Epigenetic regulation by GSK3 has been especially well-characterized in the regulation of expression of a subset of NF-κB-regulated genes (Ougolkov et al., 2007; Zhang et al., 2014). Thus, by regulating numerous transcription factors and proteins involved in epigenetic regulation, GSK3 has widespread modulatory effects on gene expression.
Figure 3. Axin associates with several GSK3 substrates.

Axin binds to several GSK3 substrates to facilitate their phosphorylation, such as (A) Smad3, (B) tuberous sclerosis complex-1 (TSC1), and (C) TSC2. (D) The GSK3 substrate Tip60 also binds to Axin, which may facilitate its phosphorylation by GSK3 that activates Tip60. Tip60-mediated regulation of p53 is also modulated by Axin-bound HIPK2 (homeodomain-interacting protein kinase 2), raising the hypothetical possibility depicted here of a multi-protein complex that regulates both Tip60 and p53. (A-D) It is not known if these GSK3 substrates compete with β-catenin to bind the same domain of Axin, or if distinct binding sites on Axin exist for each substrate. Binding of all substrates to the same region of Axin is depicted here because this places the substrates adjacent to GSK3 for phosphorylation.
Besides these classical nuclear and mitochondrial compartments, GSK3 is also compartmentalized in other subcellular structures, such as in growth cones (Eickholt et al., 2002; Zhou et al., 2004; Wood-Kaczmar et al., 2009), and in protein complexes, as discussed in the following section. Little is known about mechanisms that regulate trafficking of GSK3 within cytosolic domains, which likely has important consequences for GSK3 function, although Fluorescence Recovery after Photobleaching (FRAP) analysis indicated that 84% of cytosolic GSK3β was mobile in mouse embryonic fibroblasts (Meares & Jope, 2007). Furthermore, sustained Wnt signaling leads to sequestration of GSK3 in multi-vesicular endosomes, which affects its functions in both Wnt-mediated and Wnt-independent actions (Taelman et al., 2010). The localization of GSK3 obviously is important in regulating its actions, but much remains to be learned about mechanisms regulating GSK3 trafficking within cells and the functional consequences.
2.4. Association in protein complexes
A major mechanism that has evolved for targeting GSK3 towards specific substrates is by its incorporation into protein complexes that recruit substrates for GSK3 to phosphorylate. These include both preassembled complexes and signal-induced complexes. The classical example of a GSK3-containing preassembled complex is the β-catenin destruction complex in the Wnt signaling pathway (Figure 2). In this complex, the scaffold protein Axin, with the assistance of the associated protein APC (Adenomatous Polyposis Coli), brings β-catenin into close proximity to GSK3. GSK3 phosphorylates β-catenin on residues 41, 37, and 33 after priming phosphorylation by casein kinase 1, which targets β-catenin to the proteasomal degradation machinery. Wnt ligands induce disruption of the complex, which prevents GSK3 from phosphorylating β-catenin, allowing the accumulation and nuclear import of β-catenin. Several Axin-binding proteins have been identified that modulate the phosphorylation of β-catenin in the Axin-GSK3 complex (Harauchi et al., 2008; Kim et al., 2009; Kashikar et al., 2011; Deng et al., 2012), suggesting that much remains to be learned about how signaling pathways are integrated at this locus. Others have reviewed the details of the β-catenin destruction complex (MacDonald et al., 2009; Valvezan and Klein, 2012; Willert and Nusse, 2012), but there are several important points that are often overlooked by investigators new to the field.
Figure 2. The Axin-β-catenin destruction complex.

A simplified scheme is shown of the preassembled complex of Axin, Adenomatous Polyposis Coli (APC), casein kinase 1 (CK1), GSK3β, and β-catenin, which allows GSK3 to phosphorylate β-catenin sequentially on residues 41, 37, and 33. Phosphorylation leads to the release of β-catenin from the complex and targets it for proteasomal degradation. Activation of Wnt signaling disrupts the complex (not shown), blocking phosphorylation of β-catenin by GSK3, resulting in β-catenin stabilization (MacDonald et al., 2009; Valvezan and Klein, 2012; Willert and Nusse, 2012).
β-catenin phosphorylation by GSK3 in this complex is impervious to serine-phosphorylation of GSK3 (Ng et al., 2009). As well-reviewed by Woodgett and colleagues (Voskas et al., 2010; Kaidanovich-Beilin and Woodgett, 2011), the activity of GSK3 coupled to Axin is not controlled by the GSK3 serine-phosphorylation mechanism. This independence of regulation by GSK3 serine-phosphorylation may be due to structural constraints that make Axin-bound GSK3 inaccessible to kinases that would otherwise phosphorylate the inhibitory serine, or it may be due to the proximity of its primed substrate β-catenin, although details of the three-dimensional structure of the complex remain unclear. Taken along with the fact that only a very small portion of cellular GSK3 is bound to Axin (Voskas et al., 2010; Kaidanovich-Beilin and Woodgett, 2011), it is evident that most phosphoserine-GSK3 measured in cells is not the GSK3 that is associated with Axin. Thus, it is important to be wary of the often-stated conclusion after an experimental treatment that there is a direct relationship between increases in the serine-phosphorylation of GSK3 and increases in the level of β-catenin; these outcomes can occur in parallel but increased serine-phosphorylation of GSK3 does not inhibit GSK3 in the β-catenin destruction complex. This mechanism of insulating Wnt signaling from signaling induced by insulin and other pathways that regulate serine-phosphorylation of GSK3 is not only ingenious as only nature can be, but it is obviously critical to avoid disastrous activation of Wnt signaling by insulin and by every other signal that increases serine-phosphorylated GSK3. Thus, numerous reports that certain conditions increase the serine-phosphorylation of GSK3 and elevate β-catenin levels should not be interpreted as a cause-and-effect relationship between these two events.
In many cells, if not all, only a small portion of β-catenin is associated with the Axin destruction complex, with much of the β-catenin instead being associated in other protein complexes at the plasma membrane. Therefore, changes in total cellular levels of β-catenin often do not reflect changes in the activity of the Axin-linked β-catenin destruction complex. Rather, it is more informative to measure changes in nuclear β-catenin, which increase after the Axin-β-catenin destruction complex is inactivated.
β-catenin is not the only GSK3 substrate that binds to Axin (Figure 3). Although Wnt signaling is most often the subject of studies of Axin-GSK3-substrate interactions, Axin also brings other substrates into close proximity for phosphorylation by Axin-bound GSK3 (Kim et al., 2009), such as Smad3 (Guo et al., 2008), tuberous sclerosis complex-1 (TSC1)/hamartin and TSC2/tuberin (Mak et al., 2003; Mak et al., 2005; Rosner et al., 2008). Largely unaddressed are the regulatory consequences of the existence of multiple Axin-bound GSK3 substrates, e.g., is the phosphorylation by GSK3 of other Axin-bound substrates also impervious to regulation of GSK3 by serine-phosphorylation? Is there competition between β-catenin and other GSK3 substrates for the limited amount of Axin? Less than 10% of cellular GSK3 has been estimated to be associated with Axin (Voskas et al., 2010; Kaidanovich-Beilin and Woodgett, 2011), indicating that the level of Axin is limiting, not the GSK3 level, for substrates requiring binding to Axin to be phosphorylated by GSK3. Thus, there are likely some important regulatory interactions between different signaling pathways that utilize the Axin-GSK3-substrate route, e.g., how is priority determined between Axin-bound GSK3 substrates, and does this provide an integrative mechanism, or competition, between signaling pathways? For example, cooperative actions of Axin and p53, and several p53-regulating proteins such as Tip60 (Figure 3), have been demonstrated to regulate both p53 and β-catenin actions (Levina et al., 2004; Rui et al., 2004; Li et al., 2007b Li et al., 2009) and may contribute to the promotion of p53 actions by GSK3 (Watcharasit et al., 2002; Watcharasit et al., 2004).
Although often viewed as an independent entity in cells, the Axin-β-catenin destruction complex has a variety of interactions with Axin-independent actions of GSK3. APC not only regulates the actions of GSK3 in the Axin complex, but also regulates GSK3 activity towards other substrates not dependent on binding to Axin (Valvezan et al., 2012b). A variety of signals and proteins regulate the Axin-β-catenin destruction complex, as others have reviewed (MacDonald et al., 2009; Valvezan and Klein, 2012; Willert and Nusse, 2012). One of these is β-arrestin, which binds Axin and the GSK3-binding protein dishevelled (Bryja et al., 2007). However, β-arrestin also mediates dopamine D2 receptor-induced signaling to Akt and GSK3, as discussed in section 3. Surprisingly, the dual actions of β-arrestin in Akt and Wnt signaling have not been explored in greater detail.
In addition to Axin, a growing number of GSK3-binding proteins, have been identified that appear to direct GSK3 phosphorylation for specific functions, reflecting the importance of protein complexes in allowing signals to regulate the action of GSK3 in a substrate-selective manner. For example, members of the 14-3-3 family of proteins bind GSK3 (Agarwal-Mawal et al., 2003; Yuan et al., 2004; Liao et al., 2005), an interaction with intriguing potential consequences. 14-3-3 proteins generally bind phosphorylated proteins, raising the possibility that they interact with serine-phosphorylated GSK3 either to modulate its dephosphorylation/phosphorylation cycle or to direct its action towards specific substrates regardless of its serine-phosphorylation, similar to Axin. GSK3 also was recently found to bind to inositol hexakisphosphate-1, which enhanced GSK3 activity (Chakraborty et al., 2013). Other interactions include glycogen synthase kinase 3β interaction protein (GSKIP) that appears to function as a member of the A-kinase anchoring proteins (AKAP) family and inhibits GSK3 in the Wnt signaling complex (Chou et al., 2006; Hundsrucker et al., 2010), protein 4.1R that regulates GSK3 at microtubules (Ruiz-Saenz et al., 2013), suppressor of fused (Sufu) that is both phosphorylated by GSK3 and links GSK3 to Gli3 in the hedgehog signaling pathway (Kise et al., 2009; Chen et al., 2011) and GSK3 binds, and is activated by, the CDK5 activator protein p25 (Chow et al., 2014). Additionally, as detailed in other reviews, GSK3 is involved in protein complexes that regulate growth cones (Eickholt et al., 2002; Zhou et al., 2004), cell polarity (Etienne-Manneville and Hall, 2003), and focal adhesions (Cai et al., 2006; Kobayashi et al., 2006). The actions of GSK3 in protein complexes also extend to receptors and receptor-coupled signal transduction systems, as discussed in section 3. It is not known which of these protein binding interactions, like Axin, also insulate the activity of GSK3 from being regulated by serine-phosphorylation, i.e., does binding of GSK3 often insulate it from regulation by signals that increase the inhibitory serine-phosphorylation of GSK3? We suggest that this topic has much promise for clarifying mechanisms that regulate GSK3 and finding further substrates that are phosphorylated by GSK3 on other scaffolds.
Considering the importance of GSK3 binding proteins in regulating its actions and substrate selectivity, there is a notable gap in studies of the domains and amino acid sequences in GSK3 that associate with regulatory proteins. Although this has been studied in detail for the interacting domains of GSK3 in the Axin complex (Ferkey and Kimelman, 2002; Fraser et al., 2002; Dajani et al., 2003; Howng et al., 2010), since then detailed binding characterization has seldom been reported, such as for its interaction with Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen (Fujimuro et al., 2005) and p53 (Eom and Jope, 2009). It is likely that identification of the GSK3 domains interacting with binding partners will be crucial for developing methods to interfere with the actions of GSK3 in a manner selective to specific substrates, or at least to signaling pathways, that may be useful in therapeutics developments.
2.5. Other mechanisms regulating the actions of GSK3
2.5.1. Expression
GSK3 seldom appears to be regulated by changes in expression, as only a few conditions have been reported in which the expression or levels of GSK3 are markedly altered. However, large changes in GSK3 levels can occur, for example there is a large, ~10-fold increase in GSK3β in the Th17 subtype of T cells, relative to other T cells (Beurel et al., 2011), and large 3-4-fold changes occur in the levels of both GSK3 isoforms in mouse brain during development (Beurel et al., 2012). However these are exceptions to the more common stability of GSK3 expression and levels. Nonetheless, GSK3 levels have been reported to change in response to a variety of treatments. It is also intriguing that patients with bipolar disorder have an increased frequency of a copy number variation in the GSK3β locus (Lachman et al., 2007). Future studies directed towards identifying the molecular mechanisms regulating GSK3 expression, and its degradation, may provide important information for the development of new interventions capable of altering the cellular levels of GSK3.
2.5.2. Single nucleotide polymorphisms (SNPs)
A number of SNPs have been identified in the GSK3β gene that may be linked to disease susceptibility or responses to therapeutic drugs, particularly including links between GSK3β SNPs to susceptibility to mood disorders and to therapeutic responses to drugs used in mood disorders (Benedetti et al., 2004a; Benedetti et al., 2004b; Benedetti et al., 2005; Szczepankiewicz et al., 2006a; Adli et al., 2007; Tsai et al., 2008; Inkster et al., 2009; Inkster et al., 2010; Saus et al., 2010). However, other studies have not found mood disorder-linked associations with GSK3β SNPs (Nishiguchi et al., 2006; Szczepankiewicz et al., 2006b). SNPs may alter the expression or actions of GSK3, but this remains largely speculative as little is known about the functional outcomes of GSK3β SNPs. However, one GSK3β polymorphism associated with Parkinson’s disease was reported to alter GSK3β transcription and splicing (Kwok et al., 2005). If progress is to be made in this area, further studies of the functional effects of GSK3 SNPs should receive highest priority.
2.5.3. Alternative splicing
Alternative splicing can give rise to forms of GSK3β with altered functions (Schaffer et al., 2003; Kwok et al., 2005). Especially intriguing is the GSK3β2 variant that is particularly abundant in the brain (Mukai et al., 2002), which contains an insert within the kinase domain that is regulated by the DDX5 and DDX17 RNA helicases (Samaan et al., 2014). GSK3β2 has been reported to be targeted to growing neurites and growth cones (Wood-Kaczmar et al., 2009), to be required for axon growth (Castaño et al., 2010), and GSK3β2 has reduced activity towards some, but not all, substrates compared with GSK3β1 (Mukai et al., 2002; Soutar et al., 2010).
Altogether, in contrast to the common notion that GSK3 is only regulated by serine-phosphorylation and by binding to Axin, there is a plethora of mechanisms that guide the actions of GSK3. These mechanisms apparently provide the underpinnings for GSK3 to select among its many substrates in response to each signaling pathway that impinges upon GSK3.
3. GSK3 interactions with receptors and receptor-coupled signal transduction events
3.1. G protein-coupled receptors
Much excitement has been generated by the rapidly developing concept that GSK3 has regulatory interactions with multiple intracellular receptor-coupled signaling proteins, such as β-arrestin and G-proteins, as well as with several receptors themselves (Figure 4). Most well-understood is the dopamine D2 receptor signaling mechanism that involves GSK3 and β-arrestin (Beaulieu et al., 2004; Beaulieu et al., 2005; Beaulieu et al., 2008). As others have reviewed in detail (Beaulieu et al., 2009), D2 receptor activation leads to an initial signal that regulates cyclic AMP production. This signal is followed by β-arrestin bringing Akt and GSK3 into close proximity to protein phosphatase 2A (PP2A), which dephosphorylates Akt to deactivate it and dephosphorylates GSK3 to activate it. Furthermore, GSK3 promotes the formation of this complex, providing a feed-forward mechanism for GSK3 to promote its own activation (O’Brien et al., 2011).
Figure 4. GSK3 interactions with G protein-coupled receptor-induced signaling mechanisms.
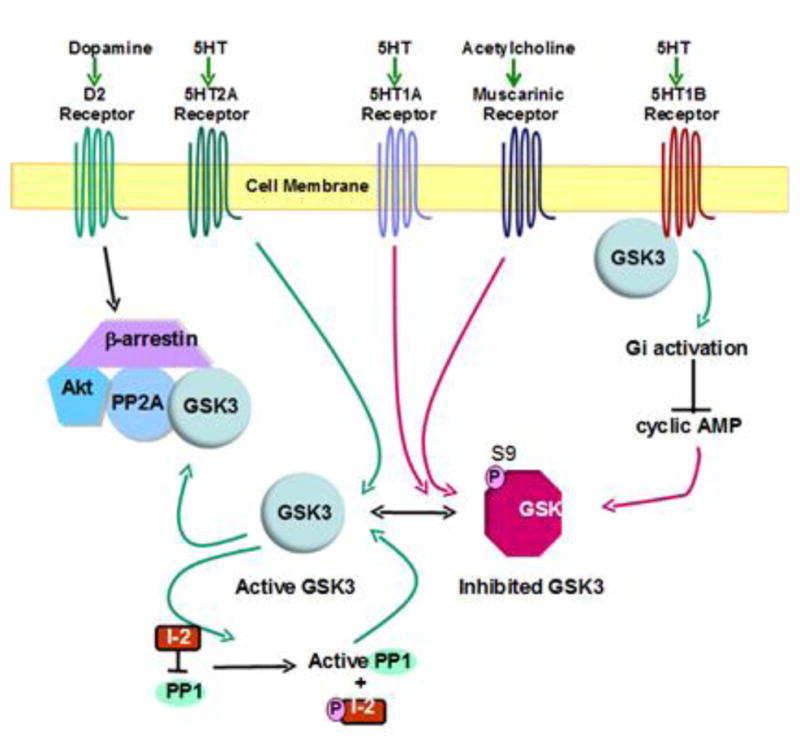
Dopaminergic D2 receptor activation induces the association of β-arrestin, Akt, GSK3, and protein phosphatase 2A (PP2A). This facilitates PP2A-mediated dephosphorylation of Akt and GSK3, deactivating Akt and activating GSK3. GSK3 facilitates the formation of this complex, providing a mechanism for GSK3 to induce its own activation. Self-activation by GSK3 is also exemplified by its phosphorylation of the protein phosphatase 1 (PP1) inhibitor I-2, resulting in increased PP1 activity, which dephosphorylates the inhibitory serine-phosphorylation of GSK3 to increase GSK3 activity. Serotonin (5HT) 2A receptor activation reduces serine-phosphorylation of GSK3, thereby increasing its activity. 5HT1A and cholinergic muscarinic receptor activation increase the inhibitory serine-phosphorylation of GSK3. GSK3 promotes 5HT1B receptor-mediated activation of the heterotrimeric G protein, Gi, which inhibits cyclic AMP production.
The β-arrestin-PP2A-Akt-GSK3 signaling interaction exemplifies two under-appreciated characteristics of GSK3, its ability to regulate Akt and its propensity to promote its own activation. There are multiple instances of bi-directional signaling between Akt and GSK3, contrary to the dogma that signals unidirectionally activate Akt, and Akt then inactivates GSK3. As noted in section 2.1, in addition to its effects in the β-arrestin complex, GSK3 can inhibit Akt and promote its own activation via GSK3-mediated activation of PP1, with active PP1 dephosphorylating Akt and GSK3, and by GSK3α-mediated inhibition of Akt via phosphorylation of threonine312-Akt (Gulen et al., 2012). Thus, active GSK3 can further drive its own activation by several processes. These multiple mechanisms for inhibition of Akt by GSK3 are also notable for explaining previous reports that inhibitors of GSK3, particularly lithium, increase the activating phosphorylation of Akt, which clearly can be a consequence of lithium’s inhibition of GSK3 rather than a direct activation of Akt by lithium. Thus, inactivation of GSK3 by lithium and other inhibitors will oppose these self-activating mechanisms, resulting in increased inhibitory serine-phosphorylation of GSK3. As discussed in section 5, impeding the GSK3 self-activating mechanisms may be critical for developing tolerable and effective therapeutic inhibitors of GSK3. Additionally, these GSK3 self-activating mechanisms have two consequences that deserve further consideration. First, GSK3 can indirectly regulate the phosphorylation of many proteins by regulating the activities of protein phosphatases and Akt. Second, these mechanisms of self-activation may be critically important in diseases that involve GSK3. If GSK3 is abnormally activated early in the development of a disease, and GSK3 further promotes its own activation, it is easy to envision how a relatively small disease-associated dysregulation of GSK3 can be amplified to promote disease processes. Equally important are the ramifications for therapeutic interventions. It appears that modest inhibition of GSK3, as provided by therapeutic levels of lithium, can be amplified by these mechanisms to increase the inhibitory serine-phosphorylation of GSK3. Thus, as previously discussed (Jope, 2006), the optimal goal for therapeutic interventions may be to achieve only modest direct drug-dependent inhibition of GSK3 that may be sufficient to dampen self-activation mechanisms, thereby attenuating GSK3 activity to pre-insult levels.
In addition to D2 receptors, GSK3 is also involved in signaling by other G protein-coupled receptors (Figure 4). Stimulation of serotonin (5HT) 5HT1A receptors increases the inhibitory serine-phosphorylation of GSK3, whereas 5HT2A receptor stimulation decreases it to activate GSK3 (Li et al., 2004). In rodent brain, the inhibitory effect of 5HT1A receptors is dominant, as in vivo serotonin deficiency induced pharmacologically or molecularly activates GSK3 (Li et al., 2004; Beaulieu et al., 2008). This is one mechanism by which GSK3 may be abnormally activated if there is a serotonin deficiency in patients with mood disorders (Jope, 2011). Conversely, increasing serotonin levels by in vivo administration of antidepressant drugs increases the inhibitory serine-phosphorylation of GSK3 in rodent brain (Li et al., 2004; Okamoto et al., 2010). GSK3 was also found to associate with 5HT1B receptors, promoting their signaling to activate the heterotrimeric G-protein Gi and activate Akt, providing another pathway by which GSK3 can regulate Akt activation (Chen et al., 2009; Chen et al., 2011). Muscarinic receptor stimulation also induces inhibitory serine-phosphorylation of GSK3 in mouse brain, although the signaling mechanisms involved remain to be identified (De Sarno et al., 2006).
Receptor-coupled, signal transducing heterotrimeric G proteins, which consist of α, β, and γ subunits, have several regulatory interactions with GSK3. Gβγ dimers released from heterotrimeric G proteins after receptor stimulation have long been known to activate phosphoinositide 3-kinase (PI3K) (Stephens et al., 1997), which leads to Akt activation and GSK3 inhibition. However, G proteins can also more directly regulate GSK3. Activated Gβ1γ2 was shown to recruit GSK3 to the plasma membrane and increase its kinase activity, which was linked to phosphorylation of the Wnt co-receptor LDL receptor-related protein 6 (LRP6) (Jernigan et al., 2011). Activated Gαq also is able to activate GSK3 (Fan et al., 2003). In contrast, Gαs displaces GSK3 from Axin, an action that inhibits the GSK3-mediated phosphorylation of β-catenin (Castellone et al., 2005). Thus, GSK3 has multiple regulatory interactions with G proteins and the receptors to which they are coupled.
3.2. Hormone receptors
Insulin signaling was the first pathway found to increase the inhibitory serine-phosphorylation of GSK3 by activation of PI3K and Akt (Cross et al., 1995). Furthermore, phosphorylation of the insulin receptor signal transducer insulin receptor substrate (IRS)-1 was the first receptor-coupled signal transducer shown to be a substrate of GSK3 (Eldar-Finkelman and Krebs, 1997). IRS-2 was subsequently also found to be phosphorylated by GSK3 (Sharfi and Eldar-Finkelman, 2008). These links between insulin and GSK3 raised the possibility that GSK3 inhibitors may be therapeutically useful in diabetes, which remains a topic of active investigation (MacAulay and Woodgett, 2008; Eldar-Finkelman et al., 2010; Amar et al., 2011).
Both subtypes of estrogen receptors (ER), ERα and ERβ, have regulatory interactions with GSK3. GSK3 phosphorylates ERα and supports ERα-mediated transcriptional activity (Medunjanin et al., 2005; Mendez and Garcia-Segura, 2006; Grisouard et al., 2007). There are conflicting reports about the regulation of ERα receptor stability by GSK3. GSK3 has been reported to down-regulate ERα levels in 293T cells (Park et al., 2008) and to stabilize ERα receptors in breast cancer cells (Grisouard et al., 2007), which may indicate a cell type-selective effect. GSK3 also phosphorylates the ERβ receptor, a modification that enhances ERβ sumoylation and stability (Picard et al., 2012). Interactions between GSK3 and the ERα, as well as androgen receptors, have been reviewed in detail (Grisouard and Mayer, 2009).
The glucocorticoid receptor is phosphorylated by GSK3, which regulates transcriptional responses to glucocorticoids, promotes nuclear export and degradation of the glucocorticoid receptor, and promotes glucocorticoid-induced apoptosis (Rogatsky et al., 1998; Galliher-Beckley et al., 2008). GSK3α, but not GSK3β, was found associated with glucocorticoid receptors, and released upon receptor activation (Spokoini et al., 2010). Conversely, glucocorticoid receptor-induced signaling activates GSK3 (Smith et al., 2002; Garza et al., 2012), which may provide a mechanism for GSK3 to subsequently down-regulate glucocorticoid receptor actions by its phosphorylation of the receptor.
Other receptors reported to be phosphorylated by GSK3 include the androgen receptor, which reduces its activation of transcription (Salas et al., 2004; Wang et al., 2004), the prolactin receptor, which targets it for proteasomal degradation (Plotnikov et al., 2008), the progesterone receptor, which regulates its stability (Wang et al., 2013b), LRP6, which promotes the recruitment of Axin to LRP6 (Zeng et al., 2005), and the IGF-1 receptor, which regulates its signaling and trafficking (Nemoto et al., 2010; Kelly et al., 2012).
3.3. Ionotropic receptors and receptor trafficking
In addition to the receptors noted above, GSK3 regulates the localization or function of several ionotropic neurotransmitter receptors. The inhibitory/excitatory neurotransmission balance is significantly influenced by the actions of GSK3 complexed with receptors and their signal transduction mediators. GSK3 phosphorylates the GABA synaptic scaffolding protein gephyrin, which diminishes its clustering and GABAergic transmission (Tyagarajan et al., 2011; Tyagarajan et al., 2013). GSK3 has also been reported to regulate dopamine-mediated regulation of cell surface GABA-A (Li et al., 2012) and NMDA (Li et al., 2009b) receptors. AMPA-mediated activity is increased by GSK3 modulating the trafficking of AMPA receptors, in part by phosphorylating guanyl nucleotide dissociation factor (GID), a regulator of Rab5 that mediates endocytosis of AMPA receptors (Wei et al., 2010; Wang et al., 2013). Regulation of kinesin is also likely important in these trafficking actions of GSK3, as GSK3 phosphorylates kinesin light chain 2, which was shown to contribute to the regulation of AMPA receptor trafficking (Morfini et al., 2002; Du et al., 2010). GSK3 also phosphorylates the postsynaptic protein PSD-95, which regulates AMPA receptor trafficking (Nelson et al., 2013). AMPA signaling, in turn, reduces GSK3 activity (Nishimoto et al., 2008), perhaps as a feed-back inhibitory mechanism for AMPA signaling, that also leads to reduced cell surface expression of the NMDA receptor subunit NR1 (Nishimoto et al., 2009). This interaction is consistent with the finding that GSK3 maintains the surface localization of NMDA receptors, including both the NR1 and NR2B subunits, by a mechanism that involves both Rab5 and PSD95 (Chen et al., 2007; Deng et al., 2014). This interaction may act as a positive feed-forward mechanism since stimulation of NR2B can activate GSK3 (Szatmari et al., 2005). These mechanisms may contribute to the earlier finding that the GSK3 inhibitor lithium diminishes NMDA receptor signaling (Nonaka et al., 1998; Hashimoto et al., 2002) and that administration of memantine, an NMDA receptor antagonist, increased the inhibitory serine-phosphorylation of both GSK3 isoforms in mouse brain (De Sarno et al., 2006).
Overall, it is now evident that GSK3 is intimately involved in numerous receptor-coupled signaling mechanisms, not only the commonly recognized signaling induced by insulin and Wnt. These interactions include regulation of receptor-coupled signal transduction proteins, direct interactions with receptors, and regulation of receptor trafficking. These accumulating findings indicate that many receptor-mediated signals are regulated by GSK3, with undoubtedly many more to be identified in the near future.
4. Differential actions and regulation of GSK3α and GSK3β
An emerging topic is the identification of differential regulation and actions of GSK3α and GSK3β. The catalytic domains of GSK3α and GSK3β are nearly identical, but their C-terminal sequences diverge and GSK3α contains a large glycine-rich N-terminal region that is absent in GSK3β (Kaidanovich-Beilin and Woodgett, 2011). Although commonly referred to as isoforms, GSK3α and GSK3β are actually paralogs, homologous proteins derived from different genes. It is surprising that little is known about mechanisms that differentially regulate their expressions, except for the novel discovery that the GSK3α gene is absent in birds (Alon et al., 2011). Reported expression differences include the selective up-regulation of GSK3β expression in the Th17 subtype of T cells, which are pathogenic in autoimmune diseases (Beurel et al., 2011; Beurel et al., 2013), and differences in the expression of GSK3α and GSK3β in mouse brain regions during development (Beurel et al., 2012) and in the suprachiasmatic nucleus during the circadian rhythm (Iwahana et al., 2004). Studies of mechanisms that differentially regulate the expression of GSK3α and GSK3β are needed to identify signals that control the expression of each isoform.
For many years GSK3α and GSK3β were often considered as identical twins, in part because all known GSK3 inhibitors inhibit both isoforms so it was difficult to identify differential effects of the two isoforms. Unfortunately, a great many studies have attributed the outcomes of treatments with GSK3 inhibitors specifically to GSK3β, leading to incorrect conclusions that GSK3β selectively mediates the processes under investigation. There is also a widely held misconception that certain commercially available GSK3 inhibitors are specific for one isoform, derived from the fact that recombinant GSK3β is often used to screen potential inhibitors. However, there is not a commercially available GSK3 inhibitor that is selective for either isoform, although there is some progress in the development of GSK3 isoform-selective inhibitors (Lo Monte et al., 2012).
Recently, different actions of the GSK3 isoforms have begun to be identified, primarily due to the advent of knockout and knockdown methods. Identifying individual actions of GSK3α and GSK3β was particularly stimulated by the finding that GSK3β knockout mice are embryonically lethal, whereas GSK3α knockout mice are viable, which provided an important demonstration that the two isoforms are not interchangeable (Hoeflich et al., 2000). Studies in GSK3α knockout mice, GSK3β+/− heterozygote knockout mice, and with partial suppression of GSK3β expression in mouse brain, have demonstrated functions regulated by each isoform (Beaulieu et al., 2004; O’Brien et al., 2004; Beaulieu et al., 2008b; Bersudsky et al., 2008; Kaidanovich-Beilin et al., 2009; Omata et al., 2011; Maurin et al., 2013). However, in most cases this approach does not prove isoform-specific actions, as it may be unclear if the outcomes of isoform reduction are due to lower total levels of GSK3, or to isoform-specific effects, unless outcomes of individually knocking down GSK3α and GSK3β are compared.
Although many substrates appear to be phosphorylated by both GSK3 isoforms, there is a growing recognition that certain substrates are preferentially linked to one isoform and that the two isoforms differ in certain regulatory functions. For example, some of the substrates reported to be differentially phosphorylated by GSK3α and GSK3β include inhibitor-2, a regulator of PP1 (Wang et al., 1994), the p62 nucleoporin protein (Miller et al., 1999), the transcription factors early growth response-1 and Smad3/4 (Liang and Chuang, 2006), and substrates in brain cortical tissue (Soutar et al., 2010). Several actions have been specifically ascribed to GSK3α, including its involvement in acute myeloid leukemia (Banerji et al., 2012), binding to the scaffold protein Receptor for Activated C-Kinase-1 as part of its regulation of the circadian clock (Zeidner et al., 2011), promotion of amyloid β-peptide production and senile plaque formation in models of Alzheimer’s disease (Phiel et al., 2003; Hurtado et al., 2012; but also see Ly et al., 2013), regulation of autophagy and age-related pathologies (Zhou et al., 2013), and GSK3α selectively modulates IL-1-mediated regulation of Th17 cells, an action that was attributed to inhibition by GSK3α of IL-1-induced activation of Akt and mTOR (Gulen et al., 2012). GSK3β selectively promotes activation of members of the STAT family of transcription factors (Beurel and Jope, 2008), and GSK3β selectively phosphorylates Mcl-1, which promotes its degradation in apoptosis signaling (Ding et al., 2007). A screen of interacting protein identified several differences between GSK3α and GSK3β (Pilot-Storck et al., 2010), which should be informative for future studies of their differential actions. Evidence has begun to be obtained indicating differential actions of GSK3a and GSK3β in synaptic plasticity, as detected by measurements of long-term potentiation (LTP) and long-term depression (LTD). In 2007, inhibition of GSK3 by serine-phosphorylation was found to be necessary for, and induced by, LTP, whereas GSK3 promotes LTD (Hooper et al., 2007; Peineau et al., 2007; Zhu et al., 2007). LTP was impaired in mice overexpressing GSK3β (Hooper et al., 2007) and in mice expressing mutant GSK3β unable to be inhibited by serine-9-phosphorylation (Dewachter et al., 2009), and GSK3β was associated with receptors thought to be important in LTP (Peineau et al., 2007). This all implied that GSK3β is an important regulator of synaptic plasticity, whereas a regulatory role for GSK3α was not specifically studied. In contrast, using mice expressing mutant GSK3α or mice deficient in GSK3α, other investigators found specific actions of GSK3α that are important in regulating synaptic plasticity (Maurin et al., 2013; Shahab et al., 2014). Thus, while it is clear that GSK3 impairs LTP and promotes LTD, which has been linked to impaired cognition in several disorders (King et al., 2014), further approaches are needed to clarify the roles of each GSK3 isoform in these processes. Additional differential actions of GSK3α and GSK3β in cell differentiation and proliferation, and cardiovascular development were comprehensively reviewed (Force and Woodgett, 2009; Cho et al., 2009; Lal et al., 2012). Altogether, there is growing evidence of differential actions of GSK3α and GSK3β, a topic that is likely to receive much attention in the near future.
As might be expected, since GSK3α and GSK3β have differences in their functional effects, there is also evidence of differences in signaling mechanisms that regulate the two isoforms. Differences in the mechanisms regulating nuclear transport of the isoforms were noted in section 2.3. Protein kinase C was reported to phosphorylate GSK3β but not GSK3α (Goode et al., 1992), whereas IκB-related kinase-1 phosphorylated GSK3α but not GSK3β (Gulen et al., 2012). All three isoforms of Akt, Akt1, Akt2, and Akt3, phosphorylate GSK3β, but only Akt 2 phosphorylated GSK3α (Brognard et al., 2007). Remarkably, treatment of rats with olanzapine was reported to reduce serine-phosphorylation of GSK3α and increase it in GSK3β (Mondelli et al., 2013). Further identification of mechanisms that differentially regulate GSK3α and GSK3β may lead the way towards the development of interventions that can predominantly affect one isoform more than the other.
5. Targeting GSK3 therapeutically
With its demonstrated participation in many signaling pathways associated with disease pathology, it is not surprising that GSK3 is being considered as a therapeutic target in multiple disorders. However, the great number of substrates that are phosphorylated by GSK3 raises the question of whether this limits its feasibility as a therapeutic target because of the potential disruption of many cellular processes. This rational skepticism is countered by the already demonstrated safe and effective long-term administration of the GSK3 inhibitor lithium. Based on over 60 years of successful treatment of bipolar disorder with lithium, it is evident that a GSK3 inhibitor can be tolerated and be effective for many years, regardless of whether or not lithium’s inhibition of GSK3 contributes to its mood stabilization effects, which remains a matter of debate. Another important consideration is the extent to which GSK3 should be inhibited therapeutically. As discussed previously in greater detail (Jope, 2006), therapeutic levels of lithium only partially inhibit GSK3, and this may be optimal for dampening GSK3’s self-activating mechanisms in disease processes while allowing GSK3 to fulfill unhindered its many other cellular actions. In addition to the potential benefit of only partial inhibition of GSK3, developing disease-selective inhibitors of GSK3 should be possible based on the many mechanisms that regulate GSK3 that were discussed in previous sections. For example, the Eldar-Finkelman laboratory has already developed an effective GSK3 inhibitor peptide derived from the sequence of the GSK3 primed substrate CREB (Licht-Murava and Eldar-Finkelman, 2012, Avrahami et al., 2013), and a peptide inhibitor of GSK3 that is primed by Akt-dependent phosphorylation was shown to be effective after insulin treatment (Kim et al., 2013). As more is learned about the mechanistic involvement of GSK3 in individual diseases, it may be possible to develop inhibitors that predominantly block the actions of GSK3 that are involved in pathological mechanisms. Thus, one goal for future developments of therapeutic GSK3 inhibitors will be to devise methods to target specific cells or signaling pathways. Even as we await these developments, GSK3 is already being considered as a therapeutic target in multiple conditions, some of which are discussed in the following sections.
5.1. Psychiatric diseases
Psychiatric diseases are likely to be one of the major classes that are amenable to GSK3 inhibitor therapeutics. Patients with bipolar disorder, previously called manic-depression, are already being effectively treated with lithium and there is no doubt that lithium is a GSK3 inhibitor (Klein and Melton, 1996; Stambolic et al., 1996). Although the therapeutic mechanism of action of lithium remains to be definitively determined, substantial evidence indicates that inhibition of GSK3 is an important component of lithium’s mood stabilizing capacity (O’Brien and Klein, 2009; Jope, 2011). Lithium directly binds and inhibits GSK3 by competing for a Mg2+ binding site (Klein and Melton, 1996; Stambolic et al., 1996). A fascinating study identified the rare characteristics of the few Mg2+-binding proteins, including GSK3, that are required for lithium to successfully compete with Mg2+ (Dudev and Lim, 2011). Besides directly inhibiting GSK3, lithium administration at therapeutically relevant levels (i.e., near 1 mM lithium in the serum) also increases the inhibitory serine-phosphorylation of GSK3 in rodent brain in vivo and in many other cell types (Jope 2003), which can result from mechanisms discussed in section 2. Peripheral blood mononuclear cells from patients treated with lithium display increased serine-phosphorylated GSK3, mirroring this response that occurs in rodent brain after lithium administration, confirming that therapeutic levels of lithium inhibit GSK3 and increase its inhibitory serine-phosphorylation in humans (Li et al., 2007). We suggested that lithium-induced serine-phosphorylation of GSK3 likely acts as an amplification mechanism for lithium’s weak direct inhibition of GSK3 at therapeutic levels of lithium (De Sarno et al., 2002). The importance of inhibition of GSK3 by serine-phosphorylation in mood disorders has been further verified in animal models by using GSK3 knockin mice developed by the Alessi laboratory (McManus et al., 2005), with the regulatory serines of GSK3 mutated to alanines. GSK3 knockin mice exhibit increased sensitivity to manic-like amphetamine-induced locomotor hyperactivity and to stress-induced depression-like behaviors (Polter et al., 2010). Importantly, this demonstrates that a single alteration, abnormal activation of GSK3, can increase susceptibility to both poles of mood displayed in bipolar disorder, further supporting the conclusion that unbridled GSK3 activity can contribute to pathological mechanisms involved in bipolar disorder.
Several ramifications of the increased serine-phosphorylation of GSK3 induced by lithium are not always fully appreciated. First, this induction will only occur with pools of GSK3 that are subject to GSK3’s self-regulation via modulating phosphatases or other mechanisms that were discussed in previous sections. Thus, the serine-phosphorylation of all cellular GSK3 may not be equally affected by lithium, a topic that deserves further investigation. Second, this serine-phosphorylation of GSK3 mechanism of action of lithium leaves untouched the activity of GSK3 in the Wnt signaling pathway, and perhaps in other protein complexes as discussed in section 2.4. However, the direct inhibitory effect of lithium on GSK3 is often sufficient to impede β-catenin degradation. Third, as noted in section 2.1, serine-phosphorylation of GSK3 only inhibits its phosphorylation of primed substrates. Furthermore, accumulated primed substrates eventually can out-compete the phosphoserine of GSK3 for binding to the primed substrate binding pocket. Thus, the lithium-induced increase of serine-phosphorylated GSK3 does not completely block its phosphorylation of primed substrates, but instead leads to a new increased steady state level of the primed substrate. We speculate that this may be crucial for allowing lithium to be therapeutic without completely blocking all of GSK3’s actions. In fact, levels of lithium slightly above the therapeutic concentration of ~1 mM are toxic, perhaps due to too great a direct inhibitory effect on GSK3 by the direct binding mechanism. Thus, the induction of inhibitory serine-phosphorylation of GSK3 by lithium may be critical to its therapeutic effectiveness and tolerability.
In addition to the substantial evidence that GSK3 inhibition is therapeutic for bipolar disorder, other reviews have discussed growing evidence that GSK3 inhibitors may contribute to therapies for depression (Jope, 2011), anxiety (Sachs et al., 2013), and schizophrenia (Beaulieu et al., 2009; Freyberg et al., 2010; Singh, 2013). Some of the mechanisms by which abnormally active GSK3 may contribute to these diseases are noted near the end of section 5.2.
5.2. Neurological diseases
There is abundant evidence that inhibition of GSK3 is likely to be therapeutic for a number of neurological disorders. The evidence is particularly strong for Alzheimer’s disease, as has been discussed in detail in multiple reviews (Martinez and Perez, 2008; Avila et al., 2010; Medina and Avila, 2010; King et al., 2014). In Alzheimer’s disease, GSK3 has been demonstrated to promote every major pathological process, including amyloid β peptide production (Phiel et al., 2003) and tau phosphorylation (Himmelstein et al., 2012), which lead to the two hallmark pathologies of Alzheimer’s disease, amyloid plaques and neurofibrillary tangles, respectively. Through these and other actions, GSK3 also promotes apoptosis, which likely contributes to neuronal loss in Alzheimer’s disease (Mines et al., 2011). Much evidence also demonstrates that GSK3 inhibitors improve many cognitive functions in rodent models of Alzheimer’s disease, which is a key outcome since dementia is the defining characteristic of Alzheimer’s disease (King et al., 2014). Encouraging findings for targeting GSK3 have been identified in examinations of dementia development in bipolar patients that have been treated with lithium for long periods of time, as these patients often have a lower prevalence of dementia than the general population (Nunes et al., 2007; Kessing et al., 2010). Clinical trials of lithium in Alzheimer’s disease have demonstrated some beneficial effects (Forlenza et al., 2012; Nunes et al., 2013), although not in all studies (Hampel et al., 2009). An initial trial of another GSK3 inhibitor, Tideglusib, did not demonstrate remarkable protective effects in Alzheimer’s disease patients, although it was designed as a safety trial, not as a therapeutic trial (del Ser et al., 2013). Tideglusib was also tolerated in patients with progressive supranuclear palsy, and although it did not alter clinical outcomes, the treated patients exhibited reduced progression of atrophy (Höglinger et al., 2014; Tolosa et al., 2014). Limited therapeutic effects obtained to date may be attributed to the requirements for treating Alzheimer’s disease patients earlier in the disease process and for more prolonged periods of time.
Studies in rodent models also have demonstrated evidence of therapeutic effects of GSK3 inhibitors in several other neurological disorders. The furthest developed of these may be Fragile X Syndrome, as previously reviewed (Mines and Jope, 2011). Fragile X syndrome is the most common known single gene mutation cause of intellectual disability. In rodent models of Fragile X syndrome, GSK3 is hyperactive in the brain (Yuskaitis et al., 2010) and GSK3 inhibitors improve behavioral measures of learning and memory and rescue abnormalities in LTP and LTD (Choi et al., 2011; King and Jope, 2013; Franklin et al., 2014). It is notable that lithium is the only drug found so far that improves any cognitive assessment in patients with Fragile X syndrome (Berry-Kravis et al., 2008).
There is also exceptionally strong evidence from mouse models that GSK3 inhibitors are able to ameliorate the autoimmune disease multiple sclerosis. Although like most neurodegenerative diseases the causes are unknown, the onset and progression of multiple sclerosis are thought to be due to a combination of the effects of inflammation and pathogenic T cells causing axonal damage and neural dysfunction that progressively worsens over time. Thus, an ideal agent may be one that reduces inflammation and increases anti-inflammatory molecules, reduces the production of the CD4+ T cell subtype that is pathogenic in multiple sclerosis, called Th17 cells, and generally promotes neuronal resilience. Remarkably, all of these criteria have been met by GSK3 inhibitors in animal models of multiple sclerosis. Long-term treatment with several different GSK3 inhibitors had remarkably strong effects in protecting mice from developing hind limb paralysis in the mouse multiple sclerosis model, and even initiating treatment with GSK3 inhibitors only after disease onset reversed the clinical symptoms of the disease in mouse models (De Sarno et al., 2008; Beurel et al., 2011; Beurel et al., 2013). These actions of GSK3 inhibitors may be due to neuroprotection, anti-inflammatory effects, and blocking the production of pathological CD4+ T cells (Beurel, 2011).
There is also evidence that GSK3 inhibitors may alleviate several other neurological diseases. The possibility that GSK3 inhibitors may counteract neuronal loss in Parkinson’s disease was first raised by the finding that GSK3 inhibitors reduced apoptosis induced by molecules modeling Parkinson’s neurotoxicity (King et al., 2001). A GSK3β polymorphism was found to be associated with Parkinson’s disease that alters GSK3β transcription and splicing (Kwok et a., 2005). Subsequent studies have further strengthened the potential therapeutic benefits of GSK3 inhibitors in several models of Parkinson’s disease (Duka et al., 2009; Lin et al., 2010b; Morales-Garcia et al., 2013). Growing evidence has been reviewed previously demonstrating that GSK3 inhibitors are effective in mouse models of Huntington’s disease (Scheuing et al., 2014), stroke (Chuang et al., 2011), traumatic brain injury (King et al., 2014; Leeds et al., 2014), and spinocerebellar ataxia type 1 (Watase et al., 2007).
This broad range of neurological diseases ameliorated by GSK3 inhibitors likely arises in part from the fact that, to some extent, most involve inflammation and neurodegeneration, and GSK3 inhibitors are strong anti-inflammatory agents (Jope et al., 2007) and neuroprotectants, reducing apoptosis after a wide range of insults (Beurel and Jope, 2006). In addition to apoptosis, GSK3 inhibitors also provide neuroprotection by other mechanisms, in particular a rapidly growing literature demonstrates that GSK3 has multiple regulatory influences on dendrite and axon growth and repair (Jiang et al., 2005; Kim et al., 2006; Dill et al., 2008; Wakatsuki et al., 2011; Chen et al., 2012; Rui et al., 2013; Saijilafu et al., 2013). Levels of the neurotrophin, brain-derived neurotrophic factor (BDNF), are reported to be deficient in several psychiatric and neurological diseases (Duman and Monteggia, 2006; Ye et al., 2012). Signaling by BDNF induces inhibition of GSK3 (Mai et al., 2002), raising the possibility that BDNF deficiency in these diseases contributes to hyperactive GSK3, which then promotes pathological processes. Improvements in cognition rendered by GSK3 inhibitor administration (King et al., 2014) may be particularly due to the intriguing actions of GSK3 to promote long-term depression and inhibit long-term potentiation (Peineau et al., 2011), and to reversal of GSK3-mediated impaired neurogenesis (Eom and Jope, 2009b; Kim et al., 2009b). In some conditions, regulation of several components of the circadian rhythm by GSK3 may also make a significant contribution (Itaka et al., 2005; Ko et al., 2010; Lavoie et al., 2013). Thus, there are multiple mechanisms by which inhibition of GSK3 may contribute to therapies of neurological and psychiatric disorders.
5.3. Inflammatory diseases
Diseases involving inflammation in the periphery have also been shown to benefit from the anti-inflammatory effects of GSK3 inhibitors (Jope et al., 2007). Remarkably, administration of GSK3 inhibitors was sufficient to afford mice protection (survival) from an otherwise lethal dose of the inflammatory stimulant lipopolysaccharide used to model sepsis (Martin et al., 2005). Administration of GSK3 inhibitors also reduced inflammation and pathology in rodent models of asthma, arthritis, colitis, and peritonitis (Cuzzocrea et al., 2006; Hu et al., 2006; Whittle et al., 2006; Bao et al., 2007), as previously reviewed (Beurel et al., 2010; Beurel, 2011). Thus, GSK3 inhibitors provide an effective intervention for all inflammatory diseases in which they have been tested in rodent models.
The strong pro-inflammatory action of GSK3 in multiple peripheral and CNS diseases raises the question of why evolutionary mechanisms did not provide the means to more adequately down-regulate the activity of GSK3 in inflammatory conditions? One possibility is that a significant survival benefit is achieved by GSK3 promoting responses to infection and stress, both of which activate inflammatory responses. Recent insights about the interactions of inflammation and depression offer an excellent example of this interpretation (Raison and Miller, 2012). We suggest that one advantage for the promotion of inflammation by GSK3 is to augment immune responses to counteract real, or stress-induced perceived, threats to survival. Thus, survival may have benefited from GSK3 promoting both innate and adaptive immune responses (Beurel et al., 2010), increasing the production of multiple inflammatory cytokines and promoting the production of Th1 and Th17 inflammatory T cells, which should increase resistance to injury and infection (Beurel et al., 2011; Beurel et al., 2013). However, we now recognize that these same innate and adaptive immune responses can exacerbate chronic conditions that have become more prevalent in modern societies, such as neurodegenerative diseases, mood disorders, asthma, arthritis, and diabetes. Thus, whereas evolutionarily a strong immune response promoted by GSK3 was likely beneficial for survival, it is detrimental in chronic conditions. Therefore, GSK3 inhibitors provide a feasible means to counteract excessive inflammation that is involved in a diverse number of chronic conditions.
5.4. Cancer
There is much interest in GSK3 as a potential therapeutic target in many types of cancer (Manoukian and Woodgett, 2002; Ougolkov and Billadeau, 2006; Mills et al., 2011; McCubrey et al., 2014). However, this application is complicated by findings that GSK3 can act as a tumor suppressor or it can promote cell proliferation in different types of cancer. The tumor suppressor action is exemplified by GSK3-mediated phosphorylation of β-catenin and its subsequent degradation, a transcriptional co-activator that often promotes cellular proliferation (Polakis, 2007). There is evidence that GSK3 acts as a tumor suppressor in some skin (Leis et al., 2002; Ma et al., 2007) and breast (Farago et al., 2005; Armanious et al., 2010; Dembowy et al., 2014) cancers, and that repression of RNA polymerase 1 transcription by GSK3 contributes to its tumor suppressor action (Vincent et al., 2008). On the other hand, the tumor promoting actions of GSK3 are indicated by the elevated levels of GSK3 present in certain types of tumors and/or by the anti-proliferative effects of GSK3 inhibitors, such as in colon (Shakoori et al., 2007) and pancreatic (Ougolkov et al., 2005; Garcea et al., 2007) cancers, and in mixed lineage leukemia (Wang et al., 2008; Wang et al., 2010), whereas conflicting data exist for prostate cancer (Mulholland et al., 2006; Darrington et al., 2012).
These differences in the actions of GSK3 in various types of cancer may have obstructed the development of GSK3 inhibitors for cancer. Examination of the mechanisms underlying the opposite actions of GSK3 in different types of cancer may provide opportunities to better understand the mechanistic cellular changes and to identify those that may safely benefit from GSK3 inhibitor administration. One of these differences may depend on which apoptotic mechanisms are involved in each cancer type. As previously reviewed (Beurel and Jope, 2006), GSK3 has opposite actions on the intrinsic and extrinsic apoptotic signaling pathways. Intrinsic apoptotic signaling, also known as the mitochondrial apoptosis pathway, is promoted by GSK3 (Beurel and Jope, 2006), so if this pathway is impaired in certain cancers, then GSK3 inhibitors would be detrimental. Oppositely, extrinsic apoptotic signaling that is mediated by so-called death receptors, such as Fas, is blocked by an anti-apoptotic protein complex containing GSK3, so GSK3 inhibitors promote death receptor-mediated apoptosis (Sun et al., 2008), a mechanism that contributes to GSK3 inhibitor-induced neuronal and motor toxicity (Gomez-Sintes and Lucas, 2010). The variable role of GSK3 in different cancers also may be related to the role of β-catenin, which can be activated by GSK3 inhibitors (Polakis, 2007; Anastas and Moon, 2012). Variable roles of GSK3 in different cancers also may be related to the different actions of the transcription factor NF-κB in individual cancer types. GSK3 has multiple regulatory interactions with NF-κB and GSK3 phosphorylates multiple components of the NF-κB signaling pathway that may result in context-dependent regulation by GSK3 (Beurel and Jope, 2006). Thus, the type of apoptosis, the role of β-catenin, and the actions of NF-κB are likely important in the actions of GSK3 and its inhibitors in specific cancers. Thus, GSK3 appears to modulate several types of cancer, but its mixed actions may make it difficult to harness potential therapeutic interventions.
5.5. Other conditions
In addition to the diseases discussed here, previous reviews have detailed potential therapeutic applications of GSK3 inhibitors in other prevalent conditions, including cardiovascular diseases (Cheng et al., 2011), AIDS (Dewhurst et al., 2007), bone disorders (Wagner et al., 2010), and diabetes (Frame and Zheleva, 2006; MacAulay and Woodgett, 2008; Eldar-Finkelman et al., 2010; Amar et al., 2011).
6. Conclusions
GSK3 has proven to be a kinase so adaptable that it has been recruited evolutionarily to phosphorylate over 100 substrates, empowering it to participate in regulating numerous cellular functions. This flexibility may stem from the multiple sophisticated mechanisms that regulate the actions of GSK3, ensuring that it phosphorylates substrates only at the proper time and in discreet subcellular compartments, often established by protein complexes. Thus, post-translational modifications, substrate priming, cellular trafficking, and protein complexes each contributes to the exquisite regulation of GSK3.
Although GSK3 has demonstrated remarkable evolutionary adaptability, every new substrate of GSK3 that evolved, as well as each new mechanism regulating GSK3 activity, provided new potential sites that may be susceptible to insults associated with diseases. We speculate that self-activating mechanisms by which GSK3 promotes its own further activation may be particularly important in some disorders and for therapeutic interventions. If a compartment of GSK3 is abnormally activated early in a disease, several mechanisms were described here by which GSK3 can further augment its own activation. Based on this, it is easy to envision how an initially small dysregulation of GSK3 can be amplified as a disease progresses to promote pathology. Thus, an early-stage insult associated with a disorder may impair one of the inhibitory mechanisms regulating GSK3 selectively in the insult-induced signaling pathway that leads to pathology. The abnormally activated GSK3 would then be able to further self-activate by one or more of the mechanisms described here, and since this occurs in the insult-induced pathological signaling pathway, GSK3 could promote pathology while maintaining normal activity in other cellular functions. This raises the possibility that a modest inhibition of GSK3 may have a particularly significant effect on disease-associated self-activating mechanisms. In other words, attenuating the disease-associated self-amplification of GSK3 activity may be sufficient to be therapeutically beneficial, and certainly more likely tolerated, compared with extensive inhibition of GSK3. There are a remarkably large number of diseases and other medical conditions that have been shown to be amenable to improvement by the administration of GSK3 inhibitors in animal models. For translation to human diseases, it appears that in most cases therapeutic goals should not be aimed at achieving a high degree of GSK3 inhibition, for this would likely be toxic due to the many actions of GSK3. Instead, the goals for the near future should be to discover drugs that target GSK3 in specific disease processes, such as drugs that specifically focus on GSK3-substrate interactions that are dysregulated in individual diseases, or drugs that weakly, but sufficiently, inhibit GSK3 enough to prevent self-activation in pathological processes.
Acknowledgments
Research in the authors’ laboratories was supported by grants from the NIMH (MH038752, MH090236, MH095380).
Abbreviations
- AKAP
A-kinase anchoring proteins
- AMPK
AMP-activated kinase
- CREB
cyclic AMP response element binding protein
- ER
estrogen receptor
- GSK3
glycogen synthase kinase-3
- HAT
histone acetyltransferase
- HDAC
histone deacetylase
- 5HT
serotonin
- IRS
insulin receptor substrate
- LRP6
LDL receptor-related protein 6
- LTD
long-term depression
- LTP
long-term potentiation
- NFAT
nuclear factor of activated T cells
- PKA
protein kinase A
- PP1
protein phosphatase-1
- PP2A
protein phosphatase 2A
- Sirt
sirtuin
- SNP
single nucleotide polymorphism
- STAT
signal transducer and activator of transcription
- TSC1
tuberous sclerosis complex-1
Footnotes
Conflict of Interest
The authors have no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adli M, Hollinde DL, Stamm T, Wiethoff K, Tsahuridu M, Kirchheiner J, et al. Response to lithium augmentation in depression is associated with the glycogen synthase kinase 3-β-50T/C single nucleotide polymorphism. Biol Psychiatry. 2007;62:1295–1302. doi: 10.1016/j.biopsych.2007.03.023. [DOI] [PubMed] [Google Scholar]
- Agarwal-Mawal A, Qureshi HY, Cafferty PW, Yuan Z, Han D, Lin R, et al. 14-3-3 connects glycogen synthase kinase-3β to tau within a brain microtubule-associated tau phosphorylation complex. J Biol Chem. 2003;278:12722–12728. doi: 10.1074/jbc.M211491200. [DOI] [PubMed] [Google Scholar]
- Alon LT, Pietrokovski S, Barkan S, Avrahami L, Kaidanovich-Beilin O, Woodgett JR, et al. Selective loss of glycogen synthase kinase-3α in birds reveals distinct roles for GSK-3 isozymes in tau phosphorylation. FEBS Lett. 2011;585:1158–1162. doi: 10.1016/j.febslet.2011.03.025. [DOI] [PubMed] [Google Scholar]
- Amar S, Belmaker RH, Agam G. The possible involvement of glycogen synthase kinase-3 (GSK-3) in diabetes, cancer and central nervous system diseases. Curr Pharm Des. 2011;17:2264–2277. doi: 10.2174/138161211797052484. [DOI] [PubMed] [Google Scholar]
- Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nat Rev Cancer. 2012;13:11–26. doi: 10.1038/nrc3419. [DOI] [PubMed] [Google Scholar]
- Armanious H, Deschenes J, Gelebart P, Ghosh S, Mackey J, Lai R. Clinical and biological significance of GSK-3β inactivation in breast cancer-an immunohistochemical study. Hum Pathol. 2010;41:1657–1663. doi: 10.1016/j.humpath.2010.04.015. [DOI] [PubMed] [Google Scholar]
- Avila J, Wandosell F, Hernández F. Role of glycogen synthase kinase-3 in Alzheimer’s disease pathogenesis and glycogen synthase kinase-3 inhibitors. Expert Rev Neurother. 2010;10:703–710. doi: 10.1586/ern.10.40. [DOI] [PubMed] [Google Scholar]
- Avrahami L, Farfara D, Shaham-Kol M, Vassar R, Frenkel D, Eldar-Finkelman H. Inhibition of glycogen synthase kinase-3 ameliorates β-amyloid pathology and restores lysosomal acidification and mammalian target of rapamycin activity in the Alzheimer disease mouse model: in vivo and in vitro studies. J Biol Chem. 2013;288:1295–1306. doi: 10.1074/jbc.M112.409250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azoulay-Alfaguter I, Yaffe Y, Licht-Murava A, Urbanska M, Jaworski J, Pietrokovski S, et al. Distinct molecular regulation of glycogen synthase kinase-3α isozyme controlled by its N-terminal region: functional role in calcium/calpain signaling. J Biol Chem. 2011;286:13470–13480. doi: 10.1074/jbc.M110.127969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji V, Frumm SM, Ross KN, Li LS, Schinzel AC, Hahn CK, et al. The intersection of genetic and chemical genomic screens identifies GSK-3α as a target in human acute myeloid leukemia. J Clin Invest. 2012;122:935–947. doi: 10.1172/JCI46465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao Z, Lim S, Liao W, Lin Y, Thiemermann C, Leung BP, et al. Glycogen synthase kinase-3β inhibition attenuates asthma in mice. Am J Respir Crit Care Med. 2007;176:431–438. doi: 10.1164/rccm.200609-1292OC. [DOI] [PubMed] [Google Scholar]
- Bardai FH, D’Mello SR. Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J Neurosc. 2011;31:1746–1751. doi: 10.1523/JNEUROSCI.5704-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellet MM, Sassone-Corsi P. Mammalian circadian clock and metabolism - the epigenetic link. J Cell Sci. 2010;123:3837–3848. doi: 10.1242/jcs.051649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti F, Bernasconi A, Lorenzi C, Pontiggia A, Serretti A, Colombo C, et al. A single nucleotide polymorphism in glycogen synthase kinase 3β promoter gene influences onset of illness in patients affected by bipolar disorder. Neurosci Lett. 2004a;355:37–40. doi: 10.1016/j.neulet.2003.10.021. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Serretti A, Colombo C, Lorenzi C, Tubazio V, Smeraldi E. A glycogen synthase kinase 3β promoter gene single nucleotide polymorphism is associated with age at onset and response to total sleep deprivation in bipolar depression. Neurosci Lett. 2004b;368:123–126. doi: 10.1016/j.neulet.2004.06.050. [DOI] [PubMed] [Google Scholar]
- Benedetti F, Serretti A, Pontiggia A, Bernasconi A, Lorenzi C, Colombo C, et al. Long-term response to lithium salts in bipolar illness is influenced by the glycogen synthase kinase 3β-50 T/C SNP. Neurosci Lett. 2005;376:51–55. doi: 10.1016/j.neulet.2004.11.022. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Yao WD, Kockeritz L, Woodgett JR, Gainetdinov RR, et al. Lithium antagonizes dopamine-dependent behaviors mediated by an AKT/glycogen synthase kinase 3 signaling cascade. Proc Natl Acad Sci USA. 2004;101:5099–5104. doi: 10.1073/pnas.0307921101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/β-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Marion S, Rodriguiz RM, Medvedev IO, Sotnikova TD, Ghisi V, et al. A β-arrestin 2 signaling complex mediates lithium action on behavior. Cell. 2008;132:125–136. doi: 10.1016/j.cell.2007.11.041. [DOI] [PubMed] [Google Scholar]
- Beaulieu JM, Zhang X, Rodriguiz RM, Sotnikova TD, Cools MJ, Wetsel WC, et al. Role of GSK3β in behavioral abnormalities induced by serotonin deficiency. Proc Natl Acad Sci USA. 2008b;105:1333–1338. doi: 10.1073/pnas.0711496105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaulieu JM, Gainetdinov RR, Caron MG. Akt/GSK3 signaling in the action of psychotropic drugs. Annu Rev Pharmacol Toxicol. 2009;49:327–347. doi: 10.1146/annurev.pharmtox.011008.145634. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Sumis A, Hervey C, Nelson M, Porges SW, Weng N, et al. Open-label treatment trial of lithium to target the underlying defect in fragile X syndrome. J Dev Behav Pediatr. 2008;29:293–302. doi: 10.1097/DBP.0b013e31817dc447. [DOI] [PubMed] [Google Scholar]
- Bersudsky Y, Shaldubina A, Kozlovsky N, Woodgett JR, Agam G, Belmaker RH. Glycogen synthase kinase-3β heterozygote knockout mice as a model of findings in postmortem schizophrenia brain or as a model of behaviors mimicking lithium action: negative results. Behav Pharmacol. 2008;19:217–224. doi: 10.1097/FBP.0b013e3282feb099. [DOI] [PubMed] [Google Scholar]
- Beurel E. Regulation by glycogen synthase kinase-3 of inflammation and T cells in CNS diseases. Front Mol Neurosci. 2011;4:18. doi: 10.3389/fnmol.2011.00018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Jope RS. The paradoxical pro- and anti-apoptotic actions of GSK3 in the intrinsic and extrinsic apoptosis signaling pathways. Prog Neurobiology. 2006;79:173–189. doi: 10.1016/j.pneurobio.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Jope RS. Differential regulation of STAT family members by glycogen synthase kinase-3. J Biol Chem. 2008;283:21934–21944. doi: 10.1074/jbc.M802481200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Michalek SM, Jope RS. Innate and adaptive immune responses regulated by glycogen synthase kinase-3 (GSK3) Trends Immunol. 2010;31:24–31. doi: 10.1016/j.it.2009.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Yeh WI, Michalek SM, Harrington LE, Jope RS. Glycogen synthase kinase-3 is an early determinant in the differentiation of pathogenic Th17 cells. J Immunol. 2011;186:1391–1398. doi: 10.4049/jimmunol.1003511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Mines MA, Song L, Jope RS. Glycogen synthase kinase-3 levels and phosphorylation undergo large fluctuations in mouse brain during development. Bipolar Disord. 2012;14:822–830. doi: 10.1111/bdi.12023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beurel E, Kaidanovich-Beilin O, Yeh WI, Song L, Palomo V, Michalek SM, et al. Regulation of Th1 cells and experimental autoimmune encephalomyelitis by glycogen synthase kinase-3. J Immunol. 2013;190:5000–5011. doi: 10.4049/jimmunol.1203057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Proapoptotic stimuli induce nuclear accumulation of glycogen synthase kinase-3β. J Biol Chem. 2001;276:37436–37442. doi: 10.1074/jbc.M105725200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bijur GN, Jope RS. Glycogen synthase kinase-3β is highly activated in nuclei and mitochondria. Neuroreport. 2003;14:2415–2419. doi: 10.1097/00001756-200312190-00025. [DOI] [PubMed] [Google Scholar]
- Bradley CA, Peineau S, Taghibiglou C, Nicolas CS, Whitcomb DJ, Bortolotto ZA, et al. A pivotal role of GSK-3 in synaptic plasticity. Front Mol Neurosci. 2012;5:13. doi: 10.3389/fnmol.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. doi: 10.1016/j.molcel.2007.02.017. [DOI] [PubMed] [Google Scholar]
- Bryja V, Gradl D, Schambony A, Arenas E, Schulte G. Beta-arrestin is a necessary component of Wnt/β-catenin signaling in vitro and in vivo. Proc Natl Acad Sci USA. 2007;104:6690–6695. doi: 10.1073/pnas.0611356104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaño Z, Gordon-Weeks PR, Kypta RM. The neuron-specific isoform of glycogen synthase kinase-3β is required for axon growth. J Neurochem. 2010;113:117–130. doi: 10.1111/j.1471-4159.2010.06581.x. [DOI] [PubMed] [Google Scholar]
- Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-β-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- Cernotta N, Clocchiatti A, Florean C, Brancolini C. Ubiquitin-dependent degradation of HDAC4, a new regulator of random cell motility. Mol Biol Cell. 2011;22:278–289. doi: 10.1091/mbc.E10-07-0616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty A, Latapy C, Xu J, Snyder SH, Beaulieu JM. Inositol hexakisphosphate kinase-1 regulates behavioral responses via GSK3 signaling pathways. Mol Psychiatry. 2014;19:284–293. doi: 10.1038/mp.2013.21. [DOI] [PubMed] [Google Scholar]
- Charvet C, Wissler M, Brauns-Schubert P, Wang SJ, Tang Y, Sigloch FC, et al. Phosphorylation of Tip60 by GSK-3 determines the induction of PUMA and apoptosis by p53. Mol Cell. 2011;42:584–596. doi: 10.1016/j.molcel.2011.03.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Gu Z, Liu W, Yan Z. Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol Pharmacol. 2007;72:40–51. doi: 10.1124/mol.107.034942. [DOI] [PubMed] [Google Scholar]
- Chen L, Salinas GD, Li X. Regulation of serotonin 1B receptor by glycogen synthase kinase-3. Mol Pharmacol. 2009;76:1150–1161. doi: 10.1124/mol.109.056994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Zhou W, Chen PC, Gaisina I, Yang S, Li X. Glycogen synthase kinase-3β is a functional modulator of serotonin-1B receptors. Mol Pharmacol. 2011;79:974–986. doi: 10.1124/mol.111.071092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Maloney JA, Kallop DY, Atwal JK, Tam SJ, Baer K, et al. Spatially coordinated kinase signaling regulates local axon degeneration. J Neurosci. 2012;32:13439–13453. doi: 10.1523/JNEUROSCI.2039-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Owens GC, Makarenkova H, Edelman DB. HDAC6 regulates mitochondrial transport in hippocampal neurons. PloS One. 2010;5:e10848. doi: 10.1371/journal.pone.0010848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Yue S, Xie L, Pu XH, Jin T, Cheng SY. Dual Phosphorylation of suppressor of fused (Sufu) by PKA and GSK3β regulates its stability and localization in the primary cilium. J Biol Chem. 2011;286:13502–13511. doi: 10.1074/jbc.M110.217604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H, Woodgett J, Maamari M, Force T. Targeting GSK-3 family members in the heart: a very sharp double-edged sword. J Mol Cell Cardiol. 2011;51:607–613. doi: 10.1016/j.yjmcc.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho J, Rameshwar P, Sadoshima J. Distinct roles of glycogen synthase kinase (GSK)-3α and GSK-3β in mediating cardiomyocyte differentiation in murine bone marrow-derived mesenchymal stem cells. J Biol Chem. 2009;284:36647–36658. doi: 10.1074/jbc.M109.019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi CH, Schoenfeld BP, Bell AJ, Hinchey P, Kollaros M, Gertner MJ, et al. Pharmacological reversal of synaptic plasticity deficits in the mouse model of Fragile X syndrome by group II mGluR antagonist or lithium treatment. Brain Res. 2011;1380:106–119. doi: 10.1016/j.brainres.2010.11.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou HY, Howng SL, Cheng TS, Hsiao YL, Lieu AS, Loh JK, et al. GSKIP is homologous to the Axin GSK3β interaction domain and functions as a negative regulator of GSK3β. Biochemistry. 2006;45:11379–11389. doi: 10.1021/bi061147r. [DOI] [PubMed] [Google Scholar]
- Chow HM, Guo D, Zhou JC, Zhang GY, Li HF, Herrup K, et al. CDK5 activator protein p25 preferentially binds and activates GSK3β. Proc Natl Acad Sci USA. 2014 doi: 10.1073/pnas.1402627111. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang DM, Wang Z, Chiu CT. GSK-3 as a Target for Lithium-Induced Neuroprotection Against Excitotoxicity in Neuronal Cultures and Animal Models of Ischemic Stroke. Front Mol Neurosci. 2011;4:15. doi: 10.3389/fnmol.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole AR, Knebel A, Morrice NA, Robertson LA, Irving AJ, Connolly CN, et al. GSK-3 phosphorylation of the Alzheimer epitope within collapsin response mediator proteins regulates axon elongation in primary neurons. J Biol Chem. 2004;279:50176–50180. doi: 10.1074/jbc.C400412200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole A, Frame S, Cohen P. Further evidence that the tyrosine phosphorylation of glycogen synthase kinase-3 (GSK3) in mammalian cells is an autophosphorylation event. Biochem J. 2004b;377:249–255. doi: 10.1042/BJ20031259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Cuzzocrea S, Mazzon E, Di Paola R, Muia C, Crisafulli C, Dugo L, et al. Glycogen synthase kinase-3β inhibition attenuates the degree of arthritis caused by type II collagen in the mouse. Clin Immunol. 2006;120:57–67. doi: 10.1016/j.clim.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Dajani R, Fraser E, Roe SM, Yeo M, Good VM, Thompson V, et al. Structural basis for recruitment of glycogen synthase kinase 3β to the axin-APC scaffold complex. EMBO J. 2003;22:494–501. doi: 10.1093/emboj/cdg068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan L, Klimenkova O, Klimiankou M, Klusman JH, van den Heuvel-Eibrink MM, Reinhardt D, et al. The role of sirtuin 2 activation by nicotinamide phosphoribosyltransferase in the aberrant proliferation and survival of myeloid leukemia cells. Haematologica. 2012;97:551–559. doi: 10.3324/haematol.2011.055236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darrington RS, Campa VM, Walker MM, Bengoa-Vergniory N, Gorrono-Etxebarria I, Uysal-Onganer P, et al. Distinct expression and activity of GSK-3α and GSK-3β in prostate cancer. Int J Cancer. 2012;131:E872–883. doi: 10.1002/ijc.27620. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Li X, Jope RS. Regulation of Akt and glycogen synthase kinase-3β phosphorylation by sodium valproate and lithium. Neuropharmacol. 2002;43:1158–1164. doi: 10.1016/s0028-3908(02)00215-0. [DOI] [PubMed] [Google Scholar]
- De Sarno P, Bijur GN, Zmijewska AA, Li X, Jope RS. In vivo regulation of GSK3 phosphorylation by cholinergic and NMDA receptors. Neurobiol Aging. 2006;27:413–422. doi: 10.1016/j.neurobiolaging.2005.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sarno P, Axtell RC, Raman C, Roth KA, Alessi DR, Jope RS. Lithium prevents and ameliorates experimental autoimmune encephalomyelitis (EAE) J Immunol. 2008;181:338–345. doi: 10.4049/jimmunol.181.1.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Ser T, Steinwachs KC, Gertz HJ, Andrés MV, Gómez-Carrillo B, Medina M, et al. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: a pilot study. J Alzheimers Dis. 2013;33:205–215. doi: 10.3233/JAD-2012-120805. [DOI] [PubMed] [Google Scholar]
- Dembowy J, Adissu HA, Liu JC, Zacksenhaus E, Woodgett JR. Effect of glycogen synthase kinase-3 inactivation on mouse mammary gland development and oncogenesis. Oncogene. 2014 Sep 8; doi: 10.1038/onc.2014.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng YZ, Yao F, Li JJ, Mao ZF, Hu PT, Long LY, et al. RACK1 suppresses gastric tumorigenesis by stabilizing the β-catenin destruction complex. Gastroenterology. 2012;142:812–823. doi: 10.1053/j.gastro.2011.12.046. [DOI] [PubMed] [Google Scholar]
- Deng Y, Xiong Z, Chen P, Wei J, Chen S, Yan Z. β-Amyloid impairs the regulation of N-methyl-D-aspartate receptors by glycogen synthase kinase 3. Neurobiol Aging. 2014;35:449–459. doi: 10.1016/j.neurobiolaging.2013.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePaoli-Roach AA. Synergistic phosphorylation and activation of ATP-Mg-dependent phosphoprotein phosphatase by F A/GSK-3 and casein kinase II (PC0.7) J Biol Chem. 1984;259:12144–12152. [PubMed] [Google Scholar]
- Dewachter I, Ris L, Jaworski T, Seymour CM, Kremer A, Borghgraef P, et al. GSK3β, a centre-staged kinase in neuropsychiatric disorders, modulates long term memory by inhibitory phosphorylation at serine-9. Neurobiol Dis. 2009;35:193–200. doi: 10.1016/j.nbd.2009.04.003. [DOI] [PubMed] [Google Scholar]
- Dewhurst S, Maggirwar SB, Schifitto G, Gendelman HE, Gelbard HA. Glycogen synthase kinase 3β (GSK-3β) as a therapeutic target in neuroAIDS. J Neuroimmune Pharmacol. 2007;2:93–96. doi: 10.1007/s11481-006-9051-1. [DOI] [PubMed] [Google Scholar]
- Dill J, Wang H, Zhou F, Li S. Inactivation of glycogen synthase kinase 3 promotes axonal growth and recovery in the CNS. J Neurosci. 2008;28:8914–8928. doi: 10.1523/JNEUROSCI.1178-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding Q, He X, Hsu JM, Xia W, Chen CT, Li LY, et al. Degradation of Mcl-1 by β-TrCP mediates glycogen synthase kinase 3-induced tumor suppression and chemosensitization. Mol Cell Biol. 2007;27:4006–4017. doi: 10.1128/MCB.00620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J, Wei Y, Liu L, Wang Y, Khairova R, Blumenthal R, et al. A kinesin signaling complex mediates the ability of GSK-3β to affect mood-associated behaviors. Proc Natl Acad Sci USA. 2010;107:11573–11578. doi: 10.1073/pnas.0913138107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudev T, Lim C. Competition between Li+ and Mg2+ in metalloproteins. Implications for lithium therapy. J Am Chem Soc. 2011;133:9506–9515. doi: 10.1021/ja201985s. [DOI] [PubMed] [Google Scholar]
- Duka T, Duka V, Joyce JN, Sidhu A. Alpha-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009;23:2820–2830. doi: 10.1096/fj.08-120410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–1127. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Eickholt BJ, Walsh FS, Doherty P. An inactive pool of GSK-3 at the leading edge of growth cones is implicated in Semaphorin 3A signaling. J Cell Biol. 2002;157:211–217. doi: 10.1083/jcb.200201098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Krebs EG. Phosphorylation of insulin receptor substrate 1 by glycogen synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA. 1997;94:9660–9664. doi: 10.1073/pnas.94.18.9660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Licht-Murava A, Pietrokovski S, Eisenstein M. Substrate competitive GSK-3 inhibitors - strategy and implications. Biochim Biophys Acta. 2010;1804:598–603. doi: 10.1016/j.bbapap.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Eldar-Finkelman H, Martinez A. GSK-3 Inhibitors: Preclinical and Clinical Focus on CNS. Front Mol Neurosci. 2011;4:32. doi: 10.3389/fnmol.2011.00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom TY, Jope RS. Blocked inhibitory serine-phosphorylation of glycogen synthase kinase-3α/β impairs in vivo neural precursor cell proliferation. Biol Psychiatry. 2009b;66:494–502. doi: 10.1016/j.biopsych.2009.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eom TY, Jope RS. GSK3β N-terminus binding to p53 promotes its acetylation. Mol Cancer. 2009;8:14. doi: 10.1186/1476-4598-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Cdc42 regulates GSK-3β and adenomatous polyposis coli to control cell polarity. Nature. 2003;421:753–756. doi: 10.1038/nature01423. [DOI] [PubMed] [Google Scholar]
- Fan G, Ballou LM, Lin RZ. Phospholipase C-independent activation of glycogen synthase kinase-3β and C-terminal Src kinase by Gαq. J Biol Chem. 2003;278:52432–52436. doi: 10.1074/jbc.M310982200. [DOI] [PubMed] [Google Scholar]
- Farago M, Dominguez I, Landesman-Bollag E, Xu X, Rosner A, Cardiff RD, et al. Kinase-inactive glycogen synthase kinase 3β promotes Wnt signaling and mammary tumorigenesis. Cancer Res. 2005;65:5792–5801. doi: 10.1158/0008-5472.CAN-05-1021. [DOI] [PubMed] [Google Scholar]
- Feijs KL, Kleine H, Braczynski A, Forst AH, Herzog N, Verheugd P, et al. ARTD10 substrate identification on protein microarrays: regulation of GSK3β by mono-ADP-ribosylation. Cell Commun Signal. 2013;11:5. doi: 10.1186/1478-811X-11-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferkey DM, Kimelman D. Glycogen synthase kinase-3β mutagenesis identifies a common binding domain for GBP and Axin. J Biol Chem. 2002;277:16147–16152. doi: 10.1074/jbc.M112363200. [DOI] [PubMed] [Google Scholar]
- Force T, Woodgett JR. Unique and overlapping functions of GSK-3 isoforms in cell differentiation and proliferation and cardiovascular development. J Biol Chem. 2009;284:9643–9647. doi: 10.1074/jbc.R800077200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forlenza OV, de Paula VJ, Machado-Vieira R, Diniz BS, Gattaz WF. Does lithium prevent Alzheimer’s disease? Drugs Aging. 2012;29:335–342. doi: 10.2165/11599180-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame S, Zheleva D. Targeting glycogen synthase kinase-3 in insulin signalling. Expert Opin Ther Targets. 2006;10:429–444. doi: 10.1517/14728222.10.3.429. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P, Biondi RM. A common phosphate binding site explains the unique substrate specificity of GSK3 and its inactivation by phosphorylation. Mol Cell. 2001;7:1321–1327. doi: 10.1016/s1097-2765(01)00253-2. [DOI] [PubMed] [Google Scholar]
- Franca-Koh J, Yeo M, Fraser E, Young N, Dale TC. The regulation of glycogen synthase kinase-3 nuclear export by Frat/GBP. J Biol Chem. 2002;277:43844–43848. doi: 10.1074/jbc.M207265200. [DOI] [PubMed] [Google Scholar]
- Franklin AV, King MK, Palomo V, Martinez A, McMahon L, Jope RS. Glycogen synthase kinase-3 inhibitors reverse deficits in long-term potentiation and cognition in Fragile X mice. Biol Psychiat. 2014;75:198–206. doi: 10.1016/j.biopsych.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser E, Young N, Dajani R, Franca-Koh J, Ryves J, Williams RS, et al. Identification of the Axin and Frat binding region of glycogen synthase kinase-3. J Biol Chem. 2002;277:2176–2185. doi: 10.1074/jbc.M109462200. [DOI] [PubMed] [Google Scholar]
- Freyberg Z, Ferrando SJ, Javitch JA. Roles of the Akt/GSK-3 and Wnt signaling pathways in schizophrenia and antipsychotic drug action. Am J Psychiat. 2010;167:388–396. doi: 10.1176/appi.ajp.2009.08121873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimuro M, Liu J, Zhu J, Yokosawa H, Hayward SD. Regulation of the interaction between glycogen synthase kinase 3 and the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen. J Virol. 2005;79:10429–10441. doi: 10.1128/JVI.79.16.10429-10441.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol Cell Biol. 2008;28:7309–7322. doi: 10.1128/MCB.00808-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcea G, Manson MM, Neal CP, Pattenden CJ, Sutton CD, Dennison AR, et al. Glycogen synthase kinase-3β; a new target in pancreatic cancer? Curr Cancer Drug Targets. 2007;7:209–215. doi: 10.2174/156800907780618266. [DOI] [PubMed] [Google Scholar]
- Garza JC, Guo M, Zhang W, Lu XY. Leptin restores adult hippocampal neurogenesis in a chronic unpredictable stress model of depression and reverses glucocorticoid-induced inhibition of GSK-3β/β-catenin signaling. Mol Psychiatry. 2012;17:790–808. doi: 10.1038/mp.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Sintes R, Lucas JJ. NFAT/Fas signaling mediates the neuronal apoptosis and motor side effects of GSK-3 inhibition in a mouse model of lithium therapy. J Clin Invest. 2010;120:2432–2445. doi: 10.1172/JCI37873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goñi-Oliver P, Lucas JJ, Avila J, Hernández F. N-terminal cleavage of GSK-3 by calpain: a new form of GSK-3 regulation. J Biol Chem. 2007;282:22406–22413. doi: 10.1074/jbc.M702793200. [DOI] [PubMed] [Google Scholar]
- Goñi-Oliver P, Avila J, Hernández F. Calpain-mediated truncation of GSK-3 in postmortem brain samples. J Neurosci Res. 2009;87:1156–1161. doi: 10.1002/jnr.21932. [DOI] [PubMed] [Google Scholar]
- Goode N, Hughes K, Woodgett JR, Parker PJ. Differential regulation of glycogen synthase kinase-3β by protein kinase C isotypes. J Biol Chem. 1992;267:16878–16882. [PubMed] [Google Scholar]
- Grimes CA, Jope RS. The multifaceted roles of glycogen synthase kinase 3β in cellular signaling. Prog Neurobiol. 2001;65:391–426. doi: 10.1016/s0301-0082(01)00011-9. [DOI] [PubMed] [Google Scholar]
- Grisouard J, Medunjanin S, Hermani A, Shukla A, Mayer D. Glycogen synthase kinase-3 protects estrogen receptor alpha from proteasomal degradation and is required for full transcriptional activity of the receptor. Mol Endocrinol. 2007;21:2427–2439. doi: 10.1210/me.2007-0129. [DOI] [PubMed] [Google Scholar]
- Grisouard J, Mayer D. Specific involvement of glycogen synthase kinase-3 in the function and activity of sex steroid hormone receptors reveals the complexity of their regulation. J Steroid Biochem Mol Biol. 2009;117:87–92. doi: 10.1016/j.jsbmb.2009.08.002. [DOI] [PubMed] [Google Scholar]
- Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, et al. Inactivation of the Enzyme GSK3α by the Kinase IKKi Promotes AKT-mTOR Signaling Pathway that Mediates Interleukin-1-Induced Th17 Cell Maintenance. Immunity. 2012;37:800–812. doi: 10.1016/j.immuni.2012.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Ramirez A, Waddell DS, Li Z, Liu X, Wang XF. Axin and GSK3- control Smad3 protein stability and modulate TGF- signaling. Genes Dev. 2008;22:106–120. doi: 10.1101/gad.1590908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta C, Kaur J, Tikoo K. Regulation of MDA-MB231 cell proliferation by GSK-3β involves epigenetic modifications under high glucose conditions. Exp Cell Res. 2014;324:75–83. doi: 10.1016/j.yexcr.2014.03.019. [DOI] [PubMed] [Google Scholar]
- Hampel H, Ewers M, Bürger K, Annas P, Mörtberg A, Bogstedt A, et al. Lithium trial in Alzheimer’s disease: a randomized, single-blind, placebo-controlled, multicenter 10-week study. J Clin Psychiatry. 2009;70:922–931. [PubMed] [Google Scholar]
- Hanger DP, Hughes K, Woodgett JR, Brion JP, Anderton BH. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci Lett. 1992;147:58–62. doi: 10.1016/0304-3940(92)90774-2. [DOI] [PubMed] [Google Scholar]
- Happel N, Stoldt S, Schmidt B, Doenecke D. M phase-specific phosphorylation of histone H1.5 at threonine 10 by GSK-3. J Mol Biol. 2009;386:339–350. doi: 10.1016/j.jmb.2008.12.047. [DOI] [PubMed] [Google Scholar]
- Haraguchi K, Ohsugi M, Abe Y, Semba K, Akiyama T, Yamamoto T. Ajuba negatively regulates the Wnt signaling pathway by promoting GSK-3β-mediated phosphorylation of β-catenin. Oncogene. 2008;27:274–284. doi: 10.1038/sj.onc.1210644. [DOI] [PubMed] [Google Scholar]
- Hartmaier RJ, Richter AS, Gillihan RM, Sallit JZ, McGuire SE, Wang J, et al. A SNP in steroid receptor coactivator-1 disrupts a GSK3β phosphorylation site and is associated with altered tamoxifen response in bone. Mol Endocrinol. 2012;26:220–227. doi: 10.1210/me.2011-1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto R, Hough C, Nakazawa T, Yamamoto T, Chuang DM. Lithium protection against glutamate excitotoxicity in rat cerebral cortical neurons: involvement of NMDA receptor inhibition possibly by decreasing NR2B tyrosine phosphorylation. J Neurochem. 2002;80:589–597. doi: 10.1046/j.0022-3042.2001.00728.x. [DOI] [PubMed] [Google Scholar]
- Himmelstein DS, Ward SM, Lancia JK, Patterson KR, Binder LI. Tau as a therapeutic target in neurodegenerative disease. Pharmacol Ther. 2012;136:8–22. doi: 10.1016/j.pharmthera.2012.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3β in cell survival and NF-κB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Höglinger GU, Huppertz HJ, Wagenpfeil S, Andrés MV, Belloch V, et al. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov Disord. 2014;29:479–487. doi: 10.1002/mds.25815. [DOI] [PubMed] [Google Scholar]
- Hooper C, Markevich V, Plattner F, Killick R, Schofield E, Engel T, et al. Glycogen synthase kinase-3 inhibition is integral to long-term potentiation. Eur J Neurosci. 2007;25:81–86. doi: 10.1111/j.1460-9568.2006.05245.x. [DOI] [PubMed] [Google Scholar]
- Howng SL, Hwang CC, Hsu CY, Hsu MY, Teng CY, Chou CH, et al. Involvement of the residues of GSKIP, AxinGID, and FRATtide in their binding with GSK3β to unravel a novel C-terminal scaffold-binding region. Mol Cell Biochem. 2010;339:23–33. doi: 10.1007/s11010-009-0366-0. [DOI] [PubMed] [Google Scholar]
- Hu X, Paik PK, Chen J, Yarilina A, Kockeritz L, Lu TT, et al. IFN-γ suppresses IL-10 production and synergizes with TLR2 by regulating GSK3 and CREB/AP-1 proteins. Immunity. 2006;24:563–574. doi: 10.1016/j.immuni.2006.02.014. [DOI] [PubMed] [Google Scholar]
- Hughes K, Nikolakaki E, Plyte SE, Totty NF, Woodgett JR. Modulation of the glycogen synthase kinase-3 family by tyrosine phosphorylation. EMBO J. 1993;12:803–808. doi: 10.1002/j.1460-2075.1993.tb05715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hundsrucker C, Skroblin P, Christian F, Zenn HM, Popara V, Joshi M, et al. Glycogen synthase kinase 3β interaction protein functions as an A-kinase anchoring protein. J Biol Chem. 2010;285:5507–5521. doi: 10.1074/jbc.M109.047944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurtado DE, Molina-Porcel L, Carroll JC, Macdonald C, Aboagye AK, Trojanowski JQ, et al. Selectively silencing GSK-3 isoforms reduces plaques and tangles in mouse models of Alzheimer’s disease. J Neurosci. 2012;32:7392–7402. doi: 10.1523/JNEUROSCI.0889-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iitaka C, Miyazaki K, Akaike T, Ishida N. A role for glycogen synthase kinase-3β in the mammalian circadian clock. J Biol Chem. 2005;280:29397–29402. doi: 10.1074/jbc.M503526200. [DOI] [PubMed] [Google Scholar]
- Inkster B, Nichols TE, Saemann PG, Auer DP, Holsboer F, Muglia P, et al. Association of GSK3β polymorphisms with brain structural changes in major depressive disorder. Arch Gen Psychiatry. 2009;66:721–728. doi: 10.1001/archgenpsychiatry.2009.70. [DOI] [PubMed] [Google Scholar]
- Inkster B, Nichols TE, Saemann PG, Auer DP, Holsboer F, Muglia P, et al. Pathway-based approaches to imaging genetics association studies: Wnt signaling, GSK3β substrates and major depression. Neuroimage. 2010;53:908–917. doi: 10.1016/j.neuroimage.2010.02.065. [DOI] [PubMed] [Google Scholar]
- Iwahana E, Akiyama M, Miyakawa K, Uchida A, Kasahara J, Fukunaga K, et al. Effect of lithium on the circadian rhythms of locomotor activity and glycogen synthase kinase-3 protein expression in the mouse suprachiasmatic nuclei. Eur J Neurosci. 2004;19:2281–2287. doi: 10.1111/j.0953-816X.2004.03322.x. [DOI] [PubMed] [Google Scholar]
- Jernigan KK, Cselenyi CS, Thorne CA, Hanson AJ, Tahinci E, Hajicek N, et al. Gβγ activates GSK3 to promote LRP6-mediated β-catenin transcriptional activity. Sci Signal. 2010;3:ra37. doi: 10.1126/scisignal.2000647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Guo W, Liang X, Rao Y. Both the establishment and the maintenance of neuronal polarity require active mechanisms: critical roles of GSK-3β and its upstream regulators. Cell. 2005;120:123–135. doi: 10.1016/j.cell.2004.12.033. [DOI] [PubMed] [Google Scholar]
- Jope RS. Lithium and GSK3: one inhibitor, two inhibitory actions, multiple outcomes. Trends Pharmacol Sci. 2003;24:441–443. doi: 10.1016/S0165-6147(03)00206-2. [DOI] [PubMed] [Google Scholar]
- Jope RS. Lithium, the seminal GSK3 inhibitor. In: Martinez A, Castro A, Medina M, editors. Glycogen synthase kinase 3 (GSK-3) and its inhibitors. John Wiley and Sons, Inc; 2006. pp. 223–242. [Google Scholar]
- Jope RS. Glycogen synthase kinase-3 in the etiology and treatment of mood disorders. Front Mol Neurosci. 2011;4:16–26. doi: 10.3389/fnmol.2011.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jope RS, Johnson GVW. The glamour and gloom of glycogen synthase kinase-3 (GSK3) Trends Biochem Sci. 2004;29:95–102. doi: 10.1016/j.tibs.2003.12.004. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): Inflammation, diseases and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Lipina TV, Takao K, van Eede M, Hattori S, Laliberté C, et al. Abnormalities in brain structure and behavior in GSK-3α mutant mice. Mol Brain. 2009;2:35. doi: 10.1186/1756-6606-2-35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaidanovich-Beilin O, Woodgett JR. GSK-3: Functional Insights from Cell Biology and Animal Models. Front Mol Neurosci. 2011;4:40. doi: 10.3389/fnmol.2011.00040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandasamy AD, Schulz R. Glycogen synthase kinase-3β is activated by matrix metalloproteinase-2 mediated proteolysis in cardiomyoblasts. Cardiovasc Res. 2009;83:698–706. doi: 10.1093/cvr/cvp175. [DOI] [PubMed] [Google Scholar]
- Kashikar ND, Zhang W, Massion PP, Gonzalez AL, Datta PK. Role of STRAP in regulating GSK3β function and Notch3 stabilization. Cell Cycle. 2011;10:1639–1654. doi: 10.4161/cc.10.10.15630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly GM, Buckley DA, Kiely PA, Adams DR, O’Connor R. Serine phosphorylation of the insulin-like growth factor I (IGF-1) receptor C-terminal tail restrains kinase activity and cell growth. J Biol Chem. 2012;287:28180–28194. doi: 10.1074/jbc.M112.385757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessing LV, Forman JL, Andersen PK. Does lithium protect against dementia? Bipolar Disord. 2010;12:87–94. doi: 10.1111/j.1399-5618.2009.00788.x. [DOI] [PubMed] [Google Scholar]
- Kim WY, Zhou FQ, Zhou J, Yokota Y, Wang YM, Yoshimura T, et al. Essential roles for GSK-3s and GSK-3-primed substrates in neurotrophin-induced and hippocampal axon growth. Neuron. 2006;52:981–996. doi: 10.1016/j.neuron.2006.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim NG, Xu C, Gumbiner BM. Identification of targets of the Wnt pathway destruction complex in addition to β-catenin. Proc Natl Acad Sci USA. 2009;106:5165–5170. doi: 10.1073/pnas.0810185106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim WY, Wang X, Wu Y, Doble BW, Patel S, Woodgett JR, et al. GSK-3 is a master regulator of neural progenitor homeostasis. Nat Neurosci. 2009b;12:1390–1397. doi: 10.1038/nn.2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Piao S, Lee E, Yoon BY, Moon HR, et al. Development of Akt-activated GSK3β inhibitory peptide. Biochem Biophys Res Commun. 2014;434:735–739. doi: 10.1016/j.bbrc.2013.03.103. [DOI] [PubMed] [Google Scholar]
- King TD, Bijur GN, Jope RS. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3β and attenuated by lithium. Brain Res. 2001;919:106–114. doi: 10.1016/s0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- King MK, Jope RS. Lithium treatment alleviates impaired cognition in a mouse model of fragile X syndrome. Genes Brain Behavior. 2013;12:723–731. doi: 10.1111/gbb.12071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King MK, Pardo M, Cheng Y, Downey K, Jope RS, Beurel E. Glycogen synthase kinase-3 inhibitors: Rescuers of cognitive impairments. Pharmacol Ther. 2014;141:1–12. doi: 10.1016/j.pharmthera.2013.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kise Y, Morinaka A, Teglund S, Miki H. Sufu recruits GSK3β for efficient processing of Gli3. Biochem Biophys Res Commun. 2009;387:569–574. doi: 10.1016/j.bbrc.2009.07.087. [DOI] [PubMed] [Google Scholar]
- Klein PS, Melton DA. A molecular mechanism for the effect of lithium on development. Proc Natl Acad Sci USA. 1996;93:8455–8459. doi: 10.1073/pnas.93.16.8455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko HW, Kim EY, Chiu J, Vanselow JT, Kramer A, Edery I. A hierarchical phosphorylation cascade that regulates the timing of PERIOD nuclear entry reveals novel roles for proline-directed kinases and GSK-3β/SGG in circadian clocks. J Neurosci. 2010;30:12664–12675. doi: 10.1523/JNEUROSCI.1586-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T, Hino S, Oue N, Asahara T, Zollo M, Yasui W, et al. Glycogen synthase kinase 3 and h-prune regulate cell migration by modulating focal adhesions. Mol Cell Biol. 2006;26:898–911. doi: 10.1128/MCB.26.3.898-911.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwok JB, Hallupp M, Loy CT, Chan DK, Woo J, Mellick GD, et al. GSK3B polymorphisms alter transcription and splicing in Parkinson’s disease. Ann Neurol. 2005;58:829–839. doi: 10.1002/ana.20691. [DOI] [PubMed] [Google Scholar]
- Lachman HM, Pedrosa E, Petruolo OA, Cockerham M, Papolos A, Novak T, et al. Increase in GSK3β gene copy number variation in bipolar disorder. Am J Med Genet B Neuropsychiatr Genet. 2007;144B:259–265. doi: 10.1002/ajmg.b.30498. [DOI] [PubMed] [Google Scholar]
- Lal H, Zhou J, Ahmad F, Zaka R, Vagnozzi RJ, Decaul M, et al. Glycogen synthase kinase-3α limits ischemic injury, cardiac rupture, post-myocardial infarction remodeling and death. Circulation. 2012;125:65–75. doi: 10.1161/CIRCULATIONAHA.111.050666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavoie J, Hébert M, Beaulieu JM. Glycogen synthase kinase-3β haploinsufficiency lengthens the circadian locomotor activity period in mice. Behav Brain Res. 2013;253:262–265. doi: 10.1016/j.bbr.2013.08.001. [DOI] [PubMed] [Google Scholar]
- Leeds PR, Yu F, Wang Z, Chiu CT, Zhang Y, Leng Y, et al. A New Avenue for Lithium: Intervention in Traumatic Brain Injury. ACS Chem Neurosci. 2014 Apr 11; doi: 10.1021/cn500040g. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leis H, Segrelles C, Ruiz S, Santos M, Paramio JM. Expression, localization, and activity of glycogen synthase kinase 3β during mouse skin tumorigenesis. Mol Carcinog. 2002;35:180–185. doi: 10.1002/mc.10087. [DOI] [PubMed] [Google Scholar]
- Levina E, Oren M, Ben-Ze’ev A. Downregulation of β-catenin by p53 involves changes in the rate of β-catenin phosphorylation and Axin dynamics. Oncogene. 2004;23:4444–4453. doi: 10.1038/sj.onc.1207587. [DOI] [PubMed] [Google Scholar]
- Li C, Ebert PJ, Li QJ. T cell receptor (TCR) and transforming growth factor β (TGF-β) signaling converge on DNA (cytosine-5)-methyltransferase to control forkhead box protein 3 (foxp3) locus methylation and inducible regulatory T cell differentiation. J Biol Chem. 2013;288:19127–19139. doi: 10.1074/jbc.M113.453357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhu W, Roh MS, Friedman AB, Rosborough K, Jope RS. In vivo regulation of glycogen synthase kinase-3β (GSK3β) by serotonergic activity in mouse brain. Neuropsychopharmacol. 2004;29:1426–1431. doi: 10.1038/sj.npp.1300439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Friedman AB, Zhu W, Wang L, Boswell S, May RS, et al. Lithium regulates glycogen synthase kinase-3β in human peripheral blood mononuclear cells: implication in the treatment of bipolar disorder. Biol Psychiat. 2007;61:216–222. doi: 10.1016/j.biopsych.2006.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Chen C, Tu Y, Sun HT, Zhao ML, Cheng SX, et al. Sirt1 promotes axonogenesis by deacetylation of Akt and inactivation of GSK3. Mol Neurobiol. 2013;48:490–499. doi: 10.1007/s12035-013-8437-3. [DOI] [PubMed] [Google Scholar]
- Li Q, Wang X, Wu X, Rui Y, Liu W, Wang J, et al. Daxx cooperates with the Axin/HIPK2/p53 complex to induce cell death. Cancer Res. 2007b;67:66–74. doi: 10.1158/0008-5472.CAN-06-1671. [DOI] [PubMed] [Google Scholar]
- Li Q, Lin S, Wang X, Lian G, Lu Z, Guo H, et al. Axin determines cell fate by controlling the p53 activation threshold after DNA damage. Nat Cell Biol. 2009;11:1128–1134. doi: 10.1038/ncb1927. [DOI] [PubMed] [Google Scholar]
- Li YC, Xi D, Roman J, Huang YQ, Gao WJ. Activation of glycogen synthase kinase-3β is required for hyperdopamine and D2 receptor-mediated inhibition of synaptic NMDA receptor function in the rat prefrontal cortex. J Neurosci. 2009b;29:15551–15563. doi: 10.1523/JNEUROSCI.3336-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Wang MJ, Gao WJ. Hyperdopaminergic modulation of inhibitory transmission is dependent on GSK-3β signaling-mediated trafficking of GABAA receptors. J Neurochem. 2012;122:308–320. doi: 10.1111/j.1471-4159.2012.07790.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang MH, Chuang DM. Differential roles of glycogen synthase kinase-3 isoforms in the regulation of transcriptional activation. J Biol Chem. 2006;281:30479–30484. doi: 10.1074/jbc.M607468200. [DOI] [PubMed] [Google Scholar]
- Liao W, Wang S, Han C, Zhang Y. 14-3-3 proteins regulate glycogen synthase 3β phosphorylation and inhibit cardiomyocyte hypertrophy. FEBS J. 2005;272:1845–1854. doi: 10.1111/j.1742-4658.2005.04614.x. [DOI] [PubMed] [Google Scholar]
- Licht-Murava A, Eldar-Finkelman H. Exploiting substrate recognition for selective inhibition of protein kinases. Curr Pharm Des. 2012;18:2914–2920. doi: 10.2174/138161212800672741. [DOI] [PubMed] [Google Scholar]
- Lin RK, Hsieh YS, Lin P, Hsu HS, Chen CY, Tang YA, et al. The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J Clin Invest. 2010a;120:521–532. doi: 10.1172/JCI40706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CH, Tsai PI, Wu RM, Chien CT. LRRK2 G2019S mutation induces dendrite degeneration through mislocalization and phosphorylation of tau by recruiting autoactivated GSK3ß. J Neurosci. 2010b;30:13138–13149. doi: 10.1523/JNEUROSCI.1737-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin SY, Li TY, Liu Q, Zhang C, Li X, Chen Y, et al. GSK3-TIP60-ULK1 signaling pathway links growth factor deprivation to autophagy. Science. 2012;336:477–481. doi: 10.1126/science.1217032. [DOI] [PubMed] [Google Scholar]
- Linding R, Jensen LJ, Ostheimer GJ, van Vugt MA, Jørgensen C, Miron IM, et al. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Monte F, Kramer T, Gu J, Anumala UR, Marinelli L, et al. Identification of glycogen synthase kinase-3 inhibitors with a selective sting for glycogen synthase kinase-3α. J Med Chem. 2012;55:4407–4424. doi: 10.1021/jm300309a. [DOI] [PubMed] [Google Scholar]
- Lu Y, Muller M, Smith D, Dutta B, Komurov K, Iadevaia S, et al. Kinome siRNA-phosphoproteomic screen identifies networks regulating AKT signaling. Oncogene. 2011;30:4567–4577. doi: 10.1038/onc.2011.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly PT, Wu Y, Zou H, Wang R, Zhou W, et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Invest. 2013;123:224–235. doi: 10.1172/JCI64516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma C, Wang J, Gao Y, Gao TW, Chen G, Bower KA, et al. The role of glycogen synthase kinase 3β in the transformation of epidermal cells. Cancer Res. 2007;67:7756–7764. doi: 10.1158/0008-5472.CAN-06-4665. [DOI] [PubMed] [Google Scholar]
- MacAulay K, Woodgett JR. Targeting glycogen synthase kinase-3 (GSK-3) in the treatment of Type 2 diabetes. Expert Opin Ther Targets. 2008;12:1265–1274. doi: 10.1517/14728222.12.10.1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacDonald BT, Tamai K, He X. Wnt/β-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17:9–26. doi: 10.1016/j.devcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai L, Jope RS, Li X. BDNF-mediated signal transduction is modulated by GSK3β and mood stabilizing agents. J Neurochem. 2002;82:75–83. doi: 10.1046/j.1471-4159.2002.00939.x. [DOI] [PubMed] [Google Scholar]
- Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates β-catenin signaling activity. J Biol Chem. 2003;278:5947–5951. doi: 10.1074/jbc.C200473200. [DOI] [PubMed] [Google Scholar]
- Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS. Aberrant β-catenin signaling in tuberous sclerosis. Am J Pathol. 2005;167:107–116. doi: 10.1016/s0002-9440(10)62958-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandelkow EM, Drewes G, Biernat J, Gustke N, Van Lint J, Vandenheede JR, et al. Glycogen synthase kinase-3 and the Alzheimer-like state of microtubule-associated protein tau. FEBS Lett. 1992;314:315–321. doi: 10.1016/0014-5793(92)81496-9. [DOI] [PubMed] [Google Scholar]
- Manoukian AS, Woodgett JR. Role of glycogen synthase kinase-3 in cancer: regulation by Wnts and other signaling pathways. Adv Cancer Res. 2002;84:203–229. doi: 10.1016/s0065-230x(02)84007-6. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nature Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez A, Perez DI. GSK-3 inhibitors: a ray of hope for the treatment of Alzheimer’s disease? J Alzheimers Dis. 2008;15:181–191. doi: 10.3233/jad-2008-15204. [DOI] [PubMed] [Google Scholar]
- Maurin H, Lechat B, Dewachter I, Ris L, Louis JV, Borghgraef P, et al. Neurological characterization of mice deficient in GSK3α highlight pleiotropic physiological functions in cognition and pathological activity as Tau kinase. Mol Brain. 2013;6:27. doi: 10.1186/1756-6606-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McColl G, Killilea DW, Hubbard AE, Vantipalli MC, Melov S, Lithgow GJ. Pharmacogenetic analysis of lithium-induced delayed aging in Caenorhabditis elegans. J Biol Chem. 2008;283:350–357. doi: 10.1074/jbc.M705028200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCubrey JA, Steelman LS, Bertrand FE, Davis NM, Abrams SL, et al. Multifaceted roles of GSK-3 and Wnt/β-catenin in hematopoiesis and leukemogenesis: opportunities for therapeutic intervention. Leukemia. 2014;28:15–33. doi: 10.1038/leu.2013.184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus EJ, Sakamoto K, Armit LJ, Ronaldson L, Shpiro N, Marquez R, et al. Role that phosphorylation of GSK3 plays in insulin and Wnt signalling defined by knockin analysis. EMBO J. 2005;24:1571–1583. doi: 10.1038/sj.emboj.7600633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meares GP, Jope RS. Resolution of the nuclear localization mechanism of glycogen synthase kinase-3: functional effects in apoptosis. J Biol Chem. 2007;282:16989–17001. doi: 10.1074/jbc.M700610200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina M, Avila J. Glycogen synthase kinase-3 (GSK-3) inhibitors for the treatment of Alzheimer’s disease. Curr Pharm Des. 2010;16:2790–2798. doi: 10.2174/138161210793176581. [DOI] [PubMed] [Google Scholar]
- Medunjanin S, Hermani A, De Servi B, Grisouard J, Rincke G, Mayer D. Glycogen synthase kinase-3 interacts with and phosphorylates estrogen receptor alpha and is involved in the regulation of receptor activity. J Biol Chem. 2005;280:33006–33014. doi: 10.1074/jbc.M506758200. [DOI] [PubMed] [Google Scholar]
- Mendez P, Garcia-Segura LM. Phosphatidylinositol 3-kinase and glycogen synthase kinase 3 regulate estrogen receptor-mediated transcription in neuronal cells. Endocrinology. 2006;147:3027–3039. doi: 10.1210/en.2005-1224. [DOI] [PubMed] [Google Scholar]
- Miller MW, Caracciolo MR, Berlin WK, Hanover JA. Phosphorylation and glycosylation of nucleoporins. Arch Biochem Biophys. 1999;367:51–60. doi: 10.1006/abbi.1999.1237. [DOI] [PubMed] [Google Scholar]
- Mills CN, Nowsheen S, Bonner JA, Yang ES. Emerging roles of glycogen synthase kinase 3 in the treatment of brain tumors. Front Mol Neurosci. 2011;4:47. doi: 10.3389/fnmol.2011.00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines MA, Beurel E, Jope RS. Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int J Alzheimers Dis. 2011;2011:861072. doi: 10.4061/2011/861072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mines MA, Jope RS. Glycogen synthase kinase-3: a promising therapeutic target for fragile x syndrome. Front Mol Neurosci. 2011;4:35. doi: 10.3389/fnmol.2011.00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mondelli V, Anacker C, Vernon AC, Cattaneo A, Natesan S, Modo M, et al. Haloperidol and olanzapine mediate metabolic abnormalities through different molecular pathways. Transl Psychiatry. 2013;3:e208. doi: 10.1038/tp.2012.138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteserin-Garcia J, Al-Massadi O, Seoane LM, Alvarez CV, Shan B, Stalla J, et al. Sirt1 inhibits the transcription factor CREB to regulate pituitary growth hormone synthesis. FASEB J. 2013;27:1561–1571. doi: 10.1096/fj.12-220129. [DOI] [PubMed] [Google Scholar]
- Morales-García JA, Susín C, Alonso-Gil S, Pérez DI, Palomo V, Pérez C, et al. Glycogen synthase kinase-3 inhibitors as potent therapeutic agents for the treatment of Parkinson disease. ACS Chem Neurosci. 2013;4:350–360. doi: 10.1021/cn300182g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morfini G, Szebenyi G, Elluru R, Ratner N, Brady ST. Glycogen synthase kinase 3 phosphorylates kinesin light chains and negatively regulates kinesin-based motility. EMBO J. 2002;21:281–293. doi: 10.1093/emboj/21.3.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukai F, Ishiguro K, Sano Y, Fujita SC. Alternative splicing isoform of tau protein kinase I/glycogen synthase kinase 3β. J Neurochem. 2002;81:1073–1083. doi: 10.1046/j.1471-4159.2002.00918.x. [DOI] [PubMed] [Google Scholar]
- Mulholland DJ, Dedhar S, Wu H, Nelson CC. PTEN and GSK3β: key regulators of progression to androgen-independent prostate cancer. Oncogene. 2006;25:329–337. doi: 10.1038/sj.onc.1209020. [DOI] [PubMed] [Google Scholar]
- Nelson CD, Kim MJ, Hsin H, Chen Y, Sheng M. Phosphorylation of threonine-19 of PSD-95 by GSK-3β is required for PSD-95 mobilization and long-term depression. J Neurosci. 2013;33:12122–12135. doi: 10.1523/JNEUROSCI.0131-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemoto T, Satoh S, Maruta T, Kanai T, Yoshikawa N, Miyazaki S, et al. Homologous posttranscriptional regulation of insulin-like growth factor-I receptor level via glycogen synthase kinase-3β and mammalian target of rapamycin in adrenal chromaffin cells: effect on tau phosphorylation. Neuropharmacol. 2010;58:1097–1108. doi: 10.1016/j.neuropharm.2010.01.018. [DOI] [PubMed] [Google Scholar]
- Ng SS, Mahmoudi T, Danenberg E, Bejaoui I, de Lau W, Korswagen HC, et al. Phosphatidylinositol 3-kinase signaling does not activate the wnt cascade. J Biol Chem. 2009;284:35308–35313. doi: 10.1074/jbc.M109.078261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiguchi N, Breen G, Russ C, St Clair D, Collier D. Association analysis of the glycogen synthase kinase-3β gene in bipolar disorder. Neurosci Lett. 2006;394:243–245. doi: 10.1016/j.neulet.2005.10.042. [DOI] [PubMed] [Google Scholar]
- Nishimoto T, Kihara T, Akaike A, Niidome T, Sugimoto H. alpha-Amino-3-hydroxy-5-methyl-4-isoxazole propionate attenuates glutamate-induced caspase-3 cleavage via regulation of glycogen synthase kinase 3β. J Neurosci Res. 2008;86:1096–1105. doi: 10.1002/jnr.21567. [DOI] [PubMed] [Google Scholar]
- Nishimoto T, Kihara T, Akaike A, Niidome T, Sugimoto H. AMPA reduces surface expression of NR1 through regulation of GSK3β. Neuroreport. 2009;20:161–165. doi: 10.1097/WNR.0b013e3283118450. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Hough CJ, Chuang DM. Chronic lithium treatment robustly protects neurons in the central nervous system against excitotoxicity by inhibiting N-methyl-D-aspartate receptor-mediated calcium influx. Proc Natl Acad Sci USA. 1998;95:2642–2647. doi: 10.1073/pnas.95.5.2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes PV, Forlenza OV, Gattaz WF. Lithium and risk for Alzheimer’s disease in elderly patients with bipolar disorder. Br J Psychiatry. 2007;190:359–360. doi: 10.1192/bjp.bp.106.029868. [DOI] [PubMed] [Google Scholar]
- Nunes MA, Viel TA, Buck HS. Microdose lithium treatment stabilized cognitive impairment in patients with Alzheimer’s disease. Curr Alzheimer Res. 2013;10:104–107. doi: 10.2174/1567205011310010014. [DOI] [PubMed] [Google Scholar]
- O’Brien WT, Harper AD, Jové F, Woodgett JR, Maretto S, Piccolo S, et al. Glycogen synthase kinase-3β haploinsufficiency mimics the behavioral and molecular effects of lithium. J Neurosci. 2004;24:6791–6798. doi: 10.1523/JNEUROSCI.4753-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien WT, Klein PS. Validating GSK3 as an in vivo target of lithium action. Biochem Soc Trans. 2009;37:1133–1138. doi: 10.1042/BST0371133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien WT, Haung J, Buccafusca R, Garskof J, Valvezan AJ, Berry GT, et al. Glycogen synthase kinase-3 is essential for β-arrestin-2 complex formation and lithium-sensitive behaviors in mice. J Clin Invest. 2011;121:3756–3762. doi: 10.1172/JCI45194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Voleti B, Banasr M, Sarhan M, Duric V, Girgenti MJ, et al. Wnt2 expression and signaling is increased by different classes of antidepressant treatments. Biol Psychiatry. 2010;68:521–527. doi: 10.1016/j.biopsych.2010.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ougolkov AV, Fernandez-Zapico ME, Savoy DN, Urrutia RA, Billadeau DD. Glycogen synthase kinase-3β participates in nuclear factor kappaB-mediated gene transcription and cell survival in pancreatic cancer cells. Cancer Res. 2005;65:2076–2081. doi: 10.1158/0008-5472.CAN-04-3642. [DOI] [PubMed] [Google Scholar]
- Ougolkov AV, Billadeau DD. Targeting GSK-3: a promising approach for cancer therapy? Future Oncol. 2006;2:91–100. doi: 10.2217/14796694.2.1.91. [DOI] [PubMed] [Google Scholar]
- Ougolkov AV, Bone ND, Fernandez-Zapico ME, Kay NE, Billadeau DD. Inhibition of glycogen synthase kinase-3 activity leads to epigenetic silencing of nuclear factor kappaB target genes and induction of apoptosis in chronic lymphocytic leukemia B cells. Blood. 2007;110:735–742. doi: 10.1182/blood-2006-12-060947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, Song J, Joe CO, Shin I. Akt stabilizes estrogen receptor alpha with the concomitant reduction in its transcriptional activity. Cell Signal. 2008;20:1368–1374. doi: 10.1016/j.cellsig.2008.03.004. [DOI] [PubMed] [Google Scholar]
- Peineau S, Taghibiglou C, Bradley C, Wong TP, Liu L, Lu J, et al. LTP inhibits LTD in the hippocampus via regulation of GSK3β. Neuron. 2007;53:703–717. doi: 10.1016/j.neuron.2007.01.029. [DOI] [PubMed] [Google Scholar]
- Phiel CJ, Wilson CA, Lee VM, Klein PS. GSK-3α regulates production of Alzheimer’s disease amyloid-β peptides. Nature. 2003;423:435–439. doi: 10.1038/nature01640. [DOI] [PubMed] [Google Scholar]
- Picard N, Caron V, Bilodeau S, Sanchez M, Mascle X, Aubry M, et al. Identification of estrogen receptor β as a SUMO-1 target reveals a novel phosphorylated sumoylation motif and regulation by glycogen synthase kinase 3β. Mol Cell Biol. 2012;32:2709–2721. doi: 10.1128/MCB.06624-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilot-Storck F, Chopin E, Rual JF, Baudot A, Dobrokhotov P, Robinson-Rechavi M, et al. Interactome mapping of the phosphatidylinositol 3-kinase-mammalian target of rapamycin pathway identifies deformed epidermal autoregulatory factor-1 as a new glycogen synthase kinase-3 interactor. Mol Cell Proteomics. 2010;9:1578–1593. doi: 10.1074/mcp.M900568-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotnikov A, Li Y, Tran TH, Tang W, Palazzo JP, Rui H, et al. Oncogene-mediated inhibition of glycogen synthase kinase 3β impairs degradation of prolactin receptor. Cancer Res. 2008;68:1354–1361. doi: 10.1158/0008-5472.CAN-07-6094. [DOI] [PubMed] [Google Scholar]
- Polakis P. The many ways of Wnt in cancer. Curr Opin Genet Dev. 2007;17:45–51. doi: 10.1016/j.gde.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Polter A, Beurel E, Yang S, Garner R, Song L, Miller CA, et al. Deficiency in the inhibitory serine-phosphorylation of glycogen synthase kinase-3 increases sensitivity to mood disturbances. Neuropsychopharmacology. 2010;35:1761–1774. doi: 10.1038/npp.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkie AP, Zeidner LC, Albrecht AM, D’Ippolito A, Eckardt S, Newsom DE, et al. Phosphatidylinositol 3-kinase (PI3K) signaling via glycogen synthase kinase-3 (GSK-3) regulates DNA methylation of imprinted loci. J Biol Chem. 2010;285:41337–41347. doi: 10.1074/jbc.M110.170704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogatsky I, Waase CL, Garabedian MJ. Phosphorylation and inhibition of rat glucocorticoid receptor transcriptional activation by glycogen synthase kinase-3 (GSK-3). Species-specific differences between human and rat glucocorticoid receptor signaling as revealed through GSK-3 phosphorylation. J Biol Chem. 1998;273:14315–14321. doi: 10.1074/jbc.273.23.14315. [DOI] [PubMed] [Google Scholar]
- Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol. 2013;20:502–507. doi: 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]
- Rosner M, Hanneder M, Siegel N, Valli A, Hengstschläger M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat Res. 2008;658:234–246. doi: 10.1016/j.mrrev.2008.01.001. [DOI] [PubMed] [Google Scholar]
- Rui Y, Xu Z, Lin S, Li Q, Rui H, Luo W, et al. Axin stimulates p53 functions by activation of HIPK2 kinase through multimeric complex formation. EMBO J. 2004;23:4583–4594. doi: 10.1038/sj.emboj.7600475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rui Y, Myers KR, Yu K, Wise A, De Blas AL, Hartzell HC, et al. Activity-dependent regulation of dendritic growth and maintenance by glycogen synthase kinase 3β. Nat Commun. 2013;4:2628. doi: 10.1038/ncomms3628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Saenz A, van Haren J, Sayas L, Rangel L, Demmers J, Millán J, et al. Protein 4.1R binds to CLASP2 and regulates dynamics, organization and attachment of microtubules to the cell cortex. J Cell Sci. 2013;126:4589–4601. doi: 10.1242/jcs.120840. [DOI] [PubMed] [Google Scholar]
- Sachs BD, Rodriguiz RM, Siesser WB, Kenan A, Royer EL, Jacobsen JP, et al. The effects of brain serotonin deficiency on behavioural disinhibition and anxiety-like behaviour following mild early life stress. Int J Neuropsychopharmacol. 2013;16:2081–2094. doi: 10.1017/S1461145713000321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahar S, Zocchi L, Kinoshita C, Borrelli E, Sassone-Corsi P. Regulation of BMAL1 protein stability and circadian function by GSK3β-mediated phosphorylation. PLoS One. 2010;5:e8561. doi: 10.1371/journal.pone.0008561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saijilafu, Hur EM, Liu CM, Jiao Z, Xu WL, Zhou FQ. PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nat Commun. 2013;4:2690. doi: 10.1038/ncomms3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas TR, Kim J, Vakar-Lopez F, Sabichi AL, Troncoso P, Jenster G, et al. Glycogen synthase kinase-3β is involved in the phosphorylation and suppression of androgen receptor activity. J Biol Chem. 2004;279:19191–19200. doi: 10.1074/jbc.M309560200. [DOI] [PubMed] [Google Scholar]
- Samaan S, Tranchevent LC, Dardenne E, Polay Espinoza M, Zonta E, Germann S, et al. The Ddx5 and Ddx17 RNA helicases are cornerstones in the complex regulatory array of steroid hormone-signaling pathways. Nucleic Acids Res. 2014;42:2197–2207. doi: 10.1093/nar/gkt1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saus E, Soria V, Escaramís G, Crespo JM, Valero J, Gutiérrez-Zotes A, et al. A haplotype of glycogen synthase kinase 3β is associated with early onset of unipolar major depression. Genes Brain Behav. 2010;9:799–807. doi: 10.1111/j.1601-183X.2010.00617.x. [DOI] [PubMed] [Google Scholar]
- Schaffer B, Wiedau-Pazos M, Geschwind DH. Gene structure and alternative splicing of glycogen synthase kinase 3β (GSK-3β) in neural and non-neural tissues. Gene. 2003;302:73–81. doi: 10.1016/s0378-1119(02)01092-2. [DOI] [PubMed] [Google Scholar]
- Scheuing L, Chiu CT, Liao HM, Linares GR, Chuang DM. Preclinical and clinical investigations of mood stabilizers for Huntington’s disease: What have we learned? Int J Biol Sci. 2014;10:1024–1038. doi: 10.7150/ijbs.9898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahab L, Plattner F, Irvine EE, Cummings DM, Edwards FA. Dynamic range of GSK3α not GSK3β is essential for bidirectional synaptic plasticity at hippocampal CA3-CA1 synapses. Hippocampus. 2014 doi: 10.1002/hipo.22362. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakoori A, Mai W, Miyashita K, Yasumoto K, Takahashi Y, Ooi A, et al. Inhibition of GSK-3β activity attenuates proliferation of human colon cancer cells in rodents. Cancer Sci. 2007;98:1388–1393. doi: 10.1111/j.1349-7006.2007.00545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharfi H, Eldar-Finkelman H. Sequential phosphorylation of insulin receptor substrate-2 by glycogen synthase kinase-3 and c-Jun NH2-terminal kinase plays a role in hepatic insulin signaling. Am J Physiol Endocrinol Metab. 2008;294:E307–E315. doi: 10.1152/ajpendo.00534.2007. [DOI] [PubMed] [Google Scholar]
- Si X, Chen W, Guo X, Chen L, Wang G, Xu Y, et al. Activation of GSK3β by Sirt2 is required for early lineage commitment of mouse embryonic stem cell. PLoS One. 2013;8:e76699. doi: 10.1371/journal.pone.0076699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh KK. An emerging role for Wnt and GSK3 signaling pathways in schizophrenia. Clin Genet. 2013;83:511–517. doi: 10.1111/cge.12111. [DOI] [PubMed] [Google Scholar]
- Smith E, Coetzee GA, Frenkel B. Glucocorticoids inhibit cell cycle progression in differentiating osteoblasts via glycogen synthase kinase-3β. J Biol Chem. 2002;277:18191–18197. doi: 10.1074/jbc.M109708200. [DOI] [PubMed] [Google Scholar]
- Soutar MP, Kim WY, Williamson R, Peggie M, Hastie CJ, McLauchlan H, et al. Evidence that glycogen synthase kinase-3 isoforms have distinct substrate preference in the brain. J Neurochem. 2010;115:974–983. doi: 10.1111/j.1471-4159.2010.06988.x. [DOI] [PubMed] [Google Scholar]
- Spencer TE, Jenster G, Burcin MM, Allis CD, Zhou J, Mizzen CA, et al. Steroid receptor coactivator-1 is a histone acetyltransferase. Nature. 1997;389:194–198. doi: 10.1038/38304. [DOI] [PubMed] [Google Scholar]
- Spengler ML, Kuropatwinski KK, Schumer M, Antoch MP. A serine cluster mediates BMAL1-dependent CLOCK phosphorylation and degradation. Cell Cycle. 2009;8:4138–4146. doi: 10.4161/cc.8.24.10273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spokoini R, Kfir-Erenfeld S, Yefenof E, Sionov RV. Glycogen synthase kinase-3 plays a central role in mediating glucocorticoid-induced apoptosis. Mol Endocrinol. 2010;24:1136–1150. doi: 10.1210/me.2009-0466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler SC, Vincent CT, Fedorov VD, Patsialou A, Cherrington BD, Wakshlag JJ, et al. Dysregulation of PAD4-mediated citrullination of nuclear GSK3β activates TGF-β signaling and induces epithelial-to-mesenchymal transition in breast cancer cells. Proc Natl Acad Sci USA. 2013;110:11851–11856. doi: 10.1073/pnas.1308362110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stambolic V, Ruel L, Woodgett JR. Lithium inhibits glycogen synthase kinase-3 activity and mimics wingless signalling in intact cells. Curr Biol. 1996;6:1664–1668. doi: 10.1016/s0960-9822(02)70790-2. [DOI] [PubMed] [Google Scholar]
- Stamos JL, Chu ML, Enos MD, Shah N, Weis WI. Structural basis of GSK-3 inhibition by N-terminal phosphorylation and by the Wnt receptor LRP6. Elife. 2014;3:e01998. doi: 10.7554/eLife.01998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, et al. The Gβγ sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89:105–114. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Sun L, Zhao H, Xu Z, Liu Q, Liang Y, Wang L, et al. Phosphatidylinositol 3-kinase/protein kinase B pathway stabilizes DNA methyltransferase I protein and maintains DNA methylation. Cell Signal. 2007;19:2255–2263. doi: 10.1016/j.cellsig.2007.06.014. [DOI] [PubMed] [Google Scholar]
- Sun M, Song L, Li Y, Zhou T, Jope RS. Identification of an anti-apoptotic protein complex at death receptors. Cell Death Differentiation. 2008;15:1887–1900. doi: 10.1038/cdd.2008.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland C. What are the bona fide GSK3 substrates? Int J Alzheimers Dis. 2011;2011:505–607. doi: 10.4061/2011/505607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, et al. Inhibition of AMPK Catabolic Action by GSK3. Mol Cell. 2013;50:407–419. doi: 10.1016/j.molcel.2013.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatmari E, Habas A, Yang P, Zheng JJ, Hagg T, Hetman M. A positive feedback loop between glycogen synthase kinase 3β and protein phosphatase 1 after stimulation of NR2B NMDA receptors in forebrain neurons. J Biol Chem. 2005;280:37526–37535. doi: 10.1074/jbc.M502699200. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz A, Skibinska M, Hauser J, Slopien A, Leszczynska-Rodziewicz A, Kapelski P, et al. Association analysis of the GSK-3β T-50C gene polymorphism with schizophrenia and bipolar disorder. Neuropsychobiology. 2006a;53:51–56. doi: 10.1159/000090704. [DOI] [PubMed] [Google Scholar]
- Szczepankiewicz A, Rybakowski JK, Suwalska A, Skibinska M, Leszczynska-Rodziewicz A, Dmitrzak-Weglarz M, et al. Association study of the glycogen synthase kinase-3β gene polymorphism with prophylactic lithium response in bipolar patients. World J Biol Psychiatry. 2006b;7:158–161. doi: 10.1080/15622970600554711. [DOI] [PubMed] [Google Scholar]
- Taelman VF, Dobrowolski R, Plouhinec JL, Fuentealba LC, Vorwald PP, Gumper I, et al. Wnt signaling requires sequestration of glycogen synthase kinase 3 inside multivesicular endosomes. Cell. 2010;143:1136–1148. doi: 10.1016/j.cell.2010.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ter Haar E, Coll JT, Austen DA, Hsiao HM, Swenson L, Jain J. Structure of GSK3β reveals a primed phosphorylation mechanism. Nature Structural Biology. 2001;8:593–596. doi: 10.1038/89624. [DOI] [PubMed] [Google Scholar]
- Tolosa E, Litvan I, Höglinger GU, Burn D, Lees A, et al. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov Disord. 2014;29:470–478. doi: 10.1002/mds.25824. [DOI] [PubMed] [Google Scholar]
- Tsai SJ, Liou YJ, Hong CJ, Yu YW, Chen TJ. Glycogen synthase kinase-3β gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J. 2008;8:384–390. doi: 10.1038/sj.tpj.6500486. [DOI] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yevenes GE, Nikonenko I, Ebeling C, Schwerdel C, et al. Regulation of GABAergic synapse formation and plasticity by GSK3β-dependent phosphorylation of gephyrin. Proc Natl Acad Sci USA. 2011;108:379–384. doi: 10.1073/pnas.1011824108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyagarajan SK, Ghosh H, Yevenes GE, Imanishi SY, Zeilhofer HU, Gerrits B, et al. ERK and GSK3β regulate gephyrin postsynaptic aggregation and GABAergic synaptic function in a calpain-dependent mechanism. J Biol Chem. 2013;288:9634–9647. doi: 10.1074/jbc.M112.442616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, Klein PS. GSK-3 and Wnt Signaling in Neurogenesis and Bipolar Disorder. Front Mol Neurosci. 2012;5:1. doi: 10.3389/fnmol.2012.00001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valvezan AJ, Zhang F, Diehl JA, Klein PS. Adenomatous polyposis coli (APC) regulates multiple signaling pathways by enhancing glycogen synthase kinase-3 (GSK-3) activity. J Biol Chem. 2012b;287:3823–3832. doi: 10.1074/jbc.M111.323337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincent T, Kukalev A, Andäng M, Pettersson R, Percipalle P. The glycogen synthase kinase (GSK) 3β represses RNA polymerase I transcription. Oncogene. 2008;27:5254–5259. doi: 10.1038/onc.2008.152. [DOI] [PubMed] [Google Scholar]
- Voskas D, Ling LS, Woodgett JR. Does GSK-3 provide a shortcut for PI3K activation of Wnt signalling? F1000 Biol Rep. 2010;2:82. doi: 10.3410/B2-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner ER, Zhu G, Zhang BQ, Luo Q, Shi Q, Huang E, et al. The therapeutic potential of the Wnt signaling pathway in bone disorders. Curr Mol Pharmacol. 2011;4:14–25. [PubMed] [Google Scholar]
- Wakatsuki S, Saitoh F, Araki T. ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat Cell Biol. 2011;13:1415–1423. doi: 10.1038/ncb2373. [DOI] [PubMed] [Google Scholar]
- Wang QM, Park IK, Fiol CJ, Roach PJ, DePaoli-Roach AA. Isoform differences in substrate recognition by glycogen synthase kinases 3α and 3β in the phosphorylation of phosphatase inhibitor 2. Biochemistry. 1994;33:143–147. doi: 10.1021/bi00167a018. [DOI] [PubMed] [Google Scholar]
- Wang L, Lin HK, Hu YC, Xie S, Yang L, Chang C. Suppression of androgen receptor-mediated transactivation and cell growth by the glycogen synthase kinase 3β in prostate cells. J Biol Chem. 2004;279:32444–32452. doi: 10.1074/jbc.M313963200. [DOI] [PubMed] [Google Scholar]
- Wang Z, Smith KS, Murphy M, Piloto O, Somervaille TC, Cleary ML. Glycogen synthase kinase 3 in MLL leukaemia maintenance and targeted therapy. Nature. 2008;455:1205–1209. doi: 10.1038/nature07284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Iwasaki M, Ficara F, Lin C, Matheny C, Wong SH, et al. GSK-3 promotes conditional association of CREB and its coactivators with MEIS1 to facilitate HOX-mediated transcription and oncogenesis. Cancer Cell. 2010;17:597–608. doi: 10.1016/j.ccr.2010.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MJ, Li YC, Snyder MA, Wang H, Li F, Gao WJ. Group II metabotropic glutamate receptor agonist LY379268 regulates AMPA receptor trafficking in prefrontal cortical neurons. PLoS One. 2013;8:e61787. doi: 10.1371/journal.pone.0061787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Li Y, Hsu PH, Lee SY, Kim Y, Lee EY. Progesterone Receptor A Stability Is Mediated by Glycogen Synthase Kinase-3β in the Brca1-deficient Mammary Gland. J Biol Chem. 2013b;288:26265–26274. doi: 10.1074/jbc.M113.476556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watase K, Gatchel JR, Sun Y, Emamian E, Atkinson R, Richman R, et al. Lithium therapy improves neurological function and hippocampal dendritic arborization in a spinocerebellar ataxia type 1 mouse model. PLoS Med. 2007;4:e182. doi: 10.1371/journal.pmed.0040182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Zmijewski JW, Song L, Zmijewska A, Chen X, et al. Direct, activating interaction between glycogen synthase kinase-3β and p53 after DNA damage. Proc Natl Acad Sci USA. 2002;99:7951–7955. doi: 10.1073/pnas.122062299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watcharasit P, Bijur GN, Song L, Zhu J, Chen X, Jope RS. Glycogen synthase kinase-3β (GSK3β) binds to and promotes the actions of p53. J Biol Chem. 2003;278:48872–48879. doi: 10.1074/jbc.M305870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Liu W, Yan Z. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J Biol Chem. 2010;285:26369–26376. doi: 10.1074/jbc.M110.121376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle BJ, Varga C, Posa A, Molnar A, Collin M, Thiemermann C. Reduction of experimental colitis in the rat by inhibitors of glycogen synthase kinase-3β. Br J Pharmacol. 2006;147:575–582. doi: 10.1038/sj.bjp.0706509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willert K, Nusse R. Wnt proteins. Cold Spring Harb Perspect Biol. 2012;4:a007864. doi: 10.1101/cshperspect.a007864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood-Kaczmar A, Kraus M, Ishiguro K, Philpott KL, Gordon-Weeks PR. An alternatively spliced form of glycogen synthase kinase-3β is targeted to growing neurites and growth cones. Mol Cell Neurosci. 2009;42:184–194. doi: 10.1016/j.mcn.2009.07.002. [DOI] [PubMed] [Google Scholar]
- Woodgett JR. Judging a protein by more than its name: GSK-3. Sci STKE. 2001:re12. doi: 10.1126/stke.2001.100.re12. [DOI] [PubMed] [Google Scholar]
- Wu RC, Feng Q, Lonard DM, O’Malley BW. SRC-3 coactivator functional lifetime is regulated by a phospho-dependent ubiquitin time clock. Cell. 2007;129:1125–1140. doi: 10.1016/j.cell.2007.04.039. [DOI] [PubMed] [Google Scholar]
- Ye X, Tai W, Zhang D. The early events of Alzheimer’s disease pathology: from mitochondrial dysfunction to BDNF axonal transport deficits. Neurobiol Aging. 2012;33:1122, e1–10. doi: 10.1016/j.neurobiolaging.2011.11.004. [DOI] [PubMed] [Google Scholar]
- Yuan Z, Agarwal-Mawal A, Paudel HK. 14-3-3 binds to and mediates phosphorylation of microtubule-associated tau protein by Ser9-phosphorylated glycogen synthase kinase 3β in the brain. J Biol Chem. 2004;279:26105–26114. doi: 10.1074/jbc.M308298200. [DOI] [PubMed] [Google Scholar]
- Yuskaitis CJ, Mines MA, King MK, Sweatt JD, Miller CA, Jope RS. Lithium ameliorates altered glycogen synthase kinase-3 and behavior in a mouse model of Fragile X Syndrome. Biochemical Pharmacology. 2010;79:632–646. doi: 10.1016/j.bcp.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidner LC, Buescher JL, Phiel CJ. A novel interaction between Glycogen Synthase Kinase-3α (GSK-3α) and the scaffold protein Receptor for Activated C-Kinase 1 (RACK1) regulates the circadian clock. Int J Biochem Mol Biol. 2011;2:318–327. [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Tamai K, Doble B, Li S, Huang H, Habas R, et al. A dual-kinase mechanism for Wnt co-receptor phosphorylation and activation. Nature. 2005;438:873–877. doi: 10.1038/nature04185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Phiel CJ, Spece L, Gurvich N, Klein PS. Inhibitory phosphorylation of glycogen synthase kinase-3 (GSK-3) in response to lithium. Evidence for autoregulation of GSK-3. J Biol Chem. 2003;278:33067–33077. doi: 10.1074/jbc.M212635200. [DOI] [PubMed] [Google Scholar]
- Zhang JS, Herreros-Vilanueva M, Koenig A, Deng Z, de Narvajas AA, Gomez TS, et al. Differential activity of GSK-3 isoforms regulates NF-κB and TRAIL- or TNFα induced apoptosis in pancreatic cancer cells. Cell Death Dis. 2014;5:e1142. doi: 10.1038/cddis.2014.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou FQ, Zhou J, Dedhar S, Wu YH, Snider WD. NGF-induced axon growth is mediated by localized inactivation of GSK-3β and functions of the microtubule plus end binding protein APC. Neuron. 2004;42:897–912. doi: 10.1016/j.neuron.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Zhou J, Freeman TA, Ahmad F, Shang X, Mangano E, Gao E, et al. GSK-3α is a central regulator of age-related pathologies in mice. J Clin Invest. 2013;123:1821–1832. doi: 10.1172/JCI64398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LQ, Wang SH, Liu D, Yin YY, Tian Q, Wang XC, et al. Activation of glycogen synthase kinase-3 inhibits long-term potentiation with synapse-associated impairments. J Neurosci. 2007;27:12211–12220. doi: 10.1523/JNEUROSCI.3321-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]