

Abstract
Background
Atherosclerosis, the underlying cause of heart attack and strokes, is a progresive dyslipidemic and inflammatory disease where monocyte-derived macrophage cells play a pivotal role. Although most of the mechanisms that contribute to the progression of atherosclerosis have been identified, there is limited information on those governing regression. Conjugated linoleic acid (CLA) is a group of isomers of linoleic acid that differ in the position and/or geometry of their double bonds. We have previously shown that a specific CLA blend (80:20 cis-9,trans-11:trans-10,cis-12-CLA) induces regression of pre-established atherosclerosis in vivo, via modulation of monocyte/macrophage function. However, the exact mechanisms through which CLA mediates this effect remain to be elucidated.
Methods
Here, we address if CLA primes monocytes towards an anti-inflammatory MΦ2 macrophage and examine the effect of individual CLA isomers and the atheroprotective blend on monocyte-macrophage differentiation, cytokine generation, foam cell formation and cholesterol metabolism in human peripheral blood monocyte (HPBMC)-derived macrophages.
Results
cis-9,trans-11-CLA and the atheroprotective 80:20 CLA blend regulates expression of pro-inflammatory mediators and modulates the inflammatory cytokine profile of macrophages and foam cells. In addition, cis-9,trans-11-CLA and CLA blend primes HPBMCs towards an anti-inflammatory MΦ2 phenotype, characterised by increased scavenger receptor (CD36) and efflux protein (ABCA-1) expression. Furthermore, this altered macrophage phenotype impacts on foam cell formation, inhibiting ox-LDL accumulation and promoting cholesterol efflux via both PPARγ and LXRα dependent pathways.
Conclusion
The data increases the understanding of the pathways regulated by CLA in atheroprotection, namely, inhibiting the progressive acquisition of a pro-inflammatory macrophage phenotype.
Electronic supplementary material
The online version of this article (doi:10.1186/s12950-015-0060-9) contains supplementary material, which is available to authorized users.
Keywords: Conjugated linoleic acid, Atherosclerosis, Macrophage differentiation, Foam cell formation, Scavenger receptors, Cholesterol efflux
Background
Biologically active macrophage cells play a pivotal role in atherosclerosis development from the early stages of monocyte to macrophage differentiation, intimal accumulation of macrophages and their conversion to foam cells via uptake of oxidised lipid, to the later stage of fibrous plaque development. In the context of atherosclerosis, the underlying cause of myocardial infarction and stroke, macrophages uniquely possess a dual functionality regulating and sustaining the chronic inflammatory response and regulating lipid accumulation and metabolism [1], two of the most well documented pathways associated with the pathogenesis of the disease.
At sites of atherosclerotic lesion development, LDL passively diffuses through the tight junction of the dysfunctional endothelium [2], where it becomes oxidised as a result of exposure to the oxidative stress of vascular cells thus giving rise to minimally oxidised-LDL which stimulates the overlying endothelial cells to express pro-inflammatory mediators which recruit and mediate the adhesion of leukocytes, primarily monocytes to the artery wall [3]. Monocytes adhere to the activated endothelial cells via specific interactions mediated by integrins [4] and following transendothelial migration, rapidly differentiate into macrophages in response to growth factors, such as macrophage colony-stimulating factor (M-CSF). M-CSF is required for the survival of both circulating monocytes and resident tissue macrophages [5] and potentiates a number of monocyte functions including phagocytic activity, microbial killing, tumor cell cytotoxicity, enhanced synthesis of inflammatory cytokines [6], and, of relevance to this study, differentiation of the monocyte cell into a mature macrophage. In atherosclerosis M-CSF mediates other macrophage-specific programmes, such as scavenger receptor (SR) and apo-E gene expression [7].
ox-LDL is taken up by the mature macrophage via scavenger receptor-mediated endocytosis, primarily CD36 and SR-A1, through endosomes. Endosomes-containing ox-LDL are trafficked to lysosomes where the cholesterol ester content of the ox-LDL is hydrolysed to fatty acids and free cholesterol, which is then trafficked out of the lysosome where it is re-esterified to cholesterol esters by acyl-coenzyme A:cholesterol acetyltransferase (ACAT). Cholesterol esters are stored in the cytosol or cleaved by the neutral cholesterol ester hydrolase to free cholesterol which is then effluxed from the macrophage via several transporters, including ATP-binding cassette (ABC) family (ABCA-1, ABCG-1) and SR-B1 to acceptor molecules, such as apo-A1 and HDL for subsequent metabolism in the liver [8,9]. This reverse cholesterol transport (RCT) pathway is regulated by the nuclear receptor Liver X Receptor (LXR).
During early stages of atherosclerosis, the concentrations of ox-LDL in the intima are sufficiently low for macrophage-mediated removal. However, as disease progresses and concentrations of ox-LDL increase, the balance between efflux and influx is altered, the RCT system is overwhelmed and free cholesterol is stored as cholesterol esters in the form of cytosolic lipid droplets. The accumulation of lipid droplets in the macrophage is indicative of foam cell formation and results in the fatty streak, the first clinical hallmark of atherosclerotic plaque [10].
Macrophages are heterogeneous cell populations, of which the pan macrophage marker is CD68, a glycoprotein present on the lysosomal membrane of the cell that adapts a response to environmental cytokines. Macrophages respond to stimuli from their micro-environment and, consequently, show high plasticity and heterogeneity [11]. The initial simplified classification of macrophage phenotypes is the discrimination between type-1 pro-inflammatory (MΦ1) and type-2 anti-inflammatory (MΦ2) macrophages on the basis of the cytokine environment created by two different classes of lymphocyte T helper (TH1 or TH2). Classically activated MΦ1 pro-atherogenic macrophages are primed by TH1 cytokines, such as IFN-γ and IL-1β, and function to increase and sustain the ongoing inflammatory response via production of pro-inflammatory mediators, such as TNF-α, IL-6, IL-1β and IL-12. Thus, continuous MΦ1 macrophage activity, contributes to tissue damage [1,12]. Alternatively activated MΦ2 anti-inflammatory macrophages are primed as a result of exposure to TH2 cytokines, such as IL-4 and IL-13 [13] and promote tissue repair and healing. Both MΦ1 and MΦ2 macrophages have been identified in human atherosclerotic plaque where MΦ2 macrophages are present at more stable locations [14]. More recently, it has been shown that the MΦ1 macrophage content of atherosclerotic plaques is associated with clinical incidence of ischemic stroke and increased inflammation [15]. Furthermore, it has been shown that there is an MΦ2 to MΦ1 switch during atherosclerotic plaque progression, suggesting that interventional tools which could revert the macrophage infiltrate towards the MΦ2 phenotype, may exert an atheroprotective action [16]. We have previously shown that, in a conjugated linoleic acid (CLA)-induced model of atherosclerosis regression, there is enrichment of MΦ2 genes in the aorta in vivo [17] which is in agreement with other studies which show that decreased lipid levels are associated with lower plaque lipid content and higher MΦ2 gene expression [18].
CLA is a family of naturally occurring geometric dienoic isomers of the ω6 essential fatty acid, linoleic acid (LA) [19]. CLA has a diverse range of benefits in health and diseases such as cancer [20,21], obesity [22,23], immune function [24] and atherosclerosis [25-28].
We have previously shown that dietary administration of a 1% CLA blend of the two most abundant isomers (80:20, cis-9,trans-11-CLA:trans-10,cis-12-CLA) induces regression of pre-established atherosclerosis in the apo-E−/− mouse model, despite a continuing high cholesterol challenge [25], via modulation of monocyte/macrophage function [29,30].
Moreover, we have shown that CLA inhibits foam cell formation in vitro, via regulation of the nuclear receptor coactivator, peroxisome proliferator-activated receptor (PPAR)-γ coactivator (PGC)-1α [31]. Of relevance to this study, we have also shown that CLA increased macrophage polarization toward an anti-inflammatory MΦ2 phenotype in vivo [17] and that this is mediated via PPARγ dependent and independent mechanisms.
The effects of CLA on cholesterol homeostasis and foam cell formation have also been investigated in macrophage cell lines where t-10,c-12-CLA and c-9,t-11-CLA decreased foam cell formation [32] and increased expression of genes involved in RCT, such as LXRα and its target gene ABCA-1. In addition, levels of pro-inflammatory cytokine production (in particular TNF-α, IL-6 and IL-1β) were decreased following treatment with four different CLA isomers (cis-9,trans-11; cis-9,cis-11; trans-9,trans-11; trans-10,cis-12-CLA) in RAW-264.7 macrophages via a PPARγ dependent mechanism [33]. However, to date, studies with CLA on primary HPBMCs are limited.
The aim of this study is to investigate the effects of the atheroprotective CLA isomer c-9,t-11-CLA, the atheroprotective CLA blend (80:20 c-9,t-11:t-10,c-12-CLA) and the t-10,c-12-CLA isomer on the monocyte-macrophage-foam cell axis, specifically to identify changes in macrophage phenotype, inflammatory cytokine generation and cholesterol uptake and transport using HBPMC-derived macrophages, to ultimately further understand the mechanisms through which CLA mediates regression of pre-established atheroslerosis. Here, we provide evidence that CLA shifts HPBMC-derived macrophage differentiation to an anti-inflammatory phenotype, inducing expression of MΦ2 marker receptors and suppressing production of pro-inflammatory cytokines. Additionally, we show that CLA inhibits foam cell formation by reducing ox-LDL uptake and increasing cholesterol efflux. Our data describes a novel functional role for CLA in regulating macrophage phenotype and foam cell formation in the context of atherosclerosis regression.
Methods
Isolation of human peripheral blood monocytes
All experiments were conducted in conformity with institutional guidelines and in compliance with international laws. All volunteers gave written informed consent. Whole blood from healthy volunteers was drawn into heparin-coated vacutainers (BD, UK/Ireland). All volunteers were non-smoking, aged 25–30 years and free from medication for at least 10 days. Platelet-rich plasma (PRP) was isolated by centrifugation (190 × g for 15 min) and then diluted 1:3 with PBS before addition to Lymphoprep (Nycomed, Norway) and centrifuged at 450 × g for 30 min. Buffy-coats were recovered using a pasteur pipette, washed twice with PBS and resuspended in 10 ml serum-free medium (SFM) M-199 (Thermo Scientific), supplemented with L-glutamine (6.8 mM) and antibiotics (100 U/mL penicillin, 100 μg/mL streptomycin) and 10 ng/ml polymyxin-B-sulfate (Sigma-Aldrich, Dublin, Ireland). Monocytes were purified by plastic adherence in SFM for 2 hrs at 37°C, 5%CO2.
HPBMC-derived macrophage differentiation and cell treatment
Prior to in vitro experiments, macrophage differentiation of freshly isolated HBPMCs was stimulated with 100 ng/ml M-CSF (Gibco-BRL Life Technologies Ltd, England, UK), in 10% human serum (HS) (Sigma-Aldrich, Dublin, Ireland). After four days of culture at 37°C, 5%CO2, media was changed and a further 100 ng/ml M-CSF was added in 10% HS (Additional file 1).
For experiments on differentiating macrophages, cells were washed twice with warm PBS and treated for 48 hrs at 37°C, 5%CO2, in 1% HS, with 10 μM of cis-9,trans-11-CLA, trans-10,cis-12-CLA , CLA blend (80:20 c-9,t-11:t-10,c-12); linoleic acid (LA) and oleic acid (OA) (all Cayman Chemicals, MI, USA); 5 μM PPARγ agonist, troglitazone (TROG) or dimethyl sulfoxide (DMSO) (vehicle control) (both Sigma-Aldrich, Dublin, Ireland) at day 4 of culture. At day 6 cells were fixed for immunocytochemistry or lysed for mRNA analysis.
For experiments on mature macrophages and foam cell formation, media was changed and further 100 ng/ml M-CSF were added in 10% HS after four days of culture at 37°C, 5%CO2. Cells were treated with CLA and controls as above at day 8 and fixed at day 10. Where relevant, cells were also treated with 1 μM LXRα agonist T0901317 (T1317) or 10 μM 25-hydroxycholesterol (25-OH) (both from Sigma-Aldrich, Dublin, Ireland).
HPBMC-derived foam cell formation
After ten days of culture, as above described, mature macrophage were washed twice with warm PBS and incubated for further 4 hrs with 50 μg/ml of human ox-LDL or human fluorescently labelled Dil-ox-LDL (both Intracel MD, USA).
Following CLA and control treatments lipid loading, wells were washed three times with fresh medium and fluorescent Dil emission was measured in a Spectramax M2 (Molecular devices, CA, USA) plate fluorescence reader with 550/568 nm excitation/emission wavelength. The levels of Dil-ox-LDL were adjusted per cell number by measuring the intensity of DAPI fluorescence, with a second reading at 360/460 nm. Ox-LDL uptake measurements were repeated in triplicate in three independent experiments and the mean value was expressed as a percentage of vehicle control.
THP-1 cell culture and cell treatments
Human THP-1 monocytes (ATCC) were cultured in RPMI 1640 medium supplemented with fetal bovine serum (10%), penicillin (100 U/ml), streptomycin (100 ug/ml) and L-glutamine (2 mM) (Gibco BRL, UK). THP-1 monocytes (1 × 106) were differentiated to macrophages using 100 nmol/L phorbol 12-myristate 13-acetate (PMA) (Sigma Aldrich, Dublin, Ireland) for 72 hrs.
THP-1 macrophage were treated for 18 hrs with DMSO, 25 μM of c-9,t-11-CLA and CLA blend, TROG (10 μM), T1317 (1 uM), alone or pre-incubated for 2 hrs with 10 μM of the PPARγ inhibitor, GW9662 (GW) (Cayman Chemicals, MI, USA), or 1 μM of the LXRα inhibitor, GSK2033, (GSK) (Axon).
RNA isolation and gene expression analysis
For gene expression experiments, HBPMC- or THP-1-derived macrophages and HPBMC-derived foam cells were washed twice with ice cold PBS, prior to addition of 200 μl of RLT buffer (Qiagen, UK). Total RNA was isolated from cell lysates using the RNeasy kit (Qiagen, UK) as per manufacturers’ instructions. Reverse transcription was carried out on 1 μg of total RNA using SuperscriptTM III Reverse Transcriptase (Invitrogen) according to the manufacturers’ instructions. Relative gene expression quantification by real-time PCR (RT-PCR) was performed on an ABI Prism 7900HT Sequence Detection System (Applied Biosystems Inc., UK). MR and SRA-1 expression were examined using specific Taqman assays (Applied Biosystems Inc., UK), whilst, ABCA-1, CD36, CD14, CD68 and CD163 were measured using specific Syber green assays (Applied Biosystems Inc., UK) (Additional file 2). Ct values were normalised to 18s ribosomal RNA.
Immunocytochemistry
For visualisation of HPBMC-derived macrophages and foam cells, 5x105 cells were seeded onto glass coverslips placed in 12-well plate and treated, as above, over 6 days of culture. Mature macrophages or ox-LDL loaded-foam cells were then fixed in 3% formaldehyde (Sigma-Aldrich, Dublin, Ireland), permeabilized with Triton-X-100 (Sigma-Aldrich, Dublin, Ireland). Non-specific binding was prevented by blocking with 5% BSA (Sigma-Aldrich, Dublin, Ireland). Target proteins were then labelled using 1:100 of anti-human goat polyclonal CD68 and goat polyclonal MR (Sigma-Aldrich) and fluorescently labelled-secondary antibodies (1:200 of Alexa-Fluor 488, 568 or 647) (Invitrogen, Carlsbad, California), followed by Alexa Fluor 568-phalloidin staining (Invitrogen, Carlsbad, California), for F-actin, and DAPI (Sigma-Aldrich, Dublin, Ireland). For foam cell visualization, only DAPI staining was required as fluorescently labelled ox-LDL was used (Dil emission at 568nm). Cells were imaged using a Zeiss AxioImager M1 fluorescent microscope and the images were captured using an Olympus digital camera (Optronics, Goleta, CA).
Intracellular cholesterol measurement
Intracellular cholesterol levels were measured using the Amplex® Red Cholesterol Assay kit (Molecular Probes), based on an enzyme-coupled reaction that detects both free and esterified cholesterol.
After 10 days of culture and treatment with CLA isomers and controls as described above, HPBMC-derived macrophages and foam cells were lysed using T-PER. Firstly, a cholesterol standard curve was prepared, diluting the cholesterol reference standard. The Amplex Red reagent, containing the substrate HRP and both cholesterol oxidase/esterase, enzymes was prepared according to the manufacturers’ guidelines. 50 μl Amplex Red reagent was added to each sample and the samples were then incubated for 30 mins at 37°C. Fluorescence was measured using a microplate reader, using excitation in the range of 530–560 nm and emission detection at ~590 nm. Each point was corrected for background fluorescence by subtracting the values derived from the no-cholesterol control.
ELISA for cytokine quantification
Following 10 days of differentiation firstly to macrophages, and subsequenty to foam cells following exposure to ox-LDL, in the presence of CLA isomers or appropriate controls, as described above, HBPMC-derived macrophage/foam cell supernatants were collected and cleared by centrifugation (10,000 × g for 10 min at 4°C). The concentration of IL-10, IL-6, IL-1β, INF-γ, TNF-α and IL12-p70 in conditioned media was determined by enzyme immunoassay (EIA) using commercially available human 96 well-plate multiplex kit for tissue culture samples (MSD, Gaithersburg, MD, USA) according to the manufacturers’ guidelines.
Statistical analysis
Results are expressed as mean ± SEM or fold change relative to vehicle control. Experimental points were performed in triplicate with a minimum of three independent experiments (n = 3). Statistical comparisons between controls versus treated groups were made by Student’s unpaired t-test, assuming unequal variance with a two-tailed distribution. A value of p < 0.05 or greater were considered significant.
Results
CLA primes human monocytes towards an MΦ2 phenotype
To elucidate if the atheroprotective CLA blend (80:20 c-9,t-11:t-10,c-12-CLA) impacts on monocyte-macrophage differentiation, we analysed the mRNA expression of the pan-monocyte and pan-macrophage markers, CD14 and CD68, respectively (Figure 1a), and of the MΦ2-type macrophage markers mannose receptor (MR) and CD163 (Figure 1b) in unstimulated or M-CSF-stimulated HPBMCs. Unstimulated monocyte cells were maintained in culture for 6 days, as a model of early macrophage differentiation, and M-CSF treated HPBMCs were used as a model of mature differentiated macrophages. Following 6 days of culture, unstimulated and M-CSF-stimulated macrophages were treated with 10 μM cis-9,trans-11-CLA (c-9,t-11-CLA), trans-10,cis-12-CLA (t-10,c-12-CLA), CLA blend (80:20 c-9,t-11:t-10,c-12), linoleic acid (LA), oleic acid or 5 μM of the PPARγ agonist, troglitazone (TROG) and analysed for expression of macrophage markers by RT-PCR and fluorescent microscopy.
Figure 1.
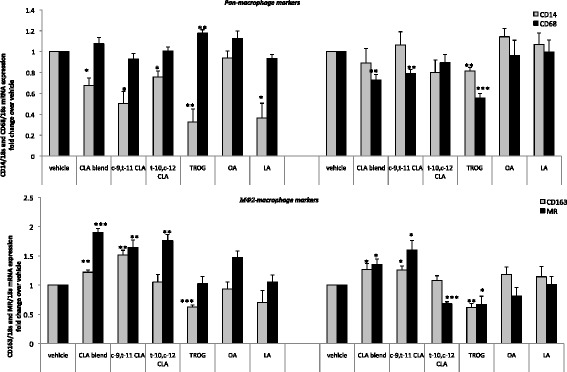
CLA primes monocytes to an anti-inflammatory MΦ2 macrophage phenotype. RT-PCR analysis of (a) CD14 and CD68 and (b) CD163 and MR mRNA expression in or M-CSF stimulated (100 ng/ml) differentiating HPBMCs following treatment with c-9,t-11-CLA, t-10,c-12-CLA, CLA blend (80:20 c-9,t-11:t-10,c-12), OA, LA or TROG. In unstimulated conditions, both CLA isomers and their blend decrease CD14 expression and increase expression of both CD163 and MR, without affecting CD68 expression. Following M-CSF stimulation, c-9,t-11-CLA and CLA blend decrease the mature macrophage marker CD68 and increase expression of both CD163 and MR (MΦ2 markers). Data are mean +/− SEM of three independent experiments. Data is expressed as fold change in gene expression relative to DMSO control, where *p < 0.05; ** p < 0.01 and ***p < 0.001 vs DMSO.
In unstimulated conditions, both the individual CLA isomers and their 80:20 blend significantly decreased CD14 mRNA expression (c-9,t-11-CLA by 2 fold, p < 0.05; t-10,c-12-CLA by 1.3 fold, p < 0.05 and 80:20 CLA blend by 1.4 fold, p < 0.05) via a PPARγ dependent mechanism, where the PPARγ agonist TROG, also decreased CD14 expression (by 3.3 fold, p < 0.01). Although the lipid control OA had no effect, the parent compound, LA, also decreased CD14 expression, suggesting a general class effect of linoleic acids on unstimulated monocytes in early differentiation. The effect of CLA in repressing CD14 expression in early differentiation, was not maintained upon M-CSF treatment. This is expected, as CD14 is a monocyte marker which is not highly expressed in mature differentiated macrophages. Interestingly, the PPARγ agonist maintained a decrease in CD14 expression (by 1.3 fold, p < 0.01) suggesting that activation of PPARγ may inhibit the process of monocyte to macrophage differentiation even in the presence of M-CSF. Although the pan-macrophage marker CD68 was not regulated by any lipid treament in unstimulated macrophages, following stimulation with M-CSF, both c-9,t-11-CLA and CLA blend decreased CD68 expression compared to DMSO control (by 1.3 fold, p < 0.01 and by 1.4 fold, p < 0.01, respectively) via a PPARγ dependent mechanism, where TROG also decreased CD68 expression (by 1.8 fold, p < 0.001). Conversely, t-10,c-12-CLA, OA and LA had no effect on CD68 expression (Figure 1a). This suggests a specificity of the atheroprotective CLA isomer (c-9,t-11) and the CLA blend in alteration of macrophage phenotype, following monocyte differentiation. This is in keeping with previous studies which show that the c-9,t-11 isomer, but not the t-10,c-12 isomer, is atheroprotective [34-36].
In these experiments, CLA was added after four days of culture, providing a model to investigate how CLA primes monocytes during their differentiation to macrophages. Therefore, this data suggests that treatment with the atheroprotective CLA blend and the c-9,t-11 isomer may prime monocytes towards an MΦ2 macrophage phenotype. This was confirmed by analysis of the expression of the MΦ2-type marker, CD163 receptor, which also indirectly contributes to an anti-inflammatory response [37]. In both the presence and absence of M-CSF, c-9,t-11-CLA and CLA blend increased CD163 expression (in unstimulated cells by 1.5 fold, p < 0.01 and by 1.2 fold, p < 0.01, respectively; in M-CSF stimulated cells by 1.3 fold, p < 0.05 for both treatments). Interestingly, this effect was independent of PPARγ, as TROG decreased CD163 expression in both the absence and presence of M-CSF. Under both conditions, t-10,c-12-CLA, OA and LA had no effect on target gene expression, again confirming specificity for the atheroprotective isomers in altering macrophage phenotype (Figure 1b).
MR mediates the endocytosis of glycoproteins by macrophages [38,39] and is a well characterised MΦ2-type phenotype marker [8,14,40].
mRNA analysis of MR showed that, in both unstimulated and M-CSF-stimulated conditions,c-9,t-11-CLA and CLA blend increased MR expression. In the absence of M-CSF, MR expression was increased by c-9,t-11-CLA and CLA blend (by 1.6 fold, p < 0.01 and 1.9 fold, p < 0.001, respectively) and this effect was maintained upon stimulation with M-CSF (c-9,t-11-CLA by 1.6 fold, p < 0.05 and the CLA blend by 1.3 fold, p < 0.05) (Figure 1b). In contrast, although t-10,c-12 increased MR expression by 1.8 fold (p < 0.01), in unstimulated conditions, this effect was not observed upon stimulation with M-CSF. Furthermore, neither OA nor LA had any effect on MR expression.
To validate the hypothesis that transcriptional changes may influence protein expression, we also performed immunocytochemistry of the pan-macrophage marker CD68 and of the MΦ2-type receptor MR, in HPBMCs either unstimulated (Figure 2a) or under M-CSF-stimulated conditions (Figure 2b). Immunofluorescence microscopy and subsequent spectrophotometric quantification of the fluorescent signals (Figure 2c) showed that, in unstimulated conditions, CLA had no effect on CD68 expression but upon M-CSF stimulation, both the c-9,t-11 isomer and CLA blend decreased CD68 (by 10 ± 1%, p < 0.01 and 18 ± 1%, p < 0.001, respectively). Importantly, in mature macrophages following M-CSF stimulation, c-9,t-11-CLA and CLA blend increased MR expression (by 57 ± 7% p < 0.01 and 55 ± 9%, p < 0.01, respectively). Finally, t-10,c-12-CLA, OA and linoleic acid had no effect on CD68 or MR expression under either condition. This confirms that the atheroprotective CLA isomer and blend primes monocytes to an MΦ2 phenotype during differentiation, thus altering the macrophage phenotype. Negative controls for immunofluorescence experiments are shown in “Additional Information” (Additional file 3).
Figure 2.
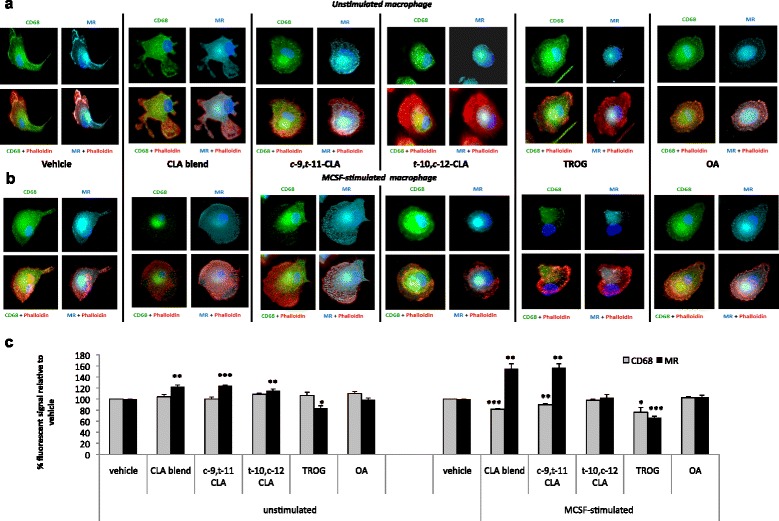
CLA inhibits CD68 and increases MR expression in HPBMC-derived macrophages. (a) Freshly isolated HBPMCs were cultured for four days in the presence or absence of M-CSF and treated with c-9,t-11-CLA, t-10,c-12-CLA, CLA blend (80:20 c-9,t-11:t-10,c-12), OA, LA or TROG for a further 48 hrs. At day 6, macrophage were fixed and stained. Immunofluorescence analysis of unstimulated macrophage shows that both CLA isomers and their blend increase MR expression but do not affect CD68 expression. (b) Differentiation of HBPMCs in the presence of 100 ng/ml M-CSF showed that c-9,t-11-CLA and CLA blend inhibits CD68 expression and increases MR expression. Epifluorescent microscope images (63× magnification) are representative of three independent experiments. Blue indicates DAPI (nuclei), green indicates CD68 (macrophage marker), cyan indicates MR and red indicates phalloidin (cytoplasmic) staining. (c) Spectrophotometry quantification of CD68 and MR fluorescent signal confirms the regulation of CD68 and MR by c-9,t-11-CLA and the atheroprotective blend. Statistical analysis of three independent experiments is expressed mean % as fluorescence ± SEM vs vehicle where *p < 0.05; **p < 0.01 and ***p < 0.001.
CLA reduces foam cell formation via a PPARγ/LXRα-dependent regulation of cholesterol metabolism
LXRα, LXRβ and PPARγ are ligand-activated nuclear receptors controlling cholesterol distribution and efflux, the inflammatory response and, in the case of PPARγ, macrophage polarization state. As described above, the effect of CLA on priming monocytes towards an MΦ2 phenotype is mediated in part via a PPARγ dependent mechanism. To understand the functional consequences of CLA in priming monocytes to alternatively activated macrophages, in the next set of experiments, we investigated the effect of CLA on foam cell formation in HPBMC-derived macrophages. Freshly isolated HPBMCs were differentiated towards a macrophage phenotype using 100 ng/ml M-CSF. After 8 days of culture cells were treated for 48 hrs with CLA or controls. To investigate the putative role of PPARγ and LXRα receptor in mediating the effect of CLA, the synthetic PPARγ agonist, TROG (10 μM) and LXRα agonist, T0901317 (also known as T1317) (1 μM) were used as controls. Following treatment, mature macrophages were loaded with 50 μg/ml Dil-ox-LDL for 4 hrs to induce foam cell formation. Similar to what we previously observed in RAW macrophage cells [31], immunofluorescence microscopy and its relative quantification clearly showed that both c-9,t-11-CLA and t-10,c-12-CLA isomers, as well as their 80:20 blend, decreased ox-LDL uptake (by 23 ± 3%, p < 0.01; 15 ± 4%, p < 0.05 and 16 ± 4%, p < 0.05, respectively), whereas LA control had no effect. Interestingly, both the PPARγ and LXRα agonists also inhibited foam cell formation (by 38 ± 2%,p < 0.001 and 36 ± 11%, p < 0.05, respectively), suggesting a dual PPARγ/LXRα-dependent mechanism, through which CLA inhibits foam cell formation (Additional file 4).
To elucidate the mechanism through which CLA, by inducing an MΦ2 phenotype, inhibits foam cell formation, we next examined expression of the scavenger receptors SR-A1 and CD36 in human macrophage-derived foam cells (Figure 3a). Although CLA had no effect on SR-A1 expression, both c-9,t-11-CLA and CLA blend increased CD36 expression in the presence of ox-LDL (by 1.3 fold, p < 0.01 for both), whereas, neither t-10,c-12-CLA isomer nor either of the two fatty acid controls modulated CD36 expression.
Figure 3.
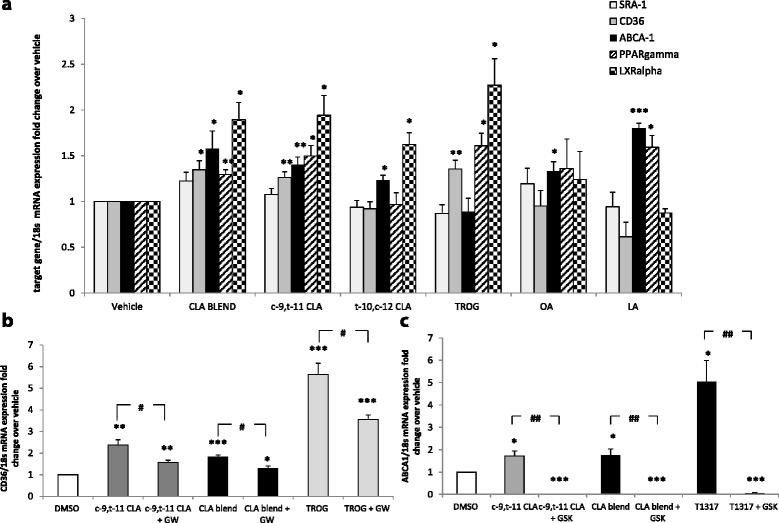
CLA increased CD36 and ABCA-1 expression via a PPARγ/LXRα mechanism. (a) RT-PCR analysis of SRA-1, CD36, ABCA-1, PPARγ and LXRα in HPBMC-derived macrophages pre-treated with c-9,t-11; t-10,c-12; CLA blend; OA; LA; TROG or T1317 and stimulated with ox-LDL for 4 hours to induce foam cell formation. CLA blend and c-9,t-11 increase CD36 and ABCA-1. Although t-10,c-12 has no effect on SR expression, it incresases ABCA-1, which is a generalized effect of linoleic acids. The same effect was observed with the parent compound LA. RT-PCR analysis of (b) CD36 and (c) ABCA-1 mRNA expression in PMA-induced macrophages treated with c-9,t-11, CLA blend and TROG alone or in combination with the PPARγ and LXRα antagonists (GW9662 and GSK2033, respectively). Pre-treatment with the antagonists attenuates or abolished the CLA-induced upregulation of both CD36 and ABCA-1, respectively. Statistical analysis of three independent experiments is expressed as fold change expression relative to DMSO control where *p < 0.05; **p < 0.01 and ***p < 0.001 or relative to the combination of the antagonist and the antagonist vs the agonist alone, where #p < 0.05 or ##p < 0.01.
Furthermore, both CLA isomers and their 80:20 blend increased ABCA-1 mRNA expression (by 1.4 fold, p < 0.01; 1.2 fold, p < 0.05 and 1.6 fold, p < 0.05, respectively). However, the fatty acid controls also increased ABCA-1 expression, suggesting that the induction of efflux proteins is a generalised fatty acid effect, whereas, the regulation of CD36 is unique to the atheroprotective CLA isomers and blend. To identify the mechanisms involved in increased CD36 and ABCA-1 levels, expression of the nuclear receptors, which regulate cholesterol transport and macrophage polarization, were investigated in both the presence and absence of CLA. In ox-LDL treated macrophages, c-9,t-11-CLA and the 80:20 CLA blend increased PPARγ expression (by 1.5 fold, p < 0.05 and by 1.3 fold, p < 0.05 respectively). As expected, the receptor agonist TROG also increased its expression by 1.6 fold (p < 0.05). Furthermore, CLA isomers and their blend, as well as the PPARγ agonist, induced a marked increase in the expression of the nuclear receptor LXRα (c-9,t-11-CLA and CLA blend by 1.9 fold, p < 0.05; t-10,c-12-CLA by 1.6 fold, p < 0.05 and TROG by 2.3 fold, p < 0.05). This is a potent and specific effect of CLA, as neither fatty acid control affected expression. As positive controls, the effects of two LXRα agonists, T1317 and 25-OH, were verified on the nuclear receptor LXRα and on its target gene ABCA-1 in HBPMCs-derived macrophages. As expected, both LXRα agonists increased LXRα expression (Additional file 5). Taken together, this data strongly suggests that the atheroprotective c-9,t-11-CLA and CLA blend-mediated regulation of CD36 and ABCA-1 expression is via PPARγ and LXRα dependent mechanisms.
To confirm our hypothesis, we extended our studies to examine the effect of the atheroprotective CLA isomer and the blend on the expression of PPARγ and LXRα target genes CD36 and ABCA-1, in the presence and absence of PPARγ or LXRα antagonists, in a distinct model of macrophage differentiation, namely PMA-induced THP-1 macrophage formation (Figure 3b and c). As confirmation of a CLA PPARγ − dependent mechanism, pre-incubation with the PPARγ antagonist, GW9662, attenuated the effect of both c-9,t-11-CLA and CLA blend (by 0.3 fold, p < 0.05, for both) on CD36 expression, similar to that observed when the PPARγ agonist, TROG and the antagonist GW9662 were combined (by 0.4 fold, p < 0.05). This provides convincing evidence that regulation of CD36 expression by c-9,t-11 and CLA blend is PPARγ dependent. Importantly, the CLA-induced increased expression of ABCA-1 was completely abolished by pre-incubation with the LXRα antagonist GSK2033 (by 1 fold, p < 0.01, for both), similar to that observed when the LXRα agonist, T1317, and the antagonist GSK2033 were combined (by 1 fold, p < 0.01). This provides convincing data that CLA mediated regulation of ABCA-1 expression is LXRα dependent. These experiment confirm the specificity of CLA in modulation of macrophage function via both PPARγ and LXR-dependent mechanisms.
CLA reduces intracellular cholesterol in foam cells
As CLA alters the macrophage to an MΦ2 MR+ve phenotype, we hypothesized that a potential functional effect of CLA on MΦ2 macrophages in response to ox-LDL and the inhibition of foam cell formation, is the reduction of intracellular cholesterol by promoting efficient cholesterol metabolism.
To address our hypothesis, we investigated the effect of CLA on intracellular cholesterol trafficking. To this end, HBPMCs-derived MCSF-induced macrophages were analysed for intracellular cholesterol content, following CLA treatment and subsequent ox-LDL loading, using the Amplex red cholesterol assay. Subsequently, we measured the fluorescent signals generated by the activity of both cholesterol oxidase and esterase, calculating the content of both free cholesterol (FC) and total cholesterol (TC). c-9,t-11-CLA significantly decreased the levels of TC by 29 ± 9% (p < 0.05) and of FC by 36 ± 5% (p < 0.01) and CLA blend reduced TC by 36 ± 11% (p < 0.05) and FC by 44 ± 7% (p < 0.01). Importantly, neither the t-10,c-12-CLA isomer nor OA had any significant effect on TC although they both reduced FC. The PPARγ agonist TROG also reduced TC by 28 ± 7% (p < 0.05) and FC by 39 ± 12% (p < 0.05) and the LXRα agonist T1317 reduced TC by 65 ± 12% (p < 0.01) and FC by 89 ± 2% (p < 0.001). This data confirms that the overall effect of the c-9,t-11-CLA isomer and the CLA blend on cholesterol trafficking is regulated by a dual mechanism: a PPARγ-dependent pathway, controlling cholesterol influx (as previously confirmed by the regulation of CD36) and an LXRα-dependent mechanism, directly influencing the cholesterol efflux (through upregulation of the efflux protein ABCA-1, as previously shown) (Figure 4).
Figure 4.
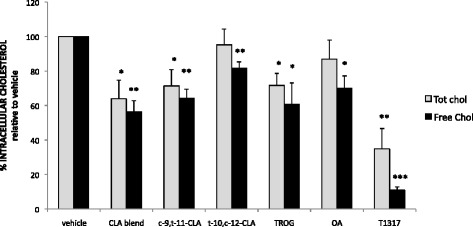
CLA reduces intracellular cholesterol content of human macrophage derived foam cells. HBPMCs-derived M-CSF stimulated macrophages, following treatment with c-9,t-11-CLA, t-10,c-12-CLA, CLA blend (80:20 c-9,t-11:t-10,c-12), OA, LA or TROG were ox-LDL loaded for 4 hours and cholesterol content measured by enzymatic assay. CLA blend and c-9,t-11-CLA significantly decrease the levels of both total cholesterol (TC) and free cholesterol (FC), whilst, t-10,c-12-CLA and OA decrease the FC fraction. The effect of CLA on cholesterol trafficking is regulated by a dual mechanism involving PPARγ/LXRα-dependent. Data are expressed as % of TC or FC content over vehicle control, where *p < 0.05, **p < 0.01 and are the mean of three independent experiments.
CLA inhibits production of pro-inflammatory cytokines in human macrophages and foam cells
It is now widely accepted that the cytokine microenvironment plays a fundamental role in the priming and switching of MΦ1/MΦ2 macrophage subtypes [41].
Using multiplex ELISA, we showed that pro-inflammatory cytokine generation was reduced in mature macrophages by CLA, in a similar manner to that of the PPARγ agonist (TROG) (Figure 5a). c-9,t-11-CLA significantly inhibited IL-6 and IL-1β generation (by 26 ± 10%,p < 0.01 and by 23 ± 2%, p < 0.05, respectively) and the CLA blend inhibited IL-1β, INF-γ and IL-12p70 (by 23 ± 5%,p < 0.05; by 20 ± 3%, p < 0.01 and by 36 ± 12%, p < 0.05, respectively). Interestingly, t-10,c-12-CLA also reduced IL-1β, INF-γ and IL-12p70 (by 30 ± 3%,p < 0.01; by 19 ± 8%, p < 0.05 and by 26 ± 11%, p < 0.05, respectively). Coincident with its role in macrophage polarisation, the PPARγ agonist inhibited all pro-inflammatory cytokines analysed. Importantly, c-9,t-11-CLA also increased anti-inflammatory cytokine IL-10 generation in macrophages (by 2.1 fold, p < 0.05), suggesting a specificity of this isomer to profoundly alter the cytokine profile of the marophage cells. Furthermore, we showed that the altered cytokine profile was maintained following stimulation of foam cell formation where pro-inflammatory cytokines generation was reduced by CLA, in a similar manner to that of the PPARγ agonist (TROG) (Figure 5b). In particular, c-9,t-11-CLA significantly inibited IL-6 and INF-γ generation (by 34 ± 15%, p < 0.05 and by 37 ± 12%, p < 0.05, respectively) and the CLA blend inibited IL-6, TNF-α and INF-γ (by 65 ± 4%,p < 0.001; by 42 ± 17%, p < 0.05; by 49 ± 6%, p < 0.01, respectively). t-10,c-12-CLA also reduced IL-6, TNF-α and IL-12p70 (by 51 ± 7%,p < 0.01; by 40 ± 16%, p < 0.05 and by 38 ± 17%, p < 0.05, respectively) and TROG reduced generation of IL-6, TNF-α, INF-γ and IL12p70.
Figure 5.
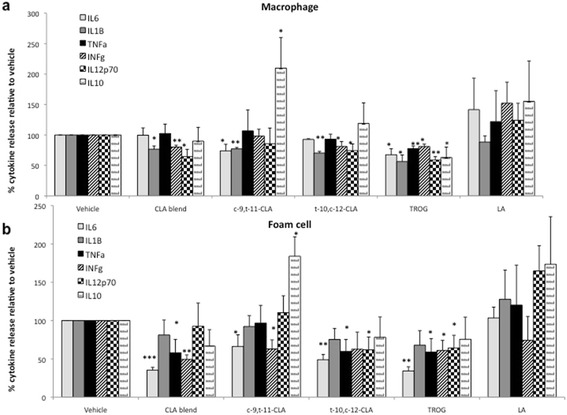
CLA inhibits production of pro-inflammatory citokine secretion in human macrophages and foam cells. Measurement of cytokine released was quantified in HPBMC-derived (a) macrophages and (b) foam cells following treatment with c-9,t-11; t-10,c-12; CLA blend; OA; LA or TROG using a multiplex ELISA. Both individual CLA isomers and the CLA bend reduced pro-inflammatory cytokine generation via a PPARγ dependent mechanisms. Uniquely, c-9,t-11-CLA increased macrophage and foam cell generation of IL10, via a PPARγ independent mechanism. Data are expressed as % cytokine release over vehicle control and are the mean of three independent experiments where *p < 0.05, **p < 0.01, ***p < 0.001.
Again, c-9,t-11-CLA increased IL-10 generation in foam cells by 1.8 fold (p < 0.05). Finally, fatty acid control LA had no effect on any of the cytokines analyzed. In summary, in both macrophage and foam cell, generation of pro-inflammatory cytokines was decreased by CLA in a similar manner to that of the PPARγ agonist (TROG) and c-9,t-11 isomer increased the generation of anti-inflammatory IL-10.
Discussion
Over the last decade, in parallel with the discovery of several anti-atherogenic properties of CLA [27,28], several studies have focused on identifying the cellular mechanism through which CLA mediates its effect with several lines of evidence, including our previous studies, suggesting that it is via the monocyte/macrophage cell [4,17,25,26,29-31]. To date, the effect of CLA on the modulation of macrophage/foam cell phenotype and function have primarily been conducted in cell lines, in particular the murine RAW-264.7 [35,36] and the human THP-1 cells [29], as well as in murine [27] and rabbit models of atherosclerosis [21,28,42]. Not surprisingly, the use of in vivo models, ex vivo cell culture systems and/or in vitro immortalised cell lines, as well as different isomeric blends of CLA, have resulted in conflicting data in relation to the effect of CLA on macrophage function.
In this study, we used a translationally relevant cell model, namely human peripheral blood monocytes, to elucidate the effect of CLA on macrophage plasticity, foam cell formation, intracellular cholesterol metabolism and cytokine generation. Importantly, we examined the effect of the 80:20 blend, previously shown by us to induce regression and inhibit foam cell formation [25,31], as well as the atheroprotective isomer c-9,t-11 which inhibits monocyte adhesion/migration [4,29]. We also examined the t-10,c-12 isomer as a control, which has previously been shown to have no effect and, in some studies, in fact, increases atherosclerosis [43]. Although CLA is a known ligand for the nuclear receptor PPARγ [44,45] and is thus a potential pathway through which CLA mediated its atheroprotective effects [33], we have recently shown that the effect of CLA on monocyte function is mediated via both PPARγ dependent and independent mechanisms [4] and, in this study, we identify that CLA mediates its effects on the macrophage/foam cell via an additional LXRα dependent mechanism.
Macrophage differentiation is diversely regulated by several growth factors [46]. A major aim of this study was to examine if CLA primes human monocytes to adopt an MΦ2 phenotype. To address this, we used M-CSF, a known inducer of macrophage polarization [47] and the effect of CLA isomers and the atheroprotective blend on the mRNA expression of macrophage markers was analysed, in the presence or absence of M-CSF stimulation. We show that, during early stages of monocyte-macrophage differentiation, modeled in vitro by the absence of M-CSF, both CLA isomers and the 80:20 blend decreases CD14, a receptor specific to the “monocytic phase” of the cell [48], whilst, as differentiation progresses, the atheroprotective CLA isomer decrease CD68 expression, a receptor which characterizes later stages of macrophage cell development [40]. Of importance is the observation that CLA also upregulates the specific MΦ2-type markers CD163 and MR [14], priming a switch towards an anti-inflammatory macrophage phenotype. Our data suggest a potential PPARγ-dependent mechanism of CLA in the regulation of the aforementioned receptors. Indeed, this is supported by Bouhlel et al., who showed that PPARγ activation primes human monocytes into alternative macrophage, characterized by high levels of MR, positively related to highly expressed PPARγ [14].
In the absence of differentiating stimulus, in early monocyte-macrophage differentiation, both c-9,t-11-CLA and the atheroprotective CLA blend promoted a CD68high/MRhigh phenotype, whilst, when M-CSF-triggered, switched to a CD68med/MRhigh and a CD68low/MRhigh phenotype, thereby also regulating CD68 protein expression. Moreover, stimulation with M-CSF, amplifies the effect of c-9,t-11-CLA on the mannose receptor, which although expressed in resting conditions, albeit at very low levels, is significantly increased following treatment. Importantly, this data identifies a putative atheroprotective role for CLA, specifically c-9,t-11 isomer, in priming monocytes towards MΦ2 macrophage phenotype.
To fully understand the functional consequences of altered macrophage phenotype on foam cell formation, HPBMC-derived macrophages were pre-treated with CLA isomers prior to the addition of the pro-atherogenic ox-LDL. c-9,t-11-CLA and CLA blend decreased foam cell formation, and t-10,c-12-CLA isomer induced a similar, although less pronounced effect. Interestingly, both agonists of PPARγ and LXRα inhibited foam cell formation, suggesting a possible dual PPARγ/LXRα-dependent mechanism, through which CLA prevents foam cell formation. This is in keeping with our recent study on RAW macrophage cells, which shows that CLA mediates its effect on foam cell formation via regulation of PGC-1α, the transcriptional activator of both PPARγ, LXR and other nuclear receptors [31].
The canonical route for ox-LDL to enter the cell is via scavenger receptor-mediated uptake and inhibition of CD36 and SRA-1 scavenger receptor expression has been shown to limit foam cell formation [49]. However, a second potential mechanism for the inhibition of foam cell formation is via the RCT system which removes cholesterol to HDL for metabolism in the liver. Primarily, the ABC transporters, including ABCA-1, regulates RCT. Previous studies have shown that LXRα regulates cholesterol trafficking via modulation of ABC efflux proteins [8]. Thus, to better understand the mechanism through which CLA inhibits foam cell formation, both uptake and efflux of ox-LDL was examined, initially by analysing the expression of SR-A1, CD36 [49,50] and ABCA-1 [9,51] following ox-LDL loading of mature HPBMC-derived macrophage. Furthermore, using the PMA-induced THP-1 macrophage cell line model, regulation of the aforementioned two nuclear receptors’ target genes CD36 and ABCA-1 expression was also verified in the presence of CLA isomers alone or in combination with PPARγ and LXRα antagonists, which, respectively, attenuates or abolished the CLA-induced upregulation of those genes. This data confirms that ox-LDL influx and efflux is controlled by a dual PPARγ/LXRα-dependent mechanism.
Our data shows that CLA inhibits foam cell formation, in the presence of an exogenous source of oxidized lipoproteins, by increasing expression of the PPARγ target CD36, thus promoting the uptake of the relatively abundant presence of circulating lipids, mimicked in vitro by loading cells using ox-LDL, which is known to activate PPARγ [52]. In parallel with the increased lipid uptake, an increased efflux of those lipids is induced by CLA via upregulation of the LXRα target ABCA-1. This is in keeping with previous studies where it has been established that increased PPARγ activity results in an increase in CD36 and a decrease in SRA-1 expression [52] and that a crosstalk between the two nuclear receptors, PPARγ and LXRα, induces a CD36-mediated positive regulation of ABCA-1 [53-55]. Interestingly, during early stages of macrophage differentiation, we show that the PPARγ agonist significantly reduced ABCA-1 (data not shown), whilst, in foam cells, ABCA-1 expression is rescued to the control level. This is likely due to the fact that ox-LDL activates the PPARγ nuclear receptor, therefore, its agonist, mediates the restoration to a basal ABCA-1 expression as previously documented [56,57].
Under normal homeostatic conditions, lipid that is taken into the macrophage can be efficiently metabolised and trafficked out of the cell via the RCT system, resulting in a balance between lipid uptake and efflux. In an atherogenic environment, however, where there are high concentrations of ox-LDL, there is a continuous influx of lipid and the increased intracellular concentration of free cholesterol overwhelms the RCT system. Excess cholesterol becomes esterified into cholesterol esters, which form lipid droplets, the distinctive features of foam cells. Therefore, to further confirm the proposed mechanism through which CLA inhibits foam cell formation, intracellular cholesterol content was analysed. c-9,t-11-CLA and CLA blend, significantly decreased the levels of both free and esterified cholesterol, confirming their specificity in atheroprotection, and reduced ox-LDL uptake, preventing intracellular accumulation of esterified cholesterol, whilst simultaneously, inducing free cholesterol efflux. The net effect of CLA, which primes the macrophages towards an MΦ2 phenotype prior to foam cell challenge, is altering cholesterol trafficking and lipid storage. Based on the data presented, it is feasible to suggest that CLA induces the macrophage to adopt an an anti-inflammatory phenotype which limits foam cell formation. To further address the inflammatory profile of CLA primed macrophages, the effect of CLA on cytokine generation was investigated.
The role of cytokine microenvironment on cholesterol metabolism has been extensively studied, playing a fundamental role in the priming of MΦ1/MΦ2 macrophage subtypes [41]. Therefore, macrophage generation of pro-inflammatory cytokines, namely, IL-1β, IL-6, IL-12p70, INF-γ and TNF-α were quantified, following CLA treatment of HPBMC-derived macrophages. Overall, in both macrophage and foam cells, the generation of most of the pro-inflammatory cytokines was prevented by CLA via a PPARγ-dependent mechanism. In general, the CLA conferred a “resolving” macrophage phenotype [11]. Moreover, it has been shown that IL-10 prevents pro-inflammatory cytokine production by activated macrophages, as part of its “deactivation” programme [58]. Supporting this is the observation that IL-10 production has been shown to be predominantly found in CD14low/CD16high or “anti-inflammatory” monocytes [59]. Indeed our recent work demonstrated that IL-10 signalling pathway was modified during CLA-induced regression in murine model [17]. In keeping with this, we show that the atheroprotective isomer c-9,t-11-CLA increases macrophage and foam cell IL-10 production. However, this is not a PPARγ-mediated effect, as TROG actually decreases IL-10 generation in HBPMC-derived macrophage and has no effect in foam cells. Similarly, it is also unlikely, that this anti-inflammatory effect is due to an LXRα activation, as it has been shown that the LXRα agonist, T1317, does not increase levels of IL-10 in CD4-positive T cells [60]. Further studies are needed to investigate the exact mechanism of action of the CLA atheroprotective effect on the anti-inflammatory cytokine generation, which will likely identify additional PPARγ/LXRα-independent pathways.
Among the pro-inflammatory cytokine panel analysed, INF-γ was the only one which was decreased in both macrophage and foam cells by the CLA blend, strongly suggesting a PPARγ-dependent mechanism regulating the inhibition of pro-inflammatory mediators. This hypothesis is supported by a study where the PPARγ antagonist GW9662 reversed the decreased expression of pro-inflammatory cytokines TNFα, IL-1β and IL-6 and increased levels of the immunosuppressive cytokine TGF-β in M2-polarized THP-1 macrophages [61]. It has been extensively shown that INF-γ increases the expression of the scavenger receptor SRA-1, in both THP-1- and HBPMC-derived macrophages [62], and that DNA binding activity is most likely responsible for the IFN-γ-dependent expression of SRA-1. In addition, INF-γ decreases ABCA-1 expression in murine peritoneal macrophages [63], decreasing cholesterol efflux, through pathways that include the upregulation of ACAT and the downregulation of efflux proteins. Based on these findings, IFN-γ can shift the equilibrium between macrophages and foam cells and, thus, impact the progression of an atherosclerotic lesion [64]. Moreover, it has been shown that LXRα activation also inhibits pro-inflammatory cytokine mRNA expression. In human lymphocytes the LXRα agonist, T1317 decreased INF-γ, TNF-α and IL-2 levels [60]. This is in keeping with our data, which shows that CLA mediates its effect via both a PPARγ and LXRα dependent mechanisms and decreases macrophage and foam cell generation of IFN-γ.
Conclusions
In summary, the data provides a novel mechanism for CLA in atherosclerosis Figure 6. The atheroprotective c-9,t-11 isomer and the CLA blend negatively regulate expression of pro-inflammatory mediators via a PPARγ and LXRα dependent mechanism. As a consequence, differentiation process is shifted towards an anti-inflammatory MΦ2 phenotype, characterised by increased CD36 and ABCA-1 expression, thus preventing macrophage lipid engulfment and promoting cholesterol efflux towards exogenous acceptors.
Figure 6.
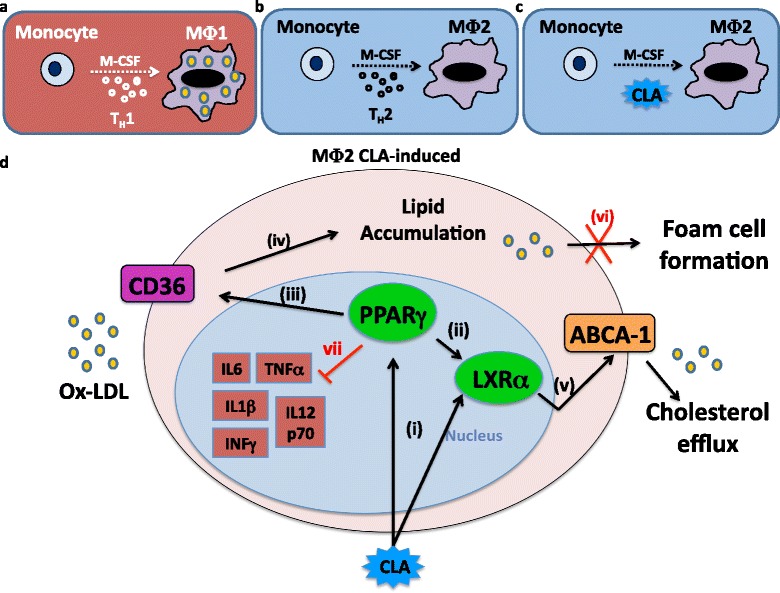
Potential atheroprotective mechanism of CLA on the macrophage/foam cell axis. (a) Th-1 cytokine environment primes M-CSF-triggered monocyte differentiation towards an MΦ1 macrophage pro-inflammatory phenotype. (b) Th-2 cytokine environment primes M-CSF-triggered monocyte differentiation towards an MΦ2 macrophage anti-inflammatory phenotype. (c) CLA action primes M-CSF-triggered monocyte differentiation towards an MΦ2-type macrophage. (d) In the presence of high levels of lipids in the extracellular matrix, CLA induces a dual mechanism PPARγ/LXRα-mediated (i-ii), by increasing CD36 levels (iii), allowing lipids to enter the cell (iv), and secondly, promoting cholesterol efflux, by increasing ABCA-1 mRNA expression (v), thus preventing lipid engulfment of the cell (vi), and the consequent foam cell formation. Moreover, CLA inhibits the secretion of pro-inflammatory cytokines (vii).
In conclusion CLA, by altering macrophage function, changes the relative distribution of macrophagic subsets, acquiring an anti-inflammatory phenotype which, ultimately, may impact on the lesion microenvironment, thus contributing to the mechanism through which CLA induces regression of pre-established atherosclerosis.
Acknowledgements
This work was supported by grants from the Health Research Board (Ireland). The authors acknowledge the support of the UCD Conway Institute Transcriptomics and Imaging Core Facilities.
Abbreviations
- ABCA1
ATP binding cassette A1
- ACAT
Acetyl-coenzyme A cholesterol acetyl-transferase
- CLA
Conjugated linoleic acid
- FC
Free cholesterol
- GSK
GSK2033
- GW
GW9662
- HPBMC
Human peripheral blood mononuclear cell
- HS
Human Serum
- IL
Interleukin
- LA
Linoleic acid
- LXRα
Liver X receptor-alpha
- M-CSF
Macrophage colony stimulating factor
- MΦ1/2
Macrophage phenotype 1/2
- MR
Mannose receptor
- OA
Oleic acid
- ox-LDL
Oxidized low density lipoprotein
- PMA
Phorbol 12-myristate 13-acetate
- PPAR
Peroxisome proliferator activated receptor
- RCT
Reverse cholesterol transport
- SFM
Serum free media
- SR
Scavenger receptor
- T1317
T0901317
- TC
Total cholesterol
- Th1/2
T helper Lymphocyte 1/2
- TNF-α
Tumor necrosis factor-α
- TROG
Troglitazone
- 25-OH
25-Hydroxy-cholesterol
Additional files
In vitro model of HBPMCs-derived macrophage differentiation.
Human Syber Green primer sequences.
Specificity of signals for co-incubation of CD68 and MR in HPBMC-derived macrophages.
CLA reduces foam cell formation in primary human cells.
LXR-a agonists increase LXRa and target gene ABCA-1 expression.
Footnotes
Competing interests
The authors declare that they have no competing interest.
Authors’ contributions
MdG participated in the design of the study, carried out the experiments and prepared the manuscript. KA and SM assisted with in vitro RT-PCR analysis. OB conceived and designed the study and prepared the manuscript. All authors read and approved the final manuscript.
Contributor Information
Monica de Gaetano, Email: monica.degaetano@ucd.ie.
Kawthar Alghamdi, Email: kawathar-saad.alghamdi@ucdconnect.ie.
Simone Marcone, Email: simone.marcone@ucd.ie.
Orina Belton, Email: orina.belton@ucd.ie.
References
- 1.Chinetti-Gbaguidi G, Staels B. Macrophage polarization in metabolic disorders: functions and regulation. Curr Opin Lipidol. 2011;22(5):365–72. doi: 10.1097/MOL.0b013e32834a77b4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabas I, Williams KJ, Boren J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: update and therapeutic implications. Circulation. 2007;116(16):1832–44. doi: 10.1161/CIRCULATIONAHA.106.676890. [DOI] [PubMed] [Google Scholar]
- 3.Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. 2006;47(8 Suppl):C7–12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- 4.de Gaetano M, Dempsey E, Marcone S, James WG, Belton O. Conjugated Linoleic Acid Targets beta2 Integrin Expression To Suppress Monocyte Adhesion. J Immunol. 2013;191(8):4326–36. doi: 10.4049/jimmunol.1300990. [DOI] [PubMed] [Google Scholar]
- 5.Tushinski RJ, Oliver IT, Guilbert LJ, Tynan PW, Warner JR, Stanley ER. Survival of mononuclear phagocytes depends on a lineage-specific growth factor that the differentiated cells selectively destroy. Cell. 1982;28(1):71–81. doi: 10.1016/0092-8674(82)90376-2. [DOI] [PubMed] [Google Scholar]
- 6.Warren MK, Ralph P. Macrophage growth factor CSF-1 stimulates human monocyte production of interferon, tumor necrosis factor, and colony stimulating activity. J Immunol. 1986;137(7):2281–5. [PubMed] [Google Scholar]
- 7.Di Gregoli K, Johnson JL. Role of colony-stimulating factors in atherosclerosis. Curr Opin Lipidol. 2012;23(5):412–21. doi: 10.1097/MOL.0b013e328357ca6e. [DOI] [PubMed] [Google Scholar]
- 8.Chinetti-Gbaguidi G, Baron M, Bouhlel MA, Vanhoutte J, Copin C, Sebti Y. Human atherosclerotic plaque alternative macrophages display low cholesterol handling but high phagocytosis because of distinct activities of the PPARgamma and LXRalpha pathways. Circ Res. 2011;108(8):985–95. doi: 10.1161/CIRCRESAHA.110.233775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jessup W, Gelissen IC, Gaus K, Kritharides L. Roles of ATP binding cassette transporters A1 and G1, scavenger receptor BI and membrane lipid domains in cholesterol export from macrophages. Curr Opin Lipidol. 2006;17(3):247–57. doi: 10.1097/01.mol.0000226116.35555.eb. [DOI] [PubMed] [Google Scholar]
- 10.Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217(5):492–502. doi: 10.1016/j.imbio.2012.02.015. [DOI] [PubMed] [Google Scholar]
- 11.Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008;8(12):958–69. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT. Macrophage infiltration in acute coronary syndromes. Implications for plaque rupture. Circulation. 1994;90(2):775–8. doi: 10.1161/01.CIR.90.2.775. [DOI] [PubMed] [Google Scholar]
- 13.Shalhoub J, Falck-Hansen MA, Davies AH, Monaco C. Innate immunity and monocyte-macrophage activation in atherosclerosis. J Inflamm (Lond) 2011;8:9. doi: 10.1186/1476-9255-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bouhlel MA, Derudas B, Rigamonti E, Dièvart R, Brozek J, Haulon S, et al. PPARgamma activation primes human monocytes into alternative M2 macrophages with anti-inflammatory properties. Cell Metab. 2007;6(2):137–43. doi: 10.1016/j.cmet.2007.06.010. [DOI] [PubMed] [Google Scholar]
- 15.Cho KY, Miyoshi H, Kuroda S, Yasuda H, Kamiyama K, Nakagawara J, et al. The phenotype of infiltrating macrophages influences arteriosclerotic plaque vulnerability in the carotid artery. J Stroke Cerebrovasc Dis. 2013;22(7):910–8. doi: 10.1016/j.jstrokecerebrovasdis.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Khallou-Laschet J, Varthaman A, Fornasa G, Compain C, Gaston AT, Clement M, et al. Macrophage plasticity in experimental atherosclerosis. PLoS One. 2010;5(1):e8852. doi: 10.1371/journal.pone.0008852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy C, Duffy MM, Mooney D, James WG, Griffin MD, Fitzgerald DJ, et al. IL-10 mediates the immunoregulatory response in conjugated linoleic acid-induced regression of atherosclerosis. FASEB J. 2012;27(2):499–510. doi: 10.1096/fj.12-215442. [DOI] [PubMed] [Google Scholar]
- 18.Feig JE, Parathath S, Rong JX, Mick SL, Vengrenyuk Y, Grauer L, et al. Reversal of hyperlipidemia with a genetic switch favorably affects the content and inflammatory state of macrophages in atherosclerotic plaques. Circulation. 2011;123(9):989–98. doi: 10.1161/CIRCULATIONAHA.110.984146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banni S. Conjugated linoleic acid metabolism. Curr Opin Lipidol. 2002;13(3):261–6. doi: 10.1097/00041433-200206000-00005. [DOI] [PubMed] [Google Scholar]
- 20.Kelley NS, Hubbard NE, Erickson KL. Conjugated linoleic acid isomers and cancer. J Nutr. 2007;137(12):2599–607. doi: 10.1093/jn/137.12.2599. [DOI] [PubMed] [Google Scholar]
- 21.Kritchevsky D. Antimutagenic and some other effects of conjugated linoleic acid. Br J Nutr. 2000;83(5):459–65. [PubMed] [Google Scholar]
- 22.Navarro V, Fernández-Quintela A, Churruca I, Portillo MP, et al. The body fat-lowering effect of conjugated linoleic acid: a comparison between animal and human studies. J Physiol Biochem. 2006;62(2):137–47. doi: 10.1007/BF03174074. [DOI] [PubMed] [Google Scholar]
- 23.Wang YW, Jones PJ. Conjugated linoleic acid and obesity control: efficacy and mechanisms. Int J Obes Relat Metab Disord. 2004;28(8):941–55. doi: 10.1038/sj.ijo.0802641. [DOI] [PubMed] [Google Scholar]
- 24.Reynolds CM, Roche HM. Conjugated linoleic acid and inflammatory cell signalling. Prostaglandins Leukot Essent Fatty Acids. 2010;82(4–6):199–204. doi: 10.1016/j.plefa.2010.02.021. [DOI] [PubMed] [Google Scholar]
- 25.Toomey S, Harhen B, Roche HM, Fitzgerald D, Belton O, et al. Profound resolution of early atherosclerosis with conjugated linoleic acid. Atherosclerosis. 2006;187(1):40–9. doi: 10.1016/j.atherosclerosis.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 26.Toomey S, Roche H, Fitzgerald D, Belton O, et al. Regression of pre-established atherosclerosis in the apoE−/− mouse by conjugated linoleic acid. Biochem Soc Trans. 2003;31(Pt 5):1075–9. doi: 10.1042/BST0311075. [DOI] [PubMed] [Google Scholar]
- 27.Arbones-Mainar JM, Navarro MA, Guzmán MA, Arnal C, Surra JC, Acín S, et al. Selective effect of conjugated linoleic acid isomers on atherosclerotic lesion development in apolipoprotein E knockout mice. Atherosclerosis. 2006;189(2):318–27. doi: 10.1016/j.atherosclerosis.2006.01.015. [DOI] [PubMed] [Google Scholar]
- 28.Kritchevsky D, Tepper SA, Wright S, Czarnecki SK, Wilson TA, Nicolosi RJ. Conjugated linoleic acid isomer effects in atherosclerosis: growth and regression of lesions. Lipids. 2004;39(7):611–6. doi: 10.1007/s11745-004-1273-8. [DOI] [PubMed] [Google Scholar]
- 29.McClelland S, Cox C, O'Connor R, de Gaetano M, McCarthy C, Cryan L, et al. Conjugated linoleic acid suppresses the migratory and inflammatory phenotype of the monocyte/macrophage cell. Atherosclerosis. 2010;211(1):96–102. doi: 10.1016/j.atherosclerosis.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 30.Mooney D, McCarthy C, Belton O. Effects of conjugated linoleic acid isomers on monocyte, macrophage and foam cell phenotype in atherosclerosis. Prostaglandins Other Lipid Mediat. 2012;98(3–4):56–62. doi: 10.1016/j.prostaglandins.2011.12.006. [DOI] [PubMed] [Google Scholar]
- 31.McCarthy C, Lieggi NT, Barry D, Mooney D, de Gaetano M, James WG, et al. Macrophage PPAR gamma Co-activator-1 alpha participates in repressing foam cell formation and atherosclerosis in response to conjugated linoleic acid. EMBO Mol Med. 2013;5(9):1443–57. doi: 10.1002/emmm.201302587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ringseis R, Wen G, Saal D, Eder K. Conjugated linoleic acid isomers reduce cholesterol accumulation in acetylated LDL-induced mouse RAW264.7 macrophage-derived foam cells. Lipids. 2008;43(10):913–23. doi: 10.1007/s11745-008-3226-x. [DOI] [PubMed] [Google Scholar]
- 33.Yu Y, Correll PH, Vanden Heuvel JP. Conjugated linoleic acid decreases production of pro-inflammatory products in macrophages: evidence for a PPAR gamma-dependent mechanism. Biochim Biophys Acta. 2002;1581(3):89–99. doi: 10.1016/S1388-1981(02)00126-9. [DOI] [PubMed] [Google Scholar]
- 34.Evans M, Brown J, McIntosh M. Isomer-specific effects of conjugated linoleic acid (CLA) on adiposity and lipid metabolism. J Nutr Biochem. 2002;13(9):508. doi: 10.1016/S0955-2863(02)00211-5. [DOI] [PubMed] [Google Scholar]
- 35.Lee Y, Thompson JT, de Lera AR, Vanden Heuvel JP. Isomer-specific effects of conjugated linoleic acid on gene expression in RAW 264.7. J Nutr Biochem. 2009;20(11):848–59. doi: 10.1016/j.jnutbio.2008.07.013. [DOI] [PubMed] [Google Scholar]
- 36.Lee Y, Thompson JT, Vanden Heuvel JP. 9E,11E-conjugated linoleic acid increases expression of the endogenous antiinflammatory factor, interleukin-1 receptor antagonist, in RAW 264.7 cells. J Nutr. 2009;139(10):1861–6. doi: 10.3945/jn.108.100461. [DOI] [PubMed] [Google Scholar]
- 37.Etzerodt A, Moestrup SK. CD163 and inflammation: biological, diagnostic, and therapeutic aspects. Antioxid Redox Signal. 2013;18(17):2352–63. doi: 10.1089/ars.2012.4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feinberg H, Park-Snyder S, Kolatkar AR, Heise CT, Taylor ME, Weis WI. Structure of a C-type carbohydrate recognition domain from the macrophage mannose receptor. J Biol Chem. 2000;275(28):21539–48. doi: 10.1074/jbc.M002366200. [DOI] [PubMed] [Google Scholar]
- 39.Taylor ME, Bezouska K, Drickamer K. Contribution to ligand binding by multiple carbohydrate-recognition domains in the macrophage mannose receptor. J Biol Chem. 1992;267(3):1719–26. [PubMed] [Google Scholar]
- 40.Boytard L, Spear R, Chinetti-Gbaguidi G, Acosta-Martin AE, Vanhoutte J, Lamblin N, et al. Role of proinflammatory CD68(+) mannose receptor(−) macrophages in peroxiredoxin-1 expression and in abdominal aortic aneurysms in humans. Arterioscler Thromb Vasc Biol. 2013;33(2):431–8. doi: 10.1161/ATVBAHA.112.300663. [DOI] [PubMed] [Google Scholar]
- 41.Wolfs IM, Donners MM, de Winther MP. Differentiation factors and cytokines in the atherosclerotic plaque micro-environment as a trigger for macrophage polarisation. Thromb Haemost. 2011;106(5):763–71. doi: 10.1160/TH11-05-0320. [DOI] [PubMed] [Google Scholar]
- 42.Lee KN, Kritchevsky D, Pariza MW. Conjugated linoleic acid and atherosclerosis in rabbits. Atherosclerosis. 1994;108(1):19–25. doi: 10.1016/0021-9150(94)90034-5. [DOI] [PubMed] [Google Scholar]
- 43.Kostogrys RB, Franczyk-Żarów M, Maślak E, Gajda M, Mateuszuk Ł, Chłopicki S. Effects of margarine supplemented with t10c12 and C9T11 CLA on atherosclerosis and steatosis in apoE/LDLR −/− mice. J Nutr Health Aging. 2012;16(5):482–90. doi: 10.1007/s12603-011-0354-4. [DOI] [PubMed] [Google Scholar]
- 44.Moya-Camarena SY, Vanden Heuvel JP, Blanchard SG, Leesnitzer LA, Belury MA. Conjugated linoleic acid is a potent naturally occurring ligand and activator of PPARalpha. J Lipid Res. 1999;40(8):1426–33. [PubMed] [Google Scholar]
- 45.Stachowska E, Kijowski J, Dziedziejko V, Siennicka A, Chlubek D. Conjugated linoleic acid regulates phosphorylation of PPARgamma by modulation of ERK 1/2 and p38 signaling in human macrophages/fatty acid-laden macrophages. J Agric Food Chem. 2011;59(21):11846–52. doi: 10.1021/jf2014233. [DOI] [PubMed] [Google Scholar]
- 46.Fleetwood AJ, Dinh H, Cook AD, Hertzog PJ, Hamilton JA. GM-CSF- and M-CSF-dependent macrophage phenotypes display differential dependence on type I interferon signaling. J Leukoc Biol. 2009;86(2):411–21. doi: 10.1189/jlb.1108702. [DOI] [PubMed] [Google Scholar]
- 47.Sierra-Filardi E, Vega MA, Sánchez-Mateos P, Corbí AL, Puig-Kröger A. Heme Oxygenase-1 expression in M-CSF-polarized M2 macrophages contributes to LPS-induced IL-10 release. Immunobiology. 2010;215(9–10):788–95. doi: 10.1016/j.imbio.2010.05.020. [DOI] [PubMed] [Google Scholar]
- 48.Estruch M, Bancells C, Beloki L, Sanchez-Quesada JL, Ordóñez-Llanos J, Benitez S. CD14 and TLR4 mediate cytokine release promoted by electronegative LDL in monocytes. Atherosclerosis. 2013;229(2):356–62. doi: 10.1016/j.atherosclerosis.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 49.Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. 2002;277(51):49982–8. doi: 10.1074/jbc.M209649200. [DOI] [PubMed] [Google Scholar]
- 50.van der Kooij MA, Morand OH, Kempen HJ, van Berkel TJ. Decrease in scavenger receptor expression in human monocyte-derived macrophages treated with granulocyte macrophage colony-stimulating factor. Arterioscler Thromb Vasc Biol. 1996;16(1):106–14. doi: 10.1161/01.ATV.16.1.106. [DOI] [PubMed] [Google Scholar]
- 51.Tall AR, Costet P, Wang N. Regulation and mechanisms of macrophage cholesterol efflux. J Clin Invest. 2002;110(7):899–904. doi: 10.1172/JCI0216391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dai Y, Su W, Ding Z, Wang X, Mercanti F, Chen M, et al. Regulation of MSR-1 and CD36 in macrophages by LOX-1 mediated through PPAR-gamma. Biochem Biophys Res Commun. 2013;431(3):496–500. doi: 10.1016/j.bbrc.2013.01.029. [DOI] [PubMed] [Google Scholar]
- 53.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, Evans RM. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nat Med. 2001;7(1):48–52. doi: 10.1038/83336. [DOI] [PubMed] [Google Scholar]
- 54.Chawla A, Boisvert WA, Lee CH, Laffitte BA, Barak Y, Joseph SB, et al. A PPAR gamma-LXR-ABCA1 pathway in macrophages is involved in cholesterol efflux and atherogenesis. Mol Cell. 2001;7(1):161–71. doi: 10.1016/S1097-2765(01)00164-2. [DOI] [PubMed] [Google Scholar]
- 55.Panzenboeck U, Kratzer I, Sovic A, Wintersperger A, Bernhart E, Hammer A, et al. Regulatory effects of synthetic liver X receptor- and peroxisome-proliferator activated receptor agonists on sterol transport pathways in polarized cerebrovascular endothelial cells. Int J Biochem Cell Biol. 2006;38(8):1314–29. doi: 10.1016/j.biocel.2006.01.013. [DOI] [PubMed] [Google Scholar]
- 56.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93(2):229–40. doi: 10.1016/S0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 57.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93(2):241–52. doi: 10.1016/S0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 58.Lang R, Rutschman RL, Greaves DR, Murray PJ. Autocrine deactivation of macrophages in transgenic mice constitutively overexpressing IL-10 under control of the human CD68 promoter. J Immunol. 2002;168(7):3402–11. doi: 10.4049/jimmunol.168.7.3402. [DOI] [PubMed] [Google Scholar]
- 59.Skrzeczynska-Moncznik J, Bzowska M, Loseke S, Grage-Griebenow E, Zembala M, Pryjma J. Peripheral blood CD14high CD16+ monocytes are main producers of IL-10. Scand J Immunol. 2008;67(2):152–9. doi: 10.1111/j.1365-3083.2007.02051.x. [DOI] [PubMed] [Google Scholar]
- 60.Walcher D, Kümmel A, Kehrle B, Bach H, Grüb M, Durst R, et al. LXR activation reduces proinflammatory cytokine expression in human CD4-positive lymphocytes. Arterioscler Thromb Vasc Biol. 2006;26(5):1022–8. doi: 10.1161/01.ATV.0000210278.67076.8f. [DOI] [PubMed] [Google Scholar]
- 61.Li CC, Hou YC, Yeh CL, Yeh SL. Effects of eicosapentaenoic acid and docosahexaenoic acid on prostate cancer cell migration and invasion induced by tumor-associated macrophages. PLoS One. 2014;9(6):e99630. doi: 10.1371/journal.pone.0099630. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 62.Grewal T, Priceputu E, Davignon J, Bernier L. Identification of a gamma-interferon-responsive element in the promoter of the human macrophage scavenger receptor A gene. Arterioscler Thromb Vasc Biol. 2001;21(5):825–31. doi: 10.1161/01.ATV.21.5.825. [DOI] [PubMed] [Google Scholar]
- 63.Panousis CG, Zuckerman SH. Interferon-gamma induces downregulation of Tangier disease gene (ATP-binding-cassette transporter 1) in macrophage-derived foam cells. Arterioscler Thromb Vasc Biol. 2000;20(6):1565–71. doi: 10.1161/01.ATV.20.6.1565. [DOI] [PubMed] [Google Scholar]
- 64.Reiss AB, Patel CA, Rahman MM, Chan ES, Hasneen K, Montesinos MC, et al. Interferon-gamma impedes reverse cholesterol transport and promotes foam cell transformation in THP-1 human monocytes/macrophages. Med Sci Monit. 2004;10(11):BR420–5. [PubMed] [Google Scholar]