

Abstract
We have developed a new strategy for antiviral peptide discovery by using lyssaviruses (rabies virus and rabies-related viruses) as models. Based on the mimicry of natural bioactive peptides, two genetically encoded combinatorial peptide libraries composed of intrinsically constrained peptides (coactamers) were designed. Proteomic knowledge concerning the functional network of interactions in the lyssavirus transcription-replication complex highlights the phosphoprotein (P) as a prime target for inhibitors of viral replication. We present an integrated, sequential drug discovery process for selection of peptides with antiviral activity directed against the P. Our approach combines (i) an exhaustive two-hybrid selection of peptides binding two phylogenetically divergent lyssavirus P's, (ii) a functional analysis of protein interaction inhibition in a viral reverse genetic assay, coupled with a physical analysis of viral nucleoprotein-P complex by protein chip mass spectrometry, and (iii) an assay for inhibition of lyssavirus infection in mammalian cells. The validity of this strategy was demonstrated by the identification of four peptides exhibiting an efficient antiviral activity. Our work highlights the importance of P as a target in anti-rabies virus drug discovery. Furthermore, the screening strategy and the coactamer libraries presented in this report could be considered, respectively, a general target validation strategy and a potential source of biologically active peptides which could also help to design pharmacologically active peptide-mimicking molecules. The strategy described here is easily applicable to other pathogens.
Our laboratory recently underscored the crucial role played by the phosphoprotein (P) in the formation of the rabies virus transcription-replication complex by using yeast two-hybrid and viral reverse genetic approaches (12). As a constituent of the viral ribonucleoprotein (RNP) complex, the P interacts with two other viral components, the nucleoprotein (N), which tightly enwraps the viral RNA genome, and the RNA-dependent RNA polymerase (L). P also binds a cellular protein implicated in retrograde transport (Dynein LC8), strongly suggesting that interfering with P functions could have deleterious effects on the viral cycle (11, 18). Thus, the pivotal roles played by P make it a prime target for inhibitors of viral transcription and replication.
A recent World Health Organization report estimated that between 40,000 to 70,000 deaths from rabies encephalomyelitis occur every year (World Health Organization Fact Sheet No. 99, 2001), primarily due to the absence of an optimal postexposure treatment protocol for human vaccination and serotherapy (22). Rabies virus immunoglobulins of human or equine origin are in short supply worldwide and completely unaffordable in many developing countries. It is therefore urgent to find alternative solutions to treat the initial phase of rabies virus exposure. Local treatment with a virucidal drug would solve this problem, and development of anti-rabies virus peptides is of great interest in this respect.
The key aspects of antiviral drug development are, successively, as follows: the selection of a target and its validation, the development of screening assays, and finally, the preliminary identification of lead compounds. Numerous studies have demonstrated the interest of combinatorial approaches in the identification of short peptide sequences able to bind proteins (5, 6, 13, 24). These studies have usually been performed with peptide aptamers (peptamers), a distinct class of molecules characterized by constrained peptidic loops displayed by a carrier protein (5). These molecules were used to counteract the conformational flexibility of linear peptides, which results otherwise in poor target binding. However, the fact that bioavailability of peptamers is determined by their scaffold-displaying protein is a critical limitation for the pharmacological potential of such peptides. In contrast, certain peptides found in nature are among the most pharmacologically active small molecules. Natural selection has favored a structurally sophisticated diversity, unified around a common characteristic: the presence of a constrained structure which decreases the conformational flexibility and thereby provides an improvement in specificity and stability. Among such autoconstrained peptides, toxins from predatory cone snail venoms (disulfide-constrained conotoxins) and insect antimicrobial proline-rich peptides (apidaecins and lebocins) can be considered the paradigms (1, 2, 10, 16, 17, 23).
The integrated antiviral drug discovery strategy developed here is based on the mimicry of these natural autoconstrained peptides. We have designed two coactamer libraries (from Latin coactus, constraint), that are rich in either cysteine or proline. The cysteine backbone mimics conotoxins-μ from Conus geographus (16, 17, 23), and the proline backbone partially overlaps with lebocin 1 and 2 from Bombyx mori (1, 10).
Both genetically encoded combinatorial peptide libraries were screened by using a yeast two-hybrid system to identify peptides binding with high affinity to the P's from two highly divergent lyssaviruses (rabies Pasteur virus [PV] and Mokola virus [Mok]) (15). To make the most exhaustive selection of P binders, the sequences of P-PV and P-Mok binding peptides were clustered into subfamilies by using a multiple-sequence alignment program. Based on these identified families, which cover a total of 755 binders, 29 representative peptides were finally selected. These interacting peptides were then submitted to a functional screening step by designing a reverse genetic viral transcription-replication interference assay. In addition, the peptide effect on the viral RNP complex formation was analyzed by surface-enhanced laser desorption ionization-time of flight (mass spectrometry) [SELDI-TOF (MS)] analysis. Finally, peptides emerging as interactors and functional inhibitors of the transcription-replication complex were tested for their capacity to inhibit PV infection of mammalian cells. A flowchart representation summarizing the sequential peptide selection process is shown in Fig. 1.
FIG. 1.
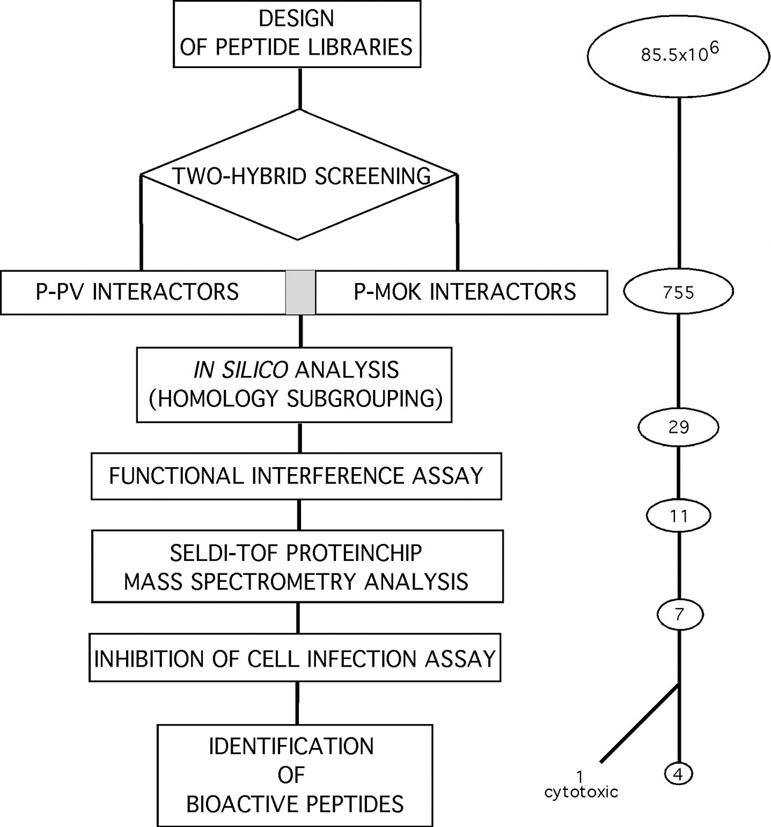
Flow chart of the peptide selection process for antiviral activity juxtaposed with the corresponding numbers of selected peptides resulting from an exhaustive two-hybrid screening of 85.5 × 106 clones.
MATERIALS AND METHODS
Combinatorial peptide libraries.
Two long oligonucleotides encoding Cys and Pro constrained peptide libraries were synthesized. In both cases, 20 random codons are incorporated in a framework generated by constant cysteine or proline codons, flanked by sequences containing NcoI and BglII restriction sites for insertion in the vector pACTII (Clontech). Oligonucleotide C5, encoding cysteine-constrained peptides, was 5′-GCGCATGCCATGGAGGGGATCCGATGT(NNK)2TGT(NNK)5TGT(NNK)6TGT(NNK)5TGT(NNK)2TGTTAATAAGATCTCGCGTG-3′. Oligonucleotide C8, encoding proline-constrained peptides, was 5′-GCGCATGCCATGGAGGGGATCCGACCACCT(NNK)5CCT(NNK)5CCTCCACCT(NNK)5CCT(NNK)5CCACCTTAATAAGATCTCGCGTG-3′.
Oligonucleotides were synthesized on an Applied Biosystems 392 synthesizer and contained triplets of the sequence NNK (where N is G, A, T, or C and K is G or C), which encodes all 20 amino acids but results in only one stop codon. To avoid synthesis bias, introduced by the chemical reactivity of each phosphoramidite, the N mix contained a final dN-CE phosphoramidite (Glen Research) concentration of 0.1 mmol/ml and a ratio of 3 dA/3 dC/2 dG/2 dT. In contrast, the K mix contained equal proportions of dG and dC.
Second-strand synthesis was performed by PCR with the primers 5′-GCGCATGCCATGGAGGGGATCC-3′ and 5′-CACGCGAGATCTTATTAA-3′. The yeast strain Y187 (Clontech) was transformed with plasmid DNA by the lithium acetate procedure with plasmid DNA from 1 × 107 and 3 × 107 primary Escherichia coli transformants, respectively, to give 2.1 × 106 (cysteine coactamers) or 3.5 × 106 (proline coactamers) individual yeast colonies, which were collected, pooled, and stored at −80°C for each library (7).
Cloning procedures.
Two-hybrid bait plasmids containing either the complete open reading frame of P-PV or P-Mok fused to the Gal4p-DNA binding domain were made by inserting the respective PCR-amplified fragments into the pAS2ΔΔ vector (laboratoire du métabolisme des ARNs, Institut Pasteur) as described previously (7). Cloning junctions and the complete open reading frames were sequenced on an ABI 377 automatic sequencer (Applied Biosystems). High-throughput cloning of PCR products encoding selected peptides was performed by recombination cloning technology with the Gateway system (Invitrogen) as described previously (21). Plasmids pDEST53 (Invitrogen) and pEGFP-C1 (Clontech) previously modified with the Gateway rf cassette were used as cloning vectors, respectively, for T7-driven peptide expression and mammalian expression of green fluorescent protein (GFP)-fused peptides. All plasmids were amplified in the E. coli strain DH5α and purified by chromatography on QIAGEN columns.
Yeast two-hybrid screening procedure.
Two-hybrid screens were performed by using a cell-to-cell mating protocol (7). Saturated screens of libraries were performed to ensure total coverage of libraries. For each bait, a test screen was performed to optimize the screening conditions. The selectivity of the His3 reporter gene was modulated with 3-aminotriazole (Sigma) to obtain a maximum of 285 histidine-positive clones for 10 million diploids screened. For all selected clones, lacZ activity was measured in a 96-well plate luminometric assay (Tropix). Inserts of all positive clones were amplified by PCR (7) and then sequenced on an ABI 3700 automatic sequencer (Applied Biosystems).
Bioinformatic analyses.
Screening results from the Cys and Pro libraries were analyzed separately. Amino acid sequences of selected P-PV and P-Mok binding peptides were compared and aligned. The clustering of these sequences into subfamilies was calculated and displayed with neighbor-joining and unweighted pair group methods with arithmetic mean dendrograms by using the Clustalw and Jalview programs (http://www.compbio.dundee.ac.uk/).
Viral transcription-replication interference assay.
To measure the effect of peptide coexpression on the formation of the functional rabies virus transcription-replication complex, we used the viral reverse genetic assay as described previously (12). T7-driven pDEST53-peptide constructs were cotransfected (1 μg) with the viral components (N, P, and L genes and luciferase negative-strand minigenomic RNA). Each of the 29 pDEST53-peptide constructs was separately tested in duplicate. Interference in the functionality of the viral complex was quantified by measuring the amount of luciferase activity which is related to the remaining transcriptional activity of the RNP. Luciferase expression was measured with a Berthold luminometer by injecting 100 μl of luciferase assay reagent (E1501; Promega) into 10 μl of each centrifuged cellular extract and counting for 10 s.
SELDI-TOF ProteinChip MS analysis.
Anti-Flag M2 monoclonal antibody (1.5 μg M2 antibody; Sigma) was covalently cross-linked to preactivated ProteinChip arrays (PS20; Ciphergen) by incubation in a humidity chamber at room temperature for 1 h. Non-cross-linked sites were inactivated twice with 4 μl of 1 M ethanolamine-HCl (pH 8.0) for 15 min. The PS20 chip array was then assembled with a loading device (Bioprocessor) to wash each spot twice for 5 min with 300 μl of 0.5% Triton X-100 in phosphate-buffered saline (PBS) followed by two washes with PBS (300 μl).
After clarification by centrifugation at 3,900 × g rpm (3 min), 200-μl aliquots of a 48-h posttransfection cellular lysate, derived from a classical viral transcription-replication interference assay where P was replaced by flagged P, were spotted on the M2 antibody modified chip array and incubated at room temperature with gentle agitation for 2 h. The whole ProteinChip array was washed with PBS-0.1% Triton X-100 (300 μl) and air dried after a brief wash with 5 mM HEPES (pH 7.5) (300 μl). Then, to facilitate the ionization process, two successive volumes of 0.5 μl of a saturated solution of sinapinic acid (3,5-dimethoxy-4-hydroxycinnamic acid; Fluka) in 50% acetonitrile-0.5% trifluoroacetic acid were spotted on the array and allowed to air dry.
Analysis of the ProteinChip array was carried out in a PBS II mass reader (Ciphergen Biosystems, Inc). The data of each spot were averaged from 240 UV laser shots (intensity of 250) and analyzed by an automated data collection in positive mode and with an external calibration by using Ciphergen's standards (14).
Determination of inhibitory effect of peptides on cell infection by rabies virus.
BSR cells were cultured in Labtek chambers (15,000 cells per chamber) for 24 h in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum at 37°C. Cells were transfected with 2 μg of peptide-encoding plasmid with FuGENE (Roche Diagnostics) according to the manufacturer's protocol. At 6 h posttransfection, cell monolayers were washed twice with DMEM and then infected with rabies virus diluted in DMEM (PV strain, 5 PFU/cell). After incubation for 1 h, the medium was discarded, and cells were washed with DMEM and then incubated at 37°C and 5% CO2 for 24 h in DMEM supplemented with 5% fetal bovine serum.
Infected cells were fixed with PBS-4% paraformaldehyde and permeabilized at 0°C with 80% acetone for 15 min. Rabies virus RNP generated by the infection were then detected by incubation of fixed cells with rabbit anti-RNP serum (incubation for 1 h at 37°C) followed by incubation with Texas Red anti-rabbit conjugate under the same experimental conditions. The detection of infected and transfected cells with the control GFP fusion expression vector was determined by immunofluorescence analysis (LEICA DMRB microscope). Under these conditions, 75 to 80% of cells were found infected, whereas 30 to 50% of cells were GFP positive, depending on the plasmid used.
The inhibition percentage of transfected cells expressing the peptide-GFP fusion was calculated as follows: [1 − (percentage of peptide-GFP rabies virus-infected cells/percentage of control-GFP rabies virus-infected cells)] × 100. For each duplicate, 50 GFP-positive cells were examined for the presence of rabies virus RNP.
RESULTS
Design of coactamer libraries.
Two libraries were constructed to direct the synthesis in yeast of various intrinsically constrained peptides fused to the Gal4p activation domain. The peptide-encoding libraries based on the mimicry of natural bioactive peptides share the presence of an invariable backbone of either cysteine or proline residues delimiting hypervariable peptide loops. In each of our libraries, 20 amino acid residues are hypervariable within a peptide total length of either 26 or 29 amino acid residues for the cysteine and proline libraries, respectively. The cysteine coactamer library sequence was C-X2-C-X5-C-X6-C-X5-C-X2-C, and the proline coactamer library sequence was PP-X5-P-X5-PPP-X5-P-X5-PP. In both sequences, X denotes a randomized position where an NNK degenerate DNA codon encodes all 20 natural amino acids. Moreover, the cysteine-rich backbone was designed to fit to a typical Cys2/Cys2 zinc finger motif which would favor organometallic complex formation and potentially increase the structural diversity of the cysteine coactamer library. The cysteine and proline libraries have an estimated diversity of 1 × 107 and 3 × 107 individual random peptides. Sequencing of 20 randomly chosen clones in each library showed 100% insertion rates. Respectively, 37 and 47% of the clones in the cysteine and proline libraries coded for full-length peptides. Other clones corresponded to shortened versions, resulting from the occurrence of frameshifts and termination codons. Yeast strain Y187 was transformed with each library, generating two yeast coactamer libraries with estimated individual complexities of 2.1 × 106 (cysteine coactamers) and 3.5 × 106 (proline coactamers).
Identification of P-binding peptides.
We screened the two coactamer libraries with two highly divergent but functionally interchangeable P's (12) derived from two phylogenetically distant lyssaviruses (PV and Mok) (15). Thus, selectivity of the screening procedure could be reinforced by cross-selection for interactors common to both baits. The results of these screening assays are presented in Table 1. The saturation of the screens was confirmed by the redundancy in the peptide sequences (see peptide occurrence in Table 2). The number of positive clones per million interactions tested shows that the P-Mok bait has selected six times more prey than the P-PV bait with both libraries (Table 1). The screening of both libraries with a Trp+ empty vector (pFL 39) (8) allowed us to discard seven false-positive prey selected in the cysteine constraint library with P-PV and P-Mok (data not shown). No such overlap was observed for the proline coactamer library screening.
TABLE 1.
Yeast two-hybrid screening results
| Bait | Library | No. of selected clones | No. of tested interactions (106) | No. of selected clones/106 tested interactions |
|---|---|---|---|---|
| P-PV | Cys | 80 | 21 | 3.8 |
| P-PV | Pro | 105 | 38 | 2.8 |
| P-Mok | Cys | 285 | 11.5 | 25 |
| P-Mok | Pro | 285 | 15 | 19 |
| Total | 755 | 85.5 |
TABLE 2.
Characteristics of selected peptides
| Library | Peptide | Sequence | No. of occurrences | Specificity |
|---|---|---|---|---|
| Cys | C1 | CKFCYGSAQCPTFLFIVRLLRFVWV | 7 | PV, Mok |
| C2 | CTMCRYQQNCFTRRLIVGGMLLVFV | 9 | PV, Mok | |
| C3 | CYSCPCERRCHKIARGLLILRSVLF | 19 | PV, Mok | |
| C4 | CQRCGWETGVGVSGFLVRILRFVVL | 14 | PV, Mok | |
| C5 | CTQCCAPSTCLNYRIFVGLLRFVVI | 15 | PV, Mok | |
| C6 | CDSCERCWYVWLILLRVRLRLLVSL | 5 | PV, Mok | |
| C7 | CKSCDTRCTCLRRRLRVGVGLPCMGC | 4 | PV, Mok | |
| C8 | CRCCELKSLCPTLMRVVRLLGLVLL | 2 | PV, Mok | |
| C9 | CLCCDKVRTCRRLLGLVMVLSVVRC | 3 | PV, Mok | |
| C10 | CGECGGGHIVGRFCMVVRFLRLVFI | 4 | PV, Mok | |
| C26 | CVTCKSTVLCDKMQHPCRRGPRCISC | 1 | PV | |
| C27 | CGRCLQRACCKYCRLKCRLILFVIF | 5 | PV, Mok | |
| Pro | P11 | PPIPIPDPPQRNRPPPRWFISLMVIRIH | 6 | PV, Mok |
| P12 | PPRLLDSPEVMVILHLGFRIGLVRLWIH | 5 | PV, Mok | |
| P13 | PPARSSPPMPPNLPPLRRRIILLRFLFH | 2 | PV, Mok | |
| P14 | PPPLPYGPNRNGEPHLRVLLRLLCIRLH | 3 | PV, Mok | |
| P15 | PPRTTPIPHLDVSLHLLILRILRVRVH | 6 | PV, Mok | |
| P16 | PPDVHTPPHALWRLHLSLRVCLVRMWIH | 5 | PV, Mok | |
| P17 | PPTSPLLPTVNLRPPPIIIVFLLRVWFH | 3 | PV, Mok | |
| P18 | PPLYGRDPTTRRMPHLLLRCCLLRLVVH | 2 | PV, Mok | |
| P19 | PPDQTTYPSAECPPPPLVSILLIGLWLH | 3 | PV, Mok | |
| P20 | PPRGAHRPNSTVVLHLVIRLCLLRFVVH | 4 | PV, Mok | |
| P21 | PPDTSLLPPVGLHLVVRLFLLRLSVH | 25 | PV, Mok | |
| P22 | PPGAPPAPFRTHTPPPRMVIVLIRVWCH | 3 | PV, Mok | |
| P23 | PPGAPPQPDSVCELHLLCVLRLLVIRIH | 2 | PV, Mok | |
| P24 | PPSHSFRPESLERLHLLRRVLLLMRIVH | 4 | PV, Mok | |
| P25 | PPCYERMPRRLIRPPPLLSVLLILRLCH | 7 | PV, Mok | |
| P28 | PPLFEDTPMVNSIPPLRVRLFLLRLVFH | 3 | PV, Mok | |
| P29 | PPRGTETPQRCRRLHLVEMLCLVRVVFH | 3 | PV, Mok |
Bioinformatic selection of peptides.
In silico analysis with multialignment programs (ClustalW with Jalview display) was used to compare the sequence data of interacting peptides selected by both P-PV and P-Mok baits from each coactamer library screen. A total of 755 selected clones were analyzed (Table 1). The preliminary output obtained by computer program analysis revealed subfamilies of peptides characterized by sequence similarity. Based on this multiple-sequence comparison, to maximize the sequence diversity of the peptides retained for further experiments, one member of each identified subfamily of similarity was selected. This selection also took into account the most frequently occurring peptides and thus the total representativity of each subfamily. Thus, 11 Cys and 17 Pro peptides were selected. In addition, one Cys peptide monospecific for P-PV was also chosen for further analysis.
Functional test of viral transcription-replication complex formation interference assay.
Each of the 29 selected peptides was subcloned in a T7 mammalian expression vector and then tested in a reverse genetic viral RNP reconstitution assay. This assay allows a quantitative analysis of the formation of a functional viral RNP by luciferase activity measurement (12). By coexpressing each individual peptide in this assay, we measured its capacity to interfere with the viral transcription-replication complex (Fig. 2). The 100% value corresponds to the control experiment realized with the peptide expression vector without the peptide-encoding sequence (empty vector), giving an average luciferase activity of about 106 arbitrary units. The background was below 100 arbitrary units.
FIG. 2.

Interference effects of selected peptides on the transcription-replication activity of rabies virus PV RNP complex (shown as means ± standard errors [error bars]). The luciferase activity was indicated as 100% in the control experiment with the empty peptide expression vector (y axis). One hundred percent corresponds to an average luciferase activity of 106 units. The experiment was performed in duplicate.
In this assay, the majority of the selected peptides was fully active, except for P11, P19, and C26. The latter was chosen as an internal control because of its monospecificity and single occurrence. This underlines the utility of a screening strategy with two different baits in yeast two-hybrid analysis. This approach provided functional data that could be superposed on the initial selection procedure and allowed us to select the 10 most active peptides plus a weak one as a control (C26). The selected peptides were submitted to a biophysical analysis of RNP complex destabilization.
SELDI-TOF ProteinChip MS analysis.
To evaluate whether functional inhibition of the RNP complex could be correlated with an alteration of the N-P interaction, we developed a method by using a Ciphergen Biosystems mass spectrometer which associates ProteinChip technology with SELDI-TOF (MS). Briefly, this assay consists of a classical coimmunoprecipitation experiment in which the anti-Flag M2 monoclonal antibody is covalently cross-linked with an epoxy preactivated ProteinChip array (PS20) instead of using protein A-Sepharose beads as the immunoabsorbant. Protein extracts are prepared from cells cotransfected with plasmids coding for N and L, the rabies virus minigenome, and a flagged P-PV, which works as a hook allowing RNP complex retention and subsequent analysis by classical SELDI-TOF (MS). Using this approach, alterations in the protein profiling of the rabies virus N-P complex in the presence of coexpressed peptide can then be measured. By comparison with a control experiment without peptide (B spectrum) (Fig. 3), the destabilization of the N-P interaction is correlated to an increase in the P/N peak intensity ratio (Table 3). Characteristic spectra observed with different peptides are shown in Fig. 3. The C2 and P16 peptides clearly destabilize the N-P interaction, whereas the C9 or C26 peptide had little or no effect on the N-P complex. As the N-P interaction plays a crucial role in the functionality of the viral replication-transcription complex, N-P interaction destabilization was used as a criterion to select six interfering peptides characterized by their increasing effects, two other peptides were kept as negative controls (C26 and C27) (Table 3). As peptides P15 and P16 displayed similar SELDI-TOF (MS) profiles, only P16 was kept. Quantification of P and peptide expression levels in the mammalian cells used in the interference assay or SELDI-TOF experiments show a peptide/P ratio of 1, with a 30 nM concentration of each in the various cell extracts (data not shown).
FIG. 3.

Representative protein profiling of rabies virus N/P complex by ProteinChip SELDI-TOF (MS) analysis. Panels A and B correspond to control (Ctl) experiments with untransfected and RNP-expressing cells, respectively, and panels C, D, E, and F show results from peptide experiments.
TABLE 3.
Evaluation of N-P interaction destabilization by ProteinChip SELDI-TOF(MS)
| Peptide | P/N peak intensity ratio |
|---|---|
| None (RNP control) | 1.05 |
| C2 | 1.70 |
| C6 | 1.64 |
| C8 | 1.52 |
| C9 | 0.82 |
| C10 | 2.53 |
| C26 | 1.09 |
| C27 | 1.00 |
| P15 | 2.47 |
| P16 | 2.49 |
| P23 | 1.08 |
| P29 | 1.60 |
Inhibition of viral replication assay.
To evaluate the antiviral activity of the 8 last selected peptides, they were subcloned into a GFP fusion mammalian expression vector (enhanced GFP-C1; Clontech). The rabies virus inhibition of replication assay consists of the transfection of BSR cells with plasmids encoding the peptides followed by infection with rabies virus (Table 4). One peptide (P29) was discarded because of its cytotoxic activity. Four peptides (C2, C6, C8, and P16) demonstrated a strong antiviral activity. Peptide C10, although slightly less active (57% inhibition), strongly reduced the size and number of virus inclusion bodies. However, this modification of the appearance of infected cells was not taken into consideration for the evaluation of inhibition, resulting in a potential underestimation of total antiviral activity.
TABLE 4.
Inhibition of rabies virus replication in PV rabies-infected BSR cells
| Peptide | % of inhibitiona |
|---|---|
| None (plasmid control) | 0 |
| C2 | 89 |
| C6 | 83 |
| C8 | 78 |
| C10 | 57 |
| C26 | 5 |
| C27 | 22 |
| P16 | 71 |
| P29 | —b |
For percentage determination; see Materials and Methods.
—, cytotoxic.
The inhibitory activity of two peptides (C2 and P16) is illustrated in Fig. 4. Whereas 75 to 80% of cells were infected under the infection conditions used (as measured by RNP production; data not shown), the majority of transfected cells expressing selected peptides (as indicated by GFP expression) were found to be negative for rabies virus RNP expression (Fig. 4A to F), indicating high levels of virus inhibitory activity. In contrast, cells transfected with the control plasmid expressing GFP (control experiment) displayed a normal expression level of rabies virus RNP (Fig. 4G to I) and no inhibition.
FIG. 4.

Inhibitory effect of peptides on infection by rabies virus. BSR cells were transfected with C2 plasmid (A to C), P16 plasmid (D to F), or empty plasmid in a control experiment (G to I). (A, D, and G) GFP fluorescence (note granular inclusions of GFP fusion with peptide C2 or P16); (B, E, and H) rabies virus RNP immunostaining with characteristic perinuclear dots (white arrows indicate transfected cells); (C, F, and I) merged GFP and rabies virus RNP immunostaining with 4′,6′-diamidino-2-phenylindole (DAPI) coloration of cell nuclei. The majority of transfected cells expressing selected peptides (indicated by GFP expression) were found to be negative for rabies virus RNP.
In summary, our results clearly indicate that selected peptides destabilize both the interaction and functionality of the lyssavirus N-P complex, an interference which could be detected in a viral reverse genetic assay, a particle-complex physical analysis, and a cell infection test.
DISCUSSION
The rabies virus P is a main component of the RNP, providing the link between the N-enwrapped genomic RNA and the viral polymerase (3, 4, 12) and playing a role in retrograde transport through its interaction with cytoplasmic dynein (11, 18). All of these data suggest that rabies virus P could be a prime antiviral target. Moreover, rabies virus P was successfully used as a bait in previous two-hybrid screens (11, 12, 18), indicating that the resulting GAL4 binding domain-P fusion folds correctly in yeast cells.
Many studies with peptide aptamers have demonstrated the interest of combinatorial approaches in the identification of short peptide sequences able to mimic the large array of structures that characterize cellular proteomes. To be biologically active, these peptide aptamers (peptamers) require conformational constraints that are induced by their carrier proteins. In contrast to such peptamers, the coactamer concept developed in this study uses intrinsically constrained peptides mimicking natural bioactive peptides. Our approach involves a sequence of selective steps of increasing stringency, going from identification of interactors to functional inhibitors. Of the 755 P-binding peptides first selected in an exhaustive two-hybrid screening, in silico analysis identified subfamilies from which the most representative peptides were chosen by using as selection requirements both their frequency and their ability to bind to two lyssavirus P's (PV and Mok). These two highly divergent P's were previously demonstrated to be functionally interchangeable in a reverse genetic assay (12) and thus were chosen to reinforce the stringency of the selection process. This assumes that the interacting peptides common to both baits were binders to conserved functional regions, given that the 47% identity between the two P's is restricted to functionally important domains (12). Thus, peptides binding to both proteins are likely to interfere more strongly with the viral infectious cycle. This double selection in the two-hybrid procedure was combined with a viral reverse genetic assay consisting of an ex vivo reconstitution of a functional transcription-replication complex. A rather high proportion of peptides selected in this study are biologically active. As previously demonstrated with single-stranded antibody selection, this is likely to be related to the use of an in vivo selection (here, the yeast two-hybrid and the viral reverse genetic assay), which should facilitate the identification of in vivo valuable peptides (20). Peptides emerging as both P-interactors and the strongest transcription-replication inhibitors were studied with ProteinChip MS analysis for their capacity to destabilize the N-P interaction. Only partial destabilization was achieved with inhibitory peptides, which is in good agreement with the peptide/target ratio close to 1 which was observed under our experimental conditions. For the peptides C9 and P23, which were inhibitory in the viral reverse genetic assay but displayed no measurable destabilization of the N-P interaction, it seems likely that they may interfere with interactions, such as that of P and L, that also play a pivotal role in rabies virus RNP complex function. Unfortunately, the size discrepancy between L (220 kDa) and P (35 kDa) invalidates further investigations with ProteinChip MS analysis. Considering the lack of mechanistic evidence for some peptides, we have favored for further analysis only six peptides essentially characterized by their different SELDI-TOF (MS) profiles with increasing effects on N-P destabilization. This highly stringent selection might possibly have resulted in missing some of the peptides that would also be effective in suppressing the viral replication. Alternatively, a more pragmatic approach could also be envisaged, consisting of the direct selection of peptides for their antiviral activities and then of the investigation of the suppressive mechanism, provided that a multiparallel inhibition of infection assay could be designed. In this work, as the ultimate validation of our overall strategy for the identification of antiviral peptides, we have performed an ex vivo inhibition of viral replication assay with rabies virus. This assay constituted the most stringent test and resulted in the identification of four strong rabies virus inhibitory peptides (C2, C6, C8, and P16) whose general features in comparison with control peptides are displayed in Table 5. The overall structural diversity of the four strongest bioactive peptides suggests that a mixture of them could potentiate their therapeutic effect by affecting different P functional domains. Furthermore, these four peptides demonstrated no inhibition of cellular infection with the unrelated human immunodeficiency virus (HIV) type 1, further underscoring the specificity of the selected peptides for the rabies virus targets (data not shown).
TABLE 5.
General features of selected bioactive and control peptides
| Peptide | No. of occurences | % RNP residual luciferase activity | N/P ratio | % Infection inhibition |
|---|---|---|---|---|
| C2 | 9 | 2.1 | 1.70 | 89 |
| C6 | 5 | 2.8 | 1.64 | 83 |
| C8 | 4 | 1.1 | 1.52 | 78 |
| C10 | 4 | 2.1 | 2.53 | 57 |
| C26 | 1 | 24.0 | 1.09 | 5 |
| C27 | 5 | 1.7 | 1.00 | 22 |
| P16 | 5 | 3.7 | 2.49 | 71 |
Interestingly, despite a larger selection of prey in the proline library, cysteine coactamers seem to be more active, suggesting that the presence of cysteine favors constraints inducing greater stability and affinity of interacting peptides. Structure prediction with the PROF algorithm within the PredictProtein program indicated that three peptides (C6, C8, and P16) have an overall propensity to assume an alpha-helical conformation. In contrast, peptide C2, which is the most active, did not exhibit significant helical content, suggesting that maximizing the helical content of a short peptide could constrain it into a rigid helix with no significant increase in inhibitory potency and binding affinity.
Finally, the peptides identified in this report may be considered molecular starting points that are likely to facilitate the design of nonpeptide mimics chemically engineered via scaffolding approaches. It should be noted that proline-rich peptides could facilitate the development of peptidomimetic approaches, which frequently use alpha-substituted proline analogues. Alternatively, these peptides, after a tailored mutagenesis to generate super binders and the addition of a protein transduction domain such as HIV TAT (19) or Antennapedia peptide, could be directly submitted to pharmacological tests. Indeed, the use of peptide-mediated transduction, through arginine-rich segments (9), is currently being investigated for therapeutic use in animals.
Interestingly, in preliminary experiments for establishing the cysteine-rich peptide library in yeast cells, we selected peptides able to bind DNA on the His3 promoter region (data not shown). Thus, the cysteine coactamer library may also be useful for the identification of a specific DNA binding peptide which could be screened in yeast one-hybrid experiments. For DNA viruses, the identification of specific binders of viral regulatory regions represents an interesting potential antiviral strategy which could complement the targeting of viral proteins.
In summary, this work shows that coactamer libraries are powerful tools for target validation as exemplified here in anti-rabies virus drug discovery with the viral P as a target. The resulting libraries are also valuable sources of novel molecules with implications for the development of a new type of anti-rabies virus treatment. Moreover, this conceptual approach and its related tools are applicable to other infectious diseases and in any context of pathway-based drug discovery.
Acknowledgments
We thank Dinh Tam Huynh, Valérie Huteau, and Catherine Gouyette for the special care given to synthesizing the long random oligonucleotides and Charles Roth and Andrew Borman for helpful discussions. We are indebted to Luc Selig for his important contribution to this work, and we thank Stephane Emiliani from INSERM U567 (Institut Cochin) for performing HIV type 1 cellular infection tests.
E.R. was a recipient of an M.E.N.R.T. fellowship.
REFERENCES
- 1.Bulet, P., C. Hetru, J. L. Dimarcq, and D. Hoffmann. 1999. Antimicrobial peptides in insects; structure and function. Dev. Comp. Immunol. 23:329-344. [DOI] [PubMed] [Google Scholar]
- 2.Casteels, P., J. Romagnolo, M. Castle, K. Casteels-Josson, H. Erdjument-Bromage, and P. Tempst. 1994. Biodiversity of Apidaecin-type peptide antibiotics. Prospects of manipulating the antibacterial spectrum and combating acquired resistance. J. Biol. Chem. 269:26107-26115. [PubMed] [Google Scholar]
- 3.Chenik, M., K. Chebli, Y. Gaudin, and D. Blondel. 1994. In vivo interaction of rabies virus phosphoprotein (P) and nucleoprotein (N): existence of two N-binding sites on P protein. J. Gen. Virol. 75:2889-2896. [DOI] [PubMed] [Google Scholar]
- 4.Chenik, M., M. Schnell, K. K. Conzelmann, and D. Blondel. 1998. Mapping the interacting domains between the rabies virus polymerase and phosphoprotein. J. Virol. 72:1925-1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Colas, P., B. Cohen, T. Jessen, I. Grishina, J. McCoy, and R. Brent. 1996. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature 380:548-550. [DOI] [PubMed] [Google Scholar]
- 6.Deshayes, K., M. L. Schaffer, N. J. Skelton, G. R. Nakamura, S. Kadkhodayan, and S. S. Sidhu. 2002. Rapid identification of small binding motifs with high-throughput phage display: discovery of peptidic antagonists of IGF-1 function. Chem. Biol. 9:495-505. [DOI] [PubMed] [Google Scholar]
- 7.Fromont-Racine, M., J. C. Rain, and P. Legrain. 2002. Building protein-protein networks by two-hybrid mating strategy. Methods Enzymol. 350:513-524. [DOI] [PubMed] [Google Scholar]
- 8.Fromont-Racine, M., J. C. Rain, and P. Legrain. 1997. Toward a functional analysis of the yeast genome through exhaustive two-hybrid screens. Nat. Genet. 16:277-282. [DOI] [PubMed] [Google Scholar]
- 9.Futaki, S., T. Suzuki, W. Ohashi, T. Yagami, S. Tanaka, K. Ueda, and Y. Sugiura. 2001. Arginine-rich peptides. An abundant source of membrane-permeable peptides having potential as carriers for intracellular protein delivery. J. Biol. Chem. 276:5836-5840. [DOI] [PubMed] [Google Scholar]
- 10.Hara, S., and M. Yamakawa. 1995. A novel antibacterial peptide family isolated from the silkworm, Bombyx mori. Biochem. J. 310:651-656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacob, Y., H. Badrane, P. E. Ceccaldi, and N. Tordo. 2000. Cytoplasmic dynein LC8 interacts with lyssavirus phosphoprotein. J. Virol. 74:10217-10222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jacob, Y., E. Real, and N. Tordo. 2001. Functional interaction map of lyssavirus phosphoprotein: identification of the minimal transcription domains. J. Virol. 75:9613-9622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolonin, M. G., and R. L. Finley, Jr. 1998. Targeting cyclin-dependent kinases in Drosophila with peptide aptamers. Proc. Natl. Acad. Sci. USA 95:14266-14271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Merchant, M., and S. R. Weinberger. 2000. Recent advancements in surface-enhanced laser desorption/ionization-time of flight-mass spectrometry. Electrophoresis 21:1164-1177. [DOI] [PubMed] [Google Scholar]
- 15.Nadin-Davis, S. A., M. Abdel-Malik, J. Armstrong, and A. I. Wandeler. 2002. Lyssavirus P gene characterisation provides insights into the phylogeny of the genus and identifies structural similarities and diversity within the encoded phosphoprotein. Virology 298:286-305. [DOI] [PubMed] [Google Scholar]
- 16.Olivera, B. M., D. R. Hillyard, M. Marsh, and D. Yoshikami. 1995. Combinatorial peptide libraries in drug design: lessons from venomous cone snails. Trends Biotechnol. 13:422-426. [DOI] [PubMed] [Google Scholar]
- 17.Olivera, B. M., J. Rivier, C. Clark, C. A. Ramilo, G. P. Corpuz, F. C. Abogadie, E. E. Mena, S. R. Woodward, D. R. Hillyard, and L. J. Cruz. 1990. Diversity of conus neuropeptides. Science 249:257-263. [DOI] [PubMed] [Google Scholar]
- 18.Raux, H., A. Flamand, and D. Blondel. 2000. Interaction of the rabies virus P protein with the LC8 dynein light chain. J. Virol. 74:10212-10216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schwarze, S. R., A. Ho, A. Vocero-Akbani, and S. F. Dowdy. 1999. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science 285:1569-1572. [DOI] [PubMed] [Google Scholar]
- 20.Visintin, M., E. Tse, H. Axelson, T. H. Rabbitts, and A. Cattaneo. 1999. Selection of antibodies for intracellular function using a two-hybrid in vivo system. Proc. Natl. Acad. Sci. USA 96:11723-11728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walhout, A. J. M., R. Sordella, X. Lu, J. L. Hartley, G. F. Temple, M. A. Brasch, N. Thierry-Mieg, and M. Vidal. 2000. Protein interaction mapping in C. elegans using proteins involved in vulval development. Science 287:116-122. [DOI] [PubMed] [Google Scholar]
- 22.Wilde, H., P. Khawplod, T. Hemachudha, and V. Sitprija. 2002. Postexposure treatment of rabies infection: can it be done without immunoglobulin? Clin. Infect. Dis. 34:477-480. [DOI] [PubMed] [Google Scholar]
- 23.Woodward, S. R., L. J. Cruz, B. M. Olivera, and D. R. Hillyard. 1990. Constant and hypervariable regions in conotoxin propeptides. EMBO. J. 9:1015-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yang, M., Z. Wu, and S. Fields. 1995. Protein-peptide interactions analyzed with the yeast two-hybrid system. Nucleic Acids Res. 23:1152-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]