

Abstract
Pain is a sensation related to potential or actual damage in some tissue of the body. The mainstay of medical pain therapy remains drugs that have been around for decades, like non-steroidal anti-inflammatory drugs (NSAIDs), or opiates. However, adverse effects of opiates, particularly tolerance, limit their clinical use. Several lines of investigations have shown that systemic (intraperitoneal) administration of NSAIDs induces antinociception with some effects of tolerance. In this review, we report that repeated microinjection of NSAIDs analgin, clodifen, ketorolac and xefocam into the central nucleus of amygdala, the midbrain periaqueductal grey matter and nucleus raphe magnus in the following 4 days result in progressively less antinociception compared to the saline control testing in the tail-flick reflex and hot plate latency tests. Hence, tolerance develops to these drugs and cross-tolerance to morphine in male rats. These findings strongly support the suggestion of endogenous opioid involvement in NSAIDs antinociception and tolerance in the descending pain-control system. Moreover, the periaqueductal grey-rostral ventro-medial part of medulla circuit should be viewed as a pain-modulation system. These data are important for human medicine. In particular, cross-tolerance between non-opioid and opioid analgesics should be important in the clinical setting.
Keywords: Analgesia, antinociception, descending pain modulation, hot plate test, non-opioid tolerance, non-steroidal anti-inflammatory drugs, tail-flick reflex, neural regeneration
Abbreviations
NSAIDs, non-steroidal anti-inflammatory drugs; PAG, periaqueductal grey matter; Ce, central nucleus of amygdale; NRM, nucleus raphe magnus
INTRODUCTION
Pain is a response of the body to the action of injuring stimuli. Notwithstanding an unpleasant experience, it appears to be an important component of the defense system of the organism and a permanent regulator of homeostatic reaction. However, when the pain signals remain over long periods, they generate chronic pain. Despite the investment of significant resources by the pharmaceutical industry to identify novel analgesic drugs, chronic pain, which is most commonly defined as pain lasting longer than 3 months (i.e., outlasting the usual healing process), still represents a difficult treatment challenge in a large sector of the population.
The role of opioids in the treatment of pain has been long known for the humankind for thousands of years. Opioid analgesics are widely to relief dull, poorly localized (usually, visceral) pain. Repeated doses may cause tolerance to these drugs and dependence so that the sudden termination of opioid analgesics may precipitate a withdrawal syndrome. Apart from the opioid drugs, non-opioid, non-steroidal anti-inflammatory drugs (NSAIDs) are the most widely used analgesics in the treatment of pain. These drugs have analgesic, antipyretic, and at higher doses, anti-inflammatory actions. Aspirin (acetylsalicylic acid) was the first NSAID but has been largely replaced by drugs that are less toxic to gastro-intestinal tract, e.g. paracetamol, ibuprophen, ketorolac, naproxen, xefocam. NSAIDs produce their effects by inhibiting cyclooxygenase, a key enzyme in the production of prostaglandins. The latter is one of the mediators released at sites of inflammation. They do not themselves cause pain but they potentiate the pain caused by other mediators, e.g. histamine, serotonin, bradykinin.
NON-OPIOID ANALGESICS INDUCED TOLERANCE
The analgesic effect of opioids is mainly induced by their action in the midbrain periaqueductal grey matter (PAG)[1,2] and this structure probably plays a crucial role in the development of tolerance too[3,4,5].
The interaction of opioids and NSAIDs is of utmost interest. NSAIDs such as dipyrone (metamizol) and aspirin [lysine-acetylsalicylate (LASA)], when microinjected to PAG, cause analgesia, whereas their repeated administration results in vivid tolerance[6,7,8,9,10,11]. This antinociception at least in part is realized by endogenous opioid peptides, since microinjections induced analgesia by NSAIDs in PAG and the rostral ventro-medial part of medulla (RVM) is impaired by opioid antagonists[8,9]. NSAIDs repeated microinjection to PAG alongside with tolerance causes cross-tolerance to morphine and drug withdrawal syndrome[6,7,9,12]. Morphine microinjection to PAG inhibits nociception on spinal level through those RVM neurons, which have direct projections to the spinal dorsal roots[12,13].
The central nucleus of amygdala is involved in tolerance to the antinociceptive effect of NSAIDs
The central nucleus of amygdala (Ce) is considered an integral component of the endogenous pain-modulatory circuit and is critical for systemic morphine-induced suppression of spinal nociceptive reflexes[14].
We have recently discovered for the first time that microinjection of NSAIDs analgine, ketorolac, and xefocam into the Ce of rats both unilaterally (Figure 1) and bilaterally (Figure 2) elicits antinociception with the development of tolerance[15,16].
Figure 1.
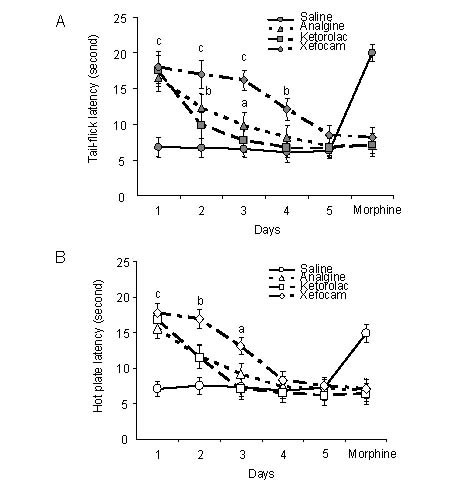
Response latencies in unilateral microinjections of non-steroidal anti-inflammatory drugs (NSAIDs) into the central nucleus of amygdala.
Mean response latencies following unilateral microinjections of each NSAIDs are plotted over the 5-day period, followed by morphine hydrochloride (3 µg/0.5 µL), for the tail-flick (A) and hot plate (B) tests.
Cutoff latency (20 seconds) was reached in some animals for xefocam and morphine microinjections.
Drugs were analgine (metamizol sodium, 150 μg/0.5 µL), ketorolac (ketorolac thromethamine, 90 μg/0.5 µL), xefocam (lornoxicam, 12 μg/0.5 µL).
Analysis of variance with post hoc Tukey-Kramer multiple comparison test were used for statistical evaluation of comparisons between treated and saline groups. aP < 0.05, bP < 0.01, cP < 0.001, vs. saline group[16].
Figure 2.
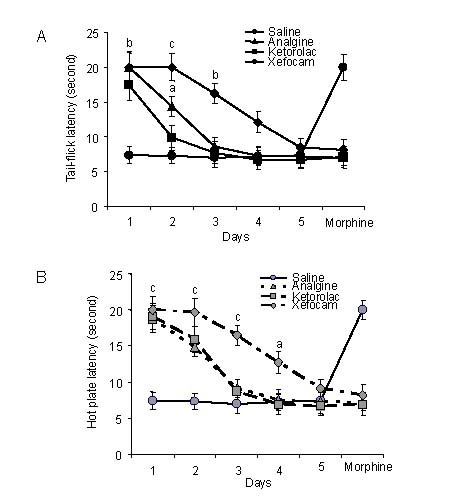
Response latencies in bilateral microinjections of non-steroidal anti-inflammatory drugs (NSAIDs) into the central nucleus of amygdale.
Mean response latencies following bilateral microinjections of each NSAIDs are plotted over the 5-day period, followed by morphine, for tail-flick (A) and hot plate (B) tests.
Cutoff latency (20 seconds) was reached in some animals for xefocam and morphine microinjections.
Analysis of variance with post hoc Tukey-Kramer multiple comparison tests were used for statistical evaluation of comparisons between treated and saline groups. aP < 0.05, bP < 0.01, cP < 0.001, vs. saline group[16].
This data confirmed our previous results with systemic (i.p.) administration of NSAIDs[17,18,19], and results of others using microinjection of the same NSAIDs into the PAG[6,9,20]. Importantly, repeated microinjection of NSAIDs into the Ce resulted in a progressive decrease in antinociceptive effectiveness (tolerance) similar to that observed with intra-PAG injections[6,9,20] and reminiscent of the effect of opiates.
A major involvement of opioidergic mechanisms in tolerance to the analgesic effect of NSAIDs was surprising, because traditionally the cellular and molecular actions of opioids were thought to differ from those of non-opioid analgesics. One interesting aspect of NSAIDs administration, namely tolerance, emphasizes their similarity to opioid analgesics. Indeed, microinjection of metamizol[9,10,20] or LASA[6,10] into PAG or into Ce[15,16], progressively led to a loss of their antinociceptive effects, i.e. produced tolerance. Furthermore, tolerance to metamizol or LASA was accompanied by cross-tolerance to morphine[6,9,20] as if opioid analgesics had been repeatedly administered. Interestingly, tolerance to the effect of PAG-microinjected metamizol can, like tolerance to morphine, be reversed by microinjection of proglumide, a cholecystokinin antagonist, at the same PAG site[20]. The latter finding constituted additional evidence that the PAG effects of non-opioid analgesics are similar to those of morphine. In addition, the data suggested that Ce should be incorporated into current models of endogenous pain control circuitry[21] and Ce integrates inputs from the limbic forebrain and deincephalon with ascending nociceptive input from dorsal horn.
There are direct projections to the PAG from a number of medial prefrontal areas including anterior cingulate and insular cortices[1,5].
It is well known that morphine injection after administration of NSAIDs or in combination, morphine plus NSAIDs usually potentiates their own analgesic effects[22]. We have recently tested each of NSAIDs for cross-tolerance to morphine given over a 5-day period in two age groups of rats. There was a significant difference between adult and juvenile rat groups for the degree of morphine analgesia, which was most marked on the first and second experimental days. Furthermore, morphine-tolerant rats exhibited cross-tolerance to analgine, ketorolac, and xefocam for both tail-flick and hot plate tests, respectively[23].
In conclusion, our data confirmed previous studies indicating that the antinociceptive action of NSAIDs may be closely related to that of endogenous opioids, including the development of tolerance. In addition, the Ce along with PAG and RVM represents an important component of the endogenous antinociceptive system.
Antinociceptive tolerance induced by non-opioid analgesics microinjected into periaqueductal grey matter
We have already noted that several lines of investigations have shown that in PAG, the microinjection of non-opioid analgesics, NSAIDs metamizol and LASA induces antinociception with some effects of tolerance[6,7]. We have just recently examined that microinjection of another type of NSAIDs clodifen, ketorolac and xefocam into the PAG leads to the development of tolerance in male rats (Figure 3).
Figure 3.
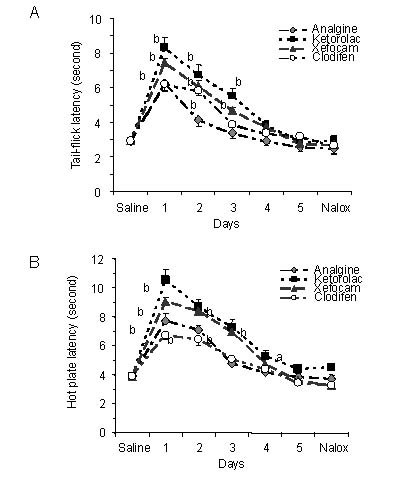
Microinjections of non-steroidal anti-inflammatory drugs into the periaqueductal grey matter for 5 consecutive days result in progressively decrease in tail-flick (A) and hot plate (B) latencies as compared to averaged saline baseline.
Intraperitoneal injections of μ-opioid antagonist naloxone (Nalox) do not change the hot plate and hot plate latencies in non-opioid tolerant rats as well as in control animals.
All data are presented as mean ± SEM. Analysis of variance with post hoc Dunnett's multiple comparison tests was used for statistical evaluation. aP < 0.05, bP < 0.01, cP < 0.001, vs. saline baseline[24].
Our obtained data add evidence to the hypothesis that, like opioids, non-opioid analgesics, particularly NSAIDs analgin, clodifen, ketorolac and xefocam induce tolerance, and also by mechanisms strongly concern to the PAG[7,8,9].
It has been shown that other non-opioid analgesic LASA also induces tolerance after repeated microinjections into the PAG[6,10]. The PAG and its descending projections to the RVM and the nucleus raphe magnus (NRM) are principal components of the descending antinociceptive pain-control system[1,2,4,11,25].
In naive rats, microinjection of morphine and dipyrone into the PAG decreases, the activity of pain-facilitating “on-cells” and increases, the activity of pain inhibiting “off-cells” thus giving rise to antinociception[8,26]. The PAG is thus crucial for tolerance to morphine as well as non-opioid NSAIDs such as metamizol, clodifen, ketorolac, LASA and xefocam[7,8,10,24]. Our findings confirm the results of other investigators that microinjection of dipyrone and LASA, and systemically dipyrone are abolished by systemically injections and/or microinjections of selective μ-opioid antagonists, naloxone and CTOP (D-phe-Cys-Tyr-D-trp-Orn-thr- Pen-thr-NH2)[6,8,22]. The latter is a cyclic analog of the neuropeptide somatostatin and is known to block the analgesic effect of morphine[8].
Taken together, the results presented here and other author's findings underscore the strong convergence of antinociceptive mechanisms of opioids and non-opioids, particularly NSAIDs in the PAG in the acute effect of and the development of tolerance to both types of analgesics. On the other hand, our data confirm the results of other authors that NSAIDs are in close relation with endogenous opioids and the tolerance to these non-opioid drugs probably depends on opioid tolerance.
Nucleus raphe magnus is opioid sensitive to analgesic effects of NSAIDs
Still 35 years ago, in classic studies carried out in the laboratory of Besson[27], electrical stimulation of the NRM completely suppressed the behavioral responses to noxious pinch of the skin and modified the jaw-opening reflex threshold to tooth pulp stimulation in the cat. Furthermore, electrical stimulation of the NRM or opioid microinjection inhibits activity of dorsal horn nociceptive neurons[28,29,30]. Subsequently, it has been shown that microinjections of dipyrone (metamizol) into the medullary NRM of lightly pentobarbital-anesthetized rats dose-dependently inhibit the nocifensive tail-flick reflex[31]. We have recently found that repeated for five-day microinjections of analgin, clodifen, ketorolac and xefocam in the NRM produce tolerance to these drugs (Figure 4).
Figure 4.
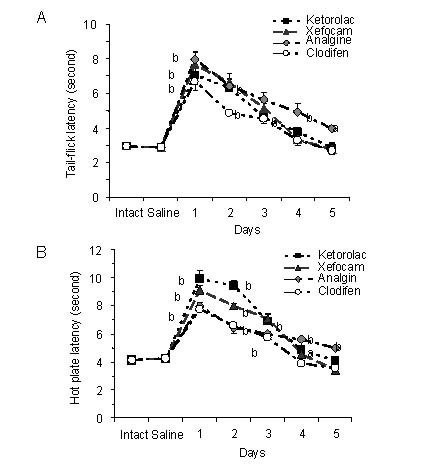
Microinjections of non-steroidal anti-inflammatory drugs into the nucleus raphe magnus for 5 consecutive days result in progressively decrease in tail-flick (A) and hot plate (B) latencies as compared to averaged saline baseline.
All data are presented as mean ± SEM. Analysis of variance with post hoc Dunnett's multiple comparison tests was used for statistical evaluation.
The statistical software utilized was InStat 3.05 (GraphPad Software, Inc, USA). aP < 0.05, bP < 0.01, cP < 0.001, vs. saline baseline[35].
It is interesting that although similar effects of antinociception can be produced by direct action of morphine upon the PAG and RVM, tolerance to morphine was not remarkably obtained by repeated microinjection into the RVM[32]. Furthermore, inactivation of the RVM did not prevent the development of tolerance to repeated morphine microinjections into the PAG, and tolerance to systemic morphine did not develop if opioid receptors were blocked in the PAG even if the RVM remained intact[33].
To examine into details, an opioid sensitivity of NSAIDs action we tested on whether post-treatment with μ-opioid antagonist naloxone in NRM diminishes NSAID-induced antinociception on the first and second experimental days. Thirty minutes after NSAIDs testing, microinjection of naloxone in NRM significantly decreased antinociceptive effects of NSAIDs at the first day in the tail-flick test for analgin (P < 0.05), clodifen (P < 0.01), ketorolac (P < 0.001), and xefocam (P < 0.001), respectively (Figure 5).
Figure 5.
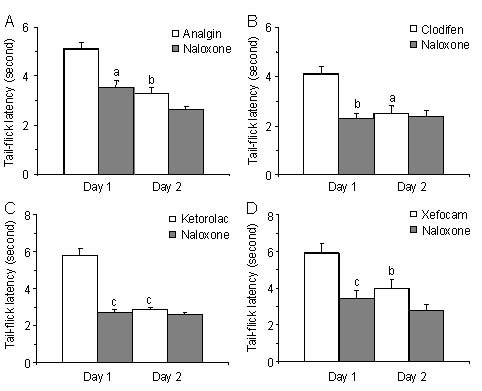
Post treatment with naloxone after microinjections of non-steroidal anti-inflammatory drugs into nucleus raphe magnus results in a significant decrease in tail-flick latency for the first day.
At the second day, naloxone has trend effects for all four non-opioid analgesics. All data are presented as mean ± SEM.
Analysis of variance with post hoc Tukey-Kramer multiple comparison tests was used for statistical evaluation of comparisons between treated and naloxone groups. aP < 0.05, bP < 0.01, cP < 0.001, vs. naloxone[35].
The same effects we discovered in the hot plate test for analgin (t = 5.701, P < 0.05), clodifen (t = 5.287, P < 0.05), ketorolac (t = 7.303, P < 0.001), and xefocam (t = 5.64, P < 0.05), respectively (Figure 6). At the second day, naloxone had generally trend effects in both tail-flick and hot plate tests (Figures 5, 6). These results strongly support the suggestion on endogenous opioid involvement in NSAIDs antinociception and tolerance[7,22,24,34,35].
Figure 6.
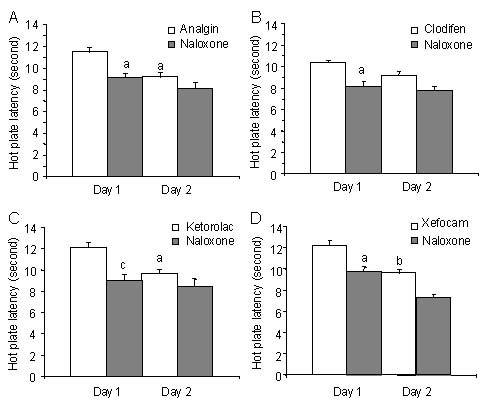
Post treatment with naloxone after microinjections of non-steroidal anti-inflammatory drugs into nucleus raphe magnus results in a significant decrease in hot plate latency for the first day.
At the second day, naloxone has trend effects for all four non-opioid analgesics. All data are presented as mean ± SEM.
Analysis of variance with post hoc Tukey-Kramer multiple comparison tests was used for statistical evaluation of comparisons between treated and saline groups. aP < 0.05, bP < 0.01, cP < 0.001, vs. naloxone[35].
The obtained data thus confirm evidence that, like opioids, non-opioid analgesics, particularly NSAIDs analgin, clodifen, ketorolac and xefocam induce tolerance. The latter should be realized by endogenous opioids, endorphins[7,22,24,36]. In our previous work, we have also shown that in the endogenous analgesic system as the RVM μ-opioid antagonist naloxone diminishes NSAID-induced antinociception on the first and second experimental days and impedes the development of tolerance to the antinociceptive effect of NSAIDs[24]. Our both findings affirm the results of other investigators that microinjection of dipyrone and LASA into PAG, and systemically dipyrone are abolished by systemically injected and/or microinjections of selective μ-opioid antagonists, naloxone and CTOP[6,8,22]. Moreover, endogenous opioids are involved in the analgesic potentiation observed with the combination of morphine plus dipyrone (metamizol). The release of endogenous opioids by dipyrone could enhance exogenous opiate effects, explaining the need for more amount of naloxone to counteract the antinociception produced by morphine plus dipyrone[22].
As mentioned above, the action of either opioid or non-opioid analgesics in the PAG leads to an excitation of PAG output neurons and this causes an activation of RVM off-cells and an inhibition of RVM on-cells, thus leading to antinociception (analgesia). When tolerance develops, PAG microinjections of morphine[37], or metamizol[8] are no longer capable of affecting RVM neurons and inducing analgesia. These results show further neuronal relationships between opioid and non-opioid analgesics as regards the descending pain-control and modulation system[38].
From a clinical point of view, the present study and evidence of other colleagues show that NSAIDs exert upon the PAG a powerful action, which could be delay and depress the establishment of inflammation-induced spinal hyperexcitability. Upon systemic administration of NSAIDs prior to persistent peripheral damage such as inflammation, the patient probably benefits from a central action at both PAG and spinal cord. When inflammation and central sensitization are fully developed, the action of NSAIDs upon PAG is sufficient to reverse spinal hyperexcitability and presumably therefore hyperalgesia and allodynia. By acting upon the PAG, NSAIDs activate the descending pain-control system, whose inhibitory function upon the spinal cord is exerted by way of the NRM and RVM[23,24,35,58].
On the other hand, the present results together with previous data using metamizol and LASA[6] are of general relevance to human medicine. The antinociceptive effect of NSAIDs in the PAG is related to endogenous opioids and implies that patients under repeated and prolonged treatment with non-opioid analgesics are, like those under opiates, at risk of tolerance to NSAIDs and a withdrawal syndrome. Finally, cross-tolerance between non-opioid and opioid analgesics and an association between systemic NSAIDs and endogenous opioids may have undesirable clinical consequences and should be important in the clinical setting[6,8,23,24].
BRAINSTEM MODULATION OF NOCICEPTIVE PROCESSING
This was an important advance in documenting that the brain itself could regulate the processing of nociceptive information. Inspired by stimulation-produced analgesia, a significant research effort led to the definition of a brainstem pain modulatory network with critical links in the RVM as well as the PAG. The antinociception resulting from stimulation in these structures is in large part due to regulation of nociceptive processing at the level of the spinal cord (Figure 7).
Figure 7.
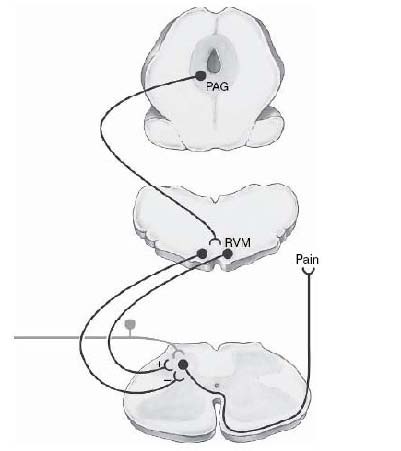
Functional organization of brainstem pain modulating system with links in midbrain periaqueductal grey matter (PAG) and rostral ventro-medial part of medulla (RVM). The PAG projects to the RVM. The RVM in turn regulates spinal nociceptive circuitry to the dorsal horn.
This system exerts bidirectional control, separates populations of RVM neurons and mediates descending inhibition and descending facilitation.
The PAG is reciprocally linked with forebrain structures including prefrontal cortex, amygdala, and hypothalamus.
These substantial interconnections provide an anatomical substrate through which emotional and cognitive variables could influence nociception via the PAG-RVM system. (Modified from Heinricher, Ingram, 2009[4]).
The PAG-RVM system mediates the analgesic actions of opioids, and it is recruited by internal and environmental challenges[4]. Accumulating evidence from neuroimaging studies supports a role for this system in top-down modulation of pain in humans, such as that produced by placebo or shifts in attention[21].
Along with the evidence that this system mediates the analgesic actions of exogenous and endogenous opioids, the fact that the net behavioral effect of nonselective experimental activation of the PAG or RVM is antinociception led to a general view of the PAG-RVM system as an analgesia system. This system is indeed activated by acute stress and opioid and non-opioid (NSAIDs) analgesics to inhibit spinal nociceptive processing. However, there is now increasing evidence that the system, especially the RVM, also facilitates nociception. The RVM has been implicated in hyperalgesia and allodynia associated with inflammation, nerve injury, acute opioid withdrawal, chronic opioid administration, and the sickness response. The PAG-RVM circuit should therefore be viewed not specifically as an analgesia system, but more generally, as a pain-modulation system[2,4,39,40]. From this perspective, the system has the potential for graded enhancement or inhibition of nociception under different external and internal conditions.
Neurotransmitters and endogenous opioids in the periaqueductal gray
All three opioid receptors, μ-, δ-, and κ-opioid receptors, are moderately to densely expressed in the PAG[41,42,43,44]. The PAG is also rich in endogenous opioids. Particularly, enkephalin-containing terminals are found apposed to both GABA-ergic and non-GABA-ergic dendrites, including those of a small percentage of PAG-RVM neurons. Endomorphin-2 (Tyr-Pro-Phe-Phe-NH2), an endogenous peptide with high selectivity for the μ-opioid receptor is concentrated in the PAG as well as the dorsal horn of the spinal cord[45,46,47]. Another endogenous opioid found in the PAG is β-endorphin. Discrete populations of β-endorphin-containing neurons in the ventromedial hypothalamus project to the PAG and have been implicated in analgesia produced by electrical stimulation and stress[48].
The μ-opioid receptor agonists produce potent antinociception when applied directly in the PAG. The behavioral antinociception produced by these agents is mediated by activation of output neurons projecting to the RVM μ-opioid receptor agonists are thought to act presynaptically to block GABA-ergic inhibition of PAG output neurons. Consistent with this hypothesis, blockade of GABA transmission within the PAG by microinjection of a GABAA receptor antagonist produces antinociception[4,49].
Postsynaptic effects of μ-opioid receptor agonists on PAG neurons include hyper-polarization via activation of a postsynaptic G-protein-activated inwardly rectifying potassium conductance (GIRK) and inhibition of calcium channels[50] (Figure 8).
Figure 8.
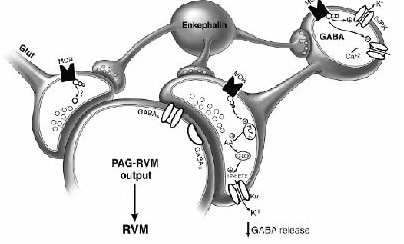
Cellular mechanisms of opioid action within the periaqueductal grey matter (PAG). Enkephalin-containing synapses are apposed to cell bodies as well as to gamma-aminobutyric acid (GABA)- and glutamate-containing terminals.
The postsynaptic μ-opioid receptor activates GIRKs and inhibits voltage-gated Ca2+ channels to hyperpolarize cells and decrease cell activity.
Presynaptic μ-opioid receptors (MOR) inhibit both GABA and glutamate release on ventrolateral PAG neurons, and apparently use different signal transduction pathways in the terminals.
MORs localized to GABA terminals are coupled to voltage-gated potassium channels via activation of the arachidonic acid/12-lipoxygenase (12-LOX) second messenger pathway.
Hyperpolarization of the terminal decreases GABA release. The signal transduction pathway for MOR inhibition of glutamate release is currently unknown. MORs are found on GABA containing interneurons in the PAG, as well as on PAG output neurons projecting to the RVM.
Activation of the descending antinociceptive pathway by opioids occurs via disinhibition (Modified from Heinricher, Ingram, 2009[4]).
The μ-opioid receptor agonists also have presynaptic effects, inhibiting GABA and glutamate release from terminals within the ventrolateral PAG[51,52,53]. Presynaptic inhibition of GABA-ergic neurotransmission is through activation of the arachidonic acid-phospholipase A2 second messenger pathway. Stimulation of this pathway results in activation of voltage-gated potassium channels (Kv channels) by metabolites of 12-lipoxygenase[52,53]. This pathway is independent of adenylyl cyclase, protein kinase A or protein kinase C activity[53]. Further research is needed to determine the relevance of the various presynaptic versus postsynaptic opioid actions to the nociceptive modulatory function of the PAG.
Opioid and non-opioid (NSAIDs) interactions
Administrations of NSAIDs systemically or microinjections into the PAG produce analgesia at the behavioral level in awake rats[6,7,8,19,23,24]. This antinociception is apparently mediated at least in part by an endogenous opioid peptide, as the analgesia produced by NSAIDs microinjected into the PAG and RVM is attenuated by the opioid antagonist naloxone[10,22,24,34,35].
In addition to stimulating release of endogenous opioids, recent evidence at the cellular level suggests that NSAIDs may also augment the signaling pathway used by opiates, potentiating the actions of exogenous opioids[4] (Figure 9).
Figure 9.
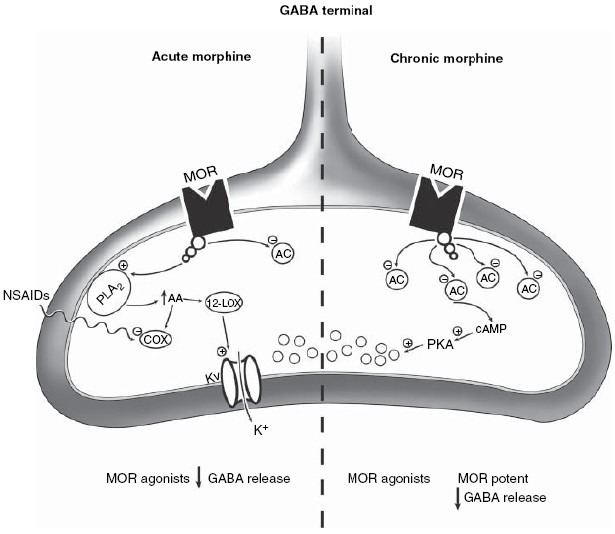
μ-opioid receptor (MOR) coupling in presynaptic GABA terminals changes with chronic morphine administration.
Acute administration of MOR agonists activates MORs coupled to phospholipase A2 (PLA2). Activation of PLA2 increases production of arachidonic acid, which is further metabolized by 12-lipoxygenase (12-LOX).
Lipoxygenase metabolites such as 12-hydroxyeicosenoic acid activate voltage-gated potassium (Kv) channels to hyperpolarize and decrease GABA release from the terminals.
Non-steroidal anti-inflammatory drugs (NSAIDs) potentiate this action of opioids by inhibiting cyclooxygenase (COX) mediated arachidonic acid metabolism, thereby shunting arachidonic acid to the 12-LOX pathway.
Activation of MORs presumably also acutely inhibits adenylyl cyclase (AC) activity in these terminals.
Chronic morphine administration upregulates AC and protein kinase A (PKA) activity. After chronic, but not acute, opioid treatment, GABA release is enhanced by increased PKA activity.
MOR agonists are more potent inhibitors of this PKA-dependent release, so that opioid removal or blockade of MORs by antagonists results in a rebound increase in GABA release.
This increased GABA release may contribute to withdrawal behaviors mediated by the PAG. (Modified from Heinricher, Ingram, 2009[4]).
Coapplication of NSAIDs potentiates the inhibition of GABA release by the μ-opioid receptor partial agonist morphine, although NSAIDs have no effect on GABA release in the absence of morphine[53,54,55,56]. NSAIDs primarily inhibit cyclooxygenases (COX-1 and COX-2), one of three types of enzymes (cyclooxygenases, 5-lipoxygenases, and 12-lipoxygenases) that metabolize arachidonic acid. One hypothesis proposed to explain the mechanism of increased analgesia with coapplication of opioids and NSAIDs is that blockade of COX-1 shunts arachidonic acid metabolism through the lipoxygenase pathways to increase the potency of opioid receptor agonists[53,54,55,57] (Figure 10).
Figure 10.
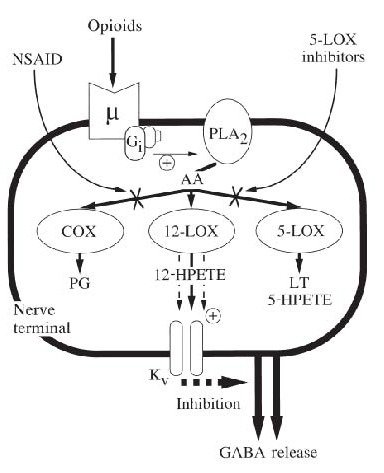
A mechanism of interaction between opioids and non-steroidal anti-inflammatory drugs (NSAIDs) in nerve terminals in the central nervous system that is independent of formation of prostanoids.
Opioids acting on μ-receptors inhibit neurotransmitter release by stimulating phospholipase A2 (PLA2) via Gi proteins.
This leads to the formation of 12-lipoxygenase (12-LOX) metabolites of arachidonic acid (AA), which enhance the activity of voltage-gated potassium (Kv) channels that inhibit neurotransmitter release.
NSAIDs and specific 5-lipoxygenase (5-LOX) inhibitors block alternative pathways COX-1, and 5-LOX of metabolism of AA, causing a shunt to enhanced formation of 12-LOX metabolites, thereby enhancing the efficacy of opioids.
This mechanism can account for opioid and NSAID synergism, and the naloxone sensitive analgesic actions of NSAIDs in the central nervous system.
PG, prostaglandins; LT, leukotrienes; HPETE, hydroperoxyeicosatetraenoic acid. (Modified from Christie et al., 2000[55]).
The fact that inhibitors of 5-lipoxygenases also appear to potentiate the effects of opioid agonists in the PAG adds further weight to this proposal[53,54,55]. These results are important in that the combined administration of NSAIDs and opiates may allow lower doses of morphine to be used to provide adequate analgesia while reducing the probability of the development of tolerance and side effects (such as respiratory depression) associated with high doses of opiates. Functional studies of NSAID/ opioid interactions in the PAG would therefore be of great interest[4].
The precise molecular mechanism of NSAIDs tolerance has not yet known. The term tolerance is sometimes used rather loosely to refer either to very short or long-term loss of agonist efficacy. In our experimental animals, ‘acute’ tolerance can be observed rapidly (minutes to days) during the course of a single episode of opioid intoxication. This type of tolerance may be more closely related to processes of rapid μ-opioid receptor desensitization and internalization that should be distinguished from the more substantial tolerance that emerges after days to weeks of opioid administration[58].
According to our and other colleagues’ data, NSAIDs tolerance mimics opioid tolerance. At present we cannot precisly determine the cellular and molecular mechanisms of such similarities. It is well established that organization of opioid adaptations in the nervous system including: (a) receptor tolerance at the μ-opioid receptor itself showing loss in the coupling of μ-opioid receptor to a major cellular effector, as the G-protein-regulated inwardly rectifying potassium channel. Several potential mechanisms could account for tolerance at this level of organization, but changes to coupling and perhaps surface expression appear to be most important. (b) Cellular tolerance and withdrawal in opioid-sensitive neurons is due to multiple adaptations to intracellular signaling cascades, but hypertrophy of cAMP signalling is the best established. (c) Systems feedback adaptations in opioid-sensitive nerve and neuroglial networks can develop and contribute to tolerance and withdrawal. (d) Synaptic plasticity and learning in opioid-sensitive nerve networks may involve changes in synaptic plasticity driven by changes in presynaptic release probability, which are well established at many opioid-sensitive GABAergic synapses, but more importantly, mechanisms resembling long-term potentiation and/or long-term depression probably involving AMPA receptor insertion in synapses may produce long-term changes in synaptic strength[59,60].
Future scientific efforts will be directed at deepening our understanding of how adaptive responses by multiple neural systems and molecular mechanisms work together to counteract the analgesic efficacy of commonly used opioids and non-opioids. Future pharmaceutical development will focus on blocking the facilitatory mechanisms that produce these adaptive changes, in the endogenous nociceptive and antinociceptive systems, in response to continual exposure to an opioid analgesic[61,62,63].
SUMMARY AND CONCLUSIONS
The present review reports that microinjection of analgin (metamizol), ketorolac, and xefocam into the central nucleus of amygdala of rats elicits antinociception with the development of tolerance. Furthermore, microinjections of these NSAIDs plus clodifen into the periaqueductal grey produced antinociception as revealed by a latency increase in tail-flick and hot plate tests compared to the baseline control with saline microinjected into the same nucleus. However, subsequent testing also took place in the following days, shows strong tolerance to these drugs. Finally, repeated administrations of these NSAIDs into the nucleus raphe magnus in the following 4 days result in progressively less antinociception compare to the saline control, i.e., tolerance develops to these medications. Special control experiments showed that post-treatment with the μ-opioid antagonist naloxone into the nucleus raphe magnus significantly decreased antinociceptive effects of NSAIDs on the first day of testing. Thus, the mechanism producing tolerance to NSAIDs can be due to the participation of endogenous opioids, endorphins. Therefore, these findings strongly support the suggestion of endogenous opioid involvement in NSAIDs antinociception and tolerance in the descending pain-control and modulation system.
Concerning the effort to understand the neural basis of nociceptive modulation by the periaqueductal grey-rostral ventro-medial part of medulla system, it highlights the importance of studying functionally identified neurons. The rostral ventro-medial medulla and nucleus raphe magnus can both facilitate and inhibit nociception. Furthermore, this region is also implicated in a number of functions other than nociceptive modulation, including reproductive behaviors, cardiovascular and respiratory control, sleep-wakefulness and arousal, thermoregulation, and behavioral suppression. A meaningful analysis of how the rostral ventro-medial medulla contributes to enhanced pain states therefore requires functional identification of the neurons under study, so that mechanisms contributing to nociceptive facilitation can be distinguished from those involved in nociceptive inhibition or other functions[2,4].
Acknowledgments:
We gratefully acknowledge Prof. E. Abzianidze for her helpful comments.
Footnotes
Funding: This research was supported by the grant from Georgian National Science Foundation, No. GNSF/ST07/6-234.
Conflicts of interest: The authors declare that that the research was conducted in the absence of any commercial or financial relationship that could be construed as a potential conflict of interest.
(Edited by Fu KY, Rácz I/Zhao LJ/Song LP)
REFERENCES
- 1.Fields HL, Basbaum AI, Heinricher MM. McMahon SB, Koltzenburg M. Wall and Mellzack's Textbook of Pain. 5th ed. London: Elsevier; 2006. Central nervous system mechanisms of pain modulation. [Google Scholar]
- 2.Ren K, Dubner R. Basbaum AI, Bushnell MC. Science of Pain. San Diego: Elsevier; 2009. Descending control mechanisms. [Google Scholar]
- 3.Lane DA, Patel PA, Morgan MM. Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neuroscience. 2005;135(1):227–234. doi: 10.1016/j.neuroscience.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 4.Heinricher MM, Ingram SL. The brainstem and nociceptive modulation. In: Basbaum AI, Bushnell MC, editors. Science of Pain. San Diego: Elsevier; 2009. [Google Scholar]
- 5.Keay KA, Bandler R. Deep and superficial noxious stimulation increases Fos-like immuno-reactivity in different regions of the midbrain periaqueductal grey of the rat. Neurosci Lett. 1993;154(1-2):23–26. doi: 10.1016/0304-3940(93)90162-e. [DOI] [PubMed] [Google Scholar]
- 6.Pernia-Andrade AJ, Tortorici V, Venegas H. Induction of opioid tolerance by lysine acetyl-salicylate in rats. Pain. 2004;111(1/2):191–200. doi: 10.1016/j.pain.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Tortorici V, Nogueira L, Aponte Y, et al. Involvement of cholecystokinin in the opioid tolerance induced by dipyrone (metamizol) microinjections into the periaqueductal gray matter of rats. Pain. 2004;112(1/2):113–120. doi: 10.1016/j.pain.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 8.Tortorici V, Aponte Y, Acevedo H, et al. Tolerance to non-opioid analgesics in PAG involves unresponsiveness of medullary pain-modulating neurons in male rats. Eur J Neurosci. 2009;29(6):1188–1196. doi: 10.1111/j.1460-9568.2009.06678.x. [DOI] [PubMed] [Google Scholar]
- 9.Tortorici V, Vanegas H. Opioid tolerance induced by metamizol (dipyrone) microinjections into the periaqueductal gray of rats. Eur J Neurosci. 2000;12(11):4074–4080. doi: 10.1046/j.1460-9568.2000.00295.x. [DOI] [PubMed] [Google Scholar]
- 10.Vanegas H, Tortorici V. Opioidergic effects of non-opioid analgesics on the central nervous system. Cell Mol Neurobiol. 2002;22(5-6):655–661. doi: 10.1023/A:1021896622089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vanegas H, Tortorici V. The periaqueductal gray as critical site for antinociception and tolerance induced by non-steroidal anti-inflammatory drugs. In: Maione S, Di Marzo V, editors. Neurotransmission in the Antinociceptive Descending Pathway. Kerala: Research Signpost; 2007. [Google Scholar]
- 12.Vanegas H, Barbaro NM, Fields HL. Tail-flick related activity in medullospinal neurons. Brain Res. 1984;321(1):135–141. doi: 10.1016/0006-8993(84)90689-9. [DOI] [PubMed] [Google Scholar]
- 13.Fields HL, Malick A, Burstein R. Dorsal horn projection targets of on- and off-cells in the rostral ventromedial medulla. J Neurophysiol. 1995;74(4):1742–1759. doi: 10.1152/jn.1995.74.4.1742. [DOI] [PubMed] [Google Scholar]
- 14.Manning BH, Mayer DJ. The central nucleus of the amygdala contributes to the production of morphine antinociception in the tail-flick test. J Neurosci. 1995;15(12):8199–8213. doi: 10.1523/JNEUROSCI.15-12-08199.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tsagareli MG, Tsiklauri N, Gurtskaia G, et al. Tolerance effects induced by NSAIDs microinjections into the central nucleus of the amygdala in rats. Neurophysiology. 2009;41(6):404–408. [Google Scholar]
- 16.Tsagareli MG, Tsiklauri N, Gurtskaia G, et al. The central nucleus of amygdala is involved in tolerance to the antinociceptive effect of NSAIDs. Inter J Health. 2010;2(1):64–68. [Google Scholar]
- 17.Tsagareli MG, Tsiklauri N, Lagidze T, et al. Tolerance induction by non-opioid analgesics in rats. Proc Georgian Acad Sci Biol Series A. 2005;31(6):903–909. [Google Scholar]
- 18.Tsiklauri N, Gurtskaia G, Tsagareli MG. Is endogenous opioid system involved in non-opioid analgesics tolerance? Georgian Med News. 2006;8(137):121–125. [PubMed] [Google Scholar]
- 19.Tsiklauri N, Tsagareli MG. Non-opioid-induced tolerance in rats. Neirofiziologia. 2006;38(4):370–373. [Google Scholar]
- 20.Tortorici V, Nogueira L, Salas R, et al. Involvement of local cholecystokinin in the tolerance induced by morphine microinjections into the periaqueductal gray of rats. Pain. 2003;102(1):9–16. doi: 10.1016/s0304-3959(02)00153-7. [DOI] [PubMed] [Google Scholar]
- 21.Price DD, Bushnell MC. Overview of pain dimensions and their psychological modulation. In: Price DD, Bushnell MC, editors. Psychological Methods of Pain Control: Basic Science and Clinical Perspectives. Seattle, WA: IASP Press; 2004. pp. 3–17. [Google Scholar]
- 22.Hernández-Delgadillo GP, Cruz SL. Endogenous opioids are involved in morphine and dipyrone analgesic potentiation in the tail flick test in rats. Eur J Pharmacol. 2006;546(1):54–59. doi: 10.1016/j.ejphar.2006.07.027. [DOI] [PubMed] [Google Scholar]
- 23.Tsiklauri N, Voitenko N, Tsagareli MG. Non-opioid tolerance in juvenile and adult rats. Eur J Pharmacol. 2010;629(1-3):68–72. doi: 10.1016/j.ejphar.2009.12.016. [DOI] [PubMed] [Google Scholar]
- 24.Tsiklauri N, Nozadze I, Gurtskaia G, et al. Tolerance induced by non-opioid analgesic microinjections into rat's periaqueductal gray and nucleus raphe. Georgian Med News. 2010;17(180):47–55. [PubMed] [Google Scholar]
- 25.Heinricher MM, Tavares I, Leith JL, et al. Descending control of nociception: Specificity, recruitment and plasticity. Brain Res Rev. 2009;60(1):214–225. doi: 10.1016/j.brainresrev.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fields H. State-dependent opioid control of pain. Nat Rev Neurosci. 2004;5(7):565–575. doi: 10.1038/nrn1431. [DOI] [PubMed] [Google Scholar]
- 27.Oliveras JL, Hosobuchi Y, Guilbaud G, et al. Analgesic electrical stimulation of the feline nucleus raphe magnus: development of tolerance and its reversal by 5-HTP. Brain Res. 1978;146(2):404–409. doi: 10.1016/0006-8993(78)90988-5. [DOI] [PubMed] [Google Scholar]
- 28.Dickenson AH, Oliveras JL, Besson JM. Role of the nucleus raphe magnus in opiate analgesia as studied by the microinjection technique in the rat. Brain Res. 1979;170(1):95–111. doi: 10.1016/0006-8993(79)90943-0. [DOI] [PubMed] [Google Scholar]
- 29.Fields HL, Anderson SD. Evidence that raphe-spinal neurons mediate opiate and midbrain stimulation produced analgesia. Pain. 1978;5(4):333–349. doi: 10.1016/0304-3959(78)90002-7. [DOI] [PubMed] [Google Scholar]
- 30.Jones SL, Light AR. Electrical stimulation in the medullary nucleus raphe magnus inhibits noxious heat evoked fos-protein-like immunoreactivity in the rat lumbar spinal cord. Brain Res. 1990;530(2):335–338. doi: 10.1016/0006-8993(90)91306-2. [DOI] [PubMed] [Google Scholar]
- 31.Jones SL. Dipyrone into the nucleus raphe magnus inhibits the rat nociceptive tail flick reflex. Eur J Pharmacol. 1996;318(1):37–40. doi: 10.1016/s0014-2999(96)00909-0. [DOI] [PubMed] [Google Scholar]
- 32.Morgan M, Clayton CC, Boyer-Quick JS. Differential susceptibility of the PAG and RVM to tolerance to the antinociceptive effect of morphine in the rat. Pain. 2005;113(1):91–98. doi: 10.1016/j.pain.2004.09.039. [DOI] [PubMed] [Google Scholar]
- 33.Lane DA, Patel PA, Morgan MM. Evidence for an intrinsic mechanism of antinociceptive tolerance within the ventrolateral periaqueductal gray of rats. Neurosci. 2005;135(1):227–234. doi: 10.1016/j.neuroscience.2005.06.014. [DOI] [PubMed] [Google Scholar]
- 34.Tsiklauri ND, Nozadze IR, Gurtskaia GP, et al. Opioid sensitivity of analgesia induced by microinjections of non-steroidal anti-inflammatory drugs into the nucleus raphe magnus. Neurophysiology. 2011;43(3):213–216. [Google Scholar]
- 35.Tsagareli MG, Nozadze I, Tsiklauri N, et al. Tolerance to non-opioid analgesics is opioid-sensitive in nucleus raphe magnus. Front Neurosci. 2011;5(7):92–96. doi: 10.3389/fnins.2011.00092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tsiklauri N, Gurtskaia G, Tsagareli MG. Is endogenous opioid system involved in non-opioid analgesics tolerance? Georgian Med News. 2006;8(137):121–125. [PubMed] [Google Scholar]
- 37.Tortorici V, Morgan MM, Vanegas H. Tolerance to repeated microinjection of morphine into the periaqueductal gray is associated with changes in the behavior of off- and on-cells in the rostral ventromedial medulla of rats. Pain. 2001;89(2-3):237–244. doi: 10.1016/s0304-3959(00)00367-5. [DOI] [PubMed] [Google Scholar]
- 38.Vanegas H, Vasques E, Tortorici V. NSAIDs, opioids, cannabinoids and the control of pain by the central nervous system. Pharmaceuticals. 2010;3:1335–1347. doi: 10.3390/ph3051335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25(6):319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- 40.Heinricher MM, Pertovaara A, Ossipov MH. Descending modulation after injury. In: Dostrovsky JO, Carr DB, Koltzenburg M, editors. Seattle, WA: Proc. 10th World Congress Pain, IASP Press; 2003. [Google Scholar]
- 41.Mansour A, Fox CA, Akil H, et al. Opioid receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18(1):22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- 42.Gutstein HB, Mansour A, Watson SJ, et al. Mu and kappa opioid receptors inperiaqueductal gray and rostral ventromedial medulla. NeuroReport. 1998;9(8):1777–1781. doi: 10.1097/00001756-199806010-00019. [DOI] [PubMed] [Google Scholar]
- 43.Kalyuzhny AE, Wessendorf MW. Relationship of mu- and delta-opioid receptors to GABAergic neurons in the central nervous system, including antinociceptive brainstem circuits. J Comp Neurol. 1998;392(4):528–547. [PubMed] [Google Scholar]
- 44.Zubieta J-K, Smith YR, Bueller JA, et al. Regional mu-opioid receptor regulation of sensory and affective dimensions of pain. Science. 2001;293(5528):311–315. doi: 10.1126/science.1060952. [DOI] [PubMed] [Google Scholar]
- 45.Schreff M, Schulz S, Wiborny D, et al. Immunofluorescent identification of endomorphin-2-containing nerve fibers and terminals in the rat brain and spinal cord. NeuroReport. 1998;9(6):1031–1034. doi: 10.1097/00001756-199804200-00014. [DOI] [PubMed] [Google Scholar]
- 46.Williams FG, Beitz AJ. Ultrastructural morphometric analysis of GABA-immunoreactive terminals in the ventrocaudal periaqueductal grey: analysis of the relationship of GABA terminals and the GABAa receptor to periaqueductal grey-raphe magnus projection neurons. J Neurocytol. 1990;19(5):686–696. doi: 10.1007/BF01188037. [DOI] [PubMed] [Google Scholar]
- 47.Zadina JE, Hackler L, Ge LJ, et al. A potent and selective endogenous agonist for the μ-opiate receptor. Nature. 1997;386(6624):499–502. doi: 10.1038/386499a0. [DOI] [PubMed] [Google Scholar]
- 48.Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66(6):355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- 49.Moreau J-L, Fields HL. Evidence for GABA involvement in midbrain control of medullary neurons that modulate nociceptive transmission. Brain Res. 1986;397(1):37–46. doi: 10.1016/0006-8993(86)91367-3. [DOI] [PubMed] [Google Scholar]
- 50.Connor M, Christie MJ. Modulation of Ca2+ channel currents of acutely dissociated rat periaqueductal grey neurons. J Physiol. 1998;509(1):47–58. doi: 10.1111/j.1469-7793.1998.047bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chieng B, Christie MJ. Inhibition by opioids acting on μ-receptors of GABAergic and glutamatergic postsynaptic potentials in single rat periaqueductal gray neurones in vitro. Brit J Pharmacol. 1994;113(1):303–309. doi: 10.1111/j.1476-5381.1994.tb16209.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. J Physiol. 1997;498(Pt 2):463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vaughan CW, Ingram SL, Connor MA, et al. How opioids inhibit GABA mediated neurotransmission. Nature. 1997;390(6660):611–614. doi: 10.1038/37610. [DOI] [PubMed] [Google Scholar]
- 54.Christie MJ, Vaughan CW, Ingram SL. Opioids, NSAIDs and 5-lipoxygenase inhibitors act synergistically in brain via arachidonic acid metabolism. Inflamm Res. 1999;48(1):1–4. doi: 10.1007/s000110050367. [DOI] [PubMed] [Google Scholar]
- 55.Christie MJ, Connor M, Vaughan CW, et al. Cellular actions of opioids and other analgesics: Implications for synergism in pain relief. Clin Exp Pharmacol Physiol. 2000;27(7):520–523. doi: 10.1046/j.1440-1681.2000.03291.x. [DOI] [PubMed] [Google Scholar]
- 56.Hack SP, Vaughan CW, Christie MJ. Modulation of GABA release during morphine withdrawal in midbrain neurons in vitro. Neuropharmacology. 2003;45(5):575–584. doi: 10.1016/s0028-3908(03)00205-3. [DOI] [PubMed] [Google Scholar]
- 57.Vaughan CW. Enhancement of opioid inhibition of GABAergic synaptic transmission by cyclo-oxygenase inhibitors in rat periaqueductal grey neurons. Br J Pharmacol. 1998;123(8):1479–1481. doi: 10.1038/sj.bjp.0701818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vazquez E, Escobar W, Ramirez C, et al. A non-opioid analgesic acts upon the PAG-RVM axis to reverse inflammatory hyperalgesia. Eur J Neurosci. 2007;25(2):471–479. doi: 10.1111/j.1460-9568.2007.05280.x. [DOI] [PubMed] [Google Scholar]
- 59.Christie MJ. Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol. 2008;154(2):384–396. doi: 10.1038/bjp.2008.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Niikura K, Narita M, Butelman ER, et al. Neuropathic and chronic pain stimuli downregulate central mu-opioid and dopaminergic transmission. Trends Pharmacol Sci. 2010;31(7):299–305. doi: 10.1016/j.tips.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 61.He L, Fong J, von Zastrow M, et al. Regulation of opioid receptor trafficking and morphine tolerance by receptor oligomerization. Cell. 2002;108(2):271–282. doi: 10.1016/s0092-8674(02)00613-x. [DOI] [PubMed] [Google Scholar]
- 62.Kieffer BL, Evans CJ. Opioid tolerance-in search of the Holy Grail. Cell. 2002;108(5):587–590. doi: 10.1016/s0092-8674(02)00666-9. [DOI] [PubMed] [Google Scholar]
- 63.DuPen A, Sheni D, Ersek M. Mechanisms of opioid- induced tolerance and hyperalgesia. Pain Manag Nurs. 2007;8(3):113–121. doi: 10.1016/j.pmn.2007.02.004. [DOI] [PubMed] [Google Scholar]