

Abstract
Studies have demonstrated that DL-3-n-butylphthalide can significantly alleviate oxygen glucose deprivation-induced injury of human umbilical vein endothelial cells at least partly associated with its enhancement on oxygen glucose deprivation -induced hypoxia inducible factor-1α expression. In this study, we hypothesized that DL-3-n-butylphthalide can protect against oxygen glucose deprivation-induced injury of newborn rat brain microvascular endothelial cells by means of upregulating hypoxia inducible factor-1α expression. MTT assay and Hoechst staining results showed that DL-3-n-butylphthalide protected brain microvascular endothelial cells against oxygen glucose deprivation-induced injury in a dose-dependent manner. Western blot and immunofluorescent staining results further confirmed that the protective effect was related to upregulation of hypoxia inducible factor-1α. Real-time RT-PCR reaction results showed that DL-3-n-butylphthalide reduced apoptosis by inhibiting downregulation of pro-apoptotic gene caspase-3 mRNA expression and upregulation of apoptosis-executive protease bcl-2 mRNA expression; however, DL-3-n-butylphthalide had no protective effects on brain microvascular endothelial cells after knockdown of hypoxia inducible factor-1α by small interfering RNA. These findings suggest that DL-3-n-butylphthalide can protect brain microvascular endothelial cells against oxygen glucose deprivation-induced injury by upregulating bcl-2 expression and downregulating caspase-3 expression though hypoxia inducible factor-1α pathway.
Keywords: DL-3-n-butylphthalide, apoptosis, brain microvascular endothelial cells, hypoxia inducible factor-1α
Abbreviations:
BMECs, brain microvascular endothelial cells; HIF-1α, hypoxia inducible factor-1α; NBP, DL-3-n-butylphthalide; OGD, oxygen glucose deprivation; NVU, neurovascular unit; PHD, prolyl hydroxylase domain
INTRODUCTION
Over the past few decades, despite the progress in molecular mechanisms of neuronal cell death, many neuroprotective therapies against ischemic stroke have failed, indicating that a purely neurocentric focus is not sufficient[1]. All cell types must be considered within the entire “neurovascular unit (NVU)” comprising neurons, glia, cerebral endothelium and the extracellular matrix[2]. Brain microvascular endothelial cells (BMECs) are the major cell type in the NVU[3].
The L-isomer of 3-n-butylphthalide is a pure component from seeds of Apium graveolens Linn, Chinese celery. DL-3-n-butylphthalide (NBP), a chiral compound containing L-and D-isomers of butylphthalide, is a synthetic compound[4]. NBP is safe and well tolerated by humans in a multi-center double-blind randomized clinical trial and has been used for improving outcomes in ischemic stroke patients since 2002 in China, and was the recommended medication in Chinese Guidelines for Management of Acute Ischemic Stroke 2010[5].
Most previous studies focused on the direct protective effect of NBP on neurons[6]. Recently, some studies found that NBP significantly reduced the incidence of experimental stroke via improvement of cerebral microvessels in vivo and could protect human umbilical vein endothelial cells (HUVECs) though reducing oxygen glucose deprivation (OGD)-induced mitochondrial reactive oxygen species and subsequent cell death in vitro, suggesting a direct action of NBP on blood vessels[4,7,8]. The protective effect of NBP seems to be associated with its enhancement of OGD-induced hypoxia inducible factor-1α (HIF-1α) expression in HUVECs[8]. BMECs are quite different from body endothelial cells in construction and function, so the objective of our present study is to confirm whether NBP can protect BMECs against the OGD-induced injury, if it can, whether it is though HIF-1α pathway.
RESULTS
Protective effect of NBP on OGD-induced injury in BMECs
To examine the effect of NBP on OGD-injured injury in BMECs, cell viability was evaluated by MTT assay and nuclear morphological analysis. NBP at a dose range of 0.1 to 1 000 μM did not affect the viability of BMECs compared with the control (data not shown). As shown in Figure 1B, the relative MTT value was 53.9 ± 4.5% in the OGD group (normal control as 100%), and was significantly increased by NBP (1, 10, 100, 1 000 μM) in a dose-dependent manner. However, the levels of relative MTT values were not significantly different between 100 μM and 1 000 μM, indicating that NBP at a concentration of 100 μM had reached its maximum protective effect. Nuclear morphological analysis yielded similar results. The nuclear morphology was normal in the normal control cells. OGD led to substantial morphological changes such as crenation and condensation in BMECs. Pretreatment with NBP significantly attenuated the number of cells with abnormal nuclear morphology induced by OGD (Figure 1A).
Figure 1.
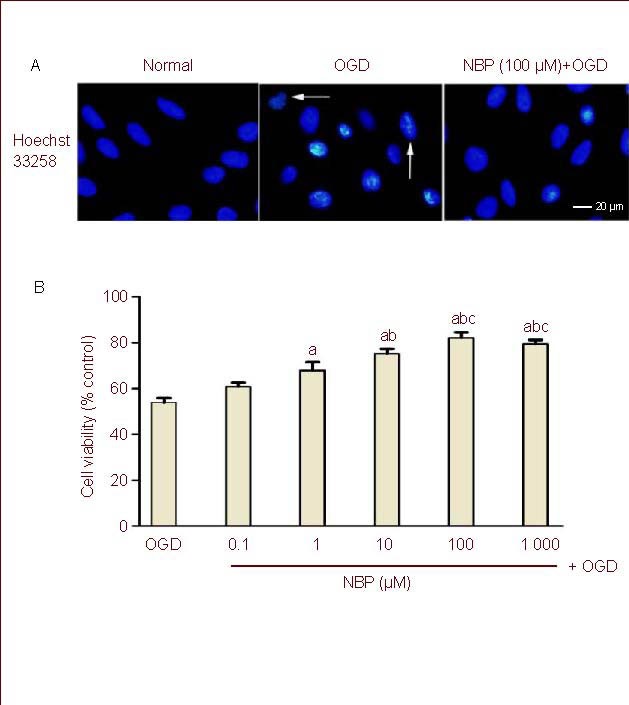
Effect of DL-3-n-butylphthalide (NBP) on cell damage induced by oxygen glucose deprivation (OGD) in rat brain microvascular endothelial cells (BMECs).
(A) BMECs were pretreatment with NBP (100 μM) for 1 hour, then underwent 2-hour OGD and 24-hour reperfusion.
BMEC nuclei were stained with Hoechst 33258. Crenation of nuclei and condensation of chromatin were evident in BMECs exposed to OGD (arrows) and OGD-induced nucleus morphological changes were obviously reduced in BMECs treated with NBP (100 μM) before exposure to OGD.
(B) Bar graph shows the effect of pretreatment with different concentrations of NBP on the viability of BMECs with OGD insult.
Each experimental group was expressed as percentage of that of the sham-OGD group (normal group).
Values were expressed as mean ± SEM. The results were obtained from five independent experiments performed in triplicate.
aP < 0.05, vs. the OGD group; bP < 0.05, vs. the NBP (1 μM) + OGD group; cP < 0.05, vs. the NBP (10 μM) + OGD group (one-way analysis of variance with the least significant difference post hoc test).
NBP enhanced OGD-induced HIF-1α expression
In order to investigate whether HIF-1α was involved in the protective effect of NBP, we examined HIF-1α expression by immunofluorescence (Figures 2A, B) and western blot (Figures 2C, D). As shown in Figure 2, little HIF-1α expression was detected in BMECs under normal culture conditions, and similar HIF-1α expression was detected when the BMECs were pretreated with NBP. After BMECs were exposed to 2-hour OGD and 24-hour reperfusion, HIF-1α expression was significantly increased compared with the control. The protein expression of HIF-1α was extraordinarily increased in the OGD + NBP group compared with the OGD group.
Figure 2.
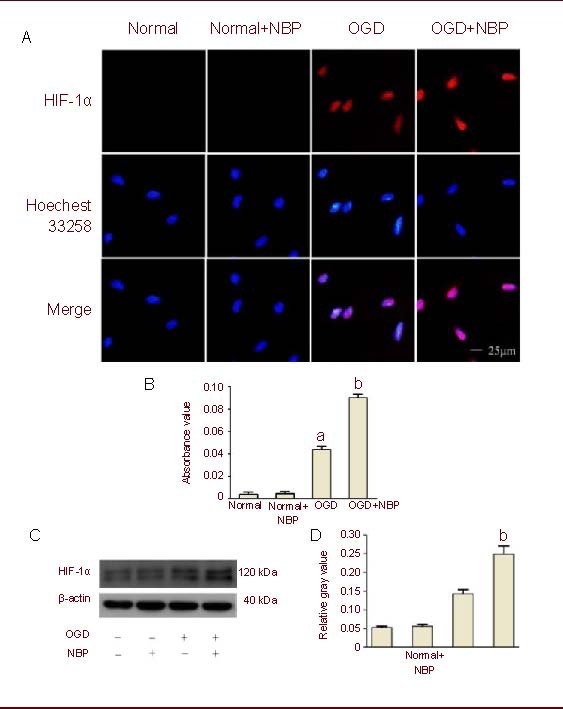
DL-3-n-butylphthalide (NBP) enhanced oxygen glucose deprivation (OGD)-induced expression of hypoxia inducible factor-1α (HIF-1α).
(A) Brain microvascular endothelial cells (BMECs) underwent 2-hour OGD and 24-hour reperfusion after 1-hour pretreatment with NBP (100 μM). Digital photomicrograph under fluorescent illumination shows the expression of HIF-1α (blue: Hoechst; red: Cy5).
(B) Bar graph shows the average quantitative absorbance of HIF-1α through Image-Pro Plus analysis of immunofluorescence. The level of HIF-1α expression was indicated by the value of absorbance over the area with fluorescence.
(C) Expression of HIF-1α was detected by western blot method. Blots were probed with antibody against HIF-1α, and β-actin was used as a control.
(D) Bar graph shows semi-quantitative densitometry from western blot analysis. HIF-1α was significantly increased in BMECs exposed to OGD insult, whereas NBP (100 μM) significantly enhanced OGD-induced HIF-1α expression.
Values were expressed mean ± SEM. The results were obtained from five independent experiments performed in triplicate. aP < 0.05, vs. the normal group (control); bP < 0.05, vs. the OGD group (one-way analysis of variance with the least significant difference post hoc test).
NBP had no protective effect on HIF-1α knockdown BMECs undergoing OGD injury
To further explore the mechanisms underlying the protective effect of NBP, knockdown of HIF-1α gene in BMECs was performed by transfection with HIF-1α small interfering RNA (siRNA). The downregulation of HIF-1α expression was confirmed by western blot analysis. As shown in Figure 3C, the protein expression of HIF-1α in BMECs was extraordinarily decreased after transfection with HIF-1α siRNA. NBP (100 μM) significantly reduced cell apoptotic rate in BMECs undergoing OGD injury (4.4% vs. 14.2%), but could not protect BMECs with HIF-1α knockdown which underwent OGD injury (the cell apoptotic rate was 28.6% and 27.4%, respectively) (Figures 3A, B).
Figure 3.
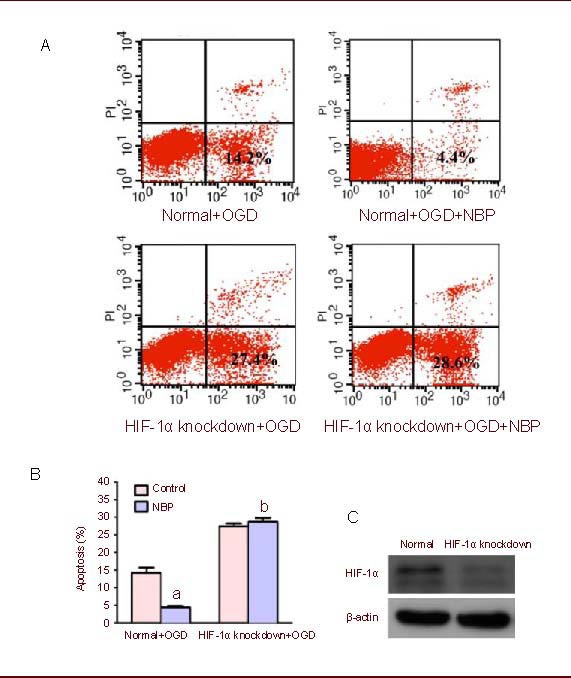
DL-3-n-butylphthalide (NBP) had no protective effect on hypoxia inducible factor-1α (HIF-1α) knockdown brain microvascular endothelial cells (BMECs) exposed to oxygen glucose deprivation (OGD).
(A) BMECs and BMECs with HIF-1α knockdown underwent 2-hour OGD and 24-hour reperfusion after pretreatment with NBP (100 μM) for 1 hour. Cell apoptosis (low right) was indicated by flow cytometric analysis.
(B) Bar graph shows that NBP has protective effect on normal BMECs, but has no protective effect on HIF-1α knockdown BMECs exposed to OGD.
Values were expressed as mean ± SEM. The results were obtained from five independent experiments performed in triplicate. Normal means non-HIF-1α knockdown. aP < 0.05, vs. the normal + OGD group; bP < 0.05, vs. normal + OGD + NBP group (chi-square test).
(C) The knockdown of HIF-1α gene in BMECs was performed by transfection with HIF-1α siRNA. Western blot analysis showed the effect of RNA interference targeting HIF-1α.
Effect of NBP and HIF-1α knockdown on the mRNA expression of bcl-2 and caspase-3 in BMECs
In order to explore the possible mechanism by which NBP inhibits OGD-mediated cell apoptosis, we investigated the effects of NBP and HIF-1α knockdown in BMECs on the mRNA expression of bcl-2 and caspase-3 by real-time RT-PCR. As shown in Figure 4, the bcl-2 mRNA expression was downregulated in BMECs undergoing OGD injury compared with the control, and the downregulation of bcl-2 mRNA expression induced by OGD could be reversed by NBP. In BMECs with knockdown of HIF-1α gene, the downregulation of bcl-2 mRNA expression induced by OGD could not be reversed by NBP. The caspase-3 mRNA expression was upregulated in BMECs exposed to OGD compared with the control, and the upregulation of caspase-3 mRNA expression induced by OGD could be reversed by NBP. In BMECs with knockdown of HIF-1α gene, the upregulation of caspase-3 mRNA expression induced by OGD could not be reversed by NBP.
Figure 4.
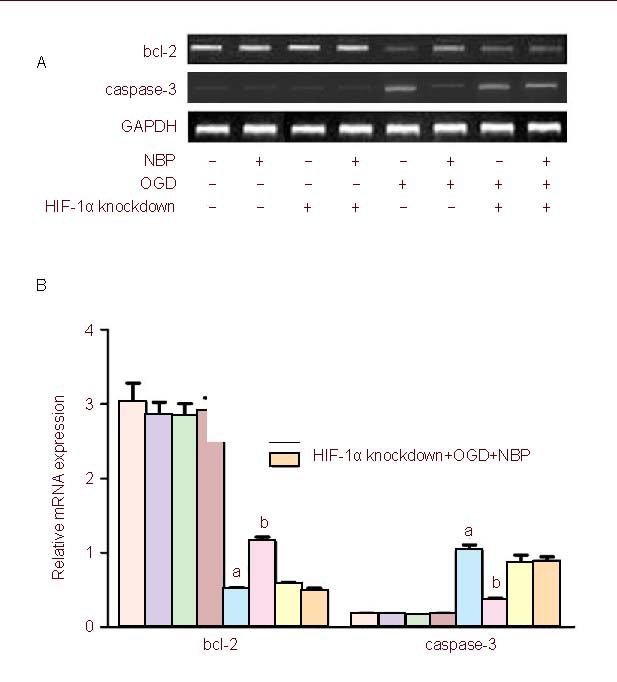
Quantitative analysis of the mRNA expression of hypoxia inducible factor-1α (HIF-1α), bcl-2 and caspase-3 by real-time RT-PCR.
Glyceraldehyde-3- phosphate dehydrogenase (GAPDH) was used as a loading control.
(A) While transfected with HIF-1α RNA interference, the upregulation of bcl-2 and downregulation of caspase-3 by DL-3-n-butylphthalide (NBP) were inhibited when brain microvascular endothelial cells were exposed to oxygen glucose deprivation (OGD).
(B) The bar graphs represent the data expressed as fold-increases over controls. Values were expressed as mean ± SEM.
The results were obtained from five independent experiments performed in triplicate. aP < 0.05, vs. the normal group; bP < 0.05, vs. normal + OGD group (one-way analysis of variance with the least significant difference post hoc test).
DISCUSSION
In order to investigate the protective mechanism of NBP for ischemic stroke, we used an OGD model, which has become an important in vitro experimental tool for investigations of ischemic stroke[9].
HIF-1α is one of the master regulators that regulate the cellular responses to hypoxia. HIF-1α protein is absent or nearly absent in most normoxic cells[10]. With the onset of hypoxia, oxygen-sensing mechanisms immediately stabilize the HIF-1α protein, in addition, hypoxia activates HIF-1α transcription, resulting in more HIF-1α protein[11,12]. Consistent with previous observations[8], we found that normal BMECs expressed little HIF-1α protein, and BMECs which underwent OGD expressed increased HIF-1α protein. In addition, NBP made no difference to the expression of HIF-1α protein in normal BMECs, but it further upregulated HIF-1α protein expression in BMECs that underwent OGD.
Elevated HIF-1α was approved to play protective roles in reducing cellular apoptosis in ischemia brain damage in many studies[13]. By controlling the transcription of hundreds of genes HIF-1α drives, many cellular processes such as angiogenesis were activated[14]. The present study found that HIF-1α protein expression was increased after BMECs underwent OGD insult, and the upregulation of HIF-1α protein expression induced by NBP was associated with the protective effect of NBP to OGD.
An interesting finding in this study is that NBP can only upregulate HIF-1α protein expression in BMECs in hypoxic condition. It is accepted that the expression of HIF-1α chains is mostly, if not exclusively, regulated at the post-translational level, by members of the prolyl hydroxylase domains (PHDs) family[15]. Hydroxylation of HIF-1α by PHDs makes HIF-1α protein degradation. In the setting of hypoxic stress, PHD activity is diminished, and HIF-1α protein is stabilized and accumulated[16]. A number of drug-like PHD antagonists have now been developed that can stabilize HIF-1α in vitro and in vivo, leading to reduction of neuronal damage and behavioral deficits, which correlate with the enhanced expression of HIF-1α protein and HIF-1-regulated gene expression in peri-infarct cortical tissue[17,18]. It is possible that NBP may upregulate HIF-1α protein expression via influencing the activity of PHDs. Since the hydroxylation of HIF-1α by PHDs is modulated by multiple factors, such as tricarboxylic acid circle, iron, ascorbate, reactive oxygen species and nitric oxide[19], NBP may affect one or more of such factors as above to modulate the activity of PHDs. Another possibility is that PHDs is relatively abundant for the hydroxylation of HIF-1α in normoxic condition[20,21]. NBP, if it can reduce the activity of PHDs, may have little effect on the expression of HIF-1α protein. In hypoxic condition, the activity of PHDs is inhibited, and reduction of PHDs activity may result in further upregulation of HIF-1α protein expression. Whether NBP upregulates HIF-1α protein expression via influencing the activity of PHDs will be detected in our future study. Our previous study demonstrated that NBP can prevent the occurrence of ischemic stroke[4], reduce OGD-induced mitochondrial fragmentation and loss of mitochondrial membrane potential in HUVECs and eventually prevent against OGD injury[8]. The intrinsic apoptotic pathway, which is also called the bcl-2-regulated or mitochondrial pathway, is strictly controlled by the bcl-2 family of proteins and by which caspases are crucial for their execution[22,23]. In this study, we demonstrated that NBP can upregulate bcl-2 expression and downregulate caspases-3 expression while BMECs are undergoing OGD injury, and finally prevent cell apoptosis. This suggests that the protective effect of NBP on OGD injury might be associated with the reduction of intrinsic apoptosis in BMECs, and this is caused by upregulation of HIF-1α expression. The central role of endothelial cells in the pathobiology of cerebral blood vessels has been encompassed under the term NVU[24]. There is increasing experimental evidence showing that BMECs are not just inert tubes for delivering blood flow, oxygen, and glucose into brain, they also secrete trophic factors that can be directly neuroprotective[25]. BMEC damage during ischemia often leads to disruption of the blood-brain barrier and dysregulation of vascular tone, eventually causing substantial neuronal death[26]. BMECs might serve as active sources of neurotrophic mediators that help defend neuronal tissue against injury and disease[27]. For this reason, NBP may protect neurons via protecting BMECs and retaining the neurotrophic mediators secreted by BMECs.
In a word, NBP can protect BMECs against OGD injury possibly by upregulation of HIF-1α and bcl-2 expression and downregulation of caspase-3 expression. Thus stimulation of angiogenesis as the novel strategy for treating ischemic stroke represents a window of opportunity for addressing and improving brain ischemia outcome[28].
MATERIALS AND METHODS
Design
A controlled observational study.
Time and setting
This study was performed in the First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, Guangdong Province, China.
Materials
Sprague-Dawley rats were purchased from the Animal Research Center of Sun Yat-Sen University (certification No. 2008A063). NBP (solution, generous gift from Shijiazhuang Pharma Group NBP Pharmaceutical Co., Ltd., Lot No. 0906251) was dissolved in dimethyl sulfoxide before dilution with the culture medium. The final concentration of dimethyl sulfoxide per well was 0.1%.
Methods
Primary BMEC culture
Primary BMECs were cultured from postnatal 3- to 4-day-old Sprague-Dawley rat pups as previously described with some modifications[29,30]. Briefly, rats were decapitated, and the cerebral cortex was dissected and homogenized in ice-cold phosphate buffered saline. The suspension was centrifuged at 400 × g, 4°C for 5 minutes. Cells were digested for 1.5 hours at 37°C in 0.1% type II collagenase (Merck, Germany) to separate microvessels from other components. Then, the supernatant was removed, and 20% bovine serum albumin was added and mixed. The mixture was centrifuged at 1 000 × g for 20 minutes and washed with phosphate buffered saline. 2 mL of 0.1% collagenase/dispase (Roche, Switzerland) was added and the suspension was incubated for 1 hour at 37°C. Then the cells were washed and resuspended in 2 mL Dulbecco's modified Eagle's medium, layered onto the top of the Percoll gradient and centrifuged at 1 000 × g for 20 minutes. The microvessel layer that contained BMECs exhibited a yellow-white band and localized primarily within the 30% (1.043 g/mL) Percoll gradients. The BMECs were collected, resuspended with Dulbecco's modified Eagle's medium supplemented with 20% heat-inactivated fetal bovine serum, 20 μg/L basic fibroblast growth factor (Millipore, Bedford, MA, USA) and 20 μg/L epidermal growth factor (Invitrogen, Carlsbad, CA, USA), and then seeded onto 1% gelatin-coated 25-cm2 tissue culture dishes. Endothelial cells were split at a ratio of 1:2 and used at passages 2 to 3. The character of the cells was routinely checked by immunocytochemical evaluation using polyclonal rat anti-human primary factor VIII antibody (1:200 dilution).
OGD and NBP intervention
The final concentrations of NBP were 0.1, 1, 10, 100 and 1 000 μM. Briefly, the culture medium was changed to fresh culture medium with different concentrations of NBP at 1 hour before OGD. For OGD, cells were exposed to glucose-free Dulbecco's modified Eagle's medium and placed into an anaerobic chamber that was flushed with 5% CO2 and 95% N2 (v/v) for 30 minutes at 37°C. The chamber was then sealed and kept at 37°C for the indicated time periods for hypoxia followed by a return to normoxic conditions with fresh normal medium for 24 hours. Control cultures were incubated in a regular incubator under normoxic conditions with fresh normal medium for the corresponding hypoxic duration.
Detection of cell viability by MTT assays and Hoechst staining
MTT assays were used to detect the cell viability. Briefly, 10 μL of the MTT labeling reagent at a final concentration of 0.5 mg/mL was added into each well for 4 hours to allow formation of purple formazan crystal. Four hours later, 100 μL of the solubilization reagent was added into each well. Finally, the spectrophotometric absorbance of the solubilized purple formazan crystals was measured using a microplate reader at an absorbance wavelength of 570 nm. All the MTT results of the OGD groups were normalized and expressed as the percentage of the average absorbance reading of the sham-OGD group (normal control)[8]. Cell viability was also evaluated using nuclear morphology following staining with the fluorescent dye, Hoechst 33258 (Sigma, St. Louis, MO, USA) after OGD. Briefly, BMECs were cultured in 4-well chamber slides till 85% confluency. After exposure to OGD, cells were stained with 1 μL of Hoechst 33258 (5 mg/mL) in 1 mL of basal medium and incubated for 10 minutes. Stained cells were washed twice with phosphate buffered saline and imaged under a fluorescent microscope (Olympus, Tokyo, Japan).
Western blot analysis for HIF-1α expression
BMECs were scraped off in lysis buffer with phenylmethanesulfonyl, centrifuged for 10 minutes at 13 000 × g (4°C). The concentration of BMEC protein was determined using a BCA Protein Assay Kit (Pierce, IL, USA). Protein samples were electrophoresed in precast SDS-polyacrylamide gel electrophoresis 6% gradient gels (Bio-Rad, Hercules, CA, USA). The proteins were transferred to polyvinylidene fluoride membranes, blocked with 5% milk diluted in Tris-buffered saline Tween-20, and immunoblotted overnight at 4°C with the mouse anti-human HIF-1α antibody (Abcam, 1:500 dilution). Anti-β-actin (Sigma; 1:1 000 dilution) antibody was used as the loading control followed by anti-mouse secondary antibody conjugated with horseradish peroxidase (1:5 000 dilution). The intensity of each band was measured and analyzed with a Chemi Doc XRS imaging system (Bio-Rad). Data were given as percentage of vehicle control and β-actin was used as an internal control.
Immunofluorescence for HIF-1α expression
Cells were fixed with 4% formaldehyde for 30 minutes and permeabilized with 0.3% Triton X-100 for 30 minutes. Following blockade of nonspecific binding sites by incubation with goat serum for 30 minutes, the cells were incubated for 2 hours at 4°C with mouse anti-human HIF-1α monoclonal antibody (1:200 dilution), followed by incubation with Cy5-coupled secondary goat anti-mouse antibody (Invitrogen). For nuclear counter staining, cells were incubated in 5 mg/mL Hoechst 33258 for 5 minutes after immunostaining. For negative control, sections stained without primary antibodies showed no signals. The slides were mounted and analyzed under an inverted fluorescence microscope (Olympus). The fluorescence intensities were measured with the software Image-Pro Plus (IPP) (Media Cybernetics, Silver Spring, MD, USA).
Knockdown of HIF-1α in BMECS
A short interfering RNA targeting rat HIF-1α was designed and synthesized by GeneChem, Shanghai, China. The sequences of HIF-1α-siRNA were as follows: 5’-CTC CAT TAC CTG CCT CTG AAA-3’ and 5’-TTT CAG AGG CAG GTA ATG Gag-3’. The lipofectins were then transfected into the BMECs (passage 3) using Lipofectamine™ 2000 (Invitrogen) according to the manufacturer's instructions. After being selected with G418 (Merck) at a working concentration of 600 μg/mL, resistant colonies were obtained. Western blot analysis was performed to identify effective RNAi against HIF-1α expression following stable transfection. The procedure of western blot analysis was shown as above.
Semi-quantitative RT-PCR assay for bcl-2 and caspase-3 mRNA expression
Total RNA was extracted from BMECs using Trizol (Invitrogen). RNA was reverse transcribed with oligo (dT) and M-MLV reverse transcriptase (Fermentas, CA). Quantitative changes in bcl-2 and caspase-3 mRNA expression of BMECs were assessed with a real-time quantitative PCR method[31]. GAPDH was chosen as an internal control. The primers are shown in Table 1. Six samples were used for each condition and each sample was performed in triplicate. The PCR mixture included 10 μL of 2 × PCR buffer, 0.6 μL of Primer F, 0.6 μL of Primer R, 2 μL of cDNA and 6.8 μL double distilled water in a final volume of 20 μL. The light cycler software version 4.0 (Roche, IN, USA) was used for instrument control, automated data collection and data analysis. Relative quantification of the mRNA expression levels of target genes were calculated using the 2−△△CT method[32].
Flow cytometric analysis using annexin V-FITC and propidium iodide staining for cell apoptosis
For apoptosis analysis, harvested cells were stained with Annexin V-FITC and propidium iodide according to the manufacturer's instruction, then analyzed with a flow cytometer (BD Biosciences, NJ, USA) as described previously[33].
Statistical analysis
Data were expressed as mean ± SEM. The comparisons of multiple quantitative variables were analyzed using one-way analysis of variance with the least significant difference post hoc test. A chi-square test was used to analyze categorical variables. The analyses were performed using SPSS software. P < 0.05 was considered to be statistically significant
Footnotes
Funding: This study was supported by the National Natural Science Foundation of China, No. 30471917 and 30770766.
Conflicts of interest: None declared.
Ethical approval: This study was approved by the Animal Ethics Committee, Sun Yat-Sen University, China.
(Edited by Tan S, Zhu QH/Song LP)
REFERENCES
- 1.Shuaib A, Lees KR, Lyden P, et al. NXY-059 for the treatment of acute ischemic stroke. N Engl J Med. 2007;357(6):562–571. doi: 10.1056/NEJMoa070240. [DOI] [PubMed] [Google Scholar]
- 2.Lo EH, Rosenberg GA. The neurovascular unit in health and disease: introduction. Stroke. 2009;40(3 Suppl):S2–3. doi: 10.1161/STROKEAHA.108.534404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- 4.Liu CL, Liao SJ, Zeng JS, et al. Dl-3n-butylphthalide prevents stroke via improvement of cerebral microvessels in RHRSP. J Neurol Sci. 2007;260(1-2):106–113. doi: 10.1016/j.jns.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 5.Zhang T, Yan W, Li Q, et al. 3-n-butylphthalide (NBP) attenuated neuronal autophagy and amyloid-beta expression in diabetic mice subjected to brain ischemia. Neurol Res. 2011;33(4):396–404. doi: 10.1179/1743132810Y.0000000006. [DOI] [PubMed] [Google Scholar]
- 6.Li J, Li Y, Ogle M, et al. DL-3-n-butylphthalide prevents neuronal cell death after focal cerebral ischemia in mice via the JNK pathway. Brain Res. 2010;1359:216–226. doi: 10.1016/j.brainres.2010.08.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao SJ, Lin JW, Pei Z, et al. Enhanced angiogenesis with dl-3n-butylphthalide treatment after focal cerebral ischemia in RHRSP. Brain Res. 2009;1289:69–78. doi: 10.1016/j.brainres.2009.06.018. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Zhang B, Tao Y, et al. DL-3-n-butylphthalide protects endothelial cells against oxidative/nitrosative stress, mitochondrial damage and subsequent cell death after oxygen glucose deprivation in vitro. Brain Res. 2009;1290:91–101. doi: 10.1016/j.brainres.2009.07.020. [DOI] [PubMed] [Google Scholar]
- 9.Iijima T. Mitochondrial membrane potential and ischemic neuronal death. Neurosci Res. 2006;55(3):234–243. doi: 10.1016/j.neures.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 10.Czibik G. Complex role of the HIF system in cardiovascular biology. J Mol Med (Berl) 2010;88(11):1101–1111. doi: 10.1007/s00109-010-0646-x. [DOI] [PubMed] [Google Scholar]
- 11.Bernaudin M, Tang Y, Reilly M, et al. Brain genomic response following hypoxia and re-oxygenation in the neonatal rat. Identification of genes that might contribute to hypoxia-induced ischemic tolerance. J Biol Chem. 2002;277(42):39728–39738. doi: 10.1074/jbc.M204619200. [DOI] [PubMed] [Google Scholar]
- 12.Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7(8):345–350. doi: 10.1016/s1471-4914(01)02090-1. [DOI] [PubMed] [Google Scholar]
- 13.Ratan RR, Siddiq A, Smirnova N, et al. Harnessing hypoxic adaptation to prevent, treat, and repair stroke. J Mol Med (Berl) 2007;85(12):1331–1338. doi: 10.1007/s00109-007-0283-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pugh CW, Ratcliffe PJ. Regulation of angiogenesis by hypoxia: role of the HIF system. Nat Med. 2003;9(6):677–684. doi: 10.1038/nm0603-677. [DOI] [PubMed] [Google Scholar]
- 15.Fong GH, Takeda K. Role and regulation of prolyl hydroxylase domain proteins. Cell Death Differ. 2008;15(4):635–641. doi: 10.1038/cdd.2008.10. [DOI] [PubMed] [Google Scholar]
- 16.Kaelin WJ, Ratcliffe PJ. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 17.Siddiq A, Ayoub IA, Chavez JC, et al. Hypoxia-inducible factor prolyl 4-hydroxylase inhibition. A target for neuroprotection in the central nervous system. J Biol Chem. 2005;280(50):41732–41743. doi: 10.1074/jbc.M504963200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ogle ME, Gu X, Espinera AR, et al. Inhibition of prolyl hydroxylases by dimethyloxaloylglycine after stroke reduces ischemic brain injury and requires hypoxia inducible factor-1alpha. Neurobiol Dis. 2012;45(2):733–742. doi: 10.1016/j.nbd.2011.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berchner-Pfannschmidt U, Yamac H, Trinidad B, et al. Nitric oxide modulates oxygen sensing by hypoxia-inducible factor 1-dependent induction of prolyl hydroxylase 2. J Biol Chem. 2007;282(3):1788–1796. doi: 10.1074/jbc.M607065200. [DOI] [PubMed] [Google Scholar]
- 20.Appelhoff RJ, Tian YM, Raval RR, et al. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem. 2004;279(37):38458–38465. doi: 10.1074/jbc.M406026200. [DOI] [PubMed] [Google Scholar]
- 21.Berra E, Ginouves A, Pouyssegur J. The hypoxia-inducible-factor hydroxylases bring fresh air into hypoxia signalling. EMBO Rep. 2006;7(1):41–45. doi: 10.1038/sj.embor.7400598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9(1):47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 23.Marsden VS, O’Connor L, O’Reilly LA, et al. Apoptosis initiated by Bcl-2-regulated caspase activation independently of the cytochrome c/Apaf-1/caspase-9 apoptosome. Nature. 2002;419(6907):634–637. doi: 10.1038/nature01101. [DOI] [PubMed] [Google Scholar]
- 24.Guo S, Lo EH. Dysfunctional cell-cell signaling in the neurovascular unit as a paradigm for central nervous system disease. Stroke. 2009;40(3 Suppl):S4–7. doi: 10.1161/STROKEAHA.108.534388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kallmann BA, Wagner S, Hummel V, et al. Characteristic gene expression profile of primary human cerebral endothelial cells. FASEB J. 2002;16(6):589–591. doi: 10.1096/fj.01-0594fje. [DOI] [PubMed] [Google Scholar]
- 26.Arai K, Jin G, Navaratna D, et al. Brain angiogenesis in developmental and pathological processes: neurovascular injury and angiogenic recovery after stroke. FEBS J. 2009;276(17):4644–4652. doi: 10.1111/j.1742-4658.2009.07176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guo S, Kim WJ, Lok J, et al. Neuroprotection via matrix-trophic coupling between cerebral endothelial cells and neurons. Proc Natl Acad Sci U S A. 2008;105(21):7582–7587. doi: 10.1073/pnas.0801105105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dirnagl U, Becker K, Meisel A. Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol. 2009;8(4):398–412. doi: 10.1016/S1474-4422(09)70054-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo Y, Shi D, Li W, et al. Proliferation and neurogenesis of neural stem cells enhanced by cerebral microvascular endothelial cells. Microsurgery. 2008;28(1):54–60. doi: 10.1002/micr.20443. [DOI] [PubMed] [Google Scholar]
- 30.Abbott NJ, Hughes CC, Revest PA, et al. Development and characterisation of a rat brain capillary endothelial culture: towards an in vitro blood-brain barrier. J Cell Sci. 1992;103(Pt 1):23–37. doi: 10.1242/jcs.103.1.23. [DOI] [PubMed] [Google Scholar]
- 31.Stephens AS, Stephens SR, Morrison NA. Internal control genes for quantitative RT-PCR expression analysis in mouse osteoblasts, osteoclasts and macrophages. BMC Res Notes. 2011;4:410. doi: 10.1186/1756-0500-4-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25(4):402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 33.Guo D, Hou X, Zhang H, et al. More expressions of BDNF and TrkB in multiple hepatocellular carcinoma and anti-BDNF or K252a induced apoptosis, supressed invasion of HepG2 and HCCLM3 cells. J Exp Clin Cancer Res. 2011;30:97. doi: 10.1186/1756-9966-30-97. [DOI] [PMC free article] [PubMed] [Google Scholar]