

Abstract
The cAMP response element (CRE)-binding protein, CREB, is a transcription factor whose activity in the brain is critical for long-term memory formation. Phosphorylation of Ser133 in the kinase-inducible domain (KID), that in turn leads to the recruitment of the transcriptional coactivator CREB-binding protein (CBP), is thought to mediate the activation of CREB. However, the importance of phosphorylation for CREB binding to DNA and subsequent gene transcription in vivo is controversial. To definitively address the role of CREB phosphorylation in gene transcription and learning and memory, we derived mutant mice lacking the Ser133 phosphorylation site. These mice exhibit normal CREB-mediated gene transcription for a number of genes implicated in learning and memory processes. Furthermore these mice have no deficits in hippocampus- or striatum-dependent learning. Strikingly, our findings show that CREB phosphorylation at Ser133 is not necessary for CREB binding to CRE sites, CREB-mediated transcription, or CREB-mediated behavioral phenotypes associated with learning and memory.
The cyclic AMP response element (CRE)-binding protein, CREB, is a member of a family of structurally related transcription factors that bind to conserved cAMP response element (CRE) sites. CREB binds to over 30,000 CRE sites and regulates >5000 target genes, roughly one quarter of the human genome (Impey et al. 2004; Zhang et al. 2005), and plays a critical role in learning, memory, addiction, and depression (Carlezon et al. 2005; Blendy 2006). It has long been held that the critical event in the activation of CREB is phosphorylation of Ser133 in the kinase-inducible domain (Gonzalez and Montminy 1989; Gonzalez et al. 1991). As CREB phosphorylation can result from the activation of multiple signaling cascades, including phosphorylation by protein kinase A, protein kinase C, and calmodulin kinases, this event has been proposed as a mechanism for the convergence of these signaling pathways (Brindle and Montminy 1992; Sassone-Corsi 1995).
Converging evidence from Aplysia, Drosophila, mice, and rats indicates that CREB is critical for long-term memory formation (Yin and Tully 1996; Silva et al. 1998; Carlezon et al. 2005). In particular, CREBα▵ and CREBIR mutant mice exhibit deficits in contextual and cued fear learning, while viral overexpression of CREB leads to enhanced fear learning (Bourtchuladze et al. 1994; Josselyn et al. 2001; Graves et al. 2002; Kida et al. 2002; Restivo et al. 2009). Similarly, alterations in CREB activity also lead to bidirectional effects on spatial memory performance (Sekeres et al. 2010). Furthermore, alterations in CREB expression are accompanied by alterations in cell excitability, synaptic plasticity, hippocampal neurogenesis, and neuronal survival (Barco et al. 2002; Nakagawa et al. 2002; Fujioka et al. 2004; Marie et al. 2005; Dong et al. 2006; Gur et al. 2007; Suzuki et al. 2011).
Because of the proposed critical role for CREB phosphorylation in the pathway activation outlined above, we investigated whether CREB phosphorylation at Ser133 is indeed necessary for fear learning. To this end, we derived a mouse with a single point mutation that prevents CREB phosphorylation at Ser133 and evaluated these mice for defects in both hippocampus- and striatum-dependent learning. Additionally, we examined whether CREB-mediated gene transcription was altered in the phospho mutant mice either at baseline or following learning. Remarkably, we find that CREB phosphorylation at Ser133 is not required for any of these processes.
Materials and Methods
Derivation of CREB S133A mice
A 15.1-kb DNA fragment, containing exon 7 of the Creb1 gene, was retrieved from C57BL/6J mouse BAC clone RP23-31C24 via bacterial recombination (Copeland et al. 2001). The TCC sequence within exon 7 encoding S133 was mutated to GCC (S133A) by site-directed mutagenesis and cloned into the retrieved genomic sequence. A silent mutation of CCT to CCC in the P132 codon was also included in order to disrupt a StuI restriction enzyme site. The self-excising selection cassette based on Bunting et al. (1999), with the exception of the replacement of the PollI promoter with the PGK promoter to allow for kanamycin resistance in bacteria, was also inserted 5′ of the mutated exon 7 to complete the targeting vector. The linearized targeting vector was electroporated into C57BL/6J-derived ES cells (Chemicon), and clones surviving G418 selection were screened for homologous recombination by Southern blot analysis. Targeted clones were injected into C57BL/6J-derived blastocyts that were then transferred to pseudopregnant females. Male offspring were mated to C57BL/6J females and ES cell-derived offspring, heterozygous for the S133A allele, were identified by PCR-based genotyping. Additional genotyping for sequences within the selection cassette was performed to ensure that proper and complete recombination between the loxP sites had occurred in the male germline. The sequence of the genotyping primers are as follows: forward: AGGGCATGGCAGTACATAG; reverse: CATGTGCTTTCCTCCATGTG. Mice were backcrossed at least five generations with C57BL/6 to eliminate any potential mutations carried by the ES cells. CREBαΔ mutant mice and wild-type littermates were bred and maintained on a F1 hybrid background (129SvEvTac/ C57BL/6NTac) as described previously (Walters and Blendy 2001). All animals were housed in a temperature and humidity controlled animal care facility with a 12 h light/dark cycle (lights on at 7:00 a.m.). All procedures were approved by the University of Pennsylvania Animal Care and Use Committee. Equal numbers of male and female mice were utilized for all experiments. No significant sex differences were seen in any of the behavioral or molecular assays and therefore the data from both sexes are combined in all analyses.
Fear conditioning
Briefly, mice were placed in a rectangular (16 in length × 6 1/4 in width × 8 3/8 in height) fear-conditioning chamber (Med Associates) and presented with two tone (85 dB, 2 kHz)/footshock (0.7 mA) pairings. The mice were given 2.5 min to explore the box prior to the onset of the first tone (conditioning stimulus; CS), which preceded the 2-sec footshock by 28 sec. The second pairing occurred 1 min later and the mice were removed from the chamber after 5 min. The chamber was washed with 70% ethanol between animals. Mice were tested for context conditioning in the same chamber at 24 h after training. For context testing, mice were placed into the unaltered chamber for 5 min. For testing of cued conditioning, the floor and sides were replaced with smooth, black panels. During the cued test, a water solution containing 1% orange extract was placed on a cotton ball underneath the floor and between subjects the chamber was wiped down with this mixture. The fan was disconnected. For cued testing, the mice were placed in the altered chamber for 5 min in total, and they were exposed to the CS for the last 3 min. Mice that were tested for contextual conditioning at 24 h after training were then tested for cued conditioning 1 h later. Freezing behavior was assessed with FreezeScan analysis software (Clever Sys) and confirmed by hand scoring the data. A separate group of mice was exposed to contextual fear conditioning alone without any presentation of tones during training. A separate group of mice was trained on fear conditioning to generate tissue for RT-PCR experiments.
Shock sensitivity
To determine whether CREB S133A mice exhibit alterations in shock sensitivity that might be masking effects on the fear-conditioning behavior, we exposed mice to a range of 2-sec footshocks (0.10–0.80 mA) over a 20-min testing period. There were three presentations of each shock intensity, with a 20-sec interstimulus interval and a 90-sec intertrial interval. We scored the animals response for each shock presentation (0 = no response, 1 = flinch, 2 = vocalization, 3 = run, 4 = jump) and averaged the three responses at each shock intensity (scoring adapted from Gulick and Gould 2009).
Instrumental learning
Mice were placed in operant chambers (Med Associates) and trained to spin a wheel manipulandum to receive a sucrose pellet. A compound cue stimulus consisting of a cue light above the active lever, a 2900-Hz tone, and house light off was concurrent with each pellet administration, followed by an additional 8-sec time-out when responding had no programmed consequences and the house light remained off. Mice were allowed to self-administer a maximum of 50 pellets per 60 min operant session. During these experiments, mice were food restricted to ∼90% of their free-feeding weight. The average percent active responding was calculated for 3 d after 7 d of training.
Western blot
Mice were taken directly from their home cages and killed by cervical dislocation. Whole hippocampi were dissected and frozen in liquid nitrogen. Tissue was homogenized in 200 μL of ice-cold extraction buffer containing PBS, 1 mM EGTA, 1 mM EDTA, 0.01% SDS, and 1 mM PMSF. Protein concentration was quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). Equal amounts of protein (30 μg for whole cell) were loaded and separated in 10% polyacrylamide Tris–Glycine gels (Invitrogen) and transferred to nitrocellulose membranes using the i-Blot dry transfer system (Invitrogen). Membranes were blocked with Li-Cor blocking buffer. Membranes were incubated for 24 h at 4°C with selective antibodies to: CREB (1:1000; Santa Cruz), pCREB (1:1000, Millipore), TORC1 (1:1000, Cell Signaling), and GAPDH (1:2000, Cell Signaling). Membranes were then incubated with fluorescent secondary antibodies (1:5000, IR-dye 680 or IR-dye 800), before being imaged on an Odyssey fluorescent scanner (Licor Biosciences). GAPDH was used as an internal loading control.
RNA extraction, cDNA synthesis, and quantitative real-time polymerase chain reaction
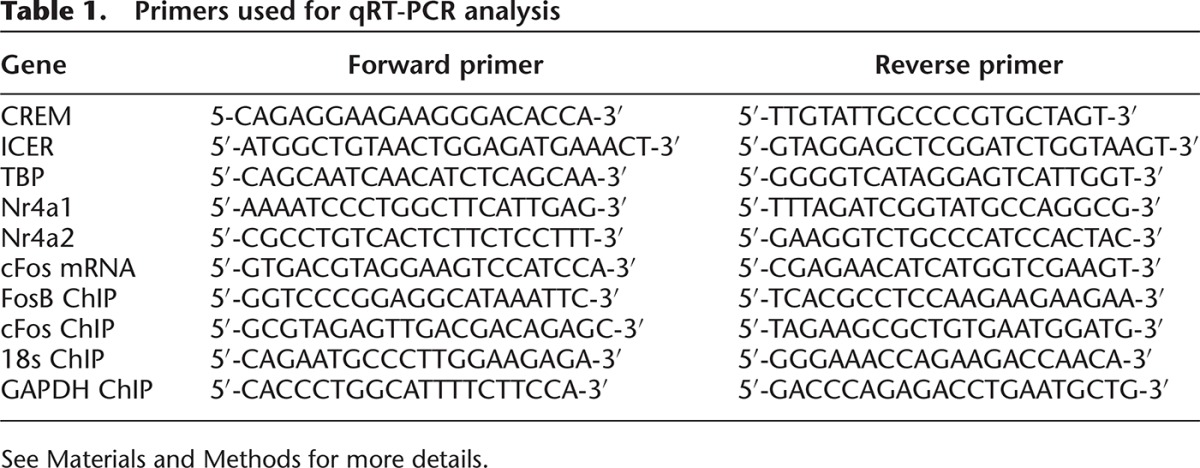
Mice were killed by cervical dislocation directly from their home cages or 30 min following fear-conditioning training for the evaluation of early gene expression changes. Brains were rapidly removed, whole hippocampi hand-dissected, and frozen in liquid nitrogen. RNA was extracted from hippocampal tissue using TRIzol/chloroform (Invitrogen) and the RNeasy Mini kit (Quiagen). cDNA was synthesized from RNA using an oligo(dT) primer (Operon) and Superscript II reverse transcriptase (Invitrogen). Quantitative real-time polymerase chain reaction (QPCR) was carried out using the SYBR-green master mix (Applied Biosystems) and 300 nM primers (final concentration) on the Stratagene MX3000 using MXPro QPCR software. Cycling parameters were 95°C for 10 min followed by 40 cycles of 95°C (30 sec) and 60°C (1 min), ending with a melting curve analysis to assess the amplification of a single amplicon. All reactions were performed in triplicate, with the median cycle time used for analysis. TATA-box binding protein (TBP) was used as a housekeeping gene against whose levels all experimental genes were normalized. Primer sequences can be found in Table 1.
Table 1.
Primers used for qRT-PCR analysis
Chromatin immunoprecipitation
Macrodissected brain tissue was chopped and suspended in 1.1% formaldehyde/PBS for 10 min at room temperature with constant agitation. Cross-linking was quenched by the addition of glycine to a final concentration of 0.125 M, after which the tissue was sedimented by centrifugation. Pellets were resuspended in ChIP cell lysis buffer (10 mM Tris–HCl, pH 8.0, 10 mM NaCl, 3 mM MgCl2, 1% NP-40, protease inhibitors) and dounce homogenized (Kontes, 18 × 150-mm tube reservoir; 8 × 220-mm pestle shaft). Chromatin was sonicated to an optimal 200-bp fragment size using a Diagenode Bioruptor for five cycles (5 min; 30-sec on, 30-sec off). Insoluble debris was removed by centrifugation at 13,000g for 15 min at 4°C, supernatant was collected and flash-frozen in liquid nitrogen. Cross-linking was reversed by the addition of NaCl to a final concentration of 192 mM, overnight incubation at 65°C, and purification using a PCR purification kit (Qiagen). Immunoprecipitations were performed as previously described (Friedman et al. 2004) with anti-CREB (sc-186, Santa Cruz), anti-trimethyl-Histone H3 (Lys4) (07-473, Millipore), or control IgG (sc-2025, Santa Cruz) antibodies. Promoter enrichment was quantified using QPCR and calculated by comparing the difference in abundance of the control DNA amplicon (18S rRNA loci) to the amplicon sequence of interest (cfos, fosB, or GAPDH) in genomic DNA (input) to the immunoprecipitated DNA. Data were analyzed using a two-way ANOVA with Bonferroni/Dunn post hoc test. Primer sequences are available upon request.
Microarray
RNA samples were labeled with the Agilent low input labeling kit in single-color mode. The resulting cRNA was then hybridized overnight to Agilent 4x44K Mouse Whole Genome v2 arrays. The arrays were scanned with an Agilent scanner and the resulting images processed with Agilent's Feature Extraction program (v10.10.1.1). The gNetSignal data values were then quantile-normalized (R package limma, normalizeBetweenArrays function) and statistical significance of differential expression assessed with the SAMR package.
Results
To specifically examine the functional role of CREB phosphorylation, we derived mutant mice with a single point mutation in exon 7 at Ser133 to prevent phosphorylation at this site (Fig. 1A). We analyzed homologous recombination and germline transmission of the mutant allele by DNA sequencing of homozygous mutant mice, confirming the presence of the Ser133Ala mutation (Fig. 1B). It should be noted that previous attempts to generate similar mice resulted in splicing around the minigene in the absence of Cre, preventing a true assessment of the contribution of the Ser133Ala mutation to mouse physiology and behavior (Wingate et al. 2009). Control mice in the current study produce normal amounts of “only” wild-type protein, allowing for the appropriate comparisons to be made. Next, we examined the effect of the Ser133Ala mutation on CREB phosphorylation by Western blot analysis. As predicted, we detected an immunoreactive band at 43 kDa corresponding to phosphorylated CREB in the hippocampus of wild-type mice, but not in Ser133Ala homozygous mutant mice (Fig. 1C,D). To assure that the Ser133Ala mutation did not alter levels of total CREB, we examined CREB protein expression by Western blot analysis. In contrast to the reduced levels of CREB protein seen in CREBαΔ mutant mice (Blendy et al. 1996), we found no differences in total CREB immunoreactivity between wild-type and Ser133Ala mice (Fig. 1E). Following deletion or reduced expression of CREB, expression of CREM is up-regulated in multiple models (Hummler et al. 1994; Mantamadiotis et al. 2002; Gundersen et al. 2013; Shin et al. 2014), and provides partial functional compensation for the loss of CREB, as phenotypes are often more severe after deletion of both CREB and CREM (Mantamadiotis et al. 2002; Lemberger et al. 2008). To determine whether the Ser133Ala mutation also causes activation of the CREM gene, we examined CREM expression (utilizing a primer that targets all isoforms) in the hippocampus of Ser133Ala homozygous mutant mice. Unlike in CREBαΔ mutant mice, in which CREM mRNA is fivefold higher than in controls mice, no increase in CREM mRNA levels occurred in Ser133Ala mice (Fig. 1F). Furthermore, ICER, the primary isoform of CREM in the brain (Molina et al. 1993), was not up-regulated in Ser133Ala mice either (Fig. 1G).
Figure 1.
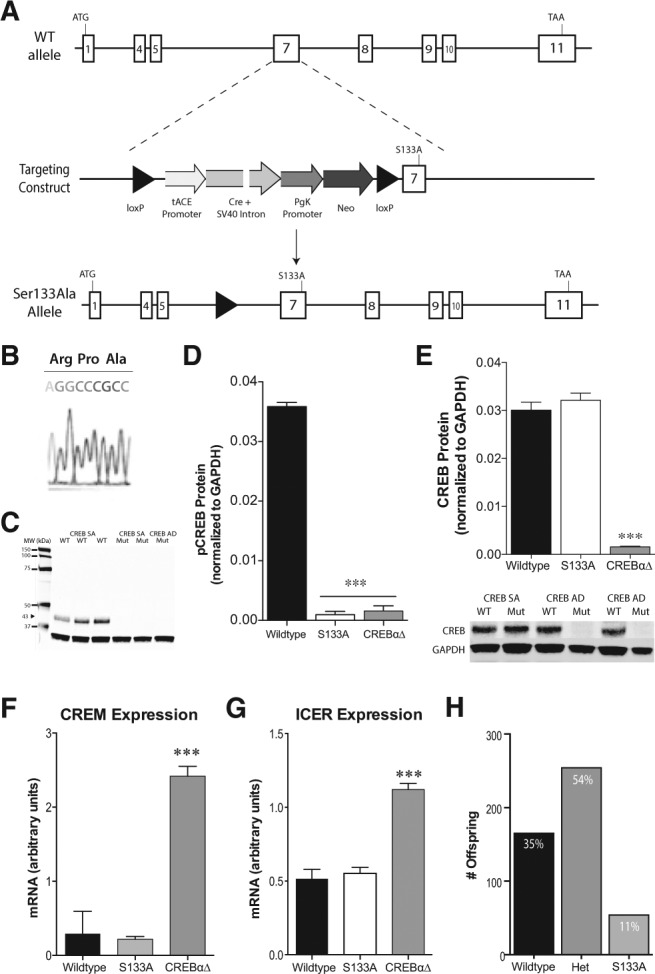
Derivation and characterization of Ser133Ala mutant mouse. (A) Schematic depicting the exon structure of the mouse Creb1 gene. Exon 7, encoding Ser133 (TCC), was replaced with a mutant sequence containing a Ser133Ala (TCC to GCC) substitution along with a self-excising selection cassette. Testes-specific expression of Cre recombinase, driven by the tACE promoter, resulted in deletion of the selection cassette and establishment of the CREBS133A allele upon germline transmission. The Pro132 codon was also changed from CCT to CCC, maintaining the Pro residue but abolishing the StuI restriction enzyme site. (B) Sequence of the PCR product confirms the presence of the Ser133Ala mutation. (C,D) Western blot analysis demonstrates that hippocampal pCREB immunoreactivity is not present in S133A mutant mice. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) served as loading control. (***) P < 0.001. (E) Western blot analysis demonstrates that hippocampal CREB immunoreactivity is normal in Ser133Ala mutant mice, as compared with CREBαΔ mutant mice. (***) P < 0.001. (F) Unlike the increase seen in CREBαΔ mutant mice, Ser133Ala homozygous mutant mice exhibited normal levels of CREM mRNA in the hippocampus, as determined by RT-PCR and normalized to the housekeeping gene TBP. (***) P < 0.001 compared with wild type. (G) There was a corresponding increase in ICER mRNA, the main CREM isoform, in the hippocampus of CREBαΔ mutant mice, but no differences were seen between Ser133Ala and wild-type mice. (***) P < 0.001 compared with wild type. (H) Analysis of 473 offspring revealed a decrease in the proportion of mice homozygous for the S133A mutation compared with expected Mendelian frequencies.
Analysis of 473 offspring from heterozygous Ser133Ala matings revealed only a 38% allelic frequency of the mutant allele (Fig. 1H) as opposed to the expected 50%, similar to the 42% frequency seen in CREBαΔ mutant mice (Hummler et al. 1994). A χ2 analysis revealed that these proportions are significantly different from the expected Mendelian frequencies [χ2 (2, N = 473) = 54.69, P < 0.0001]. Similar to CREBαΔ mutants, this deviation was driven by a decrease in the proportion of animals containing two mutant alleles (11% Ser133Ala/Ser133Ala versus 35% +/+). These data suggest that CREB phosphorylation contributes to pre- or perinatal survival, but is not critical as many homozygous mutant mice survive, in contrast to CREB null mice, which die immediately after birth from respiratory distress (Rudolph et al. 1998).
To ascertain the effects of CREB phosphorylation on learning and memory, we chose fear conditioning, because this behavioral task has been clearly identified as CREB-dependent within our laboratory, whereas tasks like the Morris water maze have yielded inconsistent results (Bourtchuladze et al. 1994; Gass et al. 1998; Graves et al. 2002). We found no genotype differences in footshock sensitivity (FGENOTYPE (1,18) = 0.078, P = 0.78; Fig. 2A) or in freezing behavior during training (FGENOTYPE (1,33) = 0.872, P = 0.36; Fig. 2B). Furthermore, no effect of genotype was seen on either contextual or cued fear memory (context: t(16) = 0.52, P = 0.88, N = 9; cued: t(19) = 0.59, P = 0.56; Fig. 2C,D).
Figure 2.
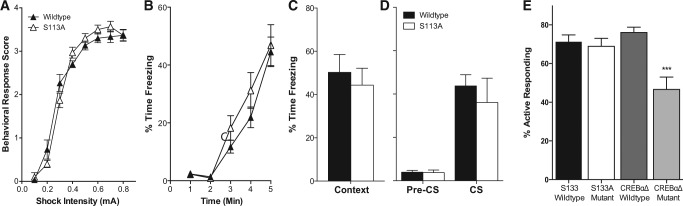
S133A mice exhibit normal aversive and instrumental learning. (A) Ser133Ala mutant mice do not exhibit alterations in shock sensitivity when exposed to increasing shock intensities (FGENOTYPE (1,18) = 0.078, P = 0.78). (B) Ser133Ala mutant mice exhibited similar levels of freezing during the 5-min fear conditioning training day (FGENOTYPE (1,33) = 0.872, P = 0.36). (C) When placed back in the conditioning context, Ser133Ala mice showed normal levels of freezing compared with wild-type mice (t(16) = 0.52, P = 0.61, N = 9). (D) Ser133Ala mice also displayed normal levels of freezing during presentation of the conditioned stimulus (CS; t(15) = 0.60, P = 0.56). (E) CREBαΔ mutant mice display a decrease in the ability to discriminate between the rewarded response and the unrewarded one in an instrumental learning task (F(3,96) = 11.80, P < 0.0001 for the CREBαΔ mutant mice), whereas Ser133Ala mutant mice do not exhibit impaired instrumental learning. (***) P < 0.001 compared with all three other groups.
Together with fear learning, disruption of CREB function has also been implicated in striatal-dependent learning (Pittenger et al. 2006; Lee et al. 2008). Therefore, we used an operant learning task, which requires mice to discriminate between active and inactive operant manipulanda. While CREBαΔ mutant mice exhibited deficits in discrimination learning compared with wild-type mice, no effects of the Ser133Ala mutation was seen in this paradigm (F(3,96) = 11.80, P < 0.0001 for the CREBαΔ mutant mice, Fig. 2C). Furthermore, despite the different background strains, no differences were seen between the F1 hybrid and C57BL/6 wild-type mice. These results demonstrate that when levels of CREB protein are constant, CREB phosphorylation at Ser133 is not necessary for hippocampus- or striatum-dependent learning tasks.
Traditionally, it has been supposed that CREB is constitutively bound to CRE sites but no gene transcription will occur until the protein is phosphorylated (Dash et al. 1991; Matthews et al. 1994). However, recent evidence suggests that CREB phosphorylation may be necessary for optimal CRE binding (Cha-Molstad et al. 2004). To determine whether CREB binding occurs in the absence of Ser133 phosphorylation, we performed chromatin immunoprecipitation (ChIP) of CREB in hippocampal lysates from naïve wild-type and Ser133Ala mice. Strikingly, we found strong CREB occupancy at the promoter regions of cfos and FosB in both wild-type and Ser133Ala mice (Fig. 3A,B). To determine whether the association of CREB with these promoters was indicative of transcriptional activation, we performed ChIP for H3K4me3 and found it enriched to the same extent at the cfos and FosB promoters in both wild-type and Ser133Ala mice (Fig. 3C,D). H3K4me3 binding is associated with transcriptionally permissive chromatin (Lauberth et al. 2013) and cfos mRNA is also up-regulated (Fig. 3G), thus confirming that CREB phosphorylation is not necessary for CREB-mediated transcription of this well-characterized CREB-target gene. Additionally, we analyzed a region of the GAPDH promoter that does not contain CREB binding sites. Binding to this fragment was <20% of that detected for the cfos promoter, consistent with what has been reported previously for this gene/promoter region (Impey et al. 2004; Wisniewska et al. 2010).
Figure 3.

Ser133Ala mice exhibit normal levels of CREB-binding and gene transcription. Chromatin immunoprecipitation revealed normal levels of CREB enrichment at the cfos (A) F(2,13) = 49.01, P < 0.0001; S133WT, S133A Mutant versus Control, P < 0.0001; S133WT versus S133A Mutant, NS) and fosB (B) F(2,13) = 22.38, P < 0.0001; S133WT, S133A Mutant versus Control, P < 0.01; S133WT versus S133A Mutant, NS) promoter regions, with no significant differences between Ser133Ala mice and controls. Normal levels of HK3K4me3 enrichment at the cfos (C) F(2,13) = 15.32, P = 0.0009; S133WT, S133A Mutant versus Control, P < 0.01; S133WT versus S133A Mutant, NS) and fosB (D) F(2,13) = 21.40, P = 0.0004; S133WT, S133A Mutant versus Control, P < 0.01; S133WT versus S133A Mutant, NS) promoter in Ser133Ala mutant mice indicate a transcriptionally permissive state. Fear conditioning leads to a significant increase in Nr4a1 (E) N = 5–8; FINTERACTION (3,42) = 6.056, P = 0.0016, S133 WT, S133A Mutant, CREBαΔ Fear versus Control, P < 0.001; CREBαΔ versus Control, NS), Nr4a2 (F) N = 5–8; FINTERACTION (3,44) = 9.566, P < 0.0001, S133 WT, S133A Mutant, CREBαΔ Fear versus Control, P < 0.0001; CREBαΔ versus Control, NS), and cfos (G) N = 5–8; FINTERACTION (3,44) = 5.87, P = 0.006, S133 WT, S133A Mutant, CREBαΔ Fear versus Control, P < 0.0001; CREBαΔ versus Control, NS) hippocampal gene expression in both wild-type and Ser133Ala mice but not CREBαΔ mutant mice. (**) P < 0.01, (***) P < 0.001 compared with tissue taken from naïve controls. Global expression profiling of hippocampal lysates from S133A versus wild-type mice revealed no significant alterations in any individual gene on the microarray (H).
To evaluate this notion further, we examined the ability of fear conditioning to induce expression of known CREB-target genes that play a role in this behavior (Hawk et al. 2012). In contrast to what is seen in CREBαΔ mutant mice, fear conditioning caused normal up-regulation of Nr4a1 (Fig. 3E), Nr4a2 (Fig. 3F), and cfos (Fig. 3G) expression in Ser133Ala mice, comparable to what was seen in both F1 hybrid (CREBαΔ wild type) and C57/Bl6 (S133 wild-type) mice.
Last, to determine whether phosphorylation-independent CREB-gene transcription was limited to genes involved in fear memory behavior, we performed microarray expression profiling, comparing the mRNA transcriptome from naïve wild type and Ser133Ala total hippocampal lysates (N = 3–4). Among the 44,000 probe sets analyzed by the array, none were downregulated even with the low threshold of a 10% false discovery rate (FDR) (Fig. 3H).
Discussion
Taken together, we derived a novel mouse model homozygous for a CREB serine to alanine mutation, preventing phosphorylation at this site. Despite the extensive literature showing that stimulation of CRE-mediated transcription by CREB correlates with phosphorylation of CREB at Ser-133, our data demonstrate clearly that, with respect to learning and memory, CREB phosphorylation at Ser133 is not necessary for CREB binding to CRE sites, CREB-mediated transcription, or CREB-mediated behavioral phenotypes.
Fear learning and memory has been firmly established as dependent upon CREB, both in our laboratory and others (Bourtchuladze et al. 1994; Kogan et al. 1997; Graves et al. 2002). The prior observations that increase in CREB Ser133 phosphorylation following fear conditioning correlate with changes in gene expression have led to the widely held belief that CREB phosphorylation must be required for these molecular and behavioral responses. Our novel mouse model enabled us to test this notion directly and demonstrate that indeed CREB, but not CREB phosphorylation, is necessary for several learning and memory behaviors (i.e., fear conditioning and operant learning).
It is clear from our findings that fear conditioning induces CREB-mediated transcription, which is not seen in naïve controls exposed to no behavioral stimulus. Having shown that CREB Ser133 phosphorylation is not required to mediate transcriptional and behavioral changes in response to these stimuli, we postulate that alternative regulators of CREB transcription must exist that mediate these signaling events. For example, other post-translational modifications to CREB, such as glycosylation (Lamarre-Vincent and Hsieh-Wilson 2003; Rexach et al. 2012) or phosphorylation at serine 142 may play a critical role in CREB-mediated gene transcription and/or behavioral response. Additionally, CREB-regulated transcriptional coactivators (CRTCs) represent a potential mechanism by which phosphorylation-independent CREB-mediated gene transcription could occur (Fimia et al. 1999; Altarejos and Montminy 2011).
Mechanistically, chromatin immunoprecipitation assays determined that CREB binds to DNA regardless of phosphorylation at Ser133, similar to what has been seen in the liver (Everett et al. 2013), and both quantitative mRNA analysis of relevant CREB targets and global expression profiling demonstrated that hippocampal CREB-mediated transcription does not rely upon the Ser133 phosphorylation event as previously believed. However, the alterations in allelic frequency in offspring from heterozygous matings described above support a role for CREB phosphorylation in early mouse development, perhaps through its established role in cell survival (Bonni et al. 1999; Riccio et al. 1999). Thus, while important biological processes require phosphorylation of CREB on Ser133, the data presented here indicate that memory consolidation is not one of them.
The role of CREB in long-term memory storage has been an area of intense investigation for over 20 yr in a variety of systems, including Aplysia, Drosophila, and rodents. CREB is referred to as an “activity-dependent” transcription factor and the phosphorylation of Ser 133 has been the “activity” most closely associated with this protein, as it is easy to detect and occurs within minutes of neuronal stimulation (Gonzalez and Montminy 1989). The novel mouse model presented here, which prevents phosphorylation of Ser 133, suggest a reevaluation of the reliance on Ser133 phosphorylation as readout of CREB activation so prevalent in the field.
Acknowledgments
We thank Jenny Xiang and Mike Spadola for technical assistance. This work was supported by R01 DA011649 (J.A.B.), K99 DA033372 (L.A.B.), P01 DA033372 (K.H.K.), and T32 GM008076 (B.G.L.).
Footnotes
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.037044.114.
References
- Altarejos JY, Montminy M 2011. CREB and the CRTC co-activators: sensors for hormonal and metabolic signals. Nat Rev Mol Cell Biol 12: 141–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barco A, Alarcon JM, Kandel ER 2002. Expression of constitutively active CREB protein facilitates the late phase of long-term potentiation by enhancing synaptic capture. Cell 108: 689–703. [DOI] [PubMed] [Google Scholar]
- Blendy JA 2006. The role of CREB in depression and antidepressant treatment. Biol Psychiatry 59: 1144–1150. [DOI] [PubMed] [Google Scholar]
- Blendy JA, Kaestner KH, Schmid W, Gass P, Schutz G 1996. Targeting of the CREB gene leads to up-regulation of a novel CREB mRNA isoform. EMBO J 15: 1098–1106. [PMC free article] [PubMed] [Google Scholar]
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME 1999. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science 286: 1358–1362. [DOI] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ 1994. Deficient long-term memory in mice with a targeted mutation of the cAMP-responsive element-binding protein. Cell 79: 59–68. [DOI] [PubMed] [Google Scholar]
- Brindle PK, Montminy MR 1992. The CREB family of transcription activators. Curr Opin Genet Dev 2: 199–204. [DOI] [PubMed] [Google Scholar]
- Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR 1999. Targeting genes for self-excision in the germ line. Genes Dev 13: 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlezon WA Jr, Duman RS, Nestler EJ 2005. The many faces of CREB. Trends Neurosci 28: 436–445. [DOI] [PubMed] [Google Scholar]
- Cha-Molstad H, Keller DM, Yochum GS, Impey S, Goodman RH 2004. Cell-type-specific binding of the transcription factor CREB to the cAMP-response element. Proc Natl Acad Sci 101: 13572–13577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland NG, Jenkins NA, Court DL 2001. Recombineering: a powerful new tool for mouse functional genomics. Nat Rev Genet 2: 769–779. [DOI] [PubMed] [Google Scholar]
- Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER 1991. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci 88: 5061–5065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong Y, Green T, Saal D, Marie H, Neve R, Nestler EJ, Malenka RC 2006. CREB modulates excitability of nucleus accumbens neurons. Nat Neurosci 9: 475–477. [DOI] [PubMed] [Google Scholar]
- Everett LJ, Le Lay J, Lukovac S, Bernstein D, Steger DJ, Lazar MA, Kaestner KH 2013. Integrative genomic analysis of CREB defines a critical role for transcription factor networks in mediating the fed/fasted switch in liver. BMC Genomics 14: 337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimia GM, De Cesare D, Sassone-Corsi P 1999. CBP-independent activation of CREM and CREB by the LIM-only protein ACT. Nature 398: 165–169. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Larris B, Le PP, Peiris TH, Arsenlis A, Schug J, Tobias JW, Kaestner KH, Greenbaum LE 2004. Orthogonal analysis of C/EBPβ targets in vivo during liver proliferation. Proc Natl Acad Sci 101: 12986–12991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka T, Fujioka A, Duman RS 2004. Activation of cAMP signaling facilitates the morphological maturation of newborn neurons in adult hippocampus. J Neurosci 24: 319–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass P, Wolfer DP, Balschun D, Rudolph D, Frey U, Lipp HP, Schütz G 1998. Deficits in memory tasks of mice with CREB mutations depend on gene dosage. Learn Mem 5: 274–288. [PMC free article] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR 1989. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell 59: 675–680. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Menzel P, Leonard J, Fischer WH, Montminy MR 1991. Characterization of motifs which are critical for activity of the cyclic AMP-responsive transcription factor CREB. Mol Cell Biol 11: 1306–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves L, Dalvi A, Lucki I, Blendy JA, Abel T 2002. Behavioral analysis of CREB αδ mutation on a B6/129 F1 hybrid background. Hippocampus 12: 18–26. [DOI] [PubMed] [Google Scholar]
- Gulick D, Gould TJ 2009. The hippocampus and cingulate cortex differentially mediate the effects of nicotine on learning versus on ethanol-induced learning deficits through different effects at nicotinic receptors. Neuropsychopharmacology 34: 2167–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen BB, Briand LA, Onksen JL, Lelay J, Kaestner KH, Blendy JA 2013. Increased hippocampal neurogenesis and accelerated response to antidepressants in mice with specific deletion of CREB in the hippocampus: role of cAMP response-element modulator τ. J Neurosci 33: 13673–13685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gur TL, Conti AC, Holden J, Bechtholt AJ, Hill TE, Lucki I, Malberg JE, Blendy JA 2007. cAMP response element-binding protein deficiency allows for increased neurogenesis and a rapid onset of antidepressant response. J Neurosci 27: 7860–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawk JD, Bookout AL, Poplawski SG, Bridi M, Rao AJ, Sulewski ME, Kroener BT, Manglesdorf DJ, Abel T 2012. NR4A nuclear receptors support memory enhancement by histone deacetylase inhibitors. J Clin Invest 122: 3593–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hummler E, Cole TJ, Blendy JA, Ganss R, Aguzzi A, Schmid W, Beermann F, Schutz G 1994. Targeted mutation of the CREB gene: compensation within the CREB/ATF family of transcription factors. Proc Natl Acad Sci 91: 5647–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH 2004. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell 119: 1041–1054. [DOI] [PubMed] [Google Scholar]
- Josselyn SA, Shi C, Carlezon WA Jr, Neve RL, Nestler EJ, Davis M 2001. Long-term memory is facilitated by cAMP response element-binding protein overexpression in the amygdala. J Neurosci 21: 2404–2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida S, Josselyn SA, Peña de Ortiz S, Kogan JH, Chevere I, Masushige S, Silva AJ 2002. CREB required for the stability of new and reactivated fear memories. Nat Neurosci 5: 348–355. [DOI] [PubMed] [Google Scholar]
- Kogan JH, Frankland PW, Blendy JA, Coblentz J, Marowitz Z, Schütz G, Silva AJ 1997. Spaced training induces normal long-term memory in CREB mutant mice. Curr Biol 7: 1–11. [DOI] [PubMed] [Google Scholar]
- Lamarre-Vincent N, Hsieh-Wilson LC 2003. Dynamic glycosylation of the transcription factor CREB: a potential role in gene regulation. J Am Chem Soc 125: 6612–6613. [DOI] [PubMed] [Google Scholar]
- Lauberth SM, Nakayama T, Wu X, Ferris AL, Tang Z, Hughes SH, Roeder RG 2013. H3K4me3 interactions with TAF3 regulate preinitiation complex assembly and selective gene activation. Cell 152: 1021–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AS, Duman RS, Pittenger C 2008. A double dissociation revealing bidirectional competition between striatum and hippocampus during learning. Proc Natl Acad Sci 105: 17163–17168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemberger T, Parkitna JR, Chai M, Schütz G, Engblom D 2008. CREB has a context-dependent role in activity-regulated transcription and maintains neuronal cholesterol homeostasis. FASEB J 22: 2872–2879. [DOI] [PubMed] [Google Scholar]
- Mantamadiotis T, Lemberger T, Bleckmann SC, Kern H, Kretz O, Martin Villalba A, Tronche F, Kellendonk C, Gau D, Kapfhammer J, et al. 2002. Disruption of CREB function in brain leads to neurodegeneration. Nat Genet 31: 47–54. [DOI] [PubMed] [Google Scholar]
- Marie H, Morishita W, Yu X, Calakos N, Malenka RC 2005. Generation of silent synapses by acute in vivo expression of CaMKIV and CREB. Neuron 45: 741–752. [DOI] [PubMed] [Google Scholar]
- Matthews RP, Guthrie CR, Wailes LM, Zhao X, Means AR, McKnight GS 1994. Calcium/calmodulin-dependent protein kinase types II and IV differentially regulate CREB-dependent gene expression. Mol Cell Biol 14: 6107–6116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina CA, Foulkes NS, Lalli E, Sassone-Corsi P 1993. Inducibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell 75: 875–886. [DOI] [PubMed] [Google Scholar]
- Nakagawa S, Kim JE, Lee R, Malberg JE, Chen J, Steffen C, Zhang YJ, Nestler EJ, Duman RS 2002. Regulation of neurogenesis in adult mouse hippocampus by cAMP and the cAMP response element-binding protein. J Neurosci 22: 3673–3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittenger C, Fasano S, Mazzocchi-Jones D, Dunnett SB, Kandel ER, Brambilla R 2006. Impaired bidirectional synaptic plasticity and procedural memory formation in striatum-specific cAMP response element-binding protein-deficient mice. J Neurosci 26: 2808–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Restivo L, Tafi E, Ammassari-Teule M, Marie H 2009. Viral-mediated expression of a constitutively active form of CREB in hippocampal neurons increases memory. Hippocampus 19: 228–234. [DOI] [PubMed] [Google Scholar]
- Rexach JE, Clark PM, Mason DE, Neve RL, Peters EC, Hsieh-Wilson LC 2012. Dynamic O-GlcNAc modification regulates CREB-mediated gene expression and memory formation. Nat Chem Biol 8: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Ahn S, Davenport CM, Blendy JA, Ginty DD 1999. Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: 2358–2361. [DOI] [PubMed] [Google Scholar]
- Rudolph D, Tafuri A, Gass P, Hammerling GJ, Arnold B, Schütz G 1998. Impaired fetal T cell development and perinatal lethality in mice lacking the cAMP response element binding protein. Proc Natl Acad Sci 95: 4481–4486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sassone-Corsi P 1995. Transcription factors responsive to cAMP. Annu Rev Cell Dev Biol 11: 355–377. [DOI] [PubMed] [Google Scholar]
- Sekeres MJ, Neve RL, Frankland PW, Josselyn SA 2010. Dorsal hippocampal CREB is both necessary and sufficient for spatial memory. Learn Mem 17: 280–283. [DOI] [PubMed] [Google Scholar]
- Shin S, Le Lay J, Everett LJ, Gupta R, Rafiq K, Kaestner KH 2014. CREB mediates the insulinotropic and anti-apoptotic effects of GLP-1 signaling in adult mouse β-cells. Mol Metab 3: 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, Kida S 1998. CREB and memory. Annu Rev Neurosci 21: 127–148. [DOI] [PubMed] [Google Scholar]
- Suzuki A, Fukushima H, Mukawa T, Toyoda H, Wu LJ, Zhao MG, Xu H, Shang Y, Endoh K, Iwamoto T, et al. 2011. Upregulation of CREB-mediated transcription enhances both short- and long-term memory. J Neurosci 31: 8786–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters CL, Blendy JA 2001. Different requirements for cAMP response element binding protein in positive and negative reinforcing properties of drugs of abuse. J Neurosci 21: 9438–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingate AD, Martin KJ, Hunter C, Carr JM, Clacher C, Arthur JS 2009. Generation of a conditional CREB Ser133Ala knockin mouse. Genesis 47: 688–696. [DOI] [PubMed] [Google Scholar]
- Wisniewska MB, Misztal K, Michowski W, Szczot M, Purta E, Lesniak W, Klejman ME, Dabrowski M, Filipkowski RK, Nagalski A, et al. 2010. LEF1/β-catenin complex regulates transcription of the Cav3.1 calcium channel gene (Cacna1g) in thalamic neurons of the adult brain. J Neurosci 30: 4957–4969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin JC, Tully T 1996. CREB and the formation of long-term memory. Curr Opin Neurobiol 6: 264–268. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, et al. 2005. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci 102: 4459–4464. [DOI] [PMC free article] [PubMed] [Google Scholar]