

Abstract
von Hippel-Lindau (VHL) gene mutations are associated with clear cell renal cell carcinoma (ccRCC). A hallmark of ccRCC is loss of the primary cilium. Loss of this key organelle in ccRCC is caused by loss of VHL and associated with increased Aurora kinase A (AURKA) and histone deacetylase 6 (HDAC6) activities, which drive disassembly of the primary cilium. However, the underlying mechanism by which VHL loss increases AURKA levels has not been clearly elucidated, although it has been suggested that hypoxia-inducible factor-1α (HIF-1α) mediates increased AURKA expression in VHL-null cells. By contrast, we found that elevated AURKA expression is not increased by HIF-1α, suggesting an alternate mechanism for AURKA dysregulation in VHL-null cells. We report here that AURKA expression is driven by β-catenin transcription in VHL-null cells. In a panel of RCC cell lines, we observed nuclear accumulation of β-catenin and increased AURKA signaling to HDAC6. Moreover, HIF-1α inhibited AURKA expression by inhibiting β-catenin transcription. VHL knockdown activated β-catenin and elevated AURKA expression, decreased primary cilia formation, and caused significant shortening of cilia length in cells that did form cilia. The β-catenin responsive transcription inhibitor iCRT14 reduced AURKA levels and rescued ciliary defects, inducing a significant increase in primary cilia formation in VHL-deficient cells. These data define a role for β-catenin in regulating AURKA and formation of primary cilia in the setting of VHL deficiency, opening new avenues for treatment with β-catenin inhibitors to rescue ciliogenesis in ccRCC.
Keywords: cell signaling, kidney cancer, renal cell biology
Mutations in the von Hippel-Lindau (VHL) gene are associated with the most aggressive histopathologic subtype of renal cell carcinoma (RCC): clear cell renal cell carcinoma (ccRCC).1 Individuals with germline defects are susceptible to second-hit mutations resulting in inactivation of both alleles of this critical tumor suppressor.2 pVHL has numerous cellular functions, with its role as the recognition component of a multiprotein ubiquitin degradation complex being most well characterized.3 Hypoxia-inducible factor-α (HIF-α) is perhaps the most well studied target of VHL’s E3 ligase activity, linking loss of pVHL with cellular proliferation and angiogenesis.4,5 In a nonproteasomal role, pVHL has been shown to stabilize microtubules6,7 and regulate cell cycle progression.8,9 More recently, loss of VHL has been linked to loss of primary cilia,10–12 which is thought to be the driver for both cyst and tumor formation in the setting of VHL deficiency.13,14
Primary cilia are microtubule-based organelles on the apical surface of renal epithelial cells that are involved in sensing environmental cues and regulating several cell signaling pathways.15–18 The appreciation for the central role of the primary cilium in cellular homeostasis has given rise to the identification of a new class of disorders, referred to as ciliopathies.19 In several ciliopathies, loss of tumor suppressors, such as VHL, results in loss of primary cilia and initiation of disease.8 The mitotic kinase Aurora kinase A (AURKA) was recently reported to have a novel nonmitotic activity. AURKA was shown to interact with enhancer of filamentation 1 (HEF1/NEDD9), to phosphorylate and activate histone deacetylase 6 (HDAC6). Activation of HDAC6, a tubulin deacetylase, causes disassembly of the microtubule axoneme of the primary cilia.20 In the context of RCC, AURKA levels are elevated in ccRCC21,22 and in high-grade renal tumors.23 In another report, AURKA was suggested to be a specific target of HIF-1α in the setting of VHL deficiency,24 although the exact mechanism linking HIF-1α and AURKA activation was not explored.
VHL has other targets in addition to HIF. For example, VHL interacts with and stabilizes Jade-1,25,26 which also functions as an E3 ligase that regulates β-catenin levels.27 In the case of VHL deficiency, Jade-1 levels are significantly reduced leading to increased levels of β-catenin.28 This led us to hypothesize that AURKA may be upregulated because of transcriptional activation by β-catenin in the setting of VHL deficiency. Our studies show that AURKA signaling to HDAC6 is modulated by β-catenin–driven transcription, and rather than increasing AURKA transcription, HIF-1α inhibits AURKA expression via repression of β-catenin itself. Increased AURKA expression after loss of VHL leads to a decrease in the number and length of primary cilia. Inhibition of AURKA expression using a β-catenin inhibitor, iCRT14, was able to rescue ciliary defects associated with VHL deficiency, suggesting that interventions targeting β-catenin could have efficacy for reversing the effects of VHL loss.
Results
AURKA Is Elevated in VHL-Deficient Cell Lines
We examined AURKA expression in a panel of VHL-proficient (Caki-1) and VHL-deficient (786-O, 769-P, and A-498) quiescent RCC cell lines. As shown in Figure 1A, AURKA mRNA levels were significantly higher in VHL-deficient cells compared with VHL-positive cells. Commensurate with elevated mRNA, we also observed higher AURKA protein levels in the VHL-null RCC cell lines (Figure 1B). Because AURKA interacts with HEF1/NEDD9 to activate HDAC6,20 we also examined levels of HEF1 and HDAC6 in these cells. Both HEF1 (3- to 40-fold) and HDAC6 (3- to 5-fold) were increased in cells lacking VHL (Figure 1B).
Figure 1.

AURKA expression and signaling to HDAC6 is elevated in RCC. (A) RT-PCR analyses of AURKA mRNA expression in VHL-proficient (Caki-1) or VHL-deficient (786-O, 769-P, and A-498) RCC cells, normalized to Caki-1 (P<0.01). (B) Lysates from VHL-proficient (Caki-1) or VHL-deficient (786-O, 769-P, and A-498) RCC cells are probed with the indicated antibodies (left). Densitometric analyses of protein expression normalized to GAPDH expression from four independent experiments, plotted as graphs showing AURKA (black bars), HEF1 (gray bars), and HDAC6 (white bars) expression compared with expression in the Caki-1 cells (P<0.05). (C) Lysates from 786-O cells expressing the siC nontargeting scrambled control or siNEDD9 are probed with NEDD9, AURKA, HDAC6, and GAPDH antibodies. (D) RT-PCR analyses of hTERT RPE-1 cells transfected with a nontargeting control siC (black bars) or siVHL (gray bars) showing fold-changes in VHL and AURKA transcript levels as indicated (P<0.01). (E) Lysates from normal hTERT RPE-1 cells expressing siC or siVHL are immunoblotted for the indicated antibodies. Densitometric analyses from at least five independent experiments are shown as graphs indicating levels of AURKA and HDAC6 in cells expressing siC (black bars) or siVHL (gray bars) (P<0.01). *Statistically significant differences. siC, scrambled nontargeting control; siVHL, VHL-specific siRNA.
Xu et al. previously reported that AURKA and HEF1 are elevated in VHL-deficient, RCC10, and RCC4 cells,24 and suggested that this increase was due to stabilization of HIF-1α. However, in contrast with the RCC cell lines in that report, HIF-1α levels were previously characterized to be virtually nonexistent in the 786-O, 769-P, and A-498 cells.29 This would suggest that an alternate mechanism exists in VHL-deficient cells to increase AURKA and HEF1 expression and cause cilia disassembly. To evaluate whether HEF1 could directly modulate AURKA expression, we knocked down HEF1 in VHL-null 786-O cells and found that AURKA levels remained unchanged after loss of HEF1 (Figure 1C). These data indicate that modulating HEF1 alone in these cells was unable to regulate AURKA expression.
Using quiescent normal human retinal pigmented epithelial (hTERT RPE-1) cells, a well characterized ciliogenesis model, we confirmed that knocking down VHL using small interfering RNA (siRNA) resulted in increased AURKA and HEF1 expression, mimicking our observations in VHL-null RCC cell lines. RT-PCR showed an 80% efficiency of knockdown in cells expressing VHL-specific siRNA compared with scrambled control (Figure 1D). With VHL knockdown, we observed a corresponding increase (80%) in AURKA mRNA (Figure 1D) and protein levels (Figure 1E). Quantitation revealed a significant (>2-fold) increase in AURKA, and a smaller but statistically significant increase in HDAC6 (Figure 1E, graph). Importantly, HDAC6 expression levels are used as an indirect measure of its activity20; thus, the modest increase in HDAC6 expression (25%) could potentially result in much higher HDAC6 activity.
HIF-1α Inhibits AURKA Expression in Normal Epithelial Cells
To further explore whether HIF-1α was involved in upregulating AURKA expression, we knocked down HIF-1α (siHIF1α) in hTERT RPE-1 cells. RT-PCR confirmed a 90% decrease in HIF-1α mRNA (Figure 2A), and protein levels (Figure 2B). We found that AURKA mRNA was significantly higher with HIF-1α knockdown compared with the nontargeting control (Figure 2A), accompanied by a 5-fold increase in AURKA protein levels (Figure 2B).
Figure 2.

HIF-1α inhibits AURKA expression in epithelial cells. (A) mRNA expression of HIF-1α and AURKA from hTERT RPE-1 cells transfected with the siC nontargeting (scrambled) control (black bars) or siHIF-1α (gray bars) (P<0.01). (B) Lysates prepared from hTERT RPE-1 cells transfected with siC (black bars) or siHIF-1α (gray bars) are analyzed by immunoblotting with the indicated antibodies. Densitometric analyses from five independent experiments showing AURKA protein expression normalized to GAPDH are denoted in the graph (P<0.01). (C) RT-PCR analyses of AURKA mRNA from hTERT RPE-1 and ACHN cells treated with water (vehicle; black bars), DMOG (1 mM; gray bars), or defroxamine (250 μM; gray bars) as indicated (P<0.01). (D) Cell lysates from hTERT RPE-1 and ACHN cells treated with vehicle (black bars), DMOG (1 mM; gray bars), or DFX (250 μM; gray bars) are immunoblotted with the indicated antibodies. Densitometric analyses showing protein expression normalized to GAPDH from at least three independent replicates are shown in the graph (right) (P<0.01). *Statistically significant differences. siC, scrambled nontargeting control; siHIF-1α, HIF-1α–specific siRNA; Veh, vehicle; DMOG, dimethyloxalylglycine; DFX, defroxamine; GLUT1, glucose transporter 1.
These data led us to hypothesize that rather than increasing AURKA expression, HIF-1α decreased expression of this kinase. We used two pharmacologic hypoxia mimetics, dimethyloxalylglycine and defroxamine, to promote accumulation of HIF-1α. Initially, dose-response experiments were conducted to optimize the concentration of the hypoxia mimetics to stabilize HIF-1α with minimal toxicity to the cells. In hTERT RPE-1 and VHL-proficient ACHN RCC cells treated with dimethyloxalylglycine (1 mM) or defroxamine (250 μM), stabilization of HIF-1α was confirmed by increased HIF-1α and Glut1 (a downstream target of HIF-1α) protein levels (Figure 2D). AURKA expression decreased at the mRNA (Figure 2C) and protein levels (Figure 2D) with both inhibitors. We also observed a significant decrease in HDAC6 and HEF1 protein expression (Figure 2D) in response to stabilized HIF-1α. These data support an alternate hypothesis that HIF-1α inhibits both AURKA and HEF1 expression.
Loss of HIF-1α and VHL Activates β-Catenin
A report in colorectal carcinoma found that HIF-1α inhibited β-catenin–driven transcription,30 and another study reported Jade-1, a protein stabilized by VHL, targeted β-catenin for proteasome-mediated degradation.27 These reports lead us to hypothesize that HIF-1α inhibited AURKA expression via modulation of β-catenin in VHL-deficient cells. First, to determine whether HIF-1α inhibited β-catenin activity, we examined mRNA levels of two well established β-catenin targets (CyclinD1 and c-myc) after HIF-1α knockdown, and we observed increased expression of both the mRNA (Figure 3A) and protein (data not shown) levels. Conversely, use of hypoxia mimetics to stabilize HIF resulted in a significant decrease in CyclinD1 and c-myc mRNA (Figure 3B), and protein expression (data not shown) in both hTERT RPE-1 and ACHN cells.
Figure 3.

Loss of HIF-1α and VHL promotes activation of β-catenin. (A) RT-PCR analyses of hTERT RPE-1 cells expressing nontargeting control siC (black bars) or siHIF-1α (gray bars), showing mRNA levels of HIF-1α, CyclinD1, and c-myc (P<0.01). (B) RT-PCR analyses showing CyclinD1 (left) and c-myc (right) transcript levels in hTERT RPE-1 and ACHN cells treated with vehicle (black bars) or hypoxia mimetics DMOG (1 mM; gray bars) or DFX (250 μM; gray bars ) as indicated (P<0.01). (C) Subcellular fractionation of VHL-proficient (Caki-1 and ACHN) and VHL-deficient (786-O, 769-P, and A-498) RCC cells, immunoblotted with the indicated antibodies (Lamin A/C, nuclear marker; LDH, cytoplasmic marker; T, whole cell extract; N, nuclear; C, cytoplasmic). Densitometric analyses (right) of the blots (left) indicating a ratio of activated β-catenin in the nuclear fraction (normalized to nuclear marker Lamin A/C) to the levels in the cytoplasmic fraction (normalized to cytoplasmic marker LDH). (D) Immunofluorescence staining of Caki-1 and 786-O cells using β-catenin (yellow) antibody. The nuclei are counterstained with DAPI (blue). (E) RT-PCR analyses of hTERT RPE-1 cells expressing nontargeting control siC (black bars) or siVHL (gray bars), showing mRNA expression levels of VHL, CyclinD1, and c-myc (P<0.01). *Statistically significant differences. siC, scrambled nontargeting control; siHIF-1α, HIF-1α-specific siRNA; Veh, vehicle; DMOG, dimethyloxalylglycine; DFX, defroxamine; T, whole cell extract; N, nuclear; C, cytoplasmic; LDH, lactate dehydrogenase; DAPI, 4′,6-diamidino-2-phenylindole; siVHL, VHL-specific siRNA. Bar, 5 μM.
Next, subcellular fractionation of VHL-proficient and VHL-deficient RCC cells revealed accumulation of nuclear β-catenin in VHL-null RCC cells compared with VHL-positive RCC cells (Figure 3C). Nuclear active β-catenin is dephosphorylated and drives transcription of its downstream targets.31 In VHL-deficient RCC cells, we found that the ratio of activated β-catenin in the nucleus (when normalized to nuclear marker expression) to that in the cytoplasm (normalized to cytoplasmic marker expression) is higher than the same ratio in VHL-proficient cells (Figure 3C, graph). Similarly, immunofluorescence showed enhanced nuclear localization of β-catenin in VHL-deficient (786-O) cells compared with the VHL-positive (Caki-1) cells (Figure 3D). We further confirmed activation of β-catenin after loss of VHL in the hTERT RPE-1 cells by measuring CyclinD1 and c-myc transcript levels, which were elevated in VHL-deficient cells (Figure 3E).
AURKA Expression Is Regulated by β-Catenin–Driven Transcription
Because we observed that β-catenin-driven transcription was repressed when HIF-1α was stabilized, as was AURKA expression, we hypothesized that AURKA transcription was regulated by β-catenin. To investigate the role of β-catenin in modulating AURKA, we overexpressed β-catenin in hTERT RPE-1 cells, and consequentially observed increased AURKA mRNA (Figure 4A), and protein levels (Figure 4B). By contrast, knocking down β-catenin or T-cell factor 1 (TCF-1) showed decreased AURKA mRNA (Figure 4, C and E) and protein (Figure 4, D and F) levels. To determine whether β-catenin was directly modulating AURKA transcription, we performed chromatin immunoprecipitation (ChIP) analyses in hTERT RPE-1 cells overexpressing β-catenin. ChIP analyses revealed that β-catenin directly recognized and immunoprecipitated regions of the AURKA promoter, similar to reports in multiple myeloma32 (Figure 4G), demonstrating that AURKA is a direct target of β-catenin. The cellular activity of β-catenin is regulated by glycogen synthase kinase-3β (GSK3β), which is part of the β-catenin destruction complex.33 In addition, AURKA was reported to phosphorylate GSK3β at S9 in gastric cancer,34 resulting in the inactivation of this kinase, thereby increasing levels of activated β-catenin. Hence, we examined phosphorylation of GSK3β (S9) in hTERT RPE-1 cells overexpressing β-catenin and elevated AURKA, and observed increased phosphorylation of GSK3β at S9 (Figure 4H). Similarly, GSK3β phosphorylation increased in cells after VHL knockdown (Figure 4I), similar to our observations in VHL-null RCC cell lines (data not shown). These data directly link elevated AURKA to β-catenin–driven transcription.
Figure 4.

β-catenin drives AURKA expression. (A) RT-PCR analyses showing AURKA mRNA levels in hTERT RPE-1 cells overexpressing β-catenin construct (gray bar) or a mock transfection control (black bar) (P<0.01). (B) Lysates from hTERT RPE-1 cells overexpressing β-catenin immunoblotted for the indicated antibodies (left), and graphical representation of densitometric analyses from five independent replicates (right) (P<0.01). Black bars indicate mock, whereas gray bars indicate β-catenin overexpression. (C) mRNA expression of AURKA and CyclinD1 from hTERT RPE-1 cells transfected with a siC nontargeting (scrambled) control (black bars) or siβ-catenin (gray bars) (P<0.01). (D) Representative blots of lysates prepared from hTERT RPE-1 cells transfected with siC or siβ-catenin are analyzed by immunoblotting with the indicated antibodies (n=4). (E) mRNA expression of AURKA and CyclinD1 from hTERT RPE-1 cells transfected with a siC nontargeting (scrambled) control (black bars) or siTCF-1 (gray bars) (P<0.05). (F) Representative blots of lysates prepared from hTERT RPE-1 cells transfected with siC or siTCF-1 are analyzed by immunoblotting with the indicated antibodies (n=3). (G) ChIP assay in hTERT RPE-1 cells transfected with a β-catenin construct or a mock transfection control demonstrating direct binding of β-catenin to the AURKA gene promoter. ChIP is performed with either IgG or β-catenin antibodies followed by RT-PCR for using primers for the regions as indicated. (H) Representative immunoblots from lysates generated by overexpressing β-catenin in hTERT RPE-1 cells probed with phospho-GSK3β (S9), GSK3β, and GAPDH antibodies. (I) Immunoblot analyses of hTERT RPE-1 cells with VHL knockdown (siVHL) probed with the indicated antibodies. GAPDH serves as the loading control. *Statistically significant differences. siC, scrambled nontargeting control; siβ-catenin, β-catenin–specific siRNA; siTCF1, T-cell factor 1–specific siRNA; TCF1, T-cell factor 1; Ab, antibody; siVHL, VHL-specific siRNA.
Elevated AURKA Expression Leads to Abnormal Primary Cilia in VHL-Deficient Cells
AURKA causes disassembly of the primary cilium via activation of HDAC6.20 Enhanced AURKA expression after loss of VHL lead us to examine primary cilia in hTERT RPE-1 cells with VHL knockdown. In contrast with a previous report by Thoma et al. in which loss of VHL in primary human cells failed to elicit a ciliary defect,11 we found that knocking down VHL in the hTERT RPE-1 cells showed a significant shortening of cilia length compared with control cells (Figure 5A). Three independent replicates revealed that in cells deficient for VHL, there was a significant nearly 2-fold increase in cells that failed to make primary cilia (Figure 5B). In cells that retained cilia after VHL knockdown, we observed a 50%–60% shortening of cilia length (representative experiment shown in Figure 5C). Importantly, these estimates are likely modest because cilia measurements were performed on a population basis, which would include both transfected and nontransfected cells.
Figure 5.
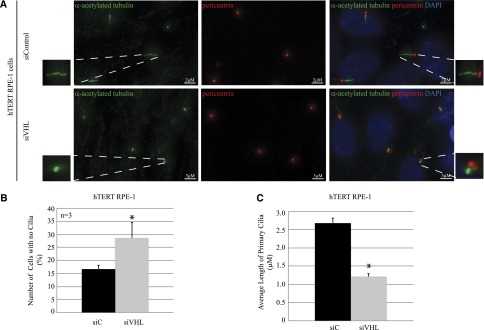
Loss of VHL results in shortening of primary cilia. (A) Immunofluorescence staining of hTERT RPE-1 cells expressing siC or siVHL using acetylated α-tubulin (cilia marker, green) and pericentrin (basal body marker, red). The nuclei are counterstained with DAPI (blue). Enlarged panels show higher magnification of primary cilia. (B) Immunofluorescence images are analyzed using Imaris software to quantitate the number of cells that failed to form primary cilia from >100 individual hTERT RPE-1 cells transfected with siC (black bars) or siVHL (gray bars). Data from three independent replicates (each >100 cells) are denoted as a percentage of cells without cilia (P<0.01). (C) Immunofluorescence images are analyzed using Imaris software to measure the length of the primary cilia from >150 individual hTERT RPE-1 cells transfected with siC (black bars) or siVHL (gray bars). A representative experiment is shown (P=3.4×10−22). *Statistically significant differences. siC, scrambled nontargeting control; siVHL, VHL-specific siRNA; DAPI, 4′,6-diamidino-2-phenylindole. Bar, 2 μM for siC in A; 3 μM for siVHL in A.
Inhibition of β-Catenin Regulated AURKA Transcription Rescues Aberrant Ciliogenesis in VHL-Deficient Cells
Because our data link β-catenin to high AURKA in VHL-deficient cells, and the resultant loss of primary cilia, we assessed the efficacy of a β-catenin inhibitor iCRT14 to rescue both elevated AURKA and cilia defects in the setting of VHL deficiency. iCRT14 is a potent and selective inhibitor of β-catenin responsive transcription with no direct effect on β-catenin or its cytoplasmic interactions with junction proteins.35,36 Initial dose-response experiments showed maximal β-catenin inhibition with minimal toxicity at a dose of 15 μM. hTERT RPE-1 cells with VHL knockdown showed higher AURKA mRNA (Figure 6A) compared with cells with scrambled control when treated with vehicle (EtOH). Treatment with iCRT14 decreased AURKA expression showing an 80% decrease in AURKA mRNA (Figure 6A). Levels of the β-catenin targets CyclinD1 and c-myc mRNA were assessed as positive controls for iCRT14 inhibition of β-catenin transcription (Figure 6A). The 3-fold decrease in AURKA expression was accompanied by a 2-fold decrease in HDAC6 protein in VHL-deficient cells treated with iCRT14 (Figure 6B).
Figure 6.
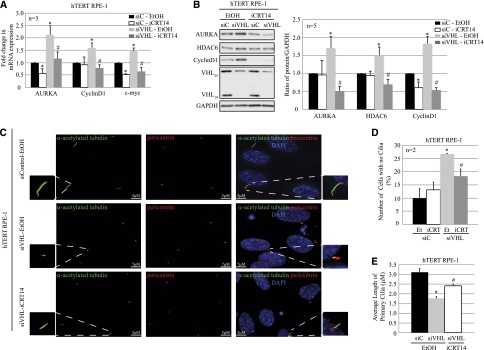
Inhibition of β-catenin responsive transcription rescues the ciliary defect in epithelial cells after acute loss of VHL. (A) RT-PCR analyses of hTERT RPE-1 cells expressing siC (scrambled) treated with vehicle (EtOH; black bars) or iCRT14 (15 μM; white bars), and siVHL treated with either vehicle (EtOH; light gray bars) or iCRT14 (15 μM, dark gray bars) showing AURKA, CyclinD1, and c-myc mRNA transcript levels as indicated (P<0.05). (B) Immunoblot analyses from hTERT RPE-1 cells transfected with either siC treated with EtOH (black bars) or iCRT14 (15 μM; white bars) and siVHL treated with EtOH (vehicle; light gray bars) or iCRT14 (15 μM; dark gray bars) probed with the indicated antibodies (left). The graph represents the densitometric analyses from five independent replicates showing protein expression normalized to GAPDH (loading control; right) (P<0.05). (C) Immunofluorescence staining of hTERT RPE-1 cells expressing siC or siVHL, treated with EtOH (vehicle) or iCRT14 (15 μM), using acetylated α-tubulin (cilia marker, green) and pericentrin (basal body marker, red). The nuclei are counterstained with DAPI (blue). A single primary cilium is shown in the enlarged panels. (D) Immunofluorescence images are analyzed using Imaris software to quantitate the number of cells that failed to make primary cilia from >100 individual hTERT RPE-1 cells transfected with siC treated with EtOH (black bars) or iCRT14 (white bars) and siVHL treated with EtOH (light gray bars) or iCRT14 (dark gray bars). Data from three independent replicates (each >100 cells) are denoted as a percentage of cells without cilia (P<0.01). (E) Immunofluorescence images are analyzed using Imaris software to measure the length of the primary cilia from >150 individual hTERT RPE-1 cells transfected with siC treated with EtOH (black bars) or siVHL treated with EtOH (gray bars) or iCRT14 (white bars). A representative experiment is shown (P<0.01). *Statistically significant differences compared with siC-EtOH; #Statistically significant differences between siVHL-EtOH and siVHL-iCRT14 treatment groups. siC, scrambled nontargeting control; siVHL, VHL-specific siRNA; DAPI, 4′,6-diamidino-2-phenylindole. Bar, 5 μM for siC-EtOH and siVHL-iCRT14 in C; 7 μM for siVHL-EtOH in C.
Importantly, we wanted to evaluate the ability of iCRT14 to rescue the ciliary defect associated with VHL deficiency. In cells with VHL knockdown, treatment with iCRT14 rescued the ciliary defect associated with VHL loss (Figure 6C). We found that fewer cells lacked primary cilia (Figure 6D), and those with cilia had longer primary cilia (Figure 6E) after iCRT14 treatment compared with the vehicle-treated controls. These data show that decreasing β-catenin–driven transcription of AURKA is sufficient to promote ciliogenesis in the setting of VHL deficiency.
Because iCRT14 successfully reduced AURKA after acute loss of VHL, we asked whether inhibition of catenin responsive transcription would also rescue AURKA signaling in the setting of RCC. We treated VHL-null 786-O and 769-P RCC cell lines (with high AURKA) with iCRT14 (15 μM), and observed a significant reduction in AURKA expression (Figure 7A) in these cells. Concomitant decrease in CyclinD1 and c-myc transcript levels served as a positive control for inhibitor treatment with HDAC6 serving as a negative control. A corresponding reduction in AURKA and HDAC6 protein levels was observed in response to iCRT14 treatment, indicating successful rescue of AURKA activation of HDAC6 (Figure 7B). As shown in Figure 7C, these data suggest a model in which inhibiting β-catenin could serve as a promising new avenue to reverse elevated AURKA and prevent loss of primary cilia in the setting of VHL-deficient ccRCC.
Figure 7.

iCRT14 rescues aberrant AURKA signaling in RCC cell lines. (A) RT-PCR analyses showing AURKA, CyclinD1, c-myc, and HDAC6 mRNA expression in VHL-null, 786-O, and 769-P RCC cell lines treated with vehicle (EtOH; black bars) or iCRT14 (15 μM; gray bars) (P<0.05). (B) Immunoblot analyses from 786-O and 769-P RCC cells treated with EtOH (black bars) or iCRT14 (gray bars), probed with the indicated antibodies (left). The graph (right) represents densitometric analyses from at least four independent replicates showing levels of protein expression normalized to GAPDH expression (loading control) (P<0.05). (C) Model for β-catenin regulation of AURKA in VHL-deficient cells. *Statistically significant differences.
Discussion
The ccRCC variant arises from loss of VHL,1 and is associated with loss of primary cilia.8,9 Ciliary defects are causally linked to renal cysts and VHL disease progression.8 Although recent studies demonstrated elevated levels of AURKA in the setting of RCC,21,23,37–39 the exact mechanism for dysregulated AURKA in this setting is not clearly understood. Our results link β-catenin–driven transcription to regulation of AURKA and loss of primary cilia in VHL-null RCC. Increased AURKA expression in the setting of VHL deficiency decreased cilia formation and inhibition of β-catenin, decreased AURKA expression, and rescued ciliogenesis in the setting of VHL deficiency.
HIF-1 was previously shown to bind HIF responsive elements in the promoter of AURKA in liver cells,40 resulting in its enhanced expression in hepatocellular carcinoma.41 Although a previous report showed a correlation between HIF-1 and AURKA levels in HIF-1–positive RCC cell lines,24 our data show that AURKA expression is inhibited by HIF-1α in normal and RCC cell lines. This could arise from reduced binding of HIF to the AURKA HIF responsive element as recently observed in breast cancer cell lines.42 Alternately, it may suggest the existence of an alternate pathway as previously reported for colorectal carcinoma, in which HIF-1α bound to β-catenin precluding its association with T cell factor to prevent activation of its downstream targets.30 High HIF-1α levels are associated with unfavorable prognosis in most cancers; however, patients with high HIF-1α expression have a better prognosis in ccRCC.43 Our studies showing inhibition of β-catenin regulated AURKA by HIF-1α validate the improved survival by upregulated HIF-1α in ccRCC. Recent studies also highlight the critical role of HIF-2α, a renal oncoprotein necessary and sufficient in the pathology of VHL, in activating β-catenin–driven transcription.44–46 Similar to a previous report that found HIF-1α and HIF-2α as having opposing functions in regulating c-myc signaling,47 it would be interesting to define the relative contributions of HIF-1α and HIF-2α in modulating β-catenin activation of AURKA and formation of primary cilia.
Loss of VHL24 and induction of HIF-1α48 regulate HEF1/NEDD9 levels. Although we observed an increase in HEF1 expression in cells that have lost VHL, stabilized HIF-1α inhibited HEF1, similar to that observed with AURKA. A recent report identified HEF1 as a novel target of Wnt signaling,49 providing a potential mechanism by which HIF-1α modulates HEF1. These data suggest that HIF-1α inhibits both AURKA and HEF1 via β-catenin–driven transcription. AURKA is a direct target of Wnt/β-catenin in multiple myeloma,32 and was reported to stabilize β-catenin by binding Axin and preventing its association with GSK3β to form the β-catenin destruction complex in glioma-initiating cells.50 Phosphorylation of β-catenin by casein kinase 1 (CK1) and GSK3β targets it for degradation,31 and GSK3β phosphorylation at S9 inhibits GSK3β’s kinase activity in gastric cancer.34 We show increased phosphorylation of GSK3β (S9) in cells that have elevated AURKA resulting from loss of VHL, highlighting a crucial positive feedback loop by which AURKA potentiates its own expression via activation of β-catenin. A recent report showed a requirement for the combined inactivation of GSK3β and VHL in destabilizing the ciliary microtubules.11 Although loss of VHL alone was sufficient to exacerbate AURKA and promote loss of primary cilia in our studies, elevated AURKA inactivated GSK3β, confirming inactivation of both proteins in driving ciliary abnormalities in our model.
Given the critical role AURKA plays in regulating primary cilia,20 its enhanced expression resulting from activation of β-catenin provides us with a potential target that can be modulated to abrogate the ciliary defects associated with VHL disease. Current efforts identifying small molecule inhibitors of β-catenin have generated several promising molecules,51 including iCRT14, specific in inhibiting catenin responsive transcription, without any effect on β-catenin degradation or β-catenin interactions with junction proteins.35 Deregulation of Wnt/β-catenin signaling was recently reported in several other ciliopathies in which loss of cystoproteins such as Jouberin,52 Meckel–Gruber syndrome,53,54 and nephrocystin55 are linked to cilia defects as well as nuclear localization and activation of β-catenin transcription. Our studies showing elevated AURKA via β-catenin activation in VHL suggest a common pathway driving the renal manifestations associated with ciliopathies. Importantly, our in vitro results showing rescue of the ciliary defect with an inhibitor of β-catenin are promising and warrant further investigation in in vivo models, and perhaps other disease settings.
Concise Methods
Cell Culture
RCC cell lines (Caki-1, 786-O, ACHN, 769-P, and A-498; ATCC) were maintained in McCoy’s 5A media (Caki-1), RPMI-1640 media (786-O), or MEM media (ACHN, 769-P, and A-498) supplemented with 10% FBS (Sigma-Aldrich, St. Louis, MO). hTERT RPE-1 cells (a kind gift from Dr. Gregory Pazour, University of Massachusetts Medical School) were maintained in DMEM/F-12 media (Life Technologies, Carlsbad, CA), supplemented with 10% FBS. All cells were maintained in 5% CO2 at 37°C, and experiments were performed in fully confluent cultures, starved (serum-free media) for 48 hours to promote ciliogenesis. All human cell lines were validated using the Characterized Cell Line Core Facility (University of Texas MD Anderson Cancer Center).
Constructs, Transfections, and Treatments
Human β-catenin construct (plasmid 16828) was purchased from Addgene, and transfected into hTERT RPE-1 cells using Lipofectamine2000 (Invitrogen) according to the manufacturer’s instructions. On-Target plus SMART pool siRNAs (nontargeting, VHL-specific siRNA, HIF-1α–specific siRNA, β-catenin–specific siRNA, and TCF-1–specific siRNA) were transfected into cells using DharmaFECT1 (Thermo Fisher Scientific, Pittsburgh, PA), per the manufacturer’s recommendations. Dimethyloxalylglycine (1 mM) and defroxamine (250 mM) (Sigma-Aldrich) were solubilized in water; cells were simultaneously treated, and were starved 6–8 hours after transfections for 48 hours. iCRT14 (Tocris Bioscience, Bristol, UK) was resuspended in ethanol, and cells were treated at a final concentration of 15 μM with serum starvation for 48 hours.
Cell Lysates and Antibodies
Cell lysates were collected in cold 1×cell lysis buffer (20 mM Tris [pH 7.5], 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 2.5 mM sodium pyrophosphate) containing 1× Complete protease inhibitor (Roche, Mannheim, Germany) and 1 mM Na3VO4. The lysates were analyzed by immunoblotting with the following primary antibodies: anti-AURKA (1:1000), anti-HDAC6 (1:1000), anti-HEF1/NEDD9 (1:500), anti–phospho-GSK3β (1:1000), anti-GSK3β (1:1000), anti-CyclinD1 (1:1000), anti-VHL (1:500), anti-lactate dehydrogenase (1:1000), and anti-TCF1 (1:1000) from Cell Signaling Technologies (Danvers, CA); anti–c-myc (1:1000), anti–glyceraldehyde-3-phosphate (GAPDH) (1:20,000), and anti-LaminA/C (1:1000) from Santa Cruz Biotechnology (Santa Cruz, CA); anti–β-catenin (1:2000) and anti–HIF-1α (1:1000) from BD Biosciences (San Jose, CA); anti-active-β-catenin (1:2000) from EMD Millipore (Billerica, MA); and anti-glucose transporter 1 (1:2500) from Abcam, Inc. (Cambridge, MA). Horseradish peroxidase–conjugated goat anti-mouse, goat anti-rabbit, and donkey anti-goat secondary antibodies were purchased from Santa Cruz Biotechnology. Immunoblots were visualized using LumiGLO (KPL, Gaithersburg, MD), Pierce ECL (Thermo Fisher Scientific, Rockford, IL), or Amersham ECL Prime (GE Life Sciences, Pittsburg, PA) substrates.
RT-PCR Analyses
mRNA was isolated from cells using the RNeasy Mini Kit (Qiagen, Valencia, CA), according to the manufacturer’s protocol. After RNA extraction, cDNA was prepared by reverse-transcribing 1 μg of RNA using the Invitrogen Superscript First-Strand Synthesis System for RT-PCR (Invitrogen, Carlsbad, CA). Real-time PCR was performed using the ABI ViiA7 Real-Time PCR system from Applied Biosystems (Foster City, CA). Fast Real-Time TaqMan assays from ABI were used to analyze gene expression of VHL, AURKA, HIF-1α, CyclinD1, and c-myc. All real-time PCR reactions were performed by mixing Universal Fast Real-Time Master Mix from ABI together with the gene assay mix first and then adding 2 μl of cDNA from each sample to make up a 25-μl volume. GAPDH was used as an endogenous control, which included probe and forward and reverse primers in a 25-μl reaction volume. The following set of conditions were used for each real-time reaction: 95°C for 20 minutes followed by 40 cycles of 1 second at 95°C and 20 seconds at 60°C. The real-time PCR reactions were all performed in triplicate and were quantified using the −ΔΔ cycle threshold (CT) method, which uses the average CT of the GAPDH subtracted from the target gene CT to obtain the average ΔCT. The siRNA/mock controls were used as calibrators from which we subtracted individual VHL, AURKA, HIF-1α, CyclinD1, or c-myc siRNA ΔCT values to obtain the −ΔΔCT. The fold change for the sample was calculated in comparison with the calibrator by taking 2−ΔΔCT.
Subcellular Fractionation
Cells were harvested, washed with ice-cold PBS, and resuspended in hypotonic buffer (10 mM HEPES [pH 7.2], 10 mM KCl, 1.5 mM MgCl2, 0.1 mM EGTA, 20 mM NaF, 100 μM Na3VO4). After disruption using a Dounce homogenizer, crude nuclei were pelleted by centrifugation and the supernatant was collected as the cytoplasmic fraction. The crude nuclei were resuspended in hypotonic buffer and any unbroken cells disrupted in the Dounce homogenizer. The pellet after centrifugation at 3000 rpm at 4°C for 5 minutes was washed with a wash buffer (10 mM Tris [pH 7.4], 0.1% NP-40, 0.05% Na-deoxycholate, 10 mM NaCl, 3 mM MgCl2), and lysed in a high-salt buffer (20 mM HEPES [pH 7.4], 0.5 M NaCl, 0.5% NP-40, 1.5 mM MgCl2). The purified nuclear fraction was collected after centrifugation at 14,000 rpm at 4°C for 10 minutes. The nuclear, cytosolic, and membrane fractions were subsequently subjected to SDS-PAGE and immunoblot analysis.
ChIP Assays
hTERT RPE-1 cells were transfected with a β-catenin overexpression construct or a mock control. After serum starvation for 48 hours, the protein-DNA complexes were cross-linked with 1% formaldehyde for 10 minutes. The cells were lysed, and the cell lysate was subsequently sonicated to shear the DNA. Then, 600 μg of DNA from each group was collected. To reduce nonspecific background, the cell lysate was first incubated with agarose beads and salmon sperm DNA. The supernatant was collected, and 20 μl of it was saved to analyze as the initial input for the reaction. The remaining lysate was incubated overnight with 2 μg of the anti–β-catenin or IgG antibody. The protocol provided with the ChIP kit (Upstate Biotechnology) was followed for immunoprecipitation, elution, and reverse cross-linking of the protein-DNA complex. The eluted DNA was purified with the PCR purification kit (Qiagen). Both the eluted and input products were then subjected to PCR analysis using the same previously published primer sets.32
Immunofluorescence Analyses
For immunofluorescence staining, hTERT RPE-1 cells were plated on glass coverslips, transfected, and starved 6 hours after transfection for 48 hours to promote ciliogenesis. Cells were fixed in 4% paraformaldehyde for 10 minutes at room temperature, after washes in 1×PBS. We used 0.05% Triton-X to permeabilize the cells, followed by blocking in 3.75% BSA solution for 1 hour at room temperature. Primary antibodies for α-acetylated tubulin (clone 6-11B-1, 1:5000; Sigma-Aldrich) and pericentrin (1:5000; Abcam, Inc.) or β-catenin (1:100; Santa Cruz Biotechnology) in blocking buffer were applied for 1 hour, followed by AlexaFluor 488 and 546 goat anti-mouse or anti-rabbit secondary antibodies (Life Technologies) for another hour. Cells were counterstained with 4′,6-diamidino-2-phenylindole (Life Technologies) and mounted using ProLong Gold antifade reagent (Life Technologies). Cells were visualized using a Deltavision deconvolution microscope (Applied Precision) at ×60 magnification. Images were analyzed using Imaris software (Bitplane). All experiments were performed in three independent replicates unless otherwise specified, and image analyses performed on at least 100 individual cells from each replicate.
Statistical Analyses
All statistical analyses were performed using the t test (one-tailed) for determination of differences between the average values of quantitation data obtained from densitometric analyses of immunoblots and average values obtained from RT-PCR analyses. The SEM was calculated and P values of P<0.01 and P<0.05 were considered statistically significant.
Disclosures
None.
Acknowledgments
The authors thank P. Chowdhury, T. Berry, and X. Tong for their technical assistance.
This work was supported by a grant from the National Institutes for Health (R01-CA143811-03) and a Robert A. Welch Endowed Chair in Chemistry (BE-0023 to C.L.W.).
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
References
- 1.Bausch B, Jilg C, Gläsker S, Vortmeyer A, Lützen N, Anton A, Eng C, Neumann HP: Renal cancer in von Hippel-Lindau disease and related syndromes. Nat Rev Nephrol 9: 529–538, 2013 [DOI] [PubMed] [Google Scholar]
- 2.Valladares Ayerbes M, Aparicio Gallego G, Díaz Prado S, Jiménez Fonseca P, García Campelo R, Antón Aparicio LM: Origin of renal cell carcinomas. Clin Transl Oncol 10: 697–712, 2008 [DOI] [PubMed] [Google Scholar]
- 3.Stebbins CE, Kaelin WG, Jr, Pavletich NP: Structure of the VHL-ElonginC-ElonginB complex: Implications for VHL tumor suppressor function. Science 284: 455–461, 1999 [DOI] [PubMed] [Google Scholar]
- 4.Haase VH: Renal cancer: Oxygen meets metabolism. Exp Cell Res 318: 1057–1067, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L, Kaelin WG, Jr: New insights into the biology of renal cell carcinoma. Hematol Oncol Clin North Am 25: 667–686, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thoma CR, Matov A, Gutbrodt KL, Hoerner CR, Smole Z, Krek W, Danuser G: Quantitative image analysis identifies pVHL as a key regulator of microtubule dynamic instability. J Cell Biol 190: 991–1003, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thoma CR, Toso A, Gutbrodt KL, Reggi SP, Frew IJ, Schraml P, Hergovich A, Moch H, Meraldi P, Krek W: VHL loss causes spindle misorientation and chromosome instability. Nat Cell Biol 11: 994–1001, 2009 [DOI] [PubMed] [Google Scholar]
- 8.Pan J, Seeger-Nukpezah T, Golemis EA: The role of the cilium in normal and abnormal cell cycles: Emphasis on renal cystic pathologies. Cell Mol Life Sci 70: 1849–1874, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Basten SG, Giles RH: Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia 2: 6, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kuehn EW, Walz G, Benzing T: von Hippel-Lindau: A tumor suppressor links microtubules to ciliogenesis and cancer development. Cancer Res 67: 4537–4540, 2007 [DOI] [PubMed] [Google Scholar]
- 11.Thoma CR, Frew IJ, Hoerner CR, Montani M, Moch H, Krek W: pVHL and GSK3beta are components of a primary cilium-maintenance signalling network. Nat Cell Biol 9: 588–595, 2007 [DOI] [PubMed] [Google Scholar]
- 12.Basten SG, Willekers S, Vermaat JS, Slaats GG, Voest EE, van Diest PJ, Giles RH: Reduced cilia frequencies in human renal cell carcinomas versus neighboring parenchymal tissue. Cilia 2: 2, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frew IJ, Thoma CR, Georgiev S, Minola A, Hitz M, Montani M, Moch H, Krek W: pVHL and PTEN tumour suppressor proteins cooperatively suppress kidney cyst formation. EMBO J 27: 1747–1757, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Montani M, Heinimann K, von Teichman A, Rudolph T, Perren A, Moch H: VHL-gene deletion in single renal tubular epithelial cells and renal tubular cysts: Further evidence for a cyst-dependent progression pathway of clear cell renal carcinoma in von Hippel-Lindau disease. Am J Surg Pathol 34: 806–815, 2010 [DOI] [PubMed] [Google Scholar]
- 15.Ezratty EJ, Stokes N, Chai S, Shah AS, Williams SE, Fuchs E: A role for the primary cilium in Notch signaling and epidermal differentiation during skin development. Cell 145: 1129–1141, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Habbig S, Bartram MP, Müller RU, Schwarz R, Andriopoulos N, Chen S, Sägmüller JG, Hoehne M, Burst V, Liebau MC, Reinhardt HC, Benzing T, Schermer B: NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J Cell Biol 193: 633–642, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST: PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol 15: 1861–1866, 2005 [DOI] [PubMed] [Google Scholar]
- 18.Simons M, Gloy J, Ganner A, Bullerkotte A, Bashkurov M, Krönig C, Schermer B, Benzing T, Cabello OA, Jenny A, Mlodzik M, Polok B, Driever W, Obara T, Walz G: Inversin, the gene product mutated in nephronophthisis type II, functions as a molecular switch between Wnt signaling pathways. Nat Genet 37: 537–543, 2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waters AM, Beales PL: Ciliopathies: An expanding disease spectrum. Pediatr Nephrol 26: 1039–1056, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pugacheva EN, Jablonski SA, Hartman TR, Henske EP, Golemis EA: HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129: 1351–1363, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ferchichi I, Kourda N, Sassi S, Romdhane KB, Balatgi S, Cremet JY, Prigent C, Elgaaied AB: Aurora A overexpression and pVHL reduced expression are correlated with a bad kidney cancer prognosis. Dis Markers 33: 333–340, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ricketts C, Zeegers MP, Lubinski J, Maher ER: Analysis of germline variants in CDH1, IGFBP3, MMP1, MMP3, STK15 and VEGF in familial and sporadic renal cell carcinoma. PLoS ONE 4: e6037, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Martin B, Chesnel F, Delcros JG, Jouan F, Couturier A, Dugay F, Le Goff X, Patard JJ, Fergelot P, Vigneau C, Rioux-Leclerq N, Arlot-Bonnemains Y: Identification of pVHL as a novel substrate for Aurora-A in clear cell renal cell carcinoma (ccRCC). PLoS ONE 8: e67071, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu J, Li H, Wang B, Xu Y, Yang J, Zhang X, Harten SK, Shukla D, Maxwell PH, Pei D, Esteban MA: VHL inactivation induces HEF1 and Aurora kinase A. J Am Soc Nephrol 21: 2041–2046, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou MI, Wang H, Foy RL, Ross JJ, Cohen HT: Tumor suppressor von Hippel-Lindau (VHL) stabilization of Jade-1 protein occurs through plant homeodomains and is VHL mutation dependent. Cancer Res 64: 1278–1286, 2004 [DOI] [PubMed] [Google Scholar]
- 26.Zhou MI, Wang H, Ross JJ, Kuzmin I, Xu C, Cohen HT: The von Hippel-Lindau tumor suppressor stabilizes novel plant homeodomain protein Jade-1. J Biol Chem 277: 39887–39898, 2002 [DOI] [PubMed] [Google Scholar]
- 27.Chitalia VC, Foy RL, Bachschmid MM, Zeng L, Panchenko MV, Zhou MI, Bharti A, Seldin DC, Lecker SH, Dominguez I, Cohen HT: Jade-1 inhibits Wnt signalling by ubiquitylating beta-catenin and mediates Wnt pathway inhibition by pVHL. Nat Cell Biol 10: 1208–1216, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lian X, Duan X, Wu X, Li C, Chen S, Wang S, Cai Y, Weng Z: Expression and clinical significance of von Hippel-Lindau downstream genes: Jade-1 and beta-catenin related to renal cell carcinoma. Urology 80: 485.e7–485.e13, 2012 [DOI] [PubMed] [Google Scholar]
- 29.Shinojima T, Oya M, Takayanagi A, Mizuno R, Shimizu N, Murai M: Renal cancer cells lacking hypoxia inducible factor (HIF)-1alpha expression maintain vascular endothelial growth factor expression through HIF-2alpha. Carcinogenesis 28: 529–536, 2007 [DOI] [PubMed] [Google Scholar]
- 30.Kaidi A, Williams AC, Paraskeva C: Interaction between beta-catenin and HIF-1 promotes cellular adaptation to hypoxia. Nat Cell Biol 9: 210–217, 2007 [DOI] [PubMed] [Google Scholar]
- 31.Saito-Diaz K, Chen TW, Wang X, Thorne CA, Wallace HA, Page-McCaw A, Lee E: The way Wnt works: Components and mechanism. Growth Factors 31: 1–31, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dutta-Simmons J, Zhang Y, Gorgun G, Gatt M, Mani M, Hideshima T, Takada K, Carlson NE, Carrasco DE, Tai YT, Raje N, Letai AG, Anderson KC, Carrasco DR: Aurora kinase A is a target of Wnt/beta-catenin involved in multiple myeloma disease progression. Blood 114: 2699–2708, 2009 [DOI] [PubMed] [Google Scholar]
- 33.Xu C, Kim NG, Gumbiner BM: Regulation of protein stability by GSK3 mediated phosphorylation. Cell Cycle 8: 4032–4039, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dar AA, Belkhiri A, El-Rifai W: The aurora kinase A regulates GSK-3beta in gastric cancer cells. Oncogene 28: 866–875, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gonsalves FC, Klein K, Carson BB, Katz S, Ekas LA, Evans S, Nagourney R, Cardozo T, Brown AM, DasGupta R: An RNAi-based chemical genetic screen identifies three small-molecule inhibitors of the Wnt/wingless signaling pathway. Proc Natl Acad Sci U S A 108: 5954–5963, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Narayanan BA, Doudican NA, Park J, Xu D, Narayanan NK, Dasgupta R, Mazumder A: Antagonistic effect of small-molecule inhibitors of Wnt/β-catenin in multiple myeloma. Anticancer Res 32: 4697–4707, 2012 [PMC free article] [PubMed] [Google Scholar]
- 37.Kurahashi T, Miyake H, Hara I, Fujisawa M: Significance of Aurora-A expression in renal cell carcinoma. Urol Oncol 25: 128–133, 2007 [DOI] [PubMed] [Google Scholar]
- 38.Li Y, Zhou W, Wei L, Jin J, Tang K, Li C, Teh BT, Chen X: The effect of Aurora kinases on cell proliferation, cell cycle regulation and metastasis in renal cell carcinoma. Int J Oncol 41: 2139–2149, 2012 [DOI] [PubMed] [Google Scholar]
- 39.Maruschke M, Hakenberg OW, Koczan D, Zimmermann W, Stief CG, Buchner A: Expression profiling of metastatic renal cell carcinoma using gene set enrichment analysis. Int J Urol 21: 46–51, 2014 [DOI] [PubMed] [Google Scholar]
- 40.Klein A, Flügel D, Kietzmann T: Transcriptional regulation of serine/threonine kinase-15 (STK15) expression by hypoxia and HIF-1. Mol Biol Cell 19: 3667–3675, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cui SY, Huang JY, Chen YT, Song HZ, Huang GC, De W, Wang R, Chen LB: The role of Aurora A in hypoxia-inducible factor 1α-promoting malignant phenotypes of hepatocelluar carcinoma. Cell Cycle 12: 2849–2866, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fanale D, Bazan V, Corsini LR, Caruso S, Insalaco L, Castiglia M, Cicero G, Bronte G, Russo A: HIF-1 is involved in the negative regulation of AURKA expression in breast cancer cell lines under hypoxic conditions. Breast Cancer Res Treat 140: 505–517, 2013 [DOI] [PubMed] [Google Scholar]
- 43.Ku JH, Park YH, Myung JK, Moon KC, Kwak C, Kim HH: Expression of hypoxia inducible factor-1α and 2α in conventional renal cell carcinoma with or without sarcomatoid differentiation. Urol Oncol 29: 731–737, 2011 [DOI] [PubMed] [Google Scholar]
- 44.Park YK, Park B, Lee S, Choi K, Moon Y, Park H: Hypoxia-inducible factor-2α-dependent hypoxic induction of Wnt10b expression in adipogenic cells. J Biol Chem 288: 26311–26322, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Choi H, Chun YS, Kim TY, Park JW: HIF-2alpha enhances beta-catenin/TCF-driven transcription by interacting with beta-catenin. Cancer Res 70: 10101–10111, 2010 [DOI] [PubMed] [Google Scholar]
- 46.Criscimanna A, Duan LJ, Rhodes JA, Fendrich V, Wickline E, Hartman DJ, Monga SP, Lotze MT, Gittes GK, Fong GH, Esni F: PanIN-specific regulation of Wnt signaling by HIF2α during early pancreatic tumorigenesis. Cancer Res 73: 4781–4790, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordan JD, Lal P, Dondeti VR, Letrero R, Parekh KN, Oquendo CE, Greenberg RA, Flaherty KT, Rathmell WK, Keith B, Simon MC, Nathanson KL: HIF-alpha effects on c-Myc distinguish two subtypes of sporadic VHL-deficient clear cell renal carcinoma. Cancer Cell 14: 435–446, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kim SH, Xia D, Kim SW, Holla V, Menter DG, Dubois RN: Human enhancer of filamentation 1 Is a mediator of hypoxia-inducible factor-1alpha-mediated migration in colorectal carcinoma cells. Cancer Res 70: 4054–4063, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li Y, Bavarva JH, Wang Z, Guo J, Qian C, Thibodeau SN, Golemis EA, Liu W: HEF1, a novel target of Wnt signaling, promotes colonic cell migration and cancer progression. Oncogene 30: 2633–2643, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xia Z, Wei P, Zhang H, Ding Z, Yang L, Huang Z, Zhang N: AURKA maintains self-renewal in glioma-initiating cells through the stabilization of beta-catenin and activation of Wnt signaling. Mol Cancer Res 11: 1101–1111, 2013 [DOI] [PubMed] [Google Scholar]
- 51.Voronkov A, Krauss S: Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des 19: 634–664, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lancaster MA, Louie CM, Silhavy JL, Sintasath L, Decambre M, Nigam SK, Willert K, Gleeson JG: Impaired Wnt-beta-catenin signaling disrupts adult renal homeostasis and leads to cystic kidney ciliopathy. Nat Med 15: 1046–1054, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wheway G, Abdelhamed Z, Natarajan S, Toomes C, Inglehearn C, Johnson CA: Aberrant Wnt signalling and cellular over-proliferation in a novel mouse model of Meckel-Gruber syndrome. Dev Biol 377: 55–66, 2013 [DOI] [PubMed] [Google Scholar]
- 54.Abdelhamed ZA, Wheway G, Szymanska K, Natarajan S, Toomes C, Inglehearn C, Johnson CA: Variable expressivity of ciliopathy neurological phenotypes that encompass Meckel-Gruber syndrome and Joubert syndrome is caused by complex de-regulated ciliogenesis, Shh and Wnt signalling defects. Hum Mol Genet 22: 1358–1372, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Borgal L, Habbig S, Hatzold J, Liebau MC, Dafinger C, Sacarea I, Hammerschmidt M, Benzing T, Schermer B: The ciliary protein nephrocystin-4 translocates the canonical Wnt regulator Jade-1 to the nucleus to negatively regulate β-catenin signaling. J Biol Chem 287: 25370–25380, 2012 [DOI] [PMC free article] [PubMed] [Google Scholar]