

Abstract
Efficient presentation of peptide-MHC class I complexes to immune T cells depends upon stable peptide-MHC class I interactions. Theoretically, determining the rate of dissociation of a peptide-MHC class I complexes is straightforward; in practical terms, however, generating the accurate and closely timed data needed to determine the rate of dissociation is not simple. Ideally, one should use a homogenous assay involving an inexhaustible and label-free assay principle. Here, we present a homogenous, high-throughput peptide-MHC class I dissociation assay, which by and large fulfill these ideal requirements. To avoid labeling of the highly variable peptide, we labeled the invariant β2m and monitored its dissociation by a scintillation proximity assay, which has no separation steps and allows for real-time quantitative measurement of dissociation. Validating this work-around to create a virtually label-free assay, we showed that rates of peptide-MHC class I dissociation measured in this assay correlated well with rates of dissociation rates measured conventionally with labeled peptides. This assay can be used to measure the stability of any peptide-MHC Class I combination, it is reproducible and it is well suited for high-throughput screening. To exemplify this, we screened a panel of 384 high-affinity peptides binding to the MHC class I molecule, HLA-A*02:01, and observed rates of dissociation that ranged from 0.1 hours to 46 hours depending on the peptide used.
Keywords: MHC-I, peptide, stability, dissociation, scintillation proximity assay
1 Introduction
Major histocompatibility complex class I (MHC-I) play a pivotal role in the generation of specific immune responses mediated by cytotoxic T cells (CTL). MHC-I molecules sample peptides derived from intracellular proteins, translocate them to the cell surface, and display them to CTLs thereby allowing immune scrutiny of the ongoing intracellular metabolism leading to the detection of the presence of any intracellular pathogens. To fulfill this crucial antigen presenting function, MHC-I molecules must be endowed with the ability not only to bind the peptides generated inside the cell, but also to retain them at the cell surface while waiting for the arrival of extremely rare circulating members of one or more CTL clones of the appropriate specificity. Therefore, some measure of stability of peptide-MHC-I (pMHC-I) complexes appears to be a fundamental requirement for the induction of specific CTL immune responses. Indeed, it has been claimed that stability, rather than affinity, of pMHC-I interactions is the better correlate of immunogenicity and immunodominance (van der Burg et al., 1996; Busch and Pamer, 1998). Nonetheless, affinity measurements account for the vast majority of the available experimental data on pMHC-I interactions (95% in the Immune Epitope Data Base, (Peters et al., 2005)), and most bioinformatics resources predicting pMHC-I interactions are aimed at affinity. One notable exception is the seminal 1994 paper by Parker et al. (Parker et al., 1994), which measured dissociation and subsequently generated corresponding predictors of dissociation (also known as the BIMAS predictors (http://www-bimas.cit.nih.gov/molbio/hla_bind/), which unfortunately have not been updated since 1997 (Kenneth C Parker, personal communication)).
One reason why pMHC-I stability has not been addressed more extensively undoubtedly relates to the cumbersome and/or low-throughput nature of current biochemical methods used to measure the dissociation of pMHC-I complexes. Traditionally, pMHC stability has been addressed by measuring the dissociation of MHC binding peptides, which have been either radioactively (Buus et al., 1987; Assarsson et al., 2007) or fluorescently (Dedier et al., 2001; Binz et al., 2003; Buchli et al., 2004) labeled. The need to label each of the peptides being studied precludes large-scale generation of stability data. Furthermore, the label itself may interfere with the binding to the MHC molecule. Surface plasmon resonance (BiaCore®) has been proposed as a label-free approach (Khilko et al., 1993; Khilko et al., 1995), however, the set-up is low-throughput, and it involves attaching each individual peptide ligand to the sensor chip; something that basically amounts to using tethering as a label. Thus, to the best of our knowledge, none of these biochemical assays are truly label-free, and none of them are high-throughput. In contrast, the original dissociation assay by Parker et al. used a work-around to provide an essentially label-free system. Exploiting that a pMHC-I complex consists of a polymorphic MHC-I heavy chain, an invariant light chain (β2m), and a peptide, they demonstrated that β2m dissociation could be used to accurately monitor peptide dissociation. Although the β2m needed to be labeled in this system, neither of the two important variable components of interest, the highly polymorphic MHC-I heavy chain and the extremely diverse peptide repertoire, needed to be labeled. Thus, this assay could be viewed as being essentially label-free, and it should be possible to address the stability of any pMHC-I complex irrespective of the identities of the peptide. However, the capacity of this assay was limited as it depended upon the separation of each pMHC-I complex and required subsequent reanalysis of these complexes at closely spaced time intervals to establish their rate of dissociation. Variants of pMHC-I dissociation assays using other β2m labeling techniques have been described (Gakamsky et al., 1999), (Joseph et al., 2007); yet, none of these techniques can been applied in a microplate format and none are suited for high-throughput experiments.
Here, we have generated a high-throughput, homogenous and microplate format version of the essentially label-free pMHC-I dissociation assay of Parker et al. This assay is based upon the scintillation proximity principle monitoring the dissociation of 125I-labeled β2m from recombinant and biotinylated MHC-I heavy chains measured in streptavidin-coated scintillation microplates (Flashplates PLUS®).
2 Materials and methods
2.1 Peptides
All peptides were purchased from Schafer-N (www.schafer-n.com). Briefly, they were synthesized by standard 9-fluorenylmethyloxycarbonyl (Fmoc)-chemistry, purified by reversed-phase high-performance liquid chromatography (to at least >80% purity, frequently 95–99% purity), validated by mass spectrometry, and quantitated by weight.
2.2 MHC-I heavy chain proteins
Synthetic genes encoding MHC-I heavy chain were cloned as previously described (Ostergaard Pedersen et al., 2001; Ferre et al., 2003; Leisner et al., 2008). Briefly, genes encoding MHC-I heavy chains truncated at position 275 (i.e. truncated before the membrane spanning domain), were fused C-terminally to a histidine-containing affinity tag, HAT, and a biotinylation signal peptide, BSP (Ferre et al., 2003; Leisner et al., 2008). The recombinant genes were inserted into the IPTG inducible pET28a expression plasmid (Novagen), and the intended DNA sequences were verified by DNA sequencing (ABI3100 Avant). An E. coli expression cell line, BL21(DE3), was transformed with the validated constructs of interest together with the IPTG inducible pACYC184 expression plasmid (Avidity, Denver) containing the BirA gene encoding a biotin-ligase.
The transformed BL21(DE3) cells were grown in a 2L fermentor (LabFors®) in media supplemented with kanamycin (50 μg/ml) and chloroamphenicol (20 μg/ml) to maintain the pET28a-derived plasmid and the BirA containing pACYC184 plasmid, respectively. When the culture reached an optical density of OD(600) = 25, the media was supplemented with biotin (Sigma #B4501, final concentration 125 μg/ml), and IPTG (1 mM) was added to induce expression of the MHC-I heavy chain gene and of the BirA gene; the induction culture was continued for 3 h. At the end of induction protease inhibitor (PMSF, 80 μg/l) was added and the cells were lysed in a cell-disrupter (basic Z, Constant Systems Ltd Daventry, UK) set at 2300 bar. The released inclusion bodies containing biotinylated MHC-I heavy chain proteins were harvested by centrifugation (Sorvall RC6, 20 min, 17000 g).
The inclusion bodies were washed in PBS, 0.5% NP-40 (Sigma), 0.1% deoxycholic acid (DOC, Sigma) and dissolved at 4°C overnight in 8 M Urea-Tris buffer (8M urea, 25mM Tris, pH 8.0) using 200 ml per 100 g wet cell paste, and cleared by centrifugation. The dissolved MHC-I proteins were then purified by Ni2+/IDA metal chelating affinity column chromatography followed by Q-Sepharose ion-exchange column chromatography, hydrophobic interaction chromatography, and eventually by Superdex-200 size exclusion chromatography. Fractions containing MHC-I heavy chain molecules were identified by A280 absorbance and SDS-PAGE, and pooled. Throughout purification and storage the MHC-I heavy chain proteins were dissolved in 8 M Urea to keep them denatured. Note that the MHC-I heavy chain proteins at no time were exposed to reducing conditions. This allowed purification of highly active pre-oxidized moieties as previously described (Ostergaard Pedersen et al., 2001). Protein concentrations were determined by BCA assay. The degree of biotinylation (usually >95%) was determined by a gel-shift assay (Leisner et al., 2008). The pre-oxidized, denatured proteins were stored at −20°C in Tris buffered 8 M urea.
2.3 MHC-I light chain (beta-2-microglobulin, β2m) protein
Native, recombinant human β2m was expressed and purified as previously described (Ostergaard Pedersen et al., 2001; Ferre et al., 2003). Briefly, a HAT followed by an FXa restriction enzyme site was cloned immediately in front (i.e. N-terminally) of a synthetic gene encoding the native, mature human β2m, the construct was verified by DNA sequencing, inserted into a IPTG-inducible pET28a expression vector (Novagen), and transformed into an E. coli expression cell line, BL21(DE3). The transformed BL21(DE3) cells were grown in a 2L fermentor (LabFors®) in media supplemented with kanamycin (50 μg/ml). When the culture reached an optical density (OD600) = 25, IPTG (1 mM) was added to induce expression of HAT-FXa-β2m; and the induction culture was continued for 3 h. Inclusion bodies containing β2m were extracted as described for MHC-I heavy chain proteins.
The urea dissolved and denatured HAT-FXa-β2m proteins were purified by Ni2+/IDA column chromatography, renatured by drop-wise dilution (100 fold final dilution) into a refolding buffer (25 mM Tris, 300 mM urea, pH 8.0), and purified by Ni2+/IDA column chromatography. Fractions containing HAT-FXa-β2m were identified and pooled as described above. Digestion for 48 h at RT with the Factor-Xa protease (≈7U/ml; enzyme-to-substrate ratio of 1/200) released native β2m. The refolded native β2m was purified by Ni2+/IDA column chromatography followed by Superdex-200 size exclusion chromatography. Fractions containing β2m protein were identified by A280 absorbance and SDS-PAGE, and pooled. Protein concentrations were determined by BCA assay. The native β2m protein was stored at −20°C.
2.4 Iodination of β2m and peptide
β2m and peptide was radio-labeled with iodine (125I) using the chloramine-T procedure (Hunter and Greenwood, 1962). Twenty μg β2m or 1 μg peptide was mixed with 1 mCi 125I and 5 μl chloramines-T (1 mg/ml) (Sigma C9887) for 1 minute. The reaction was stopped by adding 5 μl metabisulphite (1 mg/ml) (Sigma). Unreacted iodine was removed by a size exclusion chromatography using a 1 ml Sephadex G10 column equilibrated in PBS. Column fractions of 200 μl were tested for radioactivity, and the labeled fractions were identified. The radioactivity was measured on a gamma counter (Packard Cobra 5010) and diluted to 25000 cpm/μl in PBS containing 2% ethanol and 0.1% azide, and stored at 4°C.
2.5 Measuring peptide-MHC-I interactions by scintillation proximity assay
Recombinant, biotinylated MHC-I heavy chain molecules in 8 M urea were diluted at least 100-fold into PBS buffer containing β2m and peptide to initiate pMHC-I complex formation. The final concentration of MHC-I heavy chain was between 10–100nM depending on the specific activity of the MHC-I heavy chain. The reactions were carried out in the wells of streptavidin coated scintillation 384 (or 96) well microplates (Flashplate® PLUS, Perkin Elmer, Boston). In the typical format where β2m dissociation is used to indirectly monitor peptide dissociation, reactions including trace amounts of 125I-labeled β2m (approximately 1 nM, corresponding to approximately 25,000 cpm/well) and saturating concentrations (10 μM) of the peptide in question were allowed to reach steady-state by over-night incubation at 18°C. To initiate dissociation, unlabeled β2m was added to a final concentration of 1 μM, and the temperature was raised to 37°C. In the initial experiments, peptide dissociation was monitored directly using trace amounts of 125I-labeled peptide in question and saturating concentrations (1 μM) of unlabeled β2m. The refolding reactions were incubated over-night at 18°C. To initiate dissociation, the identical unlabeled peptide was added to a final concentration of 10 μM, and the temperature was raised to 37°C.
In both setups, the pMHC-I dissociation was monitored by consecutive measurement of the scintillation microplate on a scintillation multiplate counter (TopCount NTX equipped with stackers and 12 detectors, Perkin Elmer), which was modified to run at 37°C and placed in a temperature-controlled room set at 37°C. The same set of plates could automatically be read repeatedly. The TopCount instrument uses time-resolved pulse-discrimination to reject noise thus considerably increasing the counting efficiency of the scintillation proximity assay. Furthermore, the repeated measurements needed to characterize dissociation could be performed unattended. Loading a 12-detector reader with a single 96 well microtiter plate and doing one-minute/well readings, each well could be read every 9 minutes (for a single 384 well microtiter plate, each well could be read every 34 minutes).
In some cases, pMHC-I association during the 18°C complex formation was approximated using another and unmodified TopCount running at the standard instrument temperature of 18°C.
2.6 Data analysis
Association and dissociation curves were fitted using GraphPad Prism version 5.02 (GraphPad Software, San Diego, CA, USA). Association data was fitted to a one-phase association model:
Background subtracted dissociation data was fitted to a one-phase dissociation model:
3 Results
3.1 Generation of pMHC-I complexes for the dissociation assay
The complex in question must be generated before one can measure its stability. In our case, a complex consist of i) recombinant, biotinylated MHC-I heavy chain; ii) recombinant, 125I-labeled MHC-I light chain (β2m); and iii) peptide (Figure 1). Routinely, in vitro biochemical pMHC-I complex formations and affinity measurements are done at reduced temperature to avoid the confounding loss of complexes due to temperature instability (Olsen et al., 1994). Experiments establishing the conditions of complex formation were therefore conducted at 18°C incubations. All measurements were done in a TopCount instrument and used a homogenous scintillation proximity assay principle allowing on-line reading of the interactions between the radiolabeled ligand and the tagged receptor molecule. We considered setting up the reaction in a separate microtiter plate, incubate for complex formation to occur, and then transfer a sample of the formed complexes to the streptavidin-coated scintillation plate. However, setting up complex formation directly in the scintillation plate gave similar efficiency of complex formation (data not shown) and this set-up mode therefore became preferred due to the simpler and much reduced number of operations.
Figure 1.

Schematic diagram of the dissociation assay. Biotinylated MHC-I heavy chain binds to the surface of streptavidin scintillation microplates (FlashPlates PLUS). In the presence of a binding peptide, radiolabeled β2m is bound to the MHC-I heavy and the beta-radiation can reach the scintillant embedded in the microplate resulting in a scintillation signal.
To determine the concentration of recombinant biotinylated MHC-I heavy chain needed to generate pMHC-I complexes for this assay, a range of MHC-I concentrations from 0.8 nM to 100 nM were incubated with trace amounts of radiolabeled β2m in the presence, or absence, of an excess (10000 nM) of MHC-I binding peptide. The reactant were incubated for 48 hours at 18°C (conditions that allow steady-state to be reached in affinity measurements), and the resulting pMHC-I complexes were measured by scintillation proximity. In the absence of peptide, no binding of radiolabeled β2m was observed. In the presences of a saturating concentration of binding peptide, a specific signal was observed that reached a plateau at a concentration of 30 nM MHC-I (Figure 2A). The signal-to-background ratio (S/B-ratio) was approximately 30 at this MHC-I concentration. For subsequent experiments, a concentration of 30 nM recombinant, biotinylated MHC-I was used to generate pMHC-I complexes directly in the streptavidin-coated scintillation plates.
Figure 2.
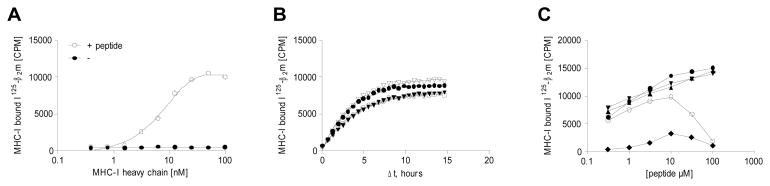
Adjusting assay conditions. A) HLA-A*02:01 heavy chain dose-response titration with or without a binding peptide (FLPSDYFPSV). B) Association of radiolabeled β2m to HLA-A*02:01 at 18°C with 4 different peptides: (▽) SLDQSVVEL, (●) GLYSLPHDL, (▼) RLTRFLSRV and (○) YLNKIQNSL. Steady-state is reached after approximately 10 hours. C) Peptide dose-response titrations of five different peptides: (●) ILYAHLHKL, (▼) ILSDENYLL, (▲) FLTSVINRV, (○) YLIDTTSREL, (◆) QVKDEKLNL. For some peptides an inhibition of the signal was observed at peptide concentrations above 10 μM.
To determine the time needed to achieve steady-state under those conditions, we exploited that the TopCount instrument can do several consecutive measurement of the same sample to monitor generation of the scintillation signal. Studying four different pMHC-I complexes, the reactions were monitored for 15 hours (Figure 2B). For these four binding peptides, steady state was reached within 10 hours and there was little variation in the rates of scintillation signal generation (doubling times between 2–4 hours). The rate of scintillation signal generation is in reality a combined measurement of the binding of radiolabeled β2m to MHC-I and the binding of the biotinylated MHC to the streptavidin coated surface of the microplate. As an approximation of the relative contribution of the latter of these two components, we measured the rate of signal generation for a radiolabeled and biotinylated β2m. The association of a biotinylated protein to the streptavidin coated surface was much faster (a doubling time of 0.4 hours in this system, data not shown) than the combined rates of pMHC-I complex formation and binding to streptavidin. Thus, pMC-I complex formation is the limiting process and the rate of scintillation signal generation is predominantly a measure of the rate of pMHC-I association in this system. For subsequent experiments, a pMHC-I incubation time of at least 12 hours at 18°C was used to generate pMHC-I complexes directly in the streptavidin-coated scintillation plates.
To determine the optimal peptide concentration during pMHC-I complex formation, different peptides were tested in a concentration range from 0.3 to 100 μM under the conditions established above. While some peptides elicited increasing signal with increasing concentration over the entire concentration range tested, others showed inhibition of the signal at peptide concentrations above 10 μM (Figure 2C). For subsequent experiments, a peptide concentration of 10 uM was used during complex formation.
3.2 Measuring dissociation of pMHC-I complexes
Re-association of any labeled ligand must be avoided once it has dissociated. This is traditionally achieved by adding an excess of unlabeled ligand at the time of initiating the dissociation. We have previously measured the affinity of β2m to MHC-I in the presence of peptides to have a KD of approximately 1 nM (Pedersen et al., 1994). Adding 1 μM unlabeled β2m to the reaction corresponds to a 1000-fold excess, both relative to the KD of the interaction and relative to the concentration of labeled β2m available, which should saturate the interaction and prevent re-association of labeled β2m. Other measures to avoid re-association include removing any unbound labeled ligand by dilution or isolation of the complexes. We investigated whether washing the scintillation microtiter plates at the time of initiating the dissociation experiment would alter the dissociation rates compared to those measured when the system was merely saturated with unlabeled β2m. Introducing a washing step did not alter the measured dissociation rates (data not shown), and we therefore chose not to include the washing step to reduce the number of operations.
At the time of initiating dissociation, the scintillation microplate was transferred to a scintillation counter thermostated at 37°C and continuously read for 48 hours. This allowed the exact same well to be measured repeatedly and since the scintillation proximity assay principle detects radiolabeled ligand being bound at the time of measurement, it should be possible to accurately determine the rate of dissociation. To demonstrate this, we selected 384 peptides that are known to bind to the human MHC-I molecule, HLA-A*02:01. The pMHC-I complexes were generated by incubating 30 nM of recombinant and biotinylated HLA-A*02:01 heavy chain with 10 μM peptide and 25000 cpm/well 125I labeled β2m over-night at 18°C in a 384 format streptavidin-coated scintillation microplate. At the end of this incubation period, dissociation was initiated as described above, and each well was read approximately every 30 min. Plotting the natural logarithm of the scintillation signal vs. the time of dissociation yielded straight lines as predicted by the first-order reaction of a unimolecular decay (four examples are given in Figure 3A). The results are readily interpretable, and the slope of the line (the dissociation rate constant, kd) and the accuracy of the fit (the linear regression of the logarithm transformed data, R2, which typically is > 0.95) can easily be done computationally (e.g. using GraphPad Prism). For this random sample of 384 binding peptides (good binding peptides with KD values better than 500 nM), the most stable pMHC-I complex had a half-life of 46 hours and the least stable pMHC-I complex had a half-life of just 0.1 hours (Figure 3B). These results show a considerable variation in the stability of different pMHC-I complexes even within a pool of high-affinity binding peptides. To examine the reproducibility of the assay, we repeated the experiment with new batches of β2m, peptides and HLA-A*02:01 heavy chain. The correlation between the half-lifes measured in the two experiments was very high (Figure 3C).
Figure 3.

A) Dissociation of four different peptides binding to HLA-A*02:01. The data was fitted to a one-phase decay function, and the calculated half-lifes were (▼, RLTRFLSRV) 26.7 hours, (○, YLNKIQNSL) 7.2 hours, (▽, SLDQSVVEL) 2.7 hours and (●, GLYSLPHDL) 1.4 hours. B) The half-life of 384 different peptides binding to HLA-A*02:01 with KD < 500nM. The half-lifes ranged from 46 hours to 0.1 hours C) Reproducibility of the assay. The stability of 384 peptides was analyzed for HLA-A*02:01 in one experiment (Experiment 1). The experiment was repeated with new batches of peptide, MHC, 125I-β2m, experiment 2, and the half-lifes for the two experiments were compared.
Since the biotin-streptavidin interaction forms part of the scintillation proximity assay principle we examined whether this interaction dissociated within the time frame of a typical experiment of up to 48 hours, and therefore might contribute to the dissociation measured. Reassuringly, we did not detect any dissociation of 125I labeled, biotinylated β2m, suggesting that the dissociation of biotin from streptavidin does not influence to the dissociation readings in this assay (data not shown).
3.3 Peptide and β2m dissociate from pMHC-I complexes with similar rates
To verify that the dissociation rates of β2m is a reliable measure of the dissociation rate of peptides, we labeled two peptides that each bind to multiple different MHC-I molecules. This allowed us to generate 16 different pMHC-I complexes and compare the dissociation rates obtained with direct measurement of peptide dissociation (Figure 4, x-axis) with those obtained indirectly by β2m dissociation (Figure 4, y-axis). There was a good correlation between the peptide and β2m dissociation rates, suggesting that peptide and β2m dissociate from the complex at the same time, i.e. that whenever one of the two components, the peptide or the β2m, dissociates off the MHC, then the complex is destabilized and falls apart releasing the other component. Thus, the dissociation of β2m can be used as a reliable measure of the dissociation of peptide.
Figure 4.

Comparison of the dissociation rates measured with either 125I-β2m or 125I-peptide. Two different peptides (FLPSDYFPSV and RLPAYAPLL) were radiolabeled. pMHC’s were generated with different MHC-I molecules (A*02:01, A*02:02, A*02:03, A*02:04, A*02:05, A*02:11, A*02:19 and A*69:01) using either labeled peptide and excess of β2m or labeled β2m and excess peptide. For each pMHC the dissociation rate measured with labeled peptide was compared to the dissociation rate measured with labeled β2m.
4 Discussion
Here, we demonstrate that β2m dissociation can be determined in a high-throughput scintillation proximity assay, and can be used as an accurate and convenient measure of peptide dissociation from pMHC-I complex.
The stability of pMHC-I complexes is an important consideration in any epitope discovery process involving MHC-I restricted CTL responses. Dissociation of peptides from pMHC-I complexes has been addressed using many different technologies spanning from low-tech separation assays (Buus et al., 1987; Parker et al., 1992) to fluorescent polarization assays (Buchli et al., 2005) and plasmon surface resonance (Khilko et al., 1993). Common to all these technologies, however, is a low throughput and/or a requirement for labeling of the peptide in question. The assay presented here measures the dissociation of radiolabeled β2m from recombinant biotinylated MHC-I heavy chains using scintillation proximity assay. This is to our knowledge the first high-throughput pMHC-I dissociation assay, which uses an essentially label-free principle previously suggested (Parker et al., 1992). In this approach the invariant β2m component is labeled to avoid labeling the highly variable peptide component of the pMHC-I complex. As a work-around to obtain a label-free detection system, this approach offers several significant theoretical and practical advantages over one that depends upon using labeled components. First, the label may in itself alter the thermodynamics of the reaction being studied. Second, not all ligands (in casu peptides) can be appropriately labeled. Third, the labeling procedure is time consuming and tends to be biochemically demanding (in particular when it involves radioactivity). In agreement with previous observations (Parker et al., 1992), we find that the dissociation rates measured with radiolabeled β2m closely reflect those obtained with radiolabeled peptides.
Whereas Parker et al. (Parker et al., 1992) used a slow and cumbersome gel filtration assay to generate the pMHC-I complexes and then repetitive gel filtration assays to monitor their stability, we have used a homogenous assay principle (i.e. a “mix-and-read” assay) based upon scintillation proximity. Scintillation is a term describing the conversion of radiation energy from radioactive isotopes into luminescence. Scintillation proximity assay (SPA), first described by (Bosworth and Towers, 1989), is based on scintillation material sensitive to beta radiation (electrons) and the short path length (μm) of beta radiation in water. Flashplates® are microplates coated on the inside with a thin layer of beta sensitive scintillant. A signal is created when a beta active ligand is bound to the scintillant material, however, beta radiation from any unbound ligand in solution is absorbed by water. Although 125I is best known as a gamma emitter, it does also emit Auger electrons (Udenfriend et al., 1985) and it can therefore be used as the radiolabel in a homogenous scintillation proximity assay (Heise et al., 2007; Khawaja, 2007). An important advantage of SPA in the context of creating a dissociation assay is that as long as the isotope is present in the proximity of scintillant the signal persists undiminished solely as a function of the radioactivity. Many other labeling systems suffer from exhaustion processes (i.e. photo bleaching, substrate depletion etc), which complicate and/or preclude accurate repetitive readings of the same sample. Instead, multiple consecutive samples must be analyzed, considerably increasing the complexity of the experimental set-up and increasing the variability of the readings. In contrast, the SPA principle allows inexhaustible repetitive readings of the same sample and reduces the variability of the readings over time. In particular, a first-order dissociation rate is independent of concentrations of the different components, MHC, β2m and peptide, and only requires that well-defined signal is present at the time of initiating dissociation. Variations in the complex set-up parameters say due to imprecise liquid handling or peptide solubility issues has little or no influence upon the dissociation rates obtained with this assay. This, and the well-defined dissociation curves, lead to the determination of highly accurate and reproducibility dissociation rates.
It should be noted that 125I-labeling of β2m must be done gently. Of the six tyrosines present in folded β2m, Parker and Strominger (Parker and Strominger, 1983) found that two could be heavily iodinated (Y63 and Y67), whereas two could only be lightly iodinated (Y10 and Y26) when β2m is iodinated in isolation (as the labeling procedure used here calls for). The ability to bind to MHC-I was intact when positions Y63 and Y67 of β2m were 125I labeled, whereas it was completely lost if it was labeled at positions Y10 or Y26. We are using a shortened version of the chloramine-T iodination procedure to label β2m and we have not experienced binding problems with β2m as a result of the labeling procedure. We have, however, noted that even this gently 125I-labeled β2m is unstable over time: even when stored cold and as dilute as possible, including 2% ethanol as a radio-scavenger and 0.1% azide to prevent contamination, the activity in terms of its ability to bind to MHC with high signal-to-background ration is lost within 2–3 weeks. We suspect that this may be the result of radiolysis of the protein; however, as we have previously noted this must be a very subtle modification without gross alterations of the molecule since the ability to bind β2m-specific antibodies is intact (Ostergaard Pedersen et al., 2001). As an alternative to iodination, other systems might benefit from the very long-lived tritium label that can be introduced into smaller molecules, and be detected by the SPA principle.
The dissociation of β2m from pMHC-I complexes does provide an indirect measurement of peptide dissociation as originally proposed by Parker et al (Parker et al., 1992). This assay serves as a convenient work-around to create a highly versatile and essentially label-free peptide dissociation assay, and the adaptation to scintillation proximity detection creates a high-throughput assay. It is also noteworthy that the assay can be used to address pMHC-I complexes from other species than humans (we have successfully measured stability of mouse and swine MHC-I with this assay and we believe that it can be used to address pMHC-I interaction in any species as long as a recombinant, biotinylated MHC-I and β2m is available). Thus, the availability of streptavidin-coated SPA microplates (Flashplate PLUS), of highly sensitive multichannel readers, of a large panel of recombinant biotinylated MHC-I molecules, and of method to 125I-label β2m; all support the development of high-throughput assays to measure pMHC-I dissociation, generate large-scale data sets representing pMHC-I dissociation events from many different isotypes and species, and eventually the corresponding bioinformatics predictors. From an immunological perspective, the long-term goal would be to predict pMHC-I stability and thereby improve the ability to identify immunogenic peptides in various studies involving vaccine and immunotherapy development, understanding autoimmunity etc. From a dissociation assay perspective, this is an easy, versatile and highly efficient high-throughput assay. It is very cost effective in terms of reagent usage since 384 dissociation curves can be obtained from a single microplate. One concern when using radioactive material in high-throughput experiments is the risk of contaminating equipment and the disposal of radioactive waste. In this assay, the radioactive component is retained in a sealed microplate and the plate can be disposed at the end of the assay.
Acknowledgments
We are grateful to Kenneth C. Parker for reviewing this manuscript. This work was supported by grants from National Institute of Health (grant HHSN266200400025C and grant HHSN272200900045C).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Assarsson E, Sidney J, Oseroff C, Pasquetto V, Bui HH, Frahm N, Brander C, Peters B, Grey H, Sette A. A quantitative analysis of the variables affecting the repertoire of T cell specificities recognized after vaccinia virus infection. J Immunol. 2007;178:7890–901. doi: 10.4049/jimmunol.178.12.7890. [DOI] [PubMed] [Google Scholar]
- Binz AK, Rodriguez RC, Biddison WE, Baker BM. Thermodynamic and kinetic analysis of a peptide-class I MHC interaction highlights the noncovalent nature and conformational dynamics of the class I heterotrimer. Biochemistry. 2003;42:4954–61. doi: 10.1021/bi034077m. [DOI] [PubMed] [Google Scholar]
- Bosworth N, Towers P. Scintillation proximity assay. Nature. 1989;341:167–8. doi: 10.1038/341167a0. [DOI] [PubMed] [Google Scholar]
- Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. Real-time measurement of in vitro peptide binding to soluble HLA-A*0201 by fluorescence polarization. Biochemistry. 2004;43:14852–63. doi: 10.1021/bi048580q. [DOI] [PubMed] [Google Scholar]
- Buchli R, VanGundy RS, Hickman-Miller HD, Giberson CF, Bardet W, Hildebrand WH. Development and validation of a fluorescence polarization-based competitive peptide-binding assay for HLA-A*0201--a new tool for epitope discovery. Biochemistry. 2005;44:12491–507. doi: 10.1021/bi050255v. [DOI] [PubMed] [Google Scholar]
- Busch DH, Pamer EG. MHC class I/peptide stability: implications for immunodominance, in vitro proliferation, and diversity of responding CTL. J Immunol. 1998;160:4441–8. [PubMed] [Google Scholar]
- Buus S, Sette A, Colon SM, Miles C, Grey HM. The relation between major histocompatibility complex (MHC) restriction and the capacity of Ia to bind immunogenic peptides. Science. 1987;235:1353–8. doi: 10.1126/science.2435001. [DOI] [PubMed] [Google Scholar]
- Dedier S, Reinelt S, Rion S, Folkers G, Rognan D. Use of fluorescence polarization to monitor MHC-peptide interactions in solution. J Immunol Methods. 2001;255:57–66. doi: 10.1016/s0022-1759(01)00423-9. [DOI] [PubMed] [Google Scholar]
- Ferre H, Ruffet E, Blicher T, Sylvester-Hvid C, Nielsen LL, Hobley TJ, Thomas OR, Buus S. Purification of correctly oxidized MHC class I heavy-chain molecules under denaturing conditions: a novel strategy exploiting disulfide assisted protein folding. Protein Sci. 2003;12:551–9. doi: 10.1110/ps.0233003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gakamsky DM, Davis DM, Haas E, Strominger JL, Pecht I. Photophysical analysis of class I major histocompatibility complex protein assembly using a xanthene-derivatized beta2-microglobulin. Biophys J. 1999;76:1552–60. doi: 10.1016/S0006-3495(99)77314-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heise CE, Sullivan SK, Crowe PD. Scintillation proximity assay as a high-throughput method to identify slowly dissociating nonpeptide ligand binding to the GnRH receptor. J Biomol Screen. 2007;12:235–9. doi: 10.1177/1087057106297362. [DOI] [PubMed] [Google Scholar]
- Hunter WM, Greenwood FC. Preparation of iodine-131 labelled human growth hormone of high specific activity. Nature. 1962;194:495–6. doi: 10.1038/194495a0. [DOI] [PubMed] [Google Scholar]
- Joseph MA, Mitchell ML, Evanseck JD, Kovacs JR, Jia L, Shen H, Meng WS. Secondary anchor substitutions in an HLA-A*0201-restricted T-cell epitope derived from Her-2/neu. Mol Immunol. 2007;44:322–31. doi: 10.1016/j.molimm.2006.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khawaja XZ. Development of a scintillation proximity assay for human insulin-like growth factor-binding protein 4 compatible with inhibitor high-throughput screening. Anal Biochem. 2007;366:80–6. doi: 10.1016/j.ab.2007.03.011. [DOI] [PubMed] [Google Scholar]
- Khilko SN, Corr M, Boyd LF, Lees A, Inman JK, Margulies DH. Direct detection of major histocompatibility complex class I binding to antigenic peptides using surface plasmon resonance. Peptide immobilization and characterization of binding specificity. J Biol Chem. 1993;268:15425–34. [PubMed] [Google Scholar]
- Khilko SN, Jelonek MT, Corr M, Boyd LF, Bothwell AL, Margulies DH. Measuring interactions of MHC class I molecules using surface plasmon resonance. J Immunol Methods. 1995;183:77–94. doi: 10.1016/0022-1759(95)00033-7. [DOI] [PubMed] [Google Scholar]
- Leisner C, Loeth N, Lamberth K, Justesen S, Sylvester-Hvid C, Schmidt EG, Claesson M, Buus S, Stryhn A. One-pot, mix-and-read peptide-MHC tetramers. PLoS ONE. 2008;3:e1678. doi: 10.1371/journal.pone.0001678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen AC, Pedersen LO, Hansen AS, Nissen MH, Olsen M, Hansen PR, Holm A, Buus S. A quantitative assay to measure the interaction between immunogenic peptides and purified class I major histocompatibility complex molecules. Eur J Immunol. 1994;24:385–92. doi: 10.1002/eji.1830240218. [DOI] [PubMed] [Google Scholar]
- Ostergaard Pedersen L, Nissen MH, Hansen NJ, Nielsen LL, Lauenmoller SL, Blicher T, Nansen A, Sylvester-Hvid C, Thromsen AR, Buus S. Efficient assembly of recombinant major histocompatibility complex class I molecules with preformed disulfide bonds. Eur J Immunol. 2001;31:2986–96. doi: 10.1002/1521-4141(2001010)31:10<2986::aid-immu2986>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Parker KC, Bednarek MA, Coligan JE. Scheme for ranking potential HLA-A2 binding peptides based on independent binding of individual peptide side-chains. J Immunol. 1994;152:163–75. [PubMed] [Google Scholar]
- Parker KC, DiBrino M, Hull L, Coligan JE. The beta 2-microglobulin dissociation rate is an accurate measure of the stability of MHC class I heterotrimers and depends on which peptide is bound. J Immunol. 1992;149:1896–904. [PubMed] [Google Scholar]
- Parker KC, Strominger JL. Localization of the sites of iodination of human beta 2-microglobulin: quaternary structure implications for histocompatibility antigens. Biochemistry. 1983;22:1145–53. doi: 10.1021/bi00274a024. [DOI] [PubMed] [Google Scholar]
- Pedersen LO, Hansen AS, Olsen AC, Gerwien J, Nissen MH, Buus S. The interaction between beta 2-microglobulin (beta 2m) and purified class-I major histocompatibility (MHC) antigen. Scand J Immunol. 1994;39:64–72. doi: 10.1111/j.1365-3083.1994.tb03341.x. [DOI] [PubMed] [Google Scholar]
- Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger S, Stewart S, Surko P, Way S, Wilson S, Sette A. The immune epitope database and analysis resource: from vision to blueprint. PLoS Biol. 2005;3:e91. doi: 10.1371/journal.pbio.0030091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Udenfriend S, Gerber LD, Brink L, Spector S. Scintillation proximity radioimmunoassay utilizing 125I-labeled ligands. Proc Natl Acad Sci U S A. 1985;82:8672–6. doi: 10.1073/pnas.82.24.8672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Burg SH, Visseren MJ, Brandt RM, Kast WM, Melief CJ. Immunogenicity of peptides bound to MHC class I molecules depends on the MHC-peptide complex stability. J Immunol. 1996;156:3308–14. [PubMed] [Google Scholar]