

Summary
Complement receptors for C3-derived fragments (CR1-4) play critical roles in innate and adaptive immune responses. Of these receptors, CR3 and CR4 are important in binding and phagocytosis of complement-opsonized pathogens including parasites. The role of CR3 and CR4 in malaria or in cerebral malaria has received little attention and remains poorly understood in both human disease and rodent models of malaria. CR3 and CR4 are members of the β2-integrin family of adhesion molecules and are expressed on all leukocytes that participate in the development of cerebral malaria (CM), most importantly as it relates to parasite phagocytosis (monocytes/macrophages) and antigen processing and presentation (dendritic cells). Thus it is possible that these receptors might play an important role in disease development. To address this question, we examined the role of CR3-/- and CR4-/- mice in experimental cerebral malaria (ECM). We found that both CR3-/- and CR4-/- mice were fully susceptible to ECM and developed disease comparable to wild type mice. Our results indicate that CR3 and CR4 are not critical to the pathogenesis of ECM despite their role in elimination of complement-opsonized pathogens. These findings support recent studies indicating the importance of the terminal complement pathway and the membrane attack complex in ECM pathogenesis.
Keywords: cerebral malaria, complement phagocytic receptors, β2-integrins
Of the complement C3 receptors, only the complement receptor 1 (CR1, CD35) has an established role in the pathophysiology of malaria. CR1 serves as a host erythrocyte receptor for Plasmodium falciparum through its binding to PfRh4 (1-3) and polymorphic variants of CR1 associate with susceptibility to, and/or resistance to, severe malaria and cerebral malaria (CM) (reviewed in (4)). By contrast, the remaining complement C3 receptors, CR2, CR3 and CR4, have poorly defined roles in the development and progression of malaria infection and CM. Based on in vitro studies, C3dg, the ligand for CR2, is generated in large amounts and deposited on red blood cells in an alternative pathway-specific mechanism in murine malaria infections (5). The relevance of this observation to human cerebral malaria remains unclear, especially in light of studies demonstrating that coupling of C3d to malaria antigens in murine vaccine studies does not provide enhanced immunogenicity (6-8). The remaining two receptors, CR3 and CR4, are well known for their role in the phagocytosis of iC3b-opsonized pathogens (reviewed in (9-11)). However, the contribution of CR3 and CR4 to parasite killing and/or clearance via phagocytosis in both human and murine uncomplicated malaria and in CM is not known.
Complement receptor 3 (a.k.a., αMβ2, CD11b/CD18) and CR4 (a.k.a., αXβ2, CD11c/CD18) are members of the β2-integrin family of adhesion molecules that play important roles in tissue-specific homing of leukocytes during inflammation, leukocyte activation in the immune response and phagocytosis (12-14). Both receptors bind multiple ligands and are widely expressed on all leukocytes (15) including neutrophils and macrophages that aid in clearance of malaria parasites and, dendritic cells which process antigen after ingesting parasite-infected red blood cells. The extent to which CR3 and CR4 contribute to these essential immune functions during malaria has received little attention. Instead CR3 and CR4 are primarily used as cell surface markers to distinguish between myeloid subsets or followed for changes in expression during the course of malaria infection (16-20). Treatment with anti-CR3 antibody reportedly had no effect on the course of experimental cerebral malaria (ECM) (21, 22). However, technical limitations of blocking antibody experiments require cautious interpretation as many variables affect experimental outcome (e.g., differing antibody affinities and avidities, and variability with respect to dosing, timing, and antibody half-life). To avoid these technical limitations and directly determine if CR3 and or CR4 are critical for the development and progression of ECM, we used mice deficient in these receptors.
We compared susceptibility and clinical severity of CR3-/-(23), CR4-/-(24) and wild type mice in P. berghei ANKA-induced ECM as previously described (25). All mice used in this study were on the C57Bl/6 background. For these studies, P. berghei ANKA was maintained by passage in BALB/c mice (26). ECM was induced by injecting mice i.p. with 5 × 105 PbA-infected RBCs. Peripheral parasitemia was monitored on day 6 post-infection by Giemsa-stained, thin-blood smears. Mice were monitored twice daily for clinical signs of neurologic disease, using the following scoring scale: 0, asymptomatic; 1, symptomatic (ruffled fur); 2, mild disease (slow righting); 3, moderate disease (difficulty righting); 4, severe disease (ataxia, seizures, coma); 5, dead. Mice observed having seizures were given a score of 4 regardless of other clinical signs of disease. Moribund animals were scored 4.5 and humanely sacrificed. Mice were classified as having ECM if they displayed these symptoms between days 6-9 post-infection, had positive thin-blood smears, and had a corresponding drop in external body temperature or succumbed to infection.
We found that CR3-/- and CR4-/-mice did not survive significantly longer than wild type mice (p>0.05, Log rank test; Figure 1a and d) and that all three groups of mice succumbed to infection at the same rate. Disease severity in CR3-/- and CR4-/-mice was identical compared to wild type mice and corresponded well to survival (Figure 1b and e). Interestingly, peripheral parasitemia was significantly elevated in CR3-/- (p=0.0028, unpaired Student's t-test), but not in CR4-/- mice compared to wild type mice (Figure 1c and f). The latter results suggest a minor role for CR3 in parasite clearance, but not in survival or disease severity. The absence of an altered disease phenotype in CR3-/- and CR4-/- mice raised questions regarding the role of other β2-integrin adhesion molecules in ECM. Previous studies have reported minimal differences in the course of ECM through day 10 in CD11d-/- (αDβ2) mice (27) not unlike what we report here for CR3 and CR4. In contrast, LFA-1 (CD11a, (αLβ2), also a member of the β2-integrin family, is thought to play a key role in the development of ECM based on studies demonstrating significant protection from the development of ECM on treatment with anti-LFA-1 antibodies (21, 22, 28). To our knowledge no one has directly assessed the role of LFA-1 in ECM using LFA-1-/- mice to verify these reports. Therefore, we performed ECM using LFA-1-/- mice (29). We found that LFA-1-/- mice were significantly protected from ECM (p=0.0001, Log rank test) developing only mild clinical disease (p<0.05, days 6-10, Wilcoxon rank-sum test) and that peripheral parasite levels were similar to wild type mice (p>0.05, Student's t-test) (Figure 2a-c).
Figure 1.
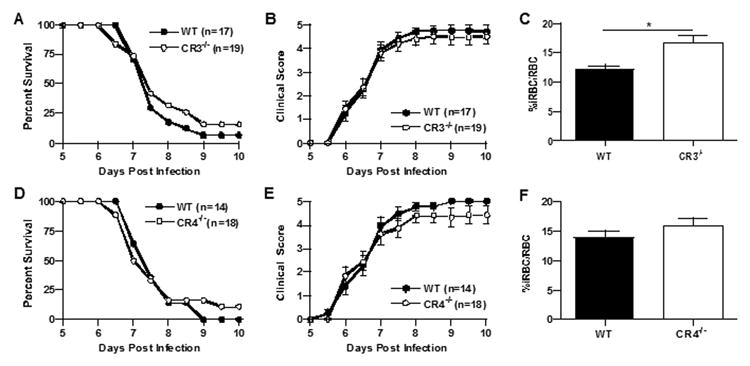
Cerebral malaria in CR3-/- and CR4-/- mice is comparable to that of wild type mice. Wild type and complement receptor-deficient mice were injected i.p. with 5 × 105 PbA-iRBC and clinical scores and survival were monitored twice daily for ten days as previously described. CR3-/- mice (n=19) were fully susceptible to disease-induced mortality (p>0.50, Log rank test) compared to wild type mice (n=17) (a) and had similar disease severity from day 6 through 10 (b). CR4-/- mice (n=18) were as susceptible to disease-induced mortality (p>0.50, Log rank test) as wild type mice (n=14) (d) and had similar disease severity from day 6 through 10 (e). Peripheral parasitemia, assessed at day 6 after infection, was significantly elevated in CR3-/- mice compared to wild type mice (12.1 vs. 16.7 %iRBCs/total RBC, wild type vs. CR3/- mice respectively, p=0.0028, Student's t-test) (c), but not for CR4-/- mice (13.9 vs. 16 %iRBCs/total RBC, wild type vs. CR4-/- mice respectively, p=0.23) (f). The data shown for all panels are the mean +/- SE pooled from four independent experiments.
Figure 2.
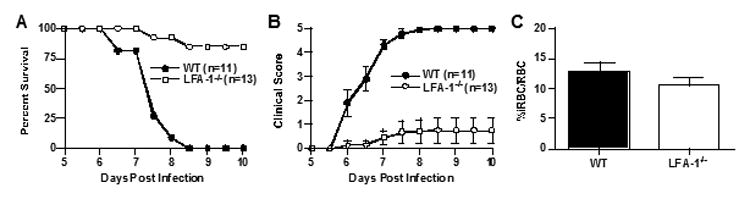
LFA-1-/- mice are highly resistant to cerebral malaria. LFA-1-/- mice (n=13) were significantly resistant to disease-induced mortality (p=0.0001, Log rank test) compared to wild type mice (n=11) (a) and had reduced disease severity clinical on days 6 through 10 (p<0.05, Wilcoxon rank-sum test) (b). There was no significant difference in peripheral parasitemia between LFA-1-/- and wild type mice (p>0.05, Student's t test) (c). The data shown for all panels are the mean +/- SE pooled from three independent experiments.
Our original hypothesis was that deletion of either CR3 or CR4 would potentiate disease development by virtue of impaired parasite clearance thus leading to a more severe course of ECM compared to wild type mice. To our surprise, there was no difference in survival or clinical disease between the complement receptor mutants and wild type mice. An alternative outcome may have been reduced disease severity due to altered leukocyte trafficking in the absence of either receptor, mostly due to loss of interaction with ICAM-1 (30-32), which is expressed at high levels on endothelial surfaces in the CNS during CM and ECM (22, 33). Thus loss of CR3 and CR4 expression on T cells and macrophages could reasonably be expected to reduce adherence and subsequent vascular occlusion, both characteristic features of CM. We cannot rule out the possibility of compensatory changes in receptor expression during ECM in either receptor-deficient mouse, however we have not observed such changes in other CNS inflammatory disease models using these mice (D.C. Bullard and S.R. Barnum, unpublished observations). The finding that LFA-1-/- mice are significantly resistant to the development of ECM, while CR3-/- and CR4-/- mice are not, indicates that, of the β2-integrin family members, LFA-1 plays the most critical role in ECM.
Regardless of the potential roles for CR3 and CR4 in ECM pathophysiology, the data we present here supports a developing story indicating that, of the complement pathways and components, the complement terminal pathway and the membrane attack complex (MAC) are most important in ECM development. Previous studies have shown that deletion of C5 results in marked increase in resistance to ECM and that inhibition of C9 (and therefore the MAC) is protective in ECM (25, 34). More recently we have shown that inhibition of the classical or alternative complement pathways does not alter the course of ECM. Furthermore, deletion of C3 does not prevent C5 cleavage indicating that the canonical C5 convertases are not wholly responsible for C5 cleavage during ECM (T.N. Ramos et al., In press). The data we present here indicate that the opsonophagocytic functions of the complement system at the level of C3-derived fragments is also not critical for the development and progression of ECM. Thus in the murine cerebral malaria model system, biological functions of the complement system derived from components and activation pathways prior to C5 cleavage play a minor role in ECM pathophysiology. Taken together, these data indicate that targeting C5 or components of the MAC may offer a new therapeutic avenue for cerebral malaria.
Acknowledgments
This work was supported by NIH grants T32 AI07051 and NS077811 (to TNR), AI08382 (to SRB). The authors gratefully acknowledge the continuing support of Drs. Julian Rayner and Oliver Billker.
Footnotes
Disclosures: None
References
- 1.Spadafora C, Awandare GA, Kopydlowski KM, et al. Complement receptor 1 is a sialic acid-independent erythrocyte receptor of Plasmodium falciparum. PLoS pathogens. 2010;6:e1000968. doi: 10.1371/journal.ppat.1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tham WH, Wilson DW, Lopaticki S, et al. Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proceedings of the National Academy of Sciences of the United States of America. doi: 10.1073/pnas.1008151107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Awandare GA, Spadafora C, Moch JK, Dutta S, Haynes JD, Stoute JA. Plasmodium falciparum field isolates use complement receptor 1 (CR1) as a receptor for invasion of erythrocytes. Molecular and biochemical parasitology. 2011;177:57–60. doi: 10.1016/j.molbiopara.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stoute JA. Complement-regulatory proteins in severe malaria: too little or too much of a good thing? Trends Parasitol. 2005;21:218–223. doi: 10.1016/j.pt.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 5.Pawluczkowycz AW, Lindorfer MA, Waitumbi JN, Taylor RP. Hematin promotes complement alternative pathway-mediated deposition of C3 activation fragments on human erythrocytes: potential implications for the pathogenesis of anemia in malaria. J Immunol. 2007;179:5543–5552. doi: 10.4049/jimmunol.179.8.5543. [DOI] [PubMed] [Google Scholar]
- 6.Bergmann-Leitner ES, Scheiblhofer S, Weiss R, et al. C3d binding to the circumsporozoite protein carboxy-terminus deviates immunity against malaria. International immunology. 2005;17:245–255. doi: 10.1093/intimm/dxh205. [DOI] [PubMed] [Google Scholar]
- 7.Bergmann-Leitner ES, Duncan EH, Leitner WW, et al. C3d-defined complement receptor-binding peptide p28 conjugated to circumsporozoite protein provides protection against Plasmodium berghei. Vaccine. 2007;25:7732–7736. doi: 10.1016/j.vaccine.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 8.Weiss R, Gabler M, Jacobs T, Gilberger TW, Thalhamer J, Scheiblhofer S. Differential effects of C3d on the immunogenicity of gene gun vaccines encoding Plasmodium falciparum and Plasmodium berghei MSP1(42) Vaccine. 2010;28:4515–4522. doi: 10.1016/j.vaccine.2010.04.054. [DOI] [PubMed] [Google Scholar]
- 9.Dale DC, Boxer L, Liles WC. The phagocytes: neutrophils and monocytes. Blood. 2008;112:935–945. doi: 10.1182/blood-2007-12-077917. [DOI] [PubMed] [Google Scholar]
- 10.Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nature immunology. 2010;11:785–797. doi: 10.1038/ni.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nature reviews Immunology. 2011;11:723–737. doi: 10.1038/nri3073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis DM. Mechanisms and functions for the duration of intercellular contacts made by lymphocytes. Nature reviews Immunology. 2009;9:543–555. doi: 10.1038/nri2602. [DOI] [PubMed] [Google Scholar]
- 13.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nature reviews Immunology. 2007;7:678–689. doi: 10.1038/nri2156. [DOI] [PubMed] [Google Scholar]
- 14.Luster AD, Alon R, von Andrain UH. Immune cell migration in inflammation: present and future therapeutics. Nature immunology. 2005;6:1182–1190. doi: 10.1038/ni1275. [DOI] [PubMed] [Google Scholar]
- 15.Hu X, Wohler JE, Dugger KJ, Barnum SR. beta2-integrins in demyelinating disease: not adhering to the paradigm. Journal of leukocyte biology. 2010;87:397–403. doi: 10.1189/jlb.1009654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schwarzer E, Alessio M, Ulliers D, Arese P. Phagocytosis of the malarial pigment, hemozoin, impairs expression of major histocompatibility complex class II antigen, CD54, and CD11c in human monocytes. Infection and immunity. 1998;66:1601–1606. doi: 10.1128/iai.66.4.1601-1606.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Perry JA, Rush A, Wilson RJ, Olver CS, Avery AC. Dendritic cells from malaria-infected mice are fully functional APC. J Immunol. 2004;172:475–482. doi: 10.4049/jimmunol.172.1.475. [DOI] [PubMed] [Google Scholar]
- 18.Van den Steen PE, Van Aelst I, Starckx S, Maskos K, Opdenakker G, Pagenstecher A. Matrix metalloproteinases, tissue inhibitors of MMPs and TACE in experimental cerebral malaria. Laboratory investigation; a journal of technical methods and pathology. 2006;86:873–888. doi: 10.1038/labinvest.3700454. [DOI] [PubMed] [Google Scholar]
- 19.Ing R, Segura M, Thawani N, Tam M, Stevenson MM. Interaction of mouse dendritic cells and malaria-infected erythrocytes: uptake, maturation, and antigen presentation. J Immunol. 2006;176:441–450. doi: 10.4049/jimmunol.176.1.441. [DOI] [PubMed] [Google Scholar]
- 20.Voisine C, Mastelic B, Sponaas AM, Langhorne J. Classical CD11c+ dendritic cells, not plasmacytoid dendritic cells, induce T cell responses to Plasmodium chabaudi malaria. International journal for parasitology. 2010;40:711–719. doi: 10.1016/j.ijpara.2009.11.005. [DOI] [PubMed] [Google Scholar]
- 21.Falanga PB, Butcher EC. Late treatment with anti-LFA-1 (CD11a) antibody prevents cerebral malaria in a mouse model. European journal of immunology. 1991;21:2259–2263. doi: 10.1002/eji.1830210938. [DOI] [PubMed] [Google Scholar]
- 22.Grau GE, Pointaire P, Piguet PF, et al. Late administration of monoclonal antibody to leukocyte function-antigen 1 abrogates incipient murine cerebral malaria. European journal of immunology. 1991;21:2265–2267. doi: 10.1002/eji.1830210939. [DOI] [PubMed] [Google Scholar]
- 23.Lu H, Smith CW, Perrard J, et al. LFA-1 is sufficient in mediating neutrophil transmigration in Mac-1 deficient mice. J Clin Invest. 1997;99:1340–1350. doi: 10.1172/JCI119293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wu H, Rodgers JR, Perrard XY, et al. Deficiency of CD11b or CD11d results in reduced staphylococcal enterotoxin-induced T cell response and T cell phenotypic changes. J Immunol. 2004;173:297–306. doi: 10.4049/jimmunol.173.1.297. [DOI] [PubMed] [Google Scholar]
- 25.Ramos TN, Darley M, Bilker O, et al. The membrane attack complex of complement is required for the development of experimental cerebral malaria. J Immun. 2011 doi: 10.4049/jimmunol.1100603. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sinden RE, Butcher GA, Beetsma AL. Maintenance of the Plasmodium berghei life cycle. Methods Mol Med. 2002;72:25–40. doi: 10.1385/1-59259-271-6:25. [DOI] [PubMed] [Google Scholar]
- 27.Miyazaki Y, Bunting M, Stafforini DM, et al. Integrin alphaDbeta2 is dynamically expressed by inflamed macrophages and alters the natural history of lethal systemic infections. J Immunol. 2008;180:590–600. doi: 10.4049/jimmunol.180.1.590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grau GE, Tacchini-Cottier F, Vesin C, et al. TNF-induced microvascular pathology: active role for platelets and importance of the LFA-1/ICAM-1 interaction. Eur Cytokine Netw. 1993;4:415–419. [PubMed] [Google Scholar]
- 29.Ding ZM, Babensee JE, Simon SI, et al. Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J Immunol. 1999;163:5029–5038. [PubMed] [Google Scholar]
- 30.Diamond MS, Staunton DE, de Fougerolles AR, et al. ICAM-1 (CD54): a counter-receptor for Mac-1 (CD11b/CD18) The Journal of cell biology. 1990;111:3129–3139. doi: 10.1083/jcb.111.6.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Diamond MS, Garcia-Aguilar J, Bickford JK, Corbi AL, Springer TA. The I domain is a major recognition site on the leukocyte integrin Mac-1 (CD11b/CD18) for four distinct adhesion ligands. The Journal of cell biology. 1993;120:1031–1043. doi: 10.1083/jcb.120.4.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Frick C, Odermatt A, Zen K, et al. Interaction of ICAM-1 with beta 2-integrin CD11c/CD18: characterization of a peptide ligand that mimics a putative binding site on domain D4 of ICAM-1. European journal of immunology. 2005;35:3610–3621. doi: 10.1002/eji.200425914. [DOI] [PubMed] [Google Scholar]
- 33.Turner GD, Morrison H, Jones M, et al. An immunohistochemical study of the pathology of fatal malaria. Evidence for widespread endothelial activation and a potential role for intercellular adhesion molecule-1 in cerebral sequestration. The American journal of pathology. 1994;145:1057–1069. [PMC free article] [PubMed] [Google Scholar]
- 34.Patel SN, Berghout J, Lovegrove FE, et al. C5 deficiency and C5a or C5aR blockade protects against cerebral malaria. J Exp Med. 2008;205:1133–1143. doi: 10.1084/jem.20072248. [DOI] [PMC free article] [PubMed] [Google Scholar]