

Abstract
A full-term female neonate presented with persistent respiratory failure and radiologic studies consistent with surfactant deficiency. Sequencing of the ATP-binding cassette transporter A3 gene (ABCA3) revealed 3 mutations: R280C, V1399M, and Q1589X. The infant underwent bilateral lung transplantation at 9 months of age and is alive at 3 years of age. Parental sequencing demonstrated that 2 of the mutations (R280C and Q1589X) were oriented on the same allele (cis) while V1399M was oriented on the opposite allele (trans). As more than one mutation in ABCA3 can be present on the same allele, parental studies are needed to determine allelic orientation to inform clinical decision making and future reproductive counseling.
INTRODUCTION
ATP Binding Cassette member A3 (ABCA3), a member of a family of transporter proteins that hydrolyze ATP to move substrates across biological membranes, is expressed in alveolar type II cells and localized to the membrane of lamellar bodies, the intracellular organelles where pulmonary surfactant is assembled and processed.1, 2 ABCA3 transports phospholipids into the lamellar bodies which then assemble with surfactant proteins B and C to form mature surfactant.3 Recessive, loss of function mutations in ABCA3 have been associated with lethal neonatal respiratory failure and childhood interstitial lung disease (chILD).4, 5 Here, we describe a full term female infant with persistent respiratory failure for whom genetic sequencing revealed 3 mutations in ABCA3 and parental studies demonstrated that 2 mutations were present on the same allele (cis). Parental studies are needed to determine whether 2 or more ABCA3 mutations in a symptomatic infant or child disrupt expression of both alleles and, therefore, result in ABCA3 deficiency.
CASE
Term female infant (birth weight 2870 grams) was born to a 30 year old G2, P1 mother via spontaneous vaginal delivery. Mother had one previous child who was healthy and both mother and father were healthy. Immediately after birth, the infant developed respiratory distress and was placed on continuous positive airway pressure (CPAP). Her physical examination was notable for bilateral coarse breath sounds, subcostal and intercostal retractions. Apgar scores were 3, 3, and 9 at 1, 5, and 10 minutes, respectively. Chest radiograph demonstrated diffuse bilateral granular opacities consistent with surfactant deficiency. Her respiratory distress persisted and prompted intubation and surfactant administration on day of life (DOL) 2. Over the next 2-3 days, she developed progressive hypoxic respiratory failure that necessitated high frequency oscillatory ventilation, FiO2 1.0, and nitric oxide administration. She developed a left-sided pneumothorax that required thoracostomy tube drainage. Repeat chest radiograph demonstrated persistent diffuse bilateral granular opacities, and she received a second dose of surfactant. At a week of life, she was transitioned to conventional ventilation and tolerated discontinuation of nitric oxide. On DOL 11, she was extubated to CPAP with FiO2 0.4-0.5, but had persistent tachypnea. An echocardiogram demonstrated no evidence of anatomic heart disease or pulmonary hypertension. Chest computed tomography at 7 weeks of life showed coarse bilateral granular opacities.
Given the clinical suspicion for a genetic disorder of surfactant dysfunction, sequencing was performed for surfactant protein B (SFTPB) and ABCA3. No mutations were identified in SFTPB, and three rare, missense and nonsense mutations were identified in ABCA3: p.R280C (c.838C>T), p.V1399M (c.4195G>A), and p.Q1589X (C.4765C>T). Parental sequencing for ABCA3 demonstrated that these mutations were present on both maternal (V1399M) and paternal (R280C and Q1589X on same allele in cis) alleles. Sequencing was performed in our research laboratory, informed consent was obtained from parents, and this study was approved by the Washington University School of Medicine Human Research Protection Office.
The infant was transferred to a lung transplant center at 2.5 months of age and underwent bilateral lung transplantation at 9 months of age. Pathologic examination of her native lungs showed diffuse interstitial fibrosis with alveolar remodeling and prominent type II pneumocyte hyperplasia. Electron microscopy demonstrated some small, dense lamellar bodies and occasional fused lamellar bodies (Figure). After postoperative tracheostomy, she was decannulated 2 months later. She was weaned to room air at approximately 1 year of age. A gastrostomy tube was placed at 18 months for nutritional supplementation. She is currently alive at 3 years of age with mildly delayed speech and motor developmental milestones.
Figure.
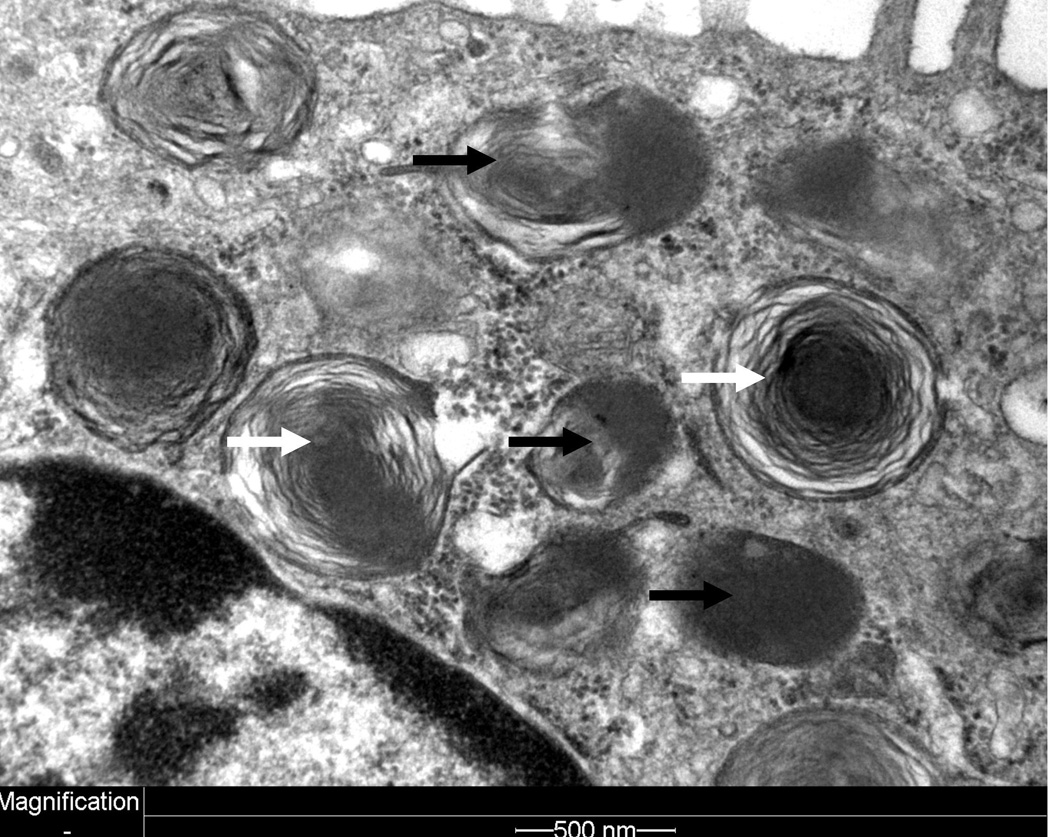
Electron microscopy image of alveolar type II cell demonstrating small, densely wound lamellar bodies and dense body (black arrows). Some relatively normal-appearing small lamellar bodies are also observed (white arrows).
DISCUSSION
ABCA3 consists of 1704 amino acids and is encoded by an 80kb gene on human chromosome 16. Recessive, loss of function mutations in ABCA3 were first identified in term neonates dying of respiratory distress syndrome,4 and later in children with interstitial lung disease.5 Mice genetically engineered to be deficient for ABCA3 (abca3-/-) die from respiratory failure within the first hour of life and demonstrate absent surfactant in the alveolar space, loss of mature lamellar bodies, and reduced phospholipid content of their lung tissue.3, 6 Lamellar bodies from patients with ABCA3 deficiency usually are small with densely packed phospholipid membranes and eccentrically-placed, dense inclusion bodies.4, 7
Over 180 ABCA3 mutations have been identified among ethnically and geographically diverse symptomatic infants and children.8 Most ABCA3 mutations are rare and private.8 Frameshift, nonsense, missense, splice site mutations, and insertions/deletions have been identified.8, 9, 10 However, phenotype and prognosis are difficult to predict based on mutation type or location, especially for individuals with missense and splice site mutations and in-frame insertion/deletions.8, 11 Approximately 1.5-3.6% of European and African-descent individuals carry single mutations in ABCA3.12 While no specific therapies exist for ABCA3 deficiency, some patients have responded to medical therapies including steroids, hydroxychloroquine, and azithromycin,13 while others have progressed to requiring lung transplantation.14
This infant was found to have 3 rare ABCA3 mutations and parental sequencing determined that 2 of the mutations were paternally inherited in cis: p.R280C and p.Q1589X. Three out of 6498 individuals of African- and European-descent in the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/, accessed 12/2014) are heterozygous for p.R280C, and this mutation has been identified both in isolation and in cis with p.Q1589X in an unrelated patient,8 likely attributable to different haplotype backgrounds. P.V1399M is rare and has been previously reported in symptomatic infants.8, 15 Neither p.V1399M nor p.Q1589X is identified among individuals in the ESP database. P.Q1589X is predicted to result in a truncated protein. Both p.R280C and p.V1399M are predicted to be damaging to ABCA3 protein function by the majority of in silico prediction programs in ANNOVAR.16 P.R280C has been studied in vitro and impairs intracellular trafficking.17
In a recent study of 185 subjects with ABCA3 deficiency, approximately 10 percent of subjects had ABCA3 mutations in cis,8 emphasizing the importance of parental DNA samples to determine mutation orientation. Symptomatic infants and children with more than 2 ABCA3 mutations have also been reported.18, 19, 20, 21 As single (monoallelic) mutations in ABCA3 have been associated with reversible respiratory distress syndrome among term and late preterm infants,12 confirming that a symptomatic infant or child has mutations on both alleles is necessary to inform clinical decision making and future reproductive counseling.
ACKNOWLEDGEMENTS
The authors would like to thank the National Heart Lung Blood Institute (NHLBI) GO Exome Sequencing Project and its ongoing studies which produced and provided exome variant calls for comparison: the Lung GO Sequencing Project (HL-102923), the WHI Sequencing Project (HL-102924), the Broad GO Sequencing Project (HL-102925), the Seattle GO Sequencing Project (HL-102926), and the Heart GO Sequencing Project (HL-103010).
Support: National Institutes of Health (K08 HL105891 (JAW), K12 HL089968 (FSC), R01 HL065174 (FSC, AH), R01 HL082747 (FSC, AH), American Lung Association (JAW), the American Thoracic Society (JAW), the Saigh Foundation (FSC, AH).
Footnotes
CONFLICT OF INTEREST: The authors declare no conflict of interest.
REFERENCES
- 1.Yamano G, Funahashi H, Kawanami O, Zhao LX, Ban N, Uchida Y, et al. ABCA3 is a lamellar body membrane protein in human lung alveolar type II cells. FEBS letters. 2001;508(2):221–225. doi: 10.1016/s0014-5793(01)03056-3. [DOI] [PubMed] [Google Scholar]
- 2.Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI, et al. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem. 2002;277(25):22147–22155. doi: 10.1074/jbc.M201812200. [DOI] [PubMed] [Google Scholar]
- 3.Ban N, Matsumura Y, Sakai H, Takanezawa Y, Sasaki M, Arai H, Inagaki N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007;282(13):9628–9634. doi: 10.1074/jbc.M611767200. [DOI] [PubMed] [Google Scholar]
- 4.Shulenin SNL, Annilo T, Wert SE, Whitsett JA, Dean M. ABCA3 Gene Mutations in Newborns with Fatal Surfactant Deficiency. N Engl J Med. 2004;350:1296–1303. doi: 10.1056/NEJMoa032178. [DOI] [PubMed] [Google Scholar]
- 5.Bullard JE, Wert SE, Whitsett JA, Dean M, Nogee LM. ABCA3 mutations associated with pediatric interstitial lung disease. American journal of respiratory and critical care medicine. 2005;172(8):1026–1031. doi: 10.1164/rccm.200503-504OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fitzgerald ML, Xavier R, Haley KJ, Welti R, Goss JL, Brown CE, Zhuang DZ, Bell SA, Lu N, Mckee M, Seed B, Freeman MW. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–632. doi: 10.1194/jlr.M600449-JLR200. [DOI] [PubMed] [Google Scholar]
- 7.Wert SE, Whitsett JA, Nogee LM. Genetic disorders of surfactant dysfunction. Pediatric and developmental pathology : the official journal of the Society for Pediatric Pathology and the Paediatric Pathology Society. 2009;12(4):253–274. doi: 10.2350/09-01-0586.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wambach JA, Casey AM, Fishman MP, Wegner DJ, Wert SE, Cole FS, et al. Genotype-phenotype correlations for infants and children with ABCA3 deficiency. American journal of respiratory and critical care medicine. 2014;189(12):1538–1543. doi: 10.1164/rccm.201402-0342OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agrawal A, Hamvas A, Cole FS, Wambach JA, Wegner D, Coghill C, et al. An intronic ABCA3 mutation that is responsible for respiratory disease. Pediatric research. 2012;71(6):633–637. doi: 10.1038/pr.2012.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Henderson LB, Melton K, Wert S, Couriel J, Bush A, Ashworth M, et al. Large ABCA3 and SFTPC deletions resulting in lung disease. Annals of the American Thoracic Society. 2013;10(6):602–607. doi: 10.1513/AnnalsATS.201306-170OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Matsumura Y, Ban N, Ueda K, Inagaki N. Characterization and classification of ATP-binding cassette transporter ABCA3 mutants in fatal surfactant deficiency. J Biol Chem. 2006;281(45):34503–34514. doi: 10.1074/jbc.M600071200. [DOI] [PubMed] [Google Scholar]
- 12.Wambach JA, Wegner DJ, Depass K, Heins H, Druley TE, Mitra RD, et al. Single ABCA3 mutations increase risk for neonatal respiratory distress syndrome. Pediatrics. 2012;130(6):e1575–e1582. doi: 10.1542/peds.2012-0918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayes D, Jr, Lloyd EA, Fitch JA, Bush A. ABCA3 transporter deficiency. American journal of respiratory and critical care medicine. 2012;186(8):807. doi: 10.1164/ajrccm.186.8.807a. [DOI] [PubMed] [Google Scholar]
- 14.Faro A HA. Lung Transplantation for Inherited Disorders of Surfactant Metabolism. Neoreviews. 2008;9(10):e468–e475. [Google Scholar]
- 15.Saugstad OD, Hansen TW, Ronnestad A, Nakstad B, Tollofsrud PA, Reinholt F, et al. Novel mutations in the gene encoding ATP binding cassette protein member A3 (ABCA3) resulting in fatal neonatal lung disease. Acta Paediatr. 2007;96(2):185–190. doi: 10.1111/j.1651-2227.2007.00016.x. [DOI] [PubMed] [Google Scholar]
- 16.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic acids research. 2010;38(16):e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Weichert N, Kaltenborn E, Hector A, Woischnik M, Schams A, Holzinger A, et al. Some ABCA3 mutations elevate ER stress and initiate apoptosis of lung epithelial cells. Respiratory research. 2011;12:4. doi: 10.1186/1465-9921-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Turcu S, Ashton E, Jenkins L, Gupta A, Mok Q. Genetic testing in children with surfactant dysfunction. Arch Dis Child. 2013;98(7):490–495. doi: 10.1136/archdischild-2012-303166. [DOI] [PubMed] [Google Scholar]
- 19.Garmany T, Moxley MA, White FV, Dean M, Nogee LM, Hamvas A. Surfactant composition and function in patients with ABCA3 mutations. Pediatr Res. 2006;59:801–805. doi: 10.1203/01.pdr.0000219311.14291.df. [DOI] [PubMed] [Google Scholar]
- 20.Young LR, Nogee LM, Barnett B, Panos RJ, Colby TV, Deutsch GH. Usual interstitial pneumonia in an adolescent with ABCA3 mutations. Chest. 2008;134(1):192–195. doi: 10.1378/chest.07-2652. [DOI] [PubMed] [Google Scholar]
- 21.Thavagnanam S, Cutz E, Manson D, Nogee LM, Dell SD. Variable clinical outcome of ABCA3 deficiency in two siblings. Pediatric pulmonology. 2013;48(10):1035–1038. doi: 10.1002/ppul.22698. [DOI] [PubMed] [Google Scholar]