

Abstract
Patient: Female, 59
Final Diagnosis: Atyipcal hemolytic uremic syndrome
Symptoms: Delirium • headache
Medication: —
Clinical Procedure: —
Specialty: Hematology
Objective:
Rare disease
Background:
Atypical hemolytic uremic syndrome (aHUS) is a rare disease characterized by hemolysis, thrombocytopenia, and renal dysfunction. It is a disease related to genetic mutations in the alternative complement pathway and has a distinct pathophysiology but is difficult to differentiate from other thrombotic microangiopathies.
Case Report:
We present a case of a 59-year-old female patient who presented with accelerated hypertension, acute renal failure, hemolysis, and encephalopathy. She was managed with antihypertensive medication, but her encephalopathy did not improve. Evaluation resulted in our impression of the disease being atypical hemolytic-uremic syndrome. The patient continued to be managed with good blood pressure control and later was started on eculizumab, but evaluation of response to therapy was hindered by the patient’s non-compliance with therapy and follow-up appointments.
Conclusions:
We have a very limited understanding of the genetics and epidemiology of atypical HUS, and the overlapping clinical features sometimes delay diagnosis and initiation of appropriate treatment of this rare disease.
MeSH Keywords: ADAM Proteins; Hemolytic-Uremic Syndrome; Purpura, Thrombotic Thrombocytopenic; Thrombotic Microangiopathies
Background
Thrombotic microangiopathies are diseases characterized by thrombocytopenia, erythrocyte fragmentation, and elevated levels of LDH. Thickening of the arterioles and capillary walls with prominent endothelial swelling and detachment and subendothelial accumulation of proteins and cell debris characterize and define the pathologic lesion seen in all thrombotic microangiopathies [1]. These can be divided into 2 broad categories of thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS), while HUS is further divided into typical and atypical. Typical HUS is a syndrome of thrombocytopenia, renal dysfunction, and microangiopathic hemolytic anemia associated with Shiga toxin producing Escherichia coli. Atypical HUS (aHUS) is not related to Shiga toxins, and accounts for around 5–10% of all cases of HUS. Compared with HUS, it has a poor prognosis [2]. Pathophysiology of TTP involves platelet aggregation with little or no fibrin involved. These platelet thrombi contain large amounts of von Willebrand factor antigen. A von Willebrand factor-cleaving metalloproteinase exists, which prevents large multimers of von Willebrand factor from entering the circulation; it is called ADAMTS13 (a disintegrin and metalloproteinase, with thrombospondin-1 like domains 13) and is produced in the liver by hepatocytes [2]. In patients with TTP, severely deficient ADAMTS13 activity has been seen in 25–79% of cases at presentation [3,4], whereas HUS is not associated with any reduction in activity or absence of ADAMTS13 [2]. Atypical HUS mostly causes renal dysfunction, but extra-renal manifestations, including neurological complications, have been reported in 10–30% of patients, while neurological complications are mostly seen in patients with TTP [5].
Case Report
A 59-year-old woman presented to the emergency department of our hospital with complaint of frontal headache for 2 days. The headache was dull in nature, with no visual, auditory, or focal neurological deficits. She admitted cocaine abuse 3 days before the presentation. She had medical comorbidities of chronic hepatitis C (never treated), benign essential hypertension (being treated with lisinopril and hydrochlorothiazide), and ischemic stroke 2 months ago, for which she was prescribed aspirin and rosuvastatin, but the she stated that she was non-compliant with the medications because she had no insurance. She also had a history of heroin abuse and was on methadone maintenance treatment. Her social history was significant for active cocaine abuse, and 30-pack/year history of smoking. She was divorced and was living with her 2 children in an apartment.
On examination she was delirious and exhibited fluctuating level of alertness. She had a blood pressure of 256/151 mmHg, pulse of 86, respiratory rate of 18, and temperature of 98°F. On auscultation of lungs, she had bilateral air entry with no adventitious sounds. On precordial exam she had normal heart sounds with no murmurs. There was no jugular venous distention. The abdominal examination result was normal. Neurological exam result was significant for residual weakness in the left hand from prior stroke. Electrocardiogram showed normal sinus rhythm with possible biatrial enlargement. She had a QTc of 428 milliseconds.
Laboratory workup showed a creatinine of 2.8 mg/dL (baseline of 1.6 mg/dL a few months ago), which gradually went up to 4.1 mg/dL. Fractional excretion of sodium (Fena) was 0.6. Complete blood count (CBC) showed hemoglobin of 11.4g/dL, hematocrit of 33.7%, reticulocytosis of 3.2%, and a white cell count of 6.0 k/uL. Her platelet count was 29 k/uL. Peripheral smear was examined, which showed anisocytosis, poikilocytosis, nucleated ; as well as few target cells and schistocytes (Figures 1 and 2), microspherocytes, rouleaux formation, and few platelet clumps. Cardiac markers were negative. Urine toxicology was positive for cocaine; opiate, and methadone. A chest x-ray did not reveal any pathology. She was admitted to the coronary care unit with impression of hypertensive emergency with acute renal failure and encephalopathy and was started on IV nitroglycerin. She did not tolerate IV nitroglycerin due to headache and was started on oral nifedipine, hydralazine, and isosorbide mononitrate with good blood pressure control.
Figure 1.
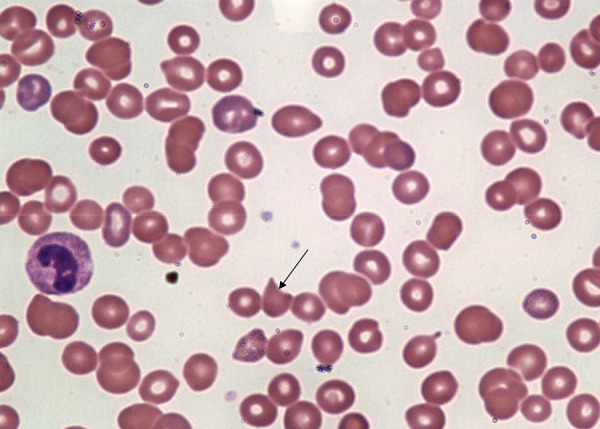
Peripheral smear showing schistocyte (arrow).
Figure 2.
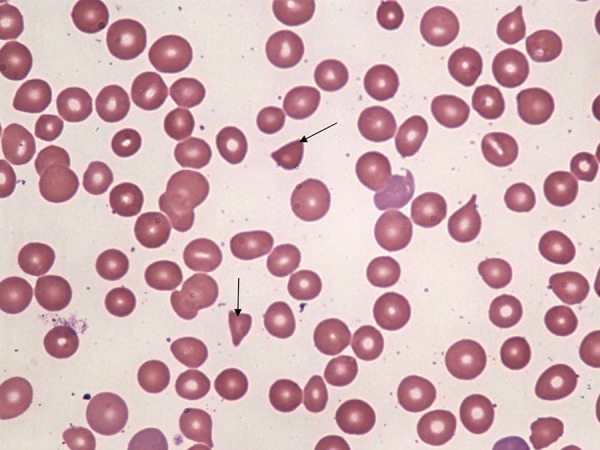
Peripheral smear showing schistocytes (arrow).
Once the blood pressure was better controlled, she was then transferred to the general medical floor. She continued to have fluctuating sensorium. In view of persistent altered mental status, a CT head without contrast was done, which showed chronic vascular ischemic changes with chronic lacunar infarcts involving the right head of the caudate nucleus and thalamus. For further evaluation of renal failure, ultrasound abdomen was done, which showed bilateral renal cortical thinning and a non-obstructing calculus in the lower pole collecting system of the left kidney. An echocardiogram showed an ejection fraction of 75%.
In view of persistent encephalopathy, thrombocytopenia, and acute renal insufficiency, our suspicion was for thrombotic thrombocytopenic purpura (TTP). Her lactate dehydrogenase (LDH) level was elevated to 516 and haptoglobin was 54. She had a normal international normalized ratio (INR), prothrombin time (PT) and partial thromboplastin time (PTT). Her ferritin was 463.5 with transferrin saturation of 25%. She had normal serum vitamin B12 and serum folate levels. Hemoglobin and serum electrophoresis were normal. She did not have any skin rash or purpura. Direct Coombs test and cold agglutinin screen were negative. She had a negative ANA, anti-DNA, myeloperoxidase, and proteinase-3 antibody assays. She was HIV-negative and had a positive anti-HCV antibody assay. She was immune to hepatitis B. Serum cryoglobulin level was normal.
A decision was made to start plasmapheresis and ADAMTS-13 was sent prior to its initiation. She underwent 5 sessions of plasmapheresis with marked improvement in her mental status, improvement in serum creatinine to 2.7; platelet count increased to 117 and LDH decreased to 277. Her ADAMTS-13 levels were 63, which was only slightly decreased and was inconsistent with TTP. Our suspicion was high for atypical hemolytic-uremic syndrome (HUS) because she showed evidence of hemolytic anemia and due to her persistent renal dysfunction. She was offered treatment with eculizumab but she refused.
Once the laboratory parameters and mental status improved, she was discharged with planned follow-up in our hematology clinic, but she did not follow up. The patient was admitted to another hospital with similar complaints and underwent plasmapheresis with improvement and was discharged. She was again admitted to our hospital, with complaints of headache and diarrhea and was found to have a hypertensive emergency with acute-on-chronic renal failure and encephalopathy. She had not been taking any medications and was actively using cocaine.
She was found to have acute renal failure with a creatinine of 11.4 with BUN of 101. Platelet count was 35. A Femoral catheter was placed for urgent plasmapheresis. She also underwent a session of hemodialysis with impression of uremic encephalopathy. There was marked improvement in her mental status. Stool was positive for Clostridium difficile GDH antigen and toxin A assay, for which she completed 10-day therapy with metronidazole every 8 hours. Blood and urine cultures were negative.
Peripheral smear showed occasional schistocytes and large platelets. The renal function and platelet count improved after 6 sessions of plasmapheresis, but the creatinine started to increase again from 4.3 mg/dl to 11 mg/dl with development of signs of fluid overload. She underwent hemodialysis regularly and underwent placement of an internal jugular dialysis catheter. She received a total of 9 sessions of plasmapheresis.
After multiple discussions with her physicians, the patient agreed to treatment with eculizumab and was given meningococcal vaccination in preparation for initiation of eculizumab. After discharge from the hospital, she followed up in our hematology clinic and refused treatment with eculizumab. She was again admitted to our hospital with accelerated hypertension, and thrombocytopenia and during this admission she agreed to be treated with eculizumab. She continued to be non-compliant with treatment and had multiple hospitalizations for uncontrolled hypertension.
Discussion
Atypical HUS is a heterogeneous group presenting with renal dysfunction, neurological dysfunction, and hypertension. Lack of appropriate treatment in a timely fashion is associated with an abysmal prognosis, and leads to chronic renal failure in 31% of patients and chronic replacement therapy in 34.5% [6]. The first attack of aHUS is associated with a mortality of around 25% and around 50% of cases receive long-term dialysis for end-stage renal disease [7].
Complement activation and dysregulation is central to the pathophysiology of aHUS but normal C3 and/or C4 levels do not rule it out, hence complement activation biomarkers cannot be reliably used for making a diagnosis of aHUS [8]. The alternative pathway is involved mostly in the pathogenesis of aHUS and unlike classical and lectin pathways, activation in this does not require initiators; in cases of aHUS it is continuously activated by hydrolysis of C3 occurring spontaneously. Regulators of this pathway involve complement factors I and H, thrombomodulin, and membrane cofactor protein [9]. Heterozygous mutations of complement factor H (CFH) [10–12], membrane cofactor protein (MCP, CD46) [13], and factor I (CFI) [2,14,15] have been associated with onset of aHUS. Gain-of-function mutations in C3 and complement factor B (complement activating factors) have also been associated with aHUS due to hyperactivation of the complement proteins [9]. In around 5–10% of patients with aHUS, autoantibodies to factor H have also been implicated as triggering factor [16,17], which is the most common mutation seen. MCP occurs at a frequency of 5–7%, CFI occurs at rate of 4–8%, and complement component 3 at 4–8%, while mutations in complement factor B and thrombomodulin occurs at 1–4% and 3–5%, respectively [18,19]. Around 35–40% of patients with aHUS will not have genetic mutation identified with current screening modalities [1]. It has also been shown that for diagnosis of aHUS or to distinguish different thrombotic microangiopathies, measurement of complement abnormalities is not reliable [20].
Male-to-female ratio is equal in children but it is more common in adult females. Patients with aHUS present with non-specific symptoms of fatigue, stupor, decreased appetite, nausea, or vomiting. They may present with oliguria and at times even anuria. Severe hypertension is also seen and can be severe enough to cause cardiac failure and encephalopathy. Neurological presentation of aHUS is diverse and patients can have blindness, diplopia, irritability, seizures, hemiparesis or hemiplegia, and even coma [1]. Cardiac microangiopathy leading to myocardial infarction has also been reported in about 3% of patients and is a possible cause of sudden death in such patients [21]. Diarrhea can also be the presenting complaint and has been found to occur in 20–30% of patients with aHUS [22]. About 20% of patients with aHUS have normal renal function [5]. In late stages of aHUS, the lungs are often involved but are never affected in cases of TTP [20].
There are certain tumors that are associated with aHUS, including breast and colon carcinomas, gastric adenocarcinoma, and small cell lung carcinoma. It can also occur with ovarian cancer, prostate cancer, lymphoma, pancreatic cancer, and, very rarely, leukemias [23,24]. It is also seen in cobalamin metabolism disorders, pregnancy (especially eclampsia), autoimmune diseases, drugs (e.g., quinine, mitomycin, cisplatin, bleomycin, tacrolimus, cyclosporine, clopidogrel, ticlopidine), and infections with Streptococcus pneumoniae and other microorganisms (eg varicella, influenza, HIV, pertussis) [9,25].
Differential diagnosis of thrombotic microangiopathies includes disseminated intravascular coagulation, malignant hypertension, and sclerodermal renal crisis. There are certain clinical situations in which aHUS can be strongly suspected: patients having recurrent HUS, recurrent HUS after kidney transplantation, family history of HUS (with no evidence of food poisoning), patients younger than 6 months of age, and the patient with no blood in stools or diarrhea [9].
Various criteria have been used to differentiate between atypical HUS and TTP. Use of clinical presentation alone or even pretreatment ADAMTS13 activity alone is problematic and can lead to delay and at times even withholding necessary therapy. Hence it is imperative to use both these and response to plasma exchange therapy to better differentiate between these 2 conditions. However, plasma exchange is given to any patient who presents with a thrombotic angiopathy, not because it is more effective in patients with aHUS, but mostly because it is difficult to differentiate between TTP and aHUS on presentation. There are no objective tests available to differentiate them accurately at the time of presentation. Although a higher platelet count and serum creatinine are not diagnostic of aHUS at presentation, patients having normal ADAMTS13 activity and poor response to plasma exchange should be considered to have aHUS in the differential diagnosis of cases presenting with thrombotic microangiopathy [8].
The Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society (JSN/JPS) has developed diagnostic criteria for aHUS to help in prompt diagnosis and management of this condition. A definitive diagnosis of aHUS is made when there is a triad of thrombocytopenia (platelets less than 150 000/uL), microangiopathic hemolytic anemia (with hemoglobin of <10g/dL with increased LDH, decreased haptoglobin and presence of fragmented red cells in peripheral smear), and acute renal failure (defined by the international guidelines group, the Kidney Disease: Improving Global Outcomes) with no association with Shiga toxins. TTP should also be excluded, in which ADAMTS13 activity has great value [9]. Stool cultures, direct detection of Shiga toxins, or detection of anti-lipopolysaccharide IgM antibodies can be used to assess for involvement of Shiga toxins (Figure 3).
Figure 3.

Thrombotic microangiopathies – differential diagnosis and evaluation.
A study done by Zuber et al. defined failure to plasma therapy when there is less than 25% reduction in the creatinine level or failure of the platelet count or LDH to normalize. Severely deficient ADAMTS13 activity with poor response to plasma exchange provides confirmation for a clinical diagnosis of TTP in which immunosuppressive therapy should be instituted. However, if ADAMTS13 is not deficient and there is no evidence of an antibody inhibitor of ADAMTS13, in patients with poor response to plasma exchange, alternative etiology for thrombotic microangiopathy including aHUS should be considered and long-term therapy with eculizumab should be initiated [26].
Since 1980, plasma exchange therapy has been the validated therapy for aHUS. Registry data reports effectiveness of this therapy in at least 70% of cases in which hematologic remission was achieved. Renal response is less certain; it should be started within 24 hours after diagnosis and performed daily until platelets, LDH, and hemoglobin are improved or alternative therapy is decided for further management. Creatinine should not be used as a guide to plasma exchange because patients may already have had irreversible renal injury. Lack of improvement in platelet count or hemolysis after 3–5 days of plasma exchange therapy should be considered as resistance to therapy, after which plasma exchange should be withheld and alternatives should be used [1]. Remission usually occurs in 7–10 days with plasma exchange, resulting in correction of the hematologic parameters mentioned above. With increase in the platelet counts, there is improvement in the urine output and serum creatinine [27].
Eculizumab is a humanized recombinant immunoglobulin G2/4 monoclonal antibody against complement factor C5. It inhibits the generation of C5b-9, a membrane attack complex, and hence inhibits the complement system. It has been approved by FDA in 2011 for the management of aHUS. Cases have been reported to have favorable outcomes with eculizumab therapy. Long-term eculizumab therapy in a patient with congenital aHUS led to persistent remission [30]. In another patient, it was used as a salvage therapy after renal replacement was initiated and resulted in good response initially in regards to hematologic parameters, but renal dysfunction continued to deteriorate with recurrent aHUS relapses. This suggests that early initiation of eculizumab therapy may be more effective in conservation of renal function and recovery from reversible insult [31]. More cases of success continue to be reported with eculizumab in patients with aHUS before end-stage renal disease and even after kidney transplantation [28,32–35].
The major concern with use of eculizumab is the risk of infections with encapsulated bacteria, especially Neisseria meningitides, because it results in blockade of the terminal complement component. Hence, prior to commencement of therapy with eculizumab, patients must receive meningococcal vaccination and can also be covered with antibiotics for 2 weeks if physicians cannot wait for immunity to develop. A recent study has shown long-term efficacy and safety with use of pulse doses of cyclophosphamide combined with plasma exchange and prednisone in aHUS associated with anti-CFH antibody, which has a very poor prognosis, leading to prevention of relapses with no need for maintenance therapy [36].
Conclusions
Our case provides a good insight into the approach to the diagnosis of thrombotic microangiopathies. Despite a vast literature on TTP and aHUS, differentiating between these 2 entities remains mostly clinical with help from hematologic parameters, and each of these parameters alone provide little help in differentiation. The combination of clinical and laboratory data, activity of ADAMTS13, and response to plasma exchange allows for better differentiation between these thrombotic microangiopathies, which itself is very important considering that both have different treatment options. Genetic testing has been shown to be important in deciding treatment options, but these tests are not readily available. The main strategy remains to continue research towards better treatment options and therapeutic regimens in patients with aHUS to prevent irreversible renal dysfunction, thereby improving quality of life.
Footnotes
Conflict of interests
We, the authors of the manuscript, do not have a direct financial relation with the commercial identities mentioned in the paper that might lead to a conflict of interest
References:
- 1.Nester CM, Thomas CP. Atypical hemolytic uremic syndrome: what is it, how is it diagnosed, and how is it treated? Hematology Am Soc Hematol Educ Program. 2012;2012:617–25. doi: 10.1182/asheducation-2012.1.617. [DOI] [PubMed] [Google Scholar]
- 2.Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347(8):589–600. doi: 10.1056/NEJMra020528. [DOI] [PubMed] [Google Scholar]
- 3.Zheng XL, 1, Kaufman RM, Goodnough LT, Sadler JE. Effect of plasma exchange on plasma ADAMTS13 metalloprotease activity, inhibitor level, and clinical outcome in patients with idiopathic and nonidiopathic thrombotic thrombocytopenic purpura. Blood. 2004;103(11):4043–9. doi: 10.1182/blood-2003-11-4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kremer Hovinga JA, Vesely SK, Terrell DR, et al. Survival and relapse in patients with thrombotic thrombocytopenic purpura. Blood. 2010;115(8):1500–11. doi: 10.1182/blood-2009-09-243790. quiz 1662. [DOI] [PubMed] [Google Scholar]
- 5.Sellier-Leclerc AL, Fremeaux-Bacchi V, Dragon-Durey MA, et al. Differential impact of complement mutations on clinical characteristics in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2007;18(8):2392–400. doi: 10.1681/ASN.2006080811. [DOI] [PubMed] [Google Scholar]
- 6.Proesmans W. Typical and atypical hemolytic uremic syndrome. Kidney Blood Press Res. 1996;19(3–4):205–8. doi: 10.1159/000174075. [DOI] [PubMed] [Google Scholar]
- 7.Noris M, Remuzzi G. Atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(17):1676–87. doi: 10.1056/NEJMra0902814. [DOI] [PubMed] [Google Scholar]
- 8.Cataland SR, Wu HM. Atypical hemolytic uremic syndrome and thrombotic thrombocytopenic purpura: clinically differentiating the thrombotic microangiopathies. Eur J Intern Med. 2013;24(6):486–91. doi: 10.1016/j.ejim.2013.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Sawai T, Nangaku M, Ashida A, et al. Diagnostic criteria for atypical hemolytic uremic syndrome proposed by the Joint Committee of the Japanese Society of Nephrology and the Japan Pediatric Society. Clin Exp Nephrol. 2014;18(1):4–9. doi: 10.1007/s10157-013-0911-8. [DOI] [PubMed] [Google Scholar]
- 10.Warwicker P, Goodship TH, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53(4):836–44. doi: 10.1111/j.1523-1755.1998.00824.x. [DOI] [PubMed] [Google Scholar]
- 11.Richards A, Buddles MR, Donne RL, et al. Factor H mutations in hemolytic uremic syndrome cluster in exons 18–20, a domain important for host cell recognition. Am J Hum Genet. 2001;68(2):485–90. doi: 10.1086/318203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Goodship TH, Liszewski MK, Kemp EJ, et al. Mutations in CD46, a complement regulatory protein, predispose to atypical HUS. Trends Mol Med. 2004;10(5):226–31. doi: 10.1016/j.molmed.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 13.Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100(22):12966–71. doi: 10.1073/pnas.2135497100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kavanagh D, Kemp EJ, Mayland E, et al. Mutations in complement factor I predispose to development of atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(7):2150–15. doi: 10.1681/ASN.2005010103. [DOI] [PubMed] [Google Scholar]
- 15.Caprioli J, Noris M, Brioschi S, et al. Genetics of HUS: the impact of MCP, CFH, and IF mutations on clinical presentation, response to treatment, and outcome. Blood. 2006;108(4):1267–79. doi: 10.1182/blood-2005-10-007252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moore I, Strain L, Pappworth I, et al. Association of factor H autoantibodies with deletions of CFHR1, CFHR3, CFHR4, and with mutations in CFH, CFI, CD46, and C3 in patients with atypical hemolytic uremic syndrome. Blood. 2010;115(2):379–87. doi: 10.1182/blood-2009-05-221549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H-related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3(3):e41. doi: 10.1371/journal.pgen.0030041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Goicoechea de Jorge E, Harris CL, Esparza-Gordillo J, et al. Gain-of-function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104(1):240–45. doi: 10.1073/pnas.0603420103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delvaeye M, Noris M, De Vriese A, et al. Thrombomodulin mutations in atypical hemolytic-uremic syndrome. N Engl J Med. 2009;361(4):345–57. doi: 10.1056/NEJMoa0810739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Laurence J. Diagnosis of atypical hemolytic uremic syndrome: a review of case studies. Clin Adv Hematol Oncol. 2012;10(10 Suppl.17):9–11. [PubMed] [Google Scholar]
- 21.Sallée M, Daniel L, Piercecchi MD, et al. Myocardial infarction is a complication of factor H-associated atypical HUS. Nephrol Dial Transplant. 2010;25(6):2028–32. doi: 10.1093/ndt/gfq160. [DOI] [PubMed] [Google Scholar]
- 22.Zuber J, Le Quintrec M, Sberro-Soussan R, et al. New insights into postrenal transplant hemolytic uremic syndrome. Nat Rev Nephrol. 2011;7(1):23–35. doi: 10.1038/nrneph.2010.155. [DOI] [PubMed] [Google Scholar]
- 23.Gordon LI, Kwaan HC. Thrombotic microangiopathy manifesting as thrombotic thrombocytopenic purpura/hemolytic uremic syndrome in the cancer patient. Semin Thromb Hemost. 1999;25(2):217–21. doi: 10.1055/s-2007-994923. [DOI] [PubMed] [Google Scholar]
- 24.Neild GH. Haemolytic-uraemic syndrome in practice. Lancet. 1994;343(8894):398–401. doi: 10.1016/s0140-6736(94)91228-9. [DOI] [PubMed] [Google Scholar]
- 25.Noris M, Remuzzi G. Hemolytic uremic syndrome. J Am Soc Nephrol. 2005;16(4):1035–50. doi: 10.1681/ASN.2004100861. [DOI] [PubMed] [Google Scholar]
- 26.Zuber J, Fakhouri F, Roumenina LT, et al. Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat Rev Nephrol. 2012;8(11):643–57. doi: 10.1038/nrneph.2012.214. [DOI] [PubMed] [Google Scholar]
- 27.Nickavar A, Sotoudeh K. Assesment, treatment and prevention of atypical hemolytic uremic syndrome. Int J Prev Med. 2013;4(1):6–14. [PMC free article] [PubMed] [Google Scholar]
- 28.Waters AM, Licht C. aHUS caused by complement dysregulation: new therapies on the horizon. Pediatr Nephrol. 2011;26(1):41–57. doi: 10.1007/s00467-010-1556-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cho HY, Lee BS, Moon KC, et al. Complete factor H deficiency-associated atypical hemolytic uremic syndrome in a neonate. Pediatr Nephrol. 2007;22(6):874–80. doi: 10.1007/s00467-007-0438-x. [DOI] [PubMed] [Google Scholar]
- 30.Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic-uremic syndrome. N Engl J Med. 2009;360(5):544–46. doi: 10.1056/NEJMc0809959. [DOI] [PubMed] [Google Scholar]
- 31.Mache CJ, Acham-Roschitz B, Frémeaux-Bacchi V, et al. Complement inhibitor eculizumab in atypical hemolytic uremic syndrome. Clin J Am Soc Nephrol. 2009;4(8):1312–16. doi: 10.2215/CJN.01090209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rosales A, Riedl M, Zimmerhackl LB. Thrombotic microangiopathy: atypical HUS: current diagnostic and therapeutic approaches. Nat Rev Nephrol. 2010;6(9):504–6. doi: 10.1038/nrneph.2010.98. [DOI] [PubMed] [Google Scholar]
- 33.Ohanian M, Cable C, Halka K. Eculizumab safely reverses neurologic impairment and eliminates need for dialysis in severe atypical hemolytic uremic syndrome. Clin Pharmacol. 2011;3:5–12. doi: 10.2147/CPAA.S17904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Prescott HC, Wu HM, Cataland SR, Baiocchi RA, et al. Eculizumab therapy in an adult with plasma exchange-refractory atypical hemolytic uremic syndrome. Am J Hematol. 2010;85(12):976–77. doi: 10.1002/ajh.21862. [DOI] [PubMed] [Google Scholar]
- 35.Vaisbich MH, Henriques Ldos S, Watanabe A, et al. [Eculizumab for the treatment of atypical hemolytic uremic syndrome: case report and revision of the literature ] J Bras Nefrol. 2013;35(3):237–41. doi: 10.5935/0101-2800.20130037. [in Portuguese] [DOI] [PubMed] [Google Scholar]
- 36.Sana G, Dragon-Durey MA, Charbit M, et al. Long-term remission of atypical HUS with anti-factor H antibodies after cyclophosphamide pulses. Pediatr Nephrol. 2014;29(1):75–83. doi: 10.1007/s00467-013-2558-9. [DOI] [PubMed] [Google Scholar]