

Abstract
Adenosine 5′-phosphosulfate reductase (APR) is an iron-sulfur enzyme that is vital for survival of Mycobacterium tuberculosis during dormancy and is an attractive target for the treatment of latent tuberculosis (TB) infection. The 4Fe-4S cluster is coordinated to APR by sulfur atoms of four cysteine residues, is proximal to substrate, adenosine 5′-phopsphosulfate (APS), and is essential for catalytic activity. Herein, we present an approach for the development of a new class of APR inhibitors. As an initial step, we have employed an improved solid-phase chemistry method to prepare a series of N6-substituted adenosine analogues and their 5′-phosphates as well as adenosine 5′-phosphate diesters bearing different Fe and S binding groups, such as thiols or carboxylic and hydroxamic acid moieties. Evaluation of the resulting compounds indicates a clearly defined spacing requirement between the Fe-S targeting group and adenosine scaffold and that smaller Fe-S targeting groups are better tolerated. Molecular docking analysis suggests that the S atom of the most potent inhibitor may establish a favorable interaction with an S atom in the cluster. In summary, this study showcases an improved solid-phase method that expedites the preparation of adenosine and related 5′-phosphate derivatives and presents a unique Fe-S targeting strategy for the development of APR inhibitors.
Introduction
Late-stage persistent tuberculosis (TB) remains a significant treatment challenge with existing antibiotics [1]. TB is caused by Mycobacterium tuberculosis (Mtb) an acid-fast bacillus that exists in two major metabolic states: an active dividing form, which is susceptible to front-line therapeutics such as isoniazid and an inactive, latent form, which resist being killed by traditional antibiotics [2, 3]. Antibiotics that target biochemical pathways in metabolically-active mycobacteria can clear the pathogen effectively from the host [2, 4]. However, mycobacteria can persist for decades in the infected host lung within granulomatous lesions by adopting a non-replicating dormant state, as a reservoir of future active infection in the event of compromised host immunity [5, 6]. Numerous studies indicate that mycobacteria confront a generally hostile, anoxic and nutrient poor environment within the granuloma [7, 8]. Mycobacteria cope with unfavorable conditions in the granuloma through upregulating stress response pathways, including the sulfate assimilation pathway that produces biological thiols employed as cofactors in fatty acid-linked energy production and to regulate redox homeostasis [7, 9].
Indeed, genes involved in sulfate assimilation are specifically upregulated during dormant TB infection [10, 11]. The pathway begins with the import of sulfate into bacilli through transporters [12]. The sulfate is then activated by adenosine 5′-phosphosulfate (APS) kinase to form APS [12], which is further reduced to sulfite by adenosine 5′-phosphosulfate reductase (APR) as shown in Figure 1 [13, 14]. The newly formed sulfite is then reduced to sulfide and incorporated into essential biomolecules, such as cofactor, coenzyme A, redox buffer, mycothiol and amino acid, cysteine [12]. APR, encoded by the cysH gene, catalyzes the first rate-limiting step in sulfate reduction and is critical for Mtb survival during chronic infection [15]. Disruption of cysH rendered Mtb auxotrophic for cysteine and methionine and attenuated virulence in immunocompetent mice [15]. The cysH mutant was significantly more susceptible to reactive oxygen species (ROS) and reactive nitrogen species (RNS) indicating that APR is important for the defense of Mtb against oxidative stress [15]. Humans do not have a cysH ortholog rendering APR as an attractive target for the development therapeutics against persistent TB.
Figure 1.
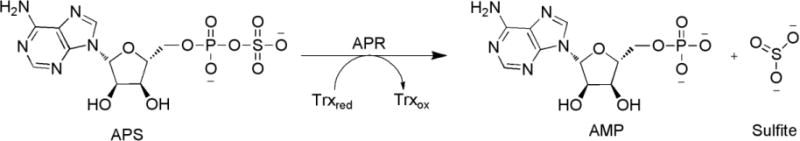
APR catalyzes the reduction of APS to sulfite and AMP with reducing equivalents from thioredoxin (Trx).
The active site of APR is distinguished by the presence of an iron-sulfur (Fe-S) cluster [14, 16]. Fe-S clusters are versatile metallo-centers involved in catalysis, radical generation, substrate activation, and maintaining protein structure [17–19]. Functional studies clearly demonstrate that an intact 4Fe-4S cluster is essential for APR catalysis [13, 20]. After substrate binding, the APR catalytic cycle is initiated through nucleophilic attack of an active site cysteine on the sulfur atom of APS to form an S-sulfocysteine intermediate. In subsequent steps the oxidoreductase, thioredoxin (Trx) release the sulfite product through thiol-disulfide exchange [14]. Structural, biochemical and spectroscopic studies demonstrate that a conserved lysine in the APR active site forms an “electrostatic bridge” that connects the Fe-S cluster to the sulfate group of APS [13, 20, 21]. In addition to functional studies, DFT modeling [22] also indicates a role for the Fe-S cluster in active site pre-organization and substrate activation.
Structural and spectroscopic studies demonstrate that Fe-S centers in enzymes like aconitase and biotin synthase directly interact with their respective citrate and S-adenosyl methionine (SAM) substrates to promote catalysis [23–25]. Pyruvate has been shown to interact with the 4Fe-4S cluster of 4-demethylwyosin synthase [26]. Moreover, 4,5-dithiohydroxyphthalic acid was shown to inhibit quinolinate synthase by binding to the Fe-S cluster [27]. In yet another report, backbone amine and carboxylate groups of methionine were shown to interact with the Fe-S cluster of molybdenum cofactor biosynthesis protein A, thereby preventing the protein from binding to SAM [28]. Most recently, the diabetic drug known as pioglitazone was shown to be inhibit the mitoNEET protein by blocking thiol-mediated reduction and installation of 2Fe-2S clusters [29]. These studies affirm that functional groups in protein ligands such as carboxylates, hydroxamates and thiols can successfully interact with Fe-S clusters and that such interactions can be exploited to block binding of the native substrate.
On the basis of these important precedents, we envisioned a new class of APR inhibitors consisting of the adenosine scaffold that would interact with residues in the substrate-binding pocket and incorporate functional groups capable of interacting with the Fe-S cluster (Figure 2). Molecular docking studies recapitulate the APS substrate pose [30] observed in the x-ray structure of APR (PDB 2GOY). APS fits into the deep active site cavity with the 5′-phosphosulfate moiety extending toward the Fe-S cluster and coordinating ligand, Cys140. The sulfate group is somewhat distal from the cluster and the shortest distances between sulfate oxygen and Fe and Sγ of Cys140 are 7.03 Å and 6.09 Å, respectively [21, 31]. Given the orientation of APS in the active site, we elected to functionalize the adenosine scaffold at the 5′-phosphate or exocyclic amine. Complex purification steps were avoided and nucleoside derivative throughput was increased using a solid-phase approach previously developed in our group [32]. Evaluation of the resulting compounds against Mtb APR indicates a clearly defined spacing requirement between the Fe-S targeting group and adenosine scaffold and that smaller Fe-S targeting groups are better tolerated. Molecular docking analysis suggests that the S atom of the most potent inhibitor may establish a favorable interaction with an S atom in the cluster. The study reported herein thereby showcases an improved solid-phase method that expedites the preparation of adenosine and related 5′-phosphate derivatives and presents a unique Fe-S targeting strategy for the future development of APR inhibitors.
Figure 2.
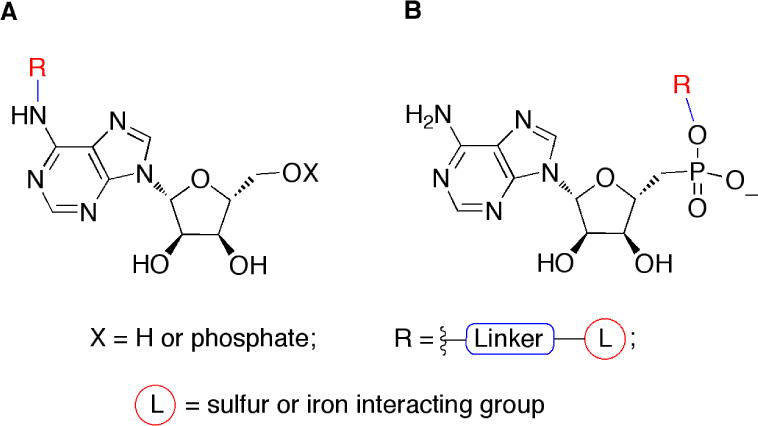
Proposed modes of inhibition of APR by substrate analogues
Results and Discussion
Fe-S clusters are more versatile and unique cofactors used by a large and diverse group of proteins. They participate in biochemical process such as electron transfer, enzyme catalysis and red-ox sensors [33]. Small molecules that harbor groups that chelate essential metal ions serve as effective inhibitors [34]. For example, carbonic anhydrase, matrix metalloproteinases, and histone deacetylases inhibitors have a classic drug-like structure and zinc-binding group. These compounds interact with protein through non-covalent interactions and zinc coordination.
To develop inhibitors with the potential to interact with the Fe-S cluster of APR we prepared a library of N6- and 5′-substituted adenosine analogues and, given the importance of the 5′-phosphate for binding [31], nucleotide derivatives linked to a variety of Fe or S binding groups. Our synthetic strategy employed polystyrene beads coupled to the ribose 2′ and 3′-hydroxyls via a p-hydroxybenzaldehyde acetal linkage, which provided for minimal protecting group manipulation and can be cleaved from the support using mild acidic conditions [32]. A solid-supported adenosine scaffold allows for selective manipulation of N6 exocyclic amine and 5′-hydroxyl groups, and facilitates analogue purification.
To prepare N6 exocyclic amine adenosine derivatives, the 5′-hydroxyl of resin-bound nucleoside 1 was protected using tert-butyldimethylsilylchloride/imidazole in high yield, thereby enabling selective alkylation of aromatic amine 2. Using N-alkylation reaction conditions previously optimized by our group [32], different types of Fe-S binding groups (carboxylate, hydroxamate, triazole, and thiol) were connected to the adenosine scaffold via methylene linkers of different length (1–5 carbons), as shown in Scheme 1. Fe-S binding groups functionalized with alkyl halide handles a–h were obtained or prepared in sufficient yield by N- or S-alkylation (Supporting Information: Synthesis of intermediates for 4a–4h and 8a–8h). Thiol-containing halides are generally amenable to acetyl protection, however the corresponding derivative of f deteriorated during isolation. Consequently, we used the bulkier benzoyl group to synthesize S-(2-bromoethyl) benzothioate f. The reaction was conducted in benzoic acid, triphenylphosphine, N-bromosuccinimide, dichloroethane and benzyltriethylammoniumtetrathiomolybdate as the sulfur transfer reagent in high yield [35]. The resulting adenosine derivatives were cleaved from the solid phase using 10% acetic acid in water. These conditions were sufficiently acidic enough to cleave the 5′-tert-butyldimethylsilyl group to yield the final products in one step, except acyl protected thiols 3e and 3f. In these two cases, acyl group cleavage and release from the solid-supported resin were simultaneously achieved in moderate yield using 50% trifluoroacetic acid at 0°C.
Scheme 1.
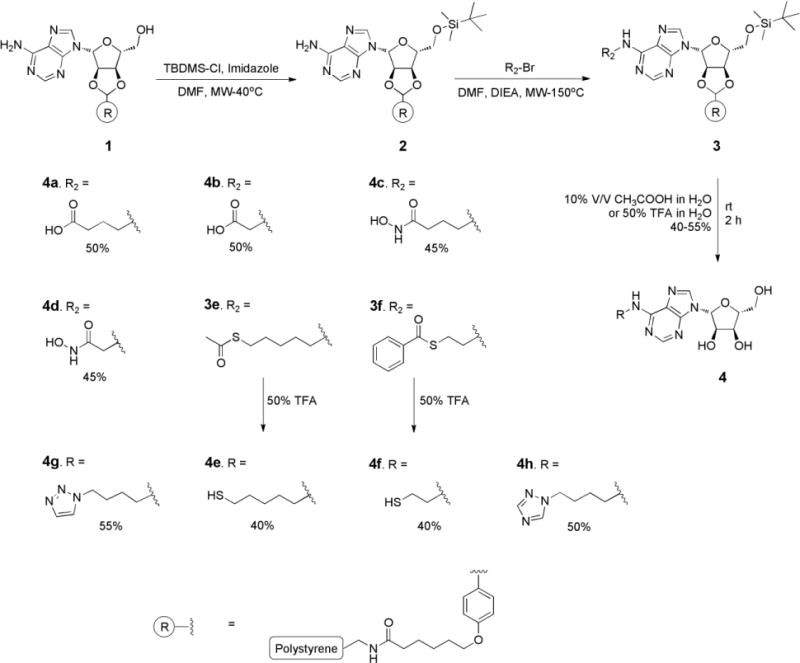
Synthesis of compounds 4a–4h.
Synthesis of N6-substituted adenosine 5′-phosphates, 8a-8h, began with solid-supported adenosine 1 (Scheme 2). The 5′-hydroxyl was protected as the diethyl phosphate using diethyl chlorophosphate and diisopropylethylamine at room temperature (rt) in high yield. Using the N-alkylation reaction conditions that were optimized in Scheme 1 different types of Fe-S binding groups (carboxylate, hydroxamate, triazole, and thiol) were connected to the adenosine scaffold as shown in Scheme 2. In the next step the phosphate was generated using trimethylsilyl bromide followed by a methanolysis step [36–38]. The first step transforms the alkyl phosphate into the corresponding trimethylsilyl phosphate, which is subsequently cleaved to the phosphoric acid under hydrolytic conditions [39]. Phosphate 6 was treated with trimethylsilylbromide at −65°C and brought to rt for 2 h, followed by the addition of methanol and finally washed with water to give 7 in high yield. The aforementioned adenosine 5′-phosphate derivatives were cleaved from solid support with 10% acetic acid in water or 50% trifluoroacetic acid in water to obtain 8 in 35–50% yields.
Scheme 2.
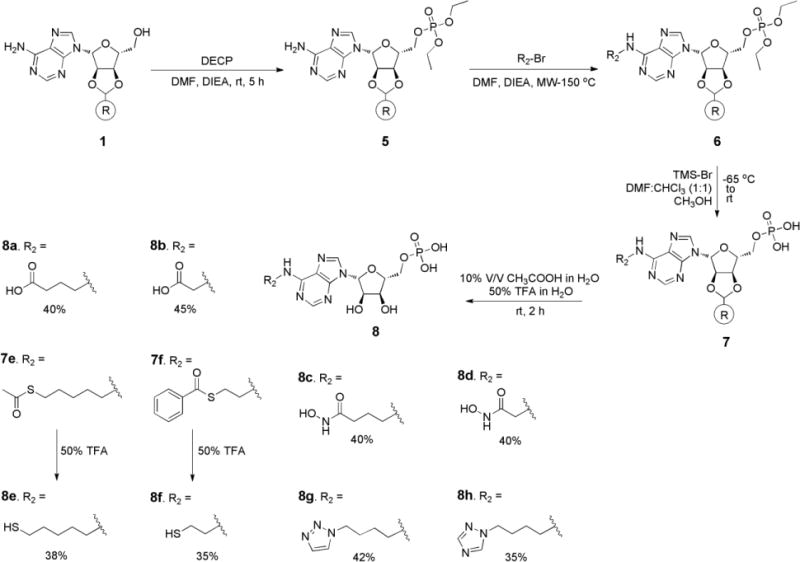
Synthesis of compounds 8a–8h.
Finally, the adenosine 5′-phosphate diesters were prepared via phosphoramidite chemistry [40, 41], as shown in Scheme 3. The solid-supported adenosine scaffold 1 was reacted with 2-cyanoethyl diisoproplyl-chlorophosphoramidite (2-CEDCP) to afford 9. A primary alcohol bearing a Fe-S binding group (Supporting information: Synthesis of intermediates for 13–16) was then used to displace diisopropylamine using 1-hydroxybenzotriazole activation to obtain 10. The phosphite 10 was then oxidized using iodine to give 11, and the 2-cyanoethyl protective group was removed under basic conditions to afford resin-bound adenosine analogue 12. The solid-supported adenosine derivative 12 was successfully cleaved from polystyrene resin under acidic conditions to afford final products 13–16.
Scheme 3.
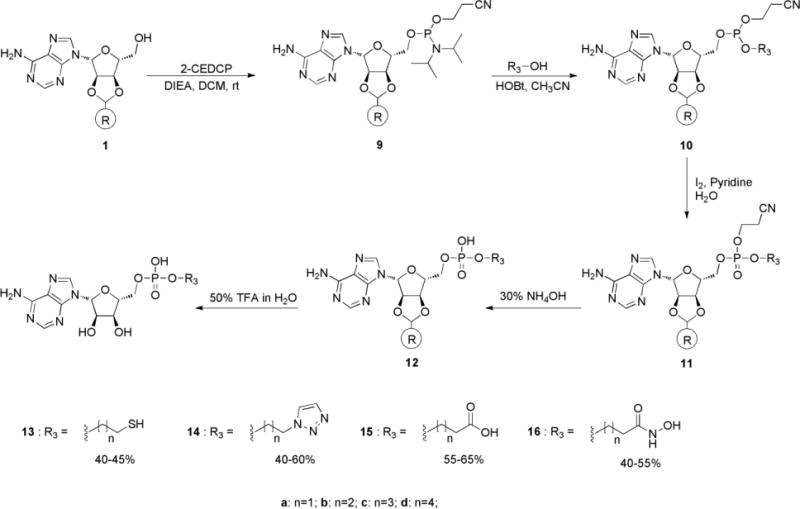
Synthesis of compounds 13–16.
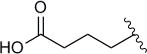
With the library of Fe-S targeted adenosine analogues in hand, we next measured their equilibrium binding constants (Kd) using our sensitive APR assay, which monitors the production of 35S-labeled sulfite from 35S-labeled APS [31, 42, 43]. The experimental Kd values are reported in Table 1. Initial inspection of the data indicates that substitution at the exocyclic amine improved binding affinity, relative to adenosine. Compounds 4a and 4c, bearing carboxylic acid and hydroxamic acid groups coupled to methylene linkers of three carbon units exhibited better affinity compared to compounds 4b and 4d harboring a shorter linker of a single methylene. Thiols 4e and 4f demonstrated two to three-fold increases in affinity. Triazoles 4g and 4h, differing not in linker length, but in the position of functionalization each showed an approximately two-fold increase in binding. When compounds were further functionalized with a 5′-phosphate (8a–8h), binding affinities increased from 70 to 375-fold, compared to adenosine (Table 2). Trends with respect to linker length were also consistent between Tables 1 and 2. However, with the exception of 8f, all 5′-phosphate derivatives exhibited a diminished affinity relative to 5′-AMP. Collectively, the data in Tables 1 and 2 suggest an optimal spacing of 5–6 atomic units for moieties extending from the N6 exocyclic amine and that the thiol group provides favorable interactions with the Fe-S cluster.
Table 1.
Equilibrium binding constants (Kd) for Fe-S targeted adenosine analogues.
| |||
|---|---|---|---|
|
| |||
| Compound | R1 | Kd (μM)a | Fold changeb |
| Adenosine | H | 3000b | (1) |
| 4a |
|
1212 | 0.4 |
| 4b |
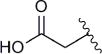
|
3069 | 1.0 |
| 4c |
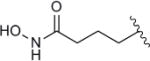
|
1726 | 0.57 |
| 4d |
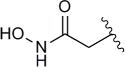
|
3011 | 1.0 |
| 4e |
|
974 | 0.32 |
| 4f |
|
1426 | 0.47 |
| 4g |
|
1414 | 0.47 |
| 4h |
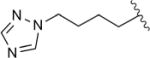
|
1758 | 0.58 |
In this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of sub saturating APS, such that Ki is equal to the Kd.
Previously reported [31].
Table 2.
Equilibrium binding constants (Kd) for Fe-S targeted AMP analogues.
| |||
|---|---|---|---|
|
| |||
| Compound | R2 | Kd (μM)a | Fold changeb |
| AMP | H | 5.4b | (1) |
| 8a |
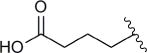
|
9 | 1.6 |
| 8b |
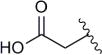
|
37 | 6.8 |
| 8c |
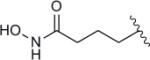
|
28 | 5.1 |
| 8d |
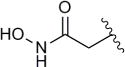
|
45 | 8.3 |
| 8e |
|
11 | 2.0 |
| 8f |
|
4 | 0.7 |
| 8g |
|
28 | 5.1 |
| 8h |
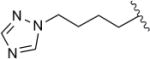
|
39 | 7.2 |
In this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of sub saturating APS, such that Ki is equal to the Kd.
Previously reported [31].
In subsequent experiments, we determined the Kds for compounds functionalized at the 5′-phosphate group (13–16, Table 3). Compared to AMP, affinities of the analogues were up to 60-fold weaker. Of compounds 13–16, thiols 13a–d exhibited the most favorable binding; 13a,c, and d were up to 8-fold less avid, whereas 13b had the same binding affinity with respect to AMP. Increasing the steric bulk of the Fe-S targeting group by substitution with a triazole (14a–d), carboxylic acid (15a–d), or hydroxamic acid (16a–d), all resulted in compounds with ≥10-fold decreased affinity, compared to AMP. Although 14–16 all interacted more weakly with APR, one interesting pattern did emerge. In these cases, compounds with three methylene linkers consistently demonstrated the most favorable binding. Together, the data in Tables 1–3 indicate that functionalization at the 5′-phosphate is much less tolerated than at the N6 exocyclic amine.
Table 3.
Equilibrium binding constants (Kd) for Fe-S targeted 5′-phosphate analogues.
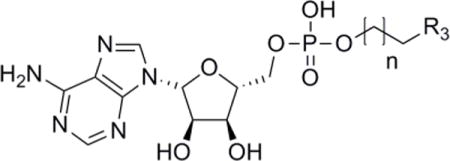
| ||||
|---|---|---|---|---|
|
| ||||
| Compound | R3 | Linker length (n) | Kd (μM)a | Fold change |
| AMP | 5.4b | (1) | ||
| 13a | –SH | 1 | 21 | 4 |
| 13b | –SH | 2 | 5 | 1 |
| 13c | –SH | 3 | 30 | 6 |
| 13d | –SH | 4 | 45 | 8 |
| 14a |
|
1 | 145 | 27 |
| 14b |
|
2 | 129 | 24 |
| 14c |
|
3 | 186 | 34 |
| 14d |
|
4 | 341 | 63 |
| 15a | –COOH | 1 | 209 | 39 |
| 15b | –COOH | 2 | 197 | 36 |
| 15c | –COOH | 3 | 249 | 46 |
| 15d | –COOH | 4 | 322 | 60 |
| 16a | –CONH2OH | 1 | 192 | 36 |
| 16b | –CONH2OH | 2 | 156 | 29 |
| 16c | –CONH2OH | 3 | 214 | 40 |
| 16d | –CONH2OH | 4 | 236 | 44 |
In this table values of Ki were determined under single turnover conditions from the dependence of the observed rate constant at a given inhibitor concentration under conditions of sub saturating APS, such that Ki is equal to the Kd.
Previously reported [31]
Among all library members, 8f exhibited the highest binding affinity for APR. To gain some molecular insight into potential interactions between APR and 8f, we performed virtual ligand docking simulations using AutoDock [44]. As shown in Figure 3, the predicted binding pose for 8f indicates that the AMP scaffold binds within the substrate-binding pocket and establishes interactions with key active site residues, as expected. In addition, the alkyl thiol portion of the compound extends out towards the Fe-S cluster. The predicted distance of ~4 Å from Fe and S atoms in the cluster would enable outer sphere electrostatic interactions. Lastly, the thiol also appears to establish hydrophobic packing interactions with residues surrounding Cys140 and this corner of the cluster.
Figure 3.
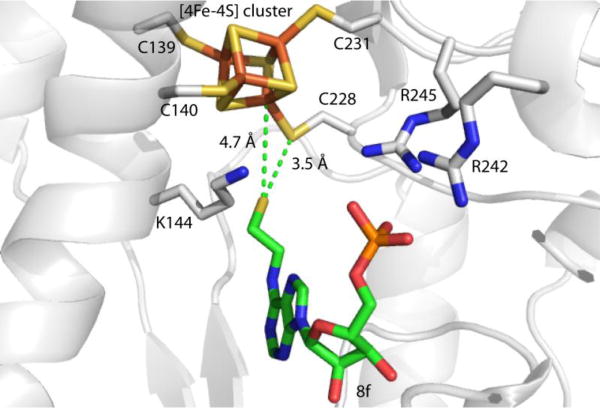
Docking model of 8f in APR (PDB: 2GOY). Green dashes indicate the distances between sulfur atom of 8f and sulfur atom of C228 and Fe atom of iron-sulfur cluster.
Most [4Fe-4S]2+ clusters within proteins are redox active, accepting or donating single electrons [45–47]. However the Fe-S cluster in APR serves a catalytic function without undergoing such redox changes [13, 14, 48, 49]. Previous work indicates that the [4Fe-4S]2+ cluster in APR can be partially converted to the [3Fe-4S]+ form by treatment with oxidant, potassium ferricyanide [20, 49]. Although, each of the Fe atoms in the [4Fe-4S] cluster is coordinated by a single cysteine ligand [21], two of these cysteine residues are adjacent to each other in the primary sequence of APR. Coordination of the CysCys motif with the Fe-S cluster in APR requires distortion away from preferred geometry within the Cys140 side chain. These collective observations indicate the presence of a labile iron site within the APR cluster, which might be exploited in future inhibitor development.
In summary, we have outlined an approach for the development of a new class of APR inhibitors. As an initial step, we have employed an improved solid-phase chemistry method to prepare a series of N6-substituted adenosine analogues and their 5′-phosphates as well as adenosine 5′-phosphate diesters bearing different Fe and S binding groups. Evaluation of the resulting compounds indicates a clearly defined spacing requirement between the Fe-S targeting group and adenosine scaffold and that smaller Fe-S targeting groups are best tolerated, particularly the N6-ethylthiol group. Molecular docking analysis of the most potent compound thiol 8f suggests that the S atom of this inhibitor is capable of establishing favorable interactions with the cluster. Strictly speaking, the compounds reported in this study do not afford a substantial improvement in affinity when compared to adenosine 5′-monophosphate (AMP); however, this work provides the conceptual framework for the design of compounds to target a protein Fe-S cluster. Detailed spectroscopic and structural characterization of interactions between the APR cluster and 8f are underway, and will be reported in due course.
Materials and Methods
Reagents and solvents were purchased from Sigma or other commercial sources and were used without further purification. Analytical thin layer chromatography (TLC) was carried out using Analtech Uniplate silica gel plates and visualized using a combination of UV and potassium permanganate staining. Flash chromatography was performed using silica gel (32–63 μM, 60Å pore size) from Sorbent Technologies Incorporated. Solid phase synthesis of adenosine analogues was done using polypropylene cartridge with 70 μ PE frit, purchased from Applied Separations. NMR spectra were obtained on a Bruker 400 (400 MHz for 1H and 13C). NMR chemical shifts are reported in parts per million (ppm) referenced to the residual solvent peak. 31P NMR chemical shift is reported in ppm referenced to the external standard, phosphoric acid as 0.00 ppm. Mass spectra were obtained with Agilent technologies 6120 quadrupole LC/MS system with porschell 120 C-18 column. Reverse-phase HPLCs were performed on agilent infinity 1200 system equipped with UV detector (λ = 260 nm) using a C18 (4.6 × 150 mm) Agilent Zorbex SB-C18 column (2 mL/min flow rate, linear 1%/min gradient from 100% A to 50% A (solvent A: 99.9% water, 0.1% TFA; solvent B: 99.9 % acetonitrile, 0.1% TFA).
Synthesis of adenosine analogues 4a-4h
Synthesis of solid phase adenosine scaffold which is 5′-tert-butyldimethylsilyl protected, 2, is prepared using previously developed protocol [32]. This protection allowed us to selectively explore the alkylation reactions on aromatic amine of adenosine. N-Alkylation reactions were also performed by following the previously optimized procedures [32].
General procedure for synthesis of 4
Resin bound 5′-TBDMS nucleoside 2 (0.35 mmol) was dried under high vacuum and swelled in DMF (6 mL) for 20 minutes, added corresponding alkyl halide (2 mmol), N,N-Diisopropylethylamine (260mg, 2mmol) at rt. The contents were stirred for 5 min then switched to microwave and conducted the reaction at 150°C, for 15minutes. Then the resin was washed with DMF followed by methanol to remove residual N,N-diisopropylethylamine and unreacted propargyl bromide. The resin was dried and under high vaccum. The resin bound 5′-TBDMS, N-alkylated nucleoside 3, was taken and washed with 10%V/V acetic acid in water or 50% trifluoroacetic acid for 2 hours and the solid phase resin was filtered with help of glass wool to release 4. Then the filtrate was lyophilized, and a small portion of obtained 4 was taken and solubilized in DMSO-d6 and NMR spectra were recorded.
4a: 1H NMR (DMSO-d6, 400 MHz): δ = 8.38 (s, 1H), 8.15 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.41 (m, J = 2.4, 2H), 4.1 (s, 1H), 3.81 (m, 2H), 3.35 (t, J = 6.9, 2H), 2.31(t, J = 6.9, 2H), 1.85 (m, 2H).13C NMR (DMSO-d6, 100 MHz): δ = 181.9, 159.8, 152.7, 151.4, 148.8, 139.8, 119.1, 97.3, 87.9, 73.6, 70.8, 61.5, 44.2, 36.1, 25.5. Mass calculated for C14H19N5O6 is 353.3306, found (M+H) 354.1; (M−H) 352.8.
4b: 1H NMR (DMSO-d6, 400 MHz): δ = 8.32 (s, 1H), 8.14 (s, 1H), 6.12 (s, 1H), 4.73(t, J = 6.82, 1H), 4.71 (m, J = 2.4, 2H), 4.51 (s, 1H), 4.01(s, 2H), 3.81 (m, 2H). 13C NMR (DMSO-d6, 100 MHz): δ = 172.8, 154.8, 152.3, 149.4, 148.8, 140.8, 119.8, 97.5, 87.2, 73.2, 70.5, 60.5, 44.2. Mass calculated for C12H15N5O6 is 325.1022, found (M+H) 326.21; (M−H) 324.8.
4c: 1H NMR (DMSO-d6, 400 MHz): δ = 8.35 (s, 1H), 8.16 (s, 1H), 8.01 (s, 1H), 6.13 (s, 1H), 4.72(t, J = 6.82, 1H), 4.71 (m, J = 2.4, 2H), 4.51 (s, 1H), 4.01(s, 2H), 3.35 (t, J = 6.9, 2H), 2.34(t, J = 6.9, 2H), 1.9 (m, 2H).13C (DMSO-d6, 100 MHz): δ = 169.9, 159.2, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 44.0, 29.9, 26.4; Mass calculated for C14H20N6O6 is 368.3452, found (M+H) 369.32; (M−H) 367.2.
4d: 1H NMR (DMSO-d6, 400 MHz): δ = 8.34 (s, 1H), 8.17 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.82, 1H), 4.45 (m, J = 2.4, 2H), 4.38 (s, 1H), 4.01 (s, 1H), 3.81 (m, 2H), 3.77 (s, 2H). 13C NMR (DMSO-d6, 100 MHz): δ = 166.0, 154.7, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 51.6; Mass calculated for C12H16N6O6 is 340.2920, found (M−H) 339.17.
4e: 1H NMR (DMSO-d6, 400 MHz): δ =8.37 (s, 1H), 8.17 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.41 (m, J = 2.4, 2H), 4.1 (s, 1H), 3.81 (m, 2H), 3.21 (t, J = 6.8, 2H), 2.51(t, J = 6.9, 2H), 1.65 (m, 4H), 1.21 (t, J = 6.6, 2H).13C NMR (DMSO-d6, 100 MHz): δ = 159.2, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 40.7, 33.9, 31.0, 25.6, 24.6; Mass calculated for C15H23N5O4S is 369.4392, found (M+H) 370.54; (M−H) 368.2.
4f: 1H NMR (DMSO-d6, 400 MHz): δ =8.36 (s, 1H), 8.14 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.49 (m, 4H), 4.0 (s, 1H), 3.35 (t, J = 6.6, 2H), 2.79 (t, J = 6.6, 2H). 13C NMR (DMSO-d6, 100 MHz): δ = 159.2, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 55.8, 26.8; Mass calculated for C12H17N5O4S is 327.3595 found (M+H) 328.54; (M−H) 326.23.
4g: 1H (DMSO-d6, 400 MHz): δ =8.36 (s, 1H), 8.14 (s, 1H), 7.63 (t, J = 6.7, 2H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.49 (m, 4H), 4.0 (s, 1H), 3.21 (t, J = 6.6, 2H), 1.79(q, J = 5.6, 2H), 1.45 (q, J = 5.6, 2H). 13C NMR (DMSO-d6, 100 MHz): δ =159.2, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 51.8, 40.4, 27.6, 25.8. Mass calculated for C16H22N8O4S is 390.3971 found (M+H) 391.43; (M−H) 389.32.
4h: 1H NMR (DMSO-d6, 400 MHz): δ = 8.77 (s, 1H), 8.36 (s, 1H), 8.14 (s, 1H), 8.06 (s, J = 6.7, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.49 (m, 4H), 4.0 (s, 1H), 3.20 (t, J = 6.6, 2H), 1.74(q, J = 5.6, 2H), 1.49 (q, J = 5.6, 2H). 13C NMR (DMSO-d6, 100 MHz): δ =159.8, 152.5, 148.8, 141.3, 118.4, 97.3, 87.4, 73.7, 70.5, 61.6, 52.8, 41.4, 26.6, 24.8. Mass calculated for C16H22N8O4S is 390.3971 found (M+H) 391.12; (M−H) 389.25.
General procedure for synthesis of 5
The solid supported adenosine scaffold 1 (225 mg, 176 μmol) was synthesized as previously reported and was allowed to swell in DMF (3 mL) for 20 minutes. To a suspension of the resin in dichloromethane (DCM) was added diethylchlorophosphate (352 μmol) followed by DIEA (142 μL, 816 μmol), and it was shaken at rt for 5 hours. The resin was then washed with, DMF, 5 mL, followed by dichloromethane (3 × 30 mL) and acetonitrile (3 × 30 mL) to obtain 5.
General procedure for synthesis of 6
Resin bound 5′-diethyl phosphate of nucleoside 2 (0.35 mmol) was dried under high vacuum and swelled in DMF (6mL) for 20 minutes, added corresponsiding alkyl halide (2 mmol), N,N-diisopropylethylamine (260mg, 2mmol) at rt. The contents were stirred for 5 min then switched to microwave and conducted the reaction at 150°C, for 15minutes. Then the resin was washed with DMF followed by methanol to remove residual N,N-diisopropylethylamine and unreacted alkyl halide. The resin was dried and under high vaccum to obtain 6.
General procedure for synthesis of 8a-8h
To a suspension of 6 (0.36mmol) in DMF and chloroform (1:1) added a solution of trimethylsilyl bromide(1.5 mmol, 5eq) in CHCl3 (5 mL) at −65°C, and the resulting mixture was stirred for 4 h. The temperature of the reaction mixture was allowed to rise to rt, and stirring was continued until the starting material had been consumed. The reaction mixture was cooled to −45°C and then MeOH (10 mL) was added. The solvent was evaporated in vacuo. Additional MeOH was added, and again the solvent was evaporated. This operation was repeated three more times. The solid phase slurry was suspended in chloroform and then filtered. Then the solid phase material was dried in vacuum to give 7 [36–38]. Then the resin bound 5′-phosphate of N-alkylated nucleoside (7), 20mg, was taken and washed with 10%V/V acetic acid in water or 50% trifluoroacetic acid for 2 hours and the solid phase resin was filtered with help of glass wool to release 8. Then the filtrate was lyophilized, and a small portion of obtained 8 was taken and solubilized in DMSO-D6 and NMR spectra were recorded.
8a: 1H NMR (DMSO-d6, 400 MHz): δ = 8.37 (s, 1H), 8.15 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.41 (m, J = 2.4, 2H), 4.0 (s, 1H), 3.81 (m, 2H), 3.35 (t, J = 6.9, 2H), 2.31(t, J = 6.9, 2H), 1.88 (t, J = 6.6, 2H).13C NMR (DMSO-d6, 100 MHz): δ = 182.7, 159.1, 152.7, 140.3, 119.4, 97.3, 87.8, 73.5, 70.4, 65.9, 44.2, 36.1, 25.4. Mass calculated for C14H20N5O9P is 433.3105, found (M−H) 432.18.
8b: 1H NMR (DMSO-d6, 400 MHz): δ = 8.32 (s, 1H), 8.14 (s, 1H), 6.12 (s, 1H), 4.73(t, J = 6.82, 1H), 4.29 (m, J = 2.4, 2H), 4.51 (s, 1H), 4.01(m, 3H). 13C NMR (DMSO-d6, 100 MHz): δ = 172.2, 154.4, 152.3, 149.4, 140.8, 119.3, 97.5, 86.2, 73.4, 70.4, 65.5, 44.9. Mass calculated for C12H16N5O9P is 405.2573, found (M+H) 406.21; (M−H) 404.8.
8c: 1H NMR (DMSO-d6, 400 MHz): δ = 8.32 (s, 1H), 8.17 (s, 1H), 8.0 (s, 1H), 6.13 (s, 1H), 4.72(t, J = 6.82, 1H), 4.29 (m, J = 2.4, 2H), 4.01(s, 1H), 3.32 (t, J = 6.9, 2H), 2.32(t, J = 6.9, 2H), 1.92 (m, 2H).13C NMR (DMSO-d6, 100 MHz): δ = 169.9, 159.2, 152.3, 149.7, 140.2, 119.3, 97.2, 88.4, 73.7, 70.5, 61.2, 44.1, 29.5, 26.3; Mass calculated for C14H21N6O9P is 448.1108, found (M+H) 449.21; (M−H) 447.23.
8d: 1H NMR (DMSO-d6, 400 MHz): δ = 8.35 (s, 1H), 8.16 (s, 1H), 8.02 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.82, 1H), 4.45 (m, J = 2.4, 2H), 4.38 (s, 1H), 4.28 (m, 2H), 3.71 (s, 2H). 13C NMR (DMSO-d6, 100 MHz): δ = 166.1, 153.7, 152.6, 148.8, 140.2, 119.3, 97.2, 87.3, 73.7, 70.4, 65.6, 51.6; Mass calculated for C12H17N6O9Pis 420.2719, found (M−H) 419.26.
8e: 1H NMR (DMSO-d6, 400 MHz): δ = 8.37 (s, 1H), 8.17 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.45 (m, J = 2.4, 2H), 4.38 (s, 1H), 4.28 (m, J = 2.4, 2H), 3.21 (t, J = 6.8, 2H), 2.55(t, J = 6.9, 2H), 1.65 (m, 5H), 1.29 (t, J = 6.6, 2H).13C NMR (DMSO-d6, 100 MHz): δ = 159.2, 151.4, 148.8, 141.3, 118.4, 97.1, 87.2, 73.5, 70.2, 61.4, 40.7, 32.9, 31.2, 24.6, 24.4; Mass calculated for C15H24N5O7PS is 449.4191, found (M−H) 448.23.
8f: 1H NMR (DMSO-d6, 400 MHz): δ = 8.36 (s, 1H), 8.14 (s, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.45 (m, J = 2.4, 2H), 4.38 (s, 1H), 4.28 (m, J = 2.4, 2H), 3.35 (t, J = 6.6, 2H), 2.79 (t, J = 6.6, 2H), 1.63 (s, 1H), 13C NMR (DMSO-d6, 100 MHz): δ = 159.2, 152.4, 149.5, 141.3, 119.3, 97.1, 86.4, 72.7, 71.5, 65.6, 55.8, 26.8; Mass calculated for C12H18N5O7PS is 407.3394 found (M+H) 408.54; (M−H) 409.23.
8g: 1H NMR (DMSO-d6, 400 MHz): δ =8.35 (s, 1H), 8.15 (s, 1H), 7.63 (m, 2H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.45 (m, J = 2.4, 2H), 4.38 (s, 1H), 4.28 (m, J = 2.4, 2H), 3.23 (t, J = 6.6, 2H), 1.75(q, J = 5.6, 2H), 1.49 (q, J = 5.6, 2H). 13C NMR (DMSO-d6, 100 MHz): δ =159.2, 152.4, 149.8, 140.3, 119.4, 97.3, 87.4, 73.7, 70.5, 61.6, 51.8, 40.4, 27.6, 25.8. Mass calculated for C16H23N8O7PS is 470.3770 found (M+H) 471.43; (M−H) 469.21.
8h: 1H NMR (DMSO-d6, 400 MHz): δ = 8.75 (s, 1H), 8.36 (s, 1H), 8.14 (s, 1H), 8.06 (s, J = 6.7, 1H), 6.16 (s, 1H), 4.75(t, J = 6.9, 1H), 4.52 (m, 6H), 3.20 (t, J = 6.6, 2H), 1.74(q, J = 5.6, 2H), 1.49 (q, J = 5.6, 2H). 13C NMR (DMSO-d6, 100 MHz): δ =159.6, 152.5, 148.8, 143.3, 140.3, 118.4, 97.1, 87.2, 73.7, 70.5, 61.6, 52.8, 41.4, 26.6, 24.8. Mass calculated for C16H23N8O7PS is 390.3971 found (M+H) 391.12; (M−H) 389.25.
Synthesis of 9
The solid supported adenosine scaffold 1 (225 mg, 176 μmol) was synthesized as previously reported and was allowed to swell in DMF (30 mL) for 20 minutes. To a suspension of the resin in DMF was added 2-cyanoethyl diisopropyl-chlorophosphoramidite (142 μL, 326 μmol) followed by DIEA (142 μL, 816 μmol), and it was shaken at rt for 40 hours. Reaction progress was monitored by shaking small fraction of solid phase resin in 50% trifluoroacetic acid to release the bound nucleoside. The clear acetic solution was analysed by LC/MS. The resin was then washed with DCM (3 × 30 mL) and acetonitrile (3 × 30 mL) to obtain 9.
General Procedure for synthesis of 10
To a suspension of the resin 2 in DMF and acetonitrile was added alcohol (5 – 10 eq) followed by HOBt (147 mg, 1.09 mmol), and the reaction mixture was shaken at rt for 65 hours. Reaction progress was monitored by shaking small fraction of solid phase resin in 50% trifluoroacetic acid to release the bound nucleoside. The clear acetic solution was analysed by LC/MS. The resin was washed with acetonitrile (3 × 30 mL) and 1:9 H2O/pyridine (3 × 30 mL) to obtain 10.
General procedure for synthesis of 11
A solution of iodine (0.5 g, 1.99 mmol) in 1:9 H2O/pyridine (30 mL) was added to the resin 10, and was allowed to shaken at rt for 24 hours. Reaction progress was monitored by shaking small fraction of solid phase resin in 50% trifluoroacetic acid to release the bound nucleoside. The clear acetic solution was analysed by LC/MS. The resin was washed with H2O (3 × 30 mL) to obtain 11.
General procedure for synthesis of 12
The resin 11 was treated with 30 mL of ammonium hydroxide (NH3 content 28 – 30%), and shaken at rt for 65 hours. Reaction progress was monitored by shaking small fraction of solid phase resin in 50% trifluoroacetic acid to release the bound nucleoside. The clear acetic solution was analysed by LC/MS. The resin was then washed with H2O (3 × 30 mL), DCM (3 × 30 mL), and MeOH (3 × 30 mL) to obtain 12.
General procedure for the synthesis of 13–16
The adenosine analogues were deprotected and cleaved off the resin by treating the resin with 8:2 TFA/H2O (10 mL) for 15 minutes. The reaction was then filtered through glass wool and the resin was washed with the cleavage solution (3 × 3 mL). The combined filtrate was then evaporated by a gentle stream of Nitrogen. The resulting residue was dissolved in water and lyophilized, and purified by C18 reversed-phase HPLC to provide the product.
13a: 1H NMR (CD3OD, 500 MHz): 8.58 (s,1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.52 (t, J = 6.4, 1H), 4.28 (m, 5H), 2.8 (t, J = 6.8, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 63.0, 24.7. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C12H18N5O7PS 407.3394, found (M+H) = 408.21; (M−H) = 406.21.
13b: 1H NMR (CD3OD, 500 MHz): 8.58 (s, 1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.52 (t, J = 6.4, 1H), 4.4 (s, 1H), 4.28 (m, 4H), 2.56 (t, J = 6.5, 2H), 1.96 (t, J = 6.2, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 63.0, 62.1, 29.6, 20.4. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C13H20N5O7PS 421.3660, found (M+H) = 422.25; (M−H) = 420.42.
13c: 1H NMR (CD3OD, 500 MHz): 8.58 (s, 1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.52 (t, J = 6.4, 1H), 4.4 (s, 1H), 4.28 (m, 4H), 2.56 (t, J = 6.5, 2H), 1.71 (t, J = 6.2, 2H), 1.51 (t, J = 6.2, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 63.0, 62.1, 28.6, 28.0, 24.4. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C14H22N5O7PS 435.3925, found (M+H) = 436.22; (M−H) = 434.12.
13d: 1H NMR (CD3OD, 500 MHz): 8.58 (s, 1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.52 (t, J = 6.4, 1H), 4.4 (s, 1H), 4.28 (m, 4H), 2.56 (t, J = 6.5, 2H), 1.72 (t, J = 6.2, 2H), 1.52 (t, J = 6.2, 2H), 1.28 (t, J = 6.2, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 66.4, 63.0, 33.8, 30.6, 24.4, 24.0. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C15H24N5O7PS 449.4191, found (M+H) = 450.23; (M−H) = 448.12.
14a: 1H NMR (CD3OD, 500 MHz): 8.58 (s, 1H), 8.35 (s, 1H), 7.61 (q, J = 6.3, 2H) 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.52 (m, 3H), 4.4 (s, 1H), 4.28 (m, 4H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 129.2, 128.8, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 66.4, 63.0, 62.4, 51.7, 24.4. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C14H19N8O7P 442.3228, found (M+H) = 443.13; (M−H) = 441.65.
14b: 1H NMR (CD3OD, 500 MHz): 8.58 (s, 1H), 8.35 (s, 1H), 7.61 (q, J = 6.3, 2H) 7.01 (s, 2H), 6.12 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.42 (m, 3H), 4.28 (m, 4H), 2.14 (q, J = 6.3, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 129.2, 128.8, 119.4, 97.0, 86.7, 73.4, 70.3, 69.7, 66.4, 63.0, 61.0, 47.9, 27.8. 31P NMR (CH3OD, 121 MHz): δ = −0.52. Mass calculate for C15H21N8O7P 456.3504, found (M+H) = 457.27; (M−H) = 455.45.
14c: 1H NMR (CH3OD, 500 MHz): δ = 8.62 (s, 1H), 8.30 (s, 1H), 7.96 (s, 1H), 7.68 (s, 1H), 6.11 (d, J = 5.5, 1H), 4.66 (t, J = 5.5, 1H), 4.39 (m, 3H), 4.23 (m, 1H), 4.12 (m, 1H), 4.06 (m, 1H), 3.90 (m, 2H), 1.89 (m, 1H), 1.64 (m, 1H), 1.37 (m, 1H); ((CD3OD, 100 MHz): δ = 156.2, 152.4, 19.8, 140.3, 129.2, 128.8, 119.4, 97.0, 86.7, 73.4, 70.3, 66.1, 63.0, 51.8, 27.2, 24.3. 31P NMR (CH3OD, 121 MHz): δ = −0.53; ESI-HRMS calcd. For C17H26N8O7P (M+H) 470.37, found (M+H) 471.37.
14d: 1H NMR (CH3OD, 500 MHz): δ = 8.63 (s, 1H), 8.31 (s, 1H), 7.97 (s, 1H), 7.70 (s, 1H), 6.10 (d, J = 5.5, 1H), 4.62 (t, J = 5.0, 1H), 4.40 (m, 3H), 4.25 (m, 1H), 4.21 (m, 1H), 4.11 (m, 1H), 3.90 (m, 2H), 1.88 (m, 1H), 1.62 (m, 1H), 1.42 (m, 1H), 1.30 (m, 1H); 13C NMR ((CD3OD, 100 MHz): δ = 152.1, 150.1, 146.3, 143.4, 134.3, 125.8, 119.9, 89.5, 85.8 (d), 76.3, 71.9, 66.9 (d), 66.2 (d), 51.1, 31.5 (d), 31.2, 27.0, 26.2; 31P NMR (CH3OD, 121 MHz): δ = −0.65; ESI-HRMS calcd. For C18H28N8O7P (M + H) 485.41, found 486.41.
15a: 1H NMR (CH3OD, 500 MHz): 8.58 (s,1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.16 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.41 (s, 1H), 4.28 (m, 3H), 2.6 (t, J = 6.5, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 177.3, 156.1, 152.0, 150.0, 140.3, 97.1, 86.7, 73.4, 70.3, 63.1, 60.3, 32.4; 31P NMR (CH3OD, 121 MHz): δ = −0.771. Mass calcd. For C13H18N5O9P 419.2839, found (M + H) 420.21.
15b: 1H NMR (CH3OD, 500 MHz): 8.58 (s,1H), 8.35 (s, 1H), 7.01 (s, 2H), 6.16 (d, J = 6.2, 1H), 4.75 (t, J = 6.5, 1H), 4.41 (s, 1H), 4.28 (m, 2H), 4.07 (m, 2h), 2.3 (t, J = 6.5, 2H), 2.03 (q, J = 6.5, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 178.3, 156.1, 152.0, 150.0, 140.3, 97.1, 86.7, 73.4, 70.3, 63.0 (2C), 31.4, 24.1. 31P NMR (CH3OD, 121 MHz): δ = −0.771. Mass calcd. For C14H20N5O9P 433.3105, found (M + H) 434.25.
15c: 1H NMR (CH3OD, 500 MHz): δ = 8.64 (s, 1H), 8.33 (s, 1H), 6.10 (d, J = 5.5, 1H), 4.63 (t, J = 5.5, 1H), 4.41 (dd, J = 4.0, 5.0, 1H), 4.25 (m, 1H), 4.23 (m, 1H), 4.13 (m, 1H), 3.93 (m, 2H), 2.30 (m, 2H), 1.67 (m, 4H); 13C NMR ((CD3OD, 100 MHz): δ = 177.3, 152.0, 150.0, 146.0, 143.5, 120.0, 89.6, 85.8 (d, J = 7.8), 76.3, 71.9, 66.8 (d, J = 6.2), 66.2 (d, J = 5.4), 34.4, 31.1 (d, J = 7.7), 22.4; 31P NMR (CH3OD, 121 MHz): δ = −0.771. ESI-HRMS calcd. For C15H23N5O9P (M + H) 448.1233, found 448.13.
15d: 1H NMR (CH3OD, 500 MHz): δ = 8.66 (s, 1H), 8.33 (s, 1H), 6.11 (d, J = 5.5, 1H), 4.63 (t, J = 5.5, 1H), 4.40 (dd, J = 3.0, 4.5, 1H), 4.24 (m, 1H), 4.15 (m, 1H), 4.09 (m, 1H), 3.87 (m, 2H), 2.25 (t, J = 7.5, 2H), 1.59 (m, 4H), 1.39 (m, 2H); 13C NMR ((CD3OD, 100 MHz): δ = 177.3, 152.0, 150.0, 146.0, 143.5, 120.0, 89.6, 85.8, 76.3, 71.9, 66.8, 66.2, 34.4, 31.1, 22.4, 21.2; 31P NMR (CH3OD, 121 MHz): δ = −2.25, −2.38. ESI-HRMS calcd. For C16H25N5O9P (M + H) 462.1390, found 462.14.
16a: 1H NMR (CH3OD, 500 MHz): δ = 8.56 (s, 1H), 8.35 (s, 1H), 7.01(s, 2h), 6.16 (d, J = 5.5, 1H), 4.73 (t, J = 5.5, 1H), 4.40 (dd, J = 3.0, 4.5, 1H), 4.31 (m, 4H), 2.55 (t, J = 6.3, 2H) 13C NMR ((CD3OD, 100 MHz): δ = 170.3, 152.0, 150.0, 146.0, 143.5, 120.0, 89.6, 85.8, 76.3, 71.9, 63.8, 61.2, 30.4. Mass calculated for C13H19N6O9P 434.2985; found (M−H) = 433.22.
16b: 1H NMR (CH3OD, 500 MHz): δ = 8.56 (s, 1H), 8.35 (s, 1H), 7.01(s, 2h), 6.16 (d, J = 5.5, 1H), 4.73 (t, J = 5.5, 1H), 4.40 (dd, J = 3.0, 4.5, 1H), 4.25 (m, 2H), 4.01 (t, J = 6.3, 2H), 2.35 (t, J = 6.3, 2H), 2.05 (t, J = 6.3, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 169.3, 156.1, 152.0, 150.0, 146.0, 143.5, 120.0, 89.6, 85.8, 76.3, 71.9, 63.8, 62.2, 28.4, 24.8. Mass calculated for C14H21N6O9P 448.3251; found (M−H) = 447.22.
16c: 1H NMR (CH3OD, 500 MHz): δ = 8.56 (s, 1H), 8.35 (s, 1H), 7.01(s, 2h), 6.16 (d, J = 5.5, 1H), 4.73 (t, J = 5.5, 1H), 4.40 (dd, J = 3.0, 4.5, 1H), 4.28 (m, 2H), 4.01 (t, J = 6.3, 2H), 2.35 (t, J = 6.3, 2H), 1.75 (t, J = 6.3, 2H), 1.71 (t, J = 6.4, 2H). 13C NMR (CD3OD, 100 MHz): δ = 169.3, 156.0, 152.0, 150.0, 140.3, 120.0, 89.6, 76.3, 71.9, 63.8, 62.2, 32.2, 28.6, 21.4. Mass calculated for C15H23N6O9P 462.3517; found (M−H) = 461.08.
16d: 1H NMR (CH3OD, 500 MHz): δ = 8.56 (s, 1H), 8.35 (s, 1H), 7.01(s, 2h), 6.16 (d, J = 5.5, 1H), 4.73 (t, J = 5.5, 1H), 4.40 (dd, J = 3.0, 4.5, 1H), 4.28 (m, 2H), 4.01 (t, J = 6.3, 2H), 2.35 (t, J = 6.3, 2H), 1.71 (t, J = 6.3, 2H), 1.53 (t, J = 6.4, 2H), 1.28 (t, J = 6.2, 2H). 13C NMR ((CD3OD, 100 MHz): δ = 169.3, 156.0, 152.0, 150.0, 140.3, 120.0, 89.6, 76.3, 71.9, 66.4, 63.8, 62.2, 32.2, 30.6, 25.6, 24.4. Mass calculated for C16H25N6O9P 476.3783; found (M−H) = 475.23.
APR assay
35S-labeled adenosine 5′-phosphosulfate (APS) was synthesized and purified as previously described with the inclusion of an additional anion exchange purification step on a 5 mL FFQ column (GE Healthcare) eluting with a linear gradient of ammonium bicarbonate, pH 8.0. The reduction of APS to sulfite and AMP was measured in a 35S-based assay as previously described [31, 42, 43, 50]. Reactions were quenched by the addition of charcoal solution (2% w/v) containing Na2SO3 (20 mM). The suspension was vortexed, clarified by centrifugation, and an aliquot of the supernatant containing the radiolabeled sulfite product was counted in scintillation fluid. APS reductase activity was measured in single turnover reactions, with trace amounts of 35S-APS (~1 nM) and excess protein. These reactions can typically be followed to ≥90% completion. Unless otherwise specified, the standard reactions conditions were 30°C with 100 mM bis-tris propane at pH 7.5, DTT (5 mM), and thioredoxin (10 μM). The affinity of APS reductase in presence of varios drug concentrations were determined by using the previously reported procedures [31].
AutoDock
Compounds were built in Chem Bio Draw ultra 12.0 and the compounds were energy minimized using MM2 module to an RMS gradient of 0.01 kcal/mol. Then the energy-minimized molecules were saved as .mol files and then converted to PDB files using pymol. APR (PDB: 2GOY) was used as stationary file and N5-position of the bound APS was detected as center. Using AutoDock [51] a grid with 40 Å, width, height and length around the center was created and docking parameter file was created by choosing generic algorithm, 200 GA runs, population size 300 and maximum number of evaluations for each run was 25 million. The compounds were allowed to dock on to APR. The docking results were evaluated by studying clusters of molecular conformations of the ligand that are grouped based on their binding energies. The lowest binding energy containing clusters were selected and evaluated the binding interactions of the conformations.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM087638 to K.S.C.).
References
- 1.Blumberg HM, Burman WJ, Chaisson RE, Daley CL, Etkind SC, Friedman LN, Fujiwara P, Grzemska M, Hopewell PC, Iseman MD, Jasmer RM, Koppaka V, Menzies RI, O’Brien RJ, Reves RR, Reichman LB, Simone PM, Starke JR, Vernon AA. American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America: treatment of tuberculosis. Am J Respir Crit Care Med. 2003;167(4):603–62. doi: 10.1164/rccm.167.4.603. [DOI] [PubMed] [Google Scholar]
- 2.Paritala H, Carroll KS. New targets and inhibitors of mycobacterial sulfur metabolism. Infect Disord.: Drug Targets. 2013;13(2):85–115. doi: 10.2174/18715265113139990022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y. The magic bullets and tuberculosis drug targets. Annu Rev Pharmacol Toxicol. 2005;45:529–64. doi: 10.1146/annurev.pharmtox.45.120403.100120. [DOI] [PubMed] [Google Scholar]
- 4.Bhave DP, Muse WB, 3rd, Carroll KS. Drug targets in mycobacterial sulfur metabolism. Infect Disord.: Drug Targets. 2007;7(2):140–58. doi: 10.2174/187152607781001772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Flynn JL, Chan J. Tuberculosis: latency and reactivation. Infect Immun. 2001;69(7):4195–201. doi: 10.1128/IAI.69.7.4195-4201.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Flynn JL, Chan J. Immunology of tuberculosis. Annu Rev Immunol. 2001;19:93–129. doi: 10.1146/annurev.immunol.19.1.93. [DOI] [PubMed] [Google Scholar]
- 7.Buchmeier NA, Newton GL, Koledin T, Fahey RC. Association of mycothiol with protection of Mycobacterium tuberculosis from toxic oxidants and antibiotics. Mol Microbiol. 2003;47(6):1723–1732. doi: 10.1046/j.1365-2958.2003.03416.x. [DOI] [PubMed] [Google Scholar]
- 8.Boshoff HI, Barry CE., 3rd Tuberculosis – metabolism and respiration in the absence of growth. Nat Rev Microbiol. 2005;3(1):70–80. doi: 10.1038/nrmicro1065. [DOI] [PubMed] [Google Scholar]
- 9.Kwon YW, Masutani H, Nakamura H, Ishii Y, Yodoi J. Redox regulation of cell growth and cell death. Biol Chem. 2003;384(7):991–6. doi: 10.1515/BC.2003.111. [DOI] [PubMed] [Google Scholar]
- 10.Schnappinger D, Ehrt S, Voskuil MI, Liu Y, Mangan JA, Monahan IM, Dolganov G, Efron B, Butcher PD, Nathan C, Schoolnik GK. Transcriptional Adaptation of Mycobacterium tuberculosis within Macrophages: Insights into the Phagosomal Environment. J Exp Med. 2003;198(5):693–704. doi: 10.1084/jem.20030846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pinto R, Tang QX, Britton WJ, Leyh TS, Triccas JA. The Mycobacterium tuberculosis cysD and cysNC genes form a stress-induced operon that encodes a tri-functional sulfate-activating complex. Microbiology. 2004;150(Pt 6):1681–6. doi: 10.1099/mic.0.26894-0. [DOI] [PubMed] [Google Scholar]
- 12.Williams SJ, Senaratne RH, Mougous JD, Riley LW, Bertozzi CR. 5′-adenosinephosphosulfate lies at a metabolic branch point in mycobacteria. J Biol Chem. 2002;277(36):32606–32615. doi: 10.1074/jbc.M204613200. [DOI] [PubMed] [Google Scholar]
- 13.Carroll KS, Gao H, Chen H, Leary JA, Bertozzi CR. Investigation of the iron-sulfur cluster in Mycobacterium tuberculosis APS reductase: implications for substrate binding and catalysis. Biochemistry. 2005;44(44):14647–57. doi: 10.1021/bi051344a. [DOI] [PubMed] [Google Scholar]
- 14.Carroll KS, Gao H, Chen H, Stout CD, Leary JA, Bertozzi CR. A Conserved Mechanism for Sulfonucleotide Reduction. PLoS Biol. 2005;3(8):e250. doi: 10.1371/journal.pbio.0030250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Senaratne RH, De Silva AD, Williams SJ, Mougous JD, Reader JR, Zhang T, Chan S, Sidders B, Lee DH, Chan J, Bertozzi CR, Riley LW. 5′-Adenosinephosphosulphate reductase (CysH) protects Mycobacterium tuberculosis against free radicals during chronic infection phase in mice. Mol Microbiol. 2006;59(6):1744–1753. doi: 10.1111/j.1365-2958.2006.05075.x. [DOI] [PubMed] [Google Scholar]
- 16.Paritala H, Carroll KS. A continuous spectrophotometric assay for adenosine 5′-phosphosulfate reductase activity with sulfite-selective probes. Anal Biochem. 2013;440(1):32–39. doi: 10.1016/j.ab.2013.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dos Santos PC, Dean DR. A newly discovered role for iron-sulfur clusters. Proceedings of the National Academy of Sciences. 2008;105(33):11589–11590. doi: 10.1073/pnas.0805713105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fontecave M. Iron-sulfur clusters: ever-expanding roles. Nat Chem Biol. 2006;2(4):171–174. doi: 10.1038/nchembio0406-171. [DOI] [PubMed] [Google Scholar]
- 19.Beinert H, Holm RH, Münck E. Iron-Sulfur Clusters: Nature’s Modular, Multipurpose Structures. Science. 1997;277(5326):653–659. doi: 10.1126/science.277.5326.653. [DOI] [PubMed] [Google Scholar]
- 20.Bhave DP, Hong JA, Lee M, Jiang W, Krebs C, Carroll KS. Spectroscopic Studies on the [4Fe-4S] Cluster in Adenosine 5′-Phosphosulfate Reductase from Mycobacterium tuberculosis. J Biol Chem. 2011;286(2):1216–1226. doi: 10.1074/jbc.M110.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chartron J, Carroll KS, Shiau C, Gao H, Leary JA, Bertozzi CR, Stout CD. Substrate Recognition, Protein Dynamics, and Iron-Sulfur Cluster in Pseudomonas aeruginosa Adenosine 5′-Phosphosulfate Reductase. J Mol Biol. 2006;364(2):152–169. doi: 10.1016/j.jmb.2006.08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhave DP, Han WG, Pazicni S, Penner-Hahn JE, Carroll KS, Noodleman L. Geometric and electrostatic study of the [4Fe-4S] cluster of adenosine-5′-phosphosulfate reductase from broken symmetry density functional calculations and extended X-ray absorption fine structure spectroscopy. Inorg Chem. 2011;50(14):6610–25. doi: 10.1021/ic200446c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beinert H, Kennedy MC, Stout CD. Aconitase as Iron−Sulfur Protein, Enzyme, and Iron-Regulatory Protein. Chem Rev (Washington, DC, U S) 1996;96(7):2335–2374. doi: 10.1021/cr950040z. [DOI] [PubMed] [Google Scholar]
- 24.Hänzelmann P, Schindelin H. Binding of 5′-GTP to the C-terminal FeS cluster of the radical S-adenosylmethionine enzyme MoaA provides insights into its mechanism. Proc Natl Acad Sci U S A. 2006;103(18):6829–6834. doi: 10.1073/pnas.0510711103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Berkovitch F, Nicolet Y, Wan JT, Jarrett JT, Drennan CL. Crystal Structure of Biotin Synthase, an S-Adenosylmethionine-Dependent Radical Enzyme. Science. 2004;303(5654):76–79. doi: 10.1126/science.1088493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Perche-Letuvée P, Kathirvelu V, Berggren G, Clemancey M, Latour JM, Maurel V, Douki T, Armengaud J, Mulliez E, Fontecave M, Garcia-Serres R, Gambarelli S, Atta M. 4-Demethylwyosine Synthase from Pyrococcus abyssi Is a Radical-S-adenosyl-l-methionine Enzyme with an Additional [4Fe-4S]+2 Cluster That Interacts with the Pyruvate Co-substrate. J Biol Chem. 2012;287(49):41174–41185. doi: 10.1074/jbc.M112.405019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan A, Clémancey M, Mouesca J-M, Amara P, Hamelin O, Latour J-M, Ollagnier de Choudens S. Studies of Inhibitor Binding to the [4Fe-4S] Cluster of Quinolinate Synthase. Angew Chem. 2012;51(31):7711–7714. doi: 10.1002/anie.201202261. [DOI] [PubMed] [Google Scholar]
- 28.Marsh ENG, Patwardhan A, Huhta MS. S-Adenosylmethionine radical enzymes. Bioorg Chem. 2004;32(5):326–340. doi: 10.1016/j.bioorg.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 29.Landry AP, Ding H. Redox Control of Human Mitochondrial Outer Membrane Protein MitoNEET [2Fe-2S] Clusters by Biological Thiols and Hydrogen Peroxide. J Biol Chem. 2014;289(7):4307–4315. doi: 10.1074/jbc.M113.542050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cosconati S, Hong JA, Novellino E, Carroll KS, Goodsell DS, Olson AJ. Structure-Based Virtual Screening and Biological Evaluation of Mycobacterium tuberculosis Adenosine 5‚Ä≤-Phosphosulfate Reductase Inhibitors. J Med Chem. 2008;51(21):6627–6630. doi: 10.1021/jm800571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hong JA, Bhave DP, Carroll KS. Identification of Critical Ligand Binding Determinants in Mycobacterium tuberculosis Adenosine-5′-phosphosulfate Reductase. J Med Chem. 2009;52(17):5485–5495. doi: 10.1021/jm900728u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paritala H, Suzuki Y, Carroll KS. Efficient microwave-assisted solid phase coupling of nucleosides, small library generation, and mild conditions for release of nucleoside derivatives. Tetrahedron Lett. 2013;54(14):1869–1872. doi: 10.1016/j.tetlet.2013.01.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lill R. Function and biogenesis of iron-sulphur proteins. Nature. 2009;460(7257):831–8. doi: 10.1038/nature08301. [DOI] [PubMed] [Google Scholar]
- 34.Jacobsen FE, Lewis JA, Cohen SM. The design of inhibitors for medicinally relevant metalloproteins. ChemMedChem. 2007;2(2):152–71. doi: 10.1002/cmdc.200600204. [DOI] [PubMed] [Google Scholar]
- 35.Gopinath P, Vidyarini RS, Chandrasekaran S. Synthesis of Thioesters from Carboxylic Acids via Acyloxyphosphonium Intermediates with Benzyltriethylammonium Tetrathiomolybdate as the Sulfur Transfer Reagent. J Org Chem. 2009;74(16):6291–6294. doi: 10.1021/jo9009694. [DOI] [PubMed] [Google Scholar]
- 36.Marma MS, Khawli LA, Harutunian V, Kashemirov BA, McKenna CE. Synthesis of α-fluorinated phosphonoacetate derivatives using electrophilic fluorine reagents: Perchloryl fluoride versus 1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate) (Selectfluor®) J Fluorine Chem. 2005;126(11–12):1467–1475. [Google Scholar]
- 37.Harris WR, Brook CE, Spilling CD, Elleppan S, Peng W, Xin M, Wyk JV. Release of iron from transferrin by phosphonocarboxylate and diphosphonate chelating agents. J Inorg Biochem. 2004;98(11):1824–1836. doi: 10.1016/j.jinorgbio.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 38.Chougrani K, Niel G, Boutevin B, David G. Regioselective ester cleavage during the preparation of bisphosphonate methacrylate monomers. Beilstein J Org Chem. 2011;7:364–368. doi: 10.3762/bjoc.7.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grison C, Coutrot P, Comoy C, Balas L, Joliez S, Lavecchia G, Oliger P, Penverne B, Serre V, Hervé G. Design, synthesis and activity of bisubstrate, transition-state analogues and competitive inhibitors of aspartate transcarbamylase. Eur J Med Chem. 2004;39(4):333–344. doi: 10.1016/j.ejmech.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 40.Bernier S, Akochy PM, Lapointe J, Chenevert R. Synthesis and aminoacyl-tRNA synthetase inhibitory activity of aspartyl adenylate analogs. Bioorg Med Chem. 2005;13(1):69–75. doi: 10.1016/j.bmc.2004.09.055. [DOI] [PubMed] [Google Scholar]
- 41.Ohkubo A, Ezawa Y, Seio K, Sekine M. O-selectivity and utility of phosphorylation mediated by phosphite triester intermediates in the N-unprotected phosphoramidite method. J Am Chem Soc. 2004;126(35):10884–10896. doi: 10.1021/ja048125h. [DOI] [PubMed] [Google Scholar]
- 42.Carroll KS, Gao H, Chen HY, Leary JA, Bertozzi CR. Investigation of the iron-sulfur cluster in Mycobacterium tuberculosis APS reductase: Implications for substrate binding and catalysis. Biochemistry. 2005;44(44):14647–14657. doi: 10.1021/bi051344a. [DOI] [PubMed] [Google Scholar]
- 43.Cosconati S, Hong JA, Novellino E, Carroll KS, Goodsell DS, Olson AJ. Structure-Based Virtual Screening and Biological Evaluation of Mycobacterium tuberculosis Adenosine 5′-Phosphosulfate Reductase Inhibitors. J Med Chem. 2008;51(21):6627–6630. doi: 10.1021/jm800571m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morris GM, Huey R, Lindstrom W, Sanner MF, Belew RK, Goodsell DS, Olson AJ. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J Comput Chem. 2009;30(16):2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gillum WO, Mortenson LE, Chen JS, Holm RH. Quantitative extrusions of the iron sulfide(Fe4S4*) cores of the active sites of ferredoxins and the hydrogenase of Clostridium pasteurianum. J Am Chem Soc. 1977;99(2):584–595. doi: 10.1021/ja00444a044. [DOI] [PubMed] [Google Scholar]
- 46.Orme-Johnson WH, Holm RH. Identification of iron–sulfur clusters in proteins. Methods Enzymol. 1978;53:268–74. doi: 10.1016/s0076-6879(78)53032-2. [DOI] [PubMed] [Google Scholar]
- 47.Volbeda A, Charon MH, Piras C, Hatchikian EC, Frey M, Fontecilla-Camps JC. Crystal structure of the nickel-iron hydrogenase from Desulfovibrio gigas. Nature. 1995;373(6515):580–587. doi: 10.1038/373580a0. [DOI] [PubMed] [Google Scholar]
- 48.Kim SK, Rahman A, Mason JT, Hirasawa M, Conover RC, Johnson MK, Miginiac-Maslow M, Keryer E, Knaff DB, Leustek T. The interaction of 5′-adenylylsulfate reductase from Pseudomonas aeruginosa with its substrates. Biochim Biophys Acta. 2005;20:2–3. doi: 10.1016/j.bbabio.2005.09.004. [DOI] [PubMed] [Google Scholar]
- 49.Kim SK, Rahman A, Bick JA, Conover RC, Johnson MK, Mason JT, Hirasawa M, Leustek T, Knaff DB. Properties of the cysteine residues and iron-sulfur cluster of the assimilatory 5′-adenylyl sulfate reductase from Pseudomonas aeruginosa. Biochemistry. 2004;43(42):13478–86. doi: 10.1021/bi048811t. [DOI] [PubMed] [Google Scholar]
- 50.Gao H, Leary J, Carroll KS, Bertozzi CR, Chen H. Noncovalent complexes of APS reductase from M. tuberculosis: delineating a mechanistic model using ESI-FTICR MS. J Am Soc Mass Spectrom. 2007;18(2):167–78. doi: 10.1016/j.jasms.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huey R, Morris GM, Olson AJ, Goodsell DS. A semiempirical free energy force field with charge-based desolvation. J Comput Chem. 2007;28(6):1145–52. doi: 10.1002/jcc.20634. [DOI] [PubMed] [Google Scholar]
- 52.Takashima Y, Osaki M, Harada A. Cyclodextrin-initiated polymerization of cyclic esters in bulk: Formation of polyester-tethered cyclodextrins. J Am Chem Soc. 2004;126(42):13588–13589. doi: 10.1021/ja047171e. [DOI] [PubMed] [Google Scholar]
- 53.Spencer TA, Onofrey TJ, Cann RO, Russel JS, Lee LE, Blanchard DE, Castro A, Gu P, Jiang GJ, Shechter I. Zwitterionic sulfobetaine inhibitors of squalene synthase. J Org Chem. 1999;64(3):807–818. doi: 10.1021/jo981617q. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.