

Abstract
The accumulation and efflux kinetics of ciprofloxacin have been examined by using murine J774 macrophages. Accumulation (at equilibrium) was increased (three- to fourfold) (i) when cells were incubated with high extracellular drug concentrations (typically 200 mg/liter) as opposed to clinically meaningful concentrations (10 mg/liter or lower), (ii) during ATP- depletion and at acid pH, and (iii) during coincubation with probenecid, gemfibrozil and the preferential multidrug resistance-related protein (MRP) inhibitor MK571. All these conditions were also associated with a marked decrease in ciprofloxacin efflux (half-lives increased from <2 min in controls to up to 10 min). Monensin (a proton ionophore), verapamil, and the preferential P-glycoprotein (P-gp) inhibitor GF120918 had no or only minimal effect, while cyclosporin A, which is not specific for P-gp but also acts on MRP, had an intermediate effect. Short-term uptake studies showed that the influence of the modulators on the apparent drug influx was almost immediate (delay of ≤1 min). Cells made resistant to probenecid and showing a marked overexpression of MRP1 (by Western blot analysis and confocal microscopy) accumulated ciprofloxacin to almost the same extent as did control cells, but efflux was inhibited less by probenecid, gemfibrozil, and MK571. We conclude that ciprofloxacin is subject to constitutive efflux in J774 macrophages through the activity of an MRP-related transporter which is probably distinct from MRP1. We also suggest that the cellular accumulation of ciprofloxacin in wild-type cells is constitutively impaired at therapeutically meaningful concentrations.
Introduced into our clinical armamentarium in the mid 1980s, fluoroquinolones are still the subjects of intense clinical interest because of their wide spectrum, intense bactericidal activity, and excellent bioavailability (21). Another key feature of fluoroquinolones is their ability to accumulate in cells (36, 50), most notably in polymorphonuclear leucocytes and macrophages, where they show useful activity against several types of intracellular bacteria (6, 7, 13, 14, 34, 35, 55). Early work showed that probenecid increases the accumulation of norfloxacin in J774 macrophages (5, 45). Subsequent studies demonstrated that both probenecid and gemfibrozil enhance the activity of ciprofloxacin against intracellular Listeria monocytogenes (41, 45). These effects have been interpreted as demonstrating the existence of an efflux mechanism for fluoroquinolones similar to that responsible for the extrusion of organic anions observed in J774 macrophages (47). No further characterization of this efflux and of the transporters involved has, however, been reported up to now. In the present study, we have examined in detail the influx and efflux processes of ciprofloxacin in J774 macrophages. We used a series of conditions to demonstrate the role of specific transporters already observed in eucaryotic cells (53, 54), namely, depletion of ATP and glutathione, exposure to acidic and basic pH and coincubation with a proton ionophore, and coincubation with various inhibitors. We also used probenecid-resistant J774 macrophages since these cells display enhanced transport of organic anions (4) and since probenecid is known to reverse multidrug resistance (17).
MATERIALS AND METHODS
Materials.
Ciprofloxacin (purity, 85.5%) and azithromycin (purity, 94.4%) were obtained as laboratory samples for microbiological evaluation from Bayer AG (Leverkusen, Germany) and Pfizer Inc. (Groton, Conn.). Probenecid, gemfibrozil, and buthionine sulfoximide were obtained from Sigma-Aldrich (St Louis, Mo); verapamil, cyclosporin A, and 2-d-deoxyglucose were obtained from Fluka AG (a division of Sigma-Aldrich, Buchs, Switzerland); MK571 (3-[[[3-[2-(7-chloro-2-quinolinyl)ethenyl]phenyl]phenyl][[3-(dimethylamino)-3-oxopropyl]thio]methyl]thio]propanoic acid) was obtained from Alexis Corp., San Diego, Calif; and GF 120918 (N-[4-[2-(3,4-dihydro-6,7-dimethoxy-2(1H)-isoquinolinyl)ethyl]phenyl]-9,10-dihydro-5-methoxy-9-oxo-4-acridinecarboxamide; Elacridar) was the kind gift of GlaxoWellcome Research and Development (Laboratoire GlaxoWellcome, Les Ulis, France). Rat monoclonal anti-MRP1 antibody was purchased from Alexis Corp; Alexa Fluor 568 anti-rat immunoglobulin G (IgG) was obtained from Molecular Probes (Eugene, Oreg.); and horseradish peroxidase-conjugated goat anti-rat antibody was obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, Calif.). [14C]sucrose and [14C]urea (615.0 and 57.0 mCi/mmol, respectively) were obtained from Amersham plc, Little Chalfont, United Kingdom. Cell culture media and sera were from Life Technologies (Rockville, Md.). Wild-type Swiss-3T3 cells and Swiss 3T3 cells overexpressing the multidrug resistance-related protein 1 (MRP1) transporter (pVD6-3) were kindly donated by V. Dhondt (Unité d'Oncologie, Université Catholique de Louvain, Brussels, Belgium). Other products were purchased from Sigma-Aldrich or from E. Merck AG (Darmstadt, Germany).
Cell culture conditions and assessment of viability.
All experiments were performed with J774 macrophages, grown as monolayers in RPMI 1640 medium supplemented with 10% fetal bovine serum in 5% CO2, as previously described (38). Cultures were initiated at a density of ca. 5 × 104 cells per cm2 and used after 2 days (3 days for probenecid-resistant cells) of culture upon reaching confluence. For critical determinations, cell viability was assessed at the end of the experiments by measuring the release of lactate dehydrogenase (with a limit set at 10% of the total cell content). For experiments in which the pH had to be adjusted to values that could not be accurately controlled by the CO2/HCO3-buffer, media were buffered with 5 mM phosphate in the absence of bicarbonate and experiments were performed with a normal atmosphere.
Cell collection and protein determination.
Cell monolayers were washed three times with ice-cold phosphate-buffered saline (PBS) scraped with a Teflon policeman, and collected in 0.1 M glycine-HCl buffer (pH 3). Samples were either sonicated for immediate analysis or frozen at −20°C for further analysis (in which case they were sonicated on thawing). Total protein was measured by the method of Lowry, using serum albumin as a standard.
Ciprofloxacin and azithromycin assays.
The amount of cell-associated ciprofloxacin was determined by a fluorimetric method (8). However, in preliminary studies, we observed that cell protein caused significant interference in this assay, and the following method was therefore developed. After sonication, each cell sample was centrifuged at 14,000 rpm for 10 min (Eppendorf 5415 C centriguge; Eppendorf Gerätgebau GmbH, Engelsdorf, Germany) at room temperature. The supernatants were collected and assayed for both protein and ciprofloxacin content (the latter after suitable dilution in 0.1 M glycine-HCl buffer [pH 3]) by fluorimetry using an LS30 fluorescence spectrophotometer (Perkin-Elmer, Beaconsfield, United Kingdom) with λexc set at 275 nm and λem set at 450 nm. In parallel, samples were prepared from cells that were not exposed to ciprofloxacin but to which known amounts of ciprofloxacin had been added after collection and before any further treatment. The readings for these samples allowed us to construct a calibration curve and to correct the readings for all samples based on their actual protein content. Under these conditions, the assay method had a lower limit of detection of 5 ng/ml, a linearity of the signal up to 200 ng/ml (r2 = 0.99), and an intra-assay reproducibility of 97%. The cell content of ciprofloxacin was then expressed by reference to the total cell protein content (i.e., the protein content of samples before centrifugation), and the apparent cellular-to-extracellular concentration ratio (accumulation factor) was computed on the basis of the ratio between the cell volume and the cell protein content. To check for the absence of interference of the other drugs used in this study on the assay of ciprofloxacin, pure solutions of the fluoroquinolone (50 μM) were mixed with each of the drugs used (at the maximal concentration used for cell experiments) and the emission spectrum of ciprofloxacin was recorded. No changes were seen. We also examined cells incubated with each of the corresponding drug but without ciprofloxacin. No additional fluorescence signal was detected compared to that of control cells (with no ciprofloxacin and no drug). Because no sensitive chemical assay of azithromycin is available and because no labeled drug could be supplied, we had to use a microbiological method to assay cell-associated azithromycin. This involved a disk plate method with Micrococcus luteus ATCC 9341 as the test organism. To obtain enough sensitivity, the agar (antibiotic medium 2; Difco, Becton Dickinson & Co., Sparks, Md.) was adjusted to pH 9.5 for samples with low drug concentration (<0.5 μg/ml) and pH 8.0 for samples with higher concentrations. The lowest levels of detection were 0.08 μg/ml at pH 9.5 and 0.5 μg/ml at pH 8, with linearity up to 2 and 32 μg/ml, respectively (r2 = 0.97 and 0.99 [n = 18]). All assays were performed with 22.5- by 22.5-cm plates, with standards of the corresponding drug run in parallel with the samples (typically six standards covering the observed range of concentration of samples and tested in triplicate were used).
Assay of total-cell ATP and thiol contents.
The total ATP in cells collected in 2% HClO4 was assayed. After rapid sonication and centrifugation, cell extracts were immediately neutralized in 3 N KOH-KHCO3. Supernatants were then processed for the ATP assay (i) by high-pressure liquid chromatography (19) with a 4.7- by 125-mm (particle size, 5 μm) anion-exchange column (Partisphere SAX, Whatman plc, Maidstone, United Kingdom) with an isocratic buffer (0.45 M NH4H2PO4 [pH 3.7] at a flow rate of 1.5 ml/min) and UV detection at 245 nm, and (ii) by an ATP-dependent oxidation of d-luciferin by luciferase (Boehringer Mannheim ATP-bioluminescence assay kit CLS II; Roche Diagnostics, F. Hoffman-la Roche Ltd., Basel, Switzerland) using a Wallac type 1410 liquid scintillation counter (Perkin-Elmer Life Science, Boston, Mass.). The total-cell thiol content was assayed with ortho-phthalodialdehyde as the substrate (9). In brief, cells were harvested by trypsinization and resuspended in 0.155 M NaCl supplemented with 3% fetal bovine serum, washed twice with the same solution, and lysed with 0.1% sodium deoxycholate in the same medium. The samples were then mixed with 25% (wt/vol) meta-phosphoric acid. After centrifugation at 3,500 rpm for 15 min (Eppendorf 5415 C centriguge), the supernatants were mixed with ortho-phthalodialdehyde (1 mg/ml in methanol) and the fluorescence was read at a λexc = 345 nm and λem = 420 nm. Calibration with pure glutathione was done by using internal standards, with a recovery of approximately 85%.
Selection of probenecid-resistant cells.
Probenecid-resistant cells were obtained as previously described (except that they were grown in RPMI 1640 medium), (4), using a stepwise approach involving a succession of passages (at least 20 passages) in media with increasing concentrations of probenecid (1, 2, and finally 3 mM) to obtain multifactorial multidrug resistance (18). Clones of cells surviving these selection steps were then propagated for 9 months in the continuing presence of 3 mM probenecid. Cells were harvested and then frozen at −80°C. Experiments were performed with cells thawed and cultivated in the presence of 3 mM probenecid for about 1 month, i.e., on reaching stable growth.
Morphological studies.
Confocal and phase-contrast microscopy studies were performed with macrophages seeded at low density (1.5 × 104 and 3.0 × 104 cells/cm2 for wild-type and resistant cells, respectively) to obtain isolated cells or islets of a few cells only after 2 to 3 days of culture. The cells were fixed with 0.8 % formaldehyde in acetone for 10 s (to avoid denaturation of the MRP1 antigen[11]). Fixed cells were then washed with PBS-Ca2+ -Mg2+. Nonspecific sites were blocked with 1% barine serum albumin-1 mg of lysine per ml-0.01% saponin-0.02% azide in PBS (Q-PBS) for 30 min. The cells were then incubated for 1 h with primary antibodies (rat anti-human MRP1 [5 μg/ml]) at room temperature, washed six times for 5 min with Q-PBS, and incubated for 1 h with the secondary antibody (Alexa Fluor 568 anti-rat IgG [red signal], 5 μg/ml), washed again six times for 5 min with PBS, postfixed for 5 min with 4% formaldehyde in PBS, washed three times with PBS, and mounted in polyvinyl alcohol-diazabicyclo[2.2.2]octane (Mowiol/DABCO) overnight at 4°C. Observations were made with MRC1024 confocal scanning equipment (Bio-Rad, Richmond, Calif.) mounted on an Axiovert confocal microscope (Zeiss, Oberkochen, Germany; λexc = 495 nm and λem = 519 nm for green signals; λexc = 578 nm and λem = 603 nm for red signals). Electron microscopy was performed on cells fixed in situ, and samples were processed by conventional techniques with en bloc staining with uranyl acetate. Thin sections (ca. 65 nm) were counterstained with uranyl acetate and lead citrate and examined in the transmission mode with a Philips CM12 microscope operating at 80 kV.
Western blotting.
Pelletted cells were washed three times in ice-cold PBS and lysed by a 30-min incubation on ice in PBS buffer containing 1% Igepal CA-630, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 1% of a 10-mg/ml phenylmethylsulfonyl fluoride solution in isopropanol, 3% aprotin, and 1% of a 100 mM sodium orthovanadate solution in water. Cell lysates were then centrifuged for 25 min at 14,000 rpm (Eppendorf Centrifuge 5810R rotor A-4-62) at 4°C. The pellets were resuspended in 1.9 M glycerol containing 141 mM Tris base, 106 mM Tris-HCl, 73 mM lithium dodecylsulfate, 0.51 mM EDTA, 0.22 mM Serva Blue G 250, and 0.75 mM phenol red at pH 8.5. Samples (15 μg of protein) were separated under in denaturating conditions on a NuPAGE Bis-Tris electrophoresis gradient (4 to 12% polyacrylamide) precast gel cassette (Novex Electrophoresis GmbH, Frankfurt/Main, Germany), using appropriate running buffers supplemented with 3.5 mM sodium dodecyl sulfate and 1 mM EDTA (55 min at room temperature in an X Cell Surelock Mini-Cell apparatus [Novex] operated at 125 V; equal loading was checked by revealing actin, with no difference being detected). Separated proteins were transferred on a nitrocellulose membrane for 1 h at 50 V, in a transfer buffer made of 25 mM N,N-bis(2-hydroxyethyl)glycine, 25 mM Bis-Tris (free base), 1 mM EDTA, and 0.05 mM chlorobutanol at pH 7.2 and supplemented with 10% (vol/vol) ethanol before use. Between each of the next steps, the membranes were rinsed in Tween saline buffer [0.05% Tween 20(T) in PBS]. The membranes were blocked in 1% nonfat milk-0.025% Tween 20 for at least 90 min and then incubated for at least 1 h with gentle shaking and at room temperature with the primary antibody (monoclonal antibody raised in the rat against a fusion protein containing the human MRP1 sequence from positions 192 to 360 [15] or rat monoclonal anti-mouse β-actin) after appropriate dilution in the blocking buffer (1:3,000; final concentration, 83 ng/ml). The membranes were then exposed to the secondary antibody (goat anti-rat HRP-conjugated IgG, diluted 1:1,500 [final concentration, 266 ng/ml]). Reactive bands were detected by chemiluminescence using an ECL kit (Amersham plc) and photographic printing. Molecular weights were estimated by comigration with proteins with known molecular weights (Mark 12 wide-range protein standard; Novex, San Diego, Calif.). Positive identification of the MRP1 band was made by analyzing in parallel lysates from wild-type swiss 3T3 cells (which have minimal expression of this transporter) and from lysates of swiss 3T3 cells transfected with a retroviral vector containing multiple copies of the MRP1 cDNA together with the long terminal repeat and a PGK promoter (12).
Determination of cell-volume-to-protein ratio.
We applied the urea-sucrose method (37) using [14C]urea and unlabeled sucrose or [14C]sucrose and unlabeled urea (1 μCi/ml in each case) in parallel. In preliminary experiments, we checked for completeness of the diffusion of urea and minimal endocytic sucrose uptake by examining the variation in the urea/sucrose ratio in samples from cells incubated with the tracers for increasing periods. Accordingly, all measures were made with cells incubated with the tracers for a minimum of 30 min and a maximum of 60 min, a period during which the urea/sucrose cell ratio was fairly constant.
DNA and protein synthesis measurements.
Cells were seeded at a density of ca. 3.75 × 104 cells/cm2 and grown up to 4 days. On each day, samples were used for measurement of thymidine and leucine incorporation by a 2-h incubation with the corresponding tracer ([3H]thymidine [25 Ci/mmol; 5 μCi/ml] and [3H]leucine [70 Ci/mmol; 1 μCi/ml]). Labeled cells were collected after extensive washing and used for measurement of total and trichloroacetic acid-precipitable (40% trichloroacetic acid; 30 min at 4°C) radioactivity.
Measurement of cell growth.
We used the CellTiter 96 AQ cell proliferation assay (Promega Corp., Madison, Wis.), which is based on the reduction of3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) into formazan in the presence of the electron-coupling agent phenazine methosulfate. In brief, cells were seeded at density of ca. 7 × 103 cells/cm2 in 96-well plates and grown for up to 4 days. On each day, MTS (2 mg/ml) and phenazine methosulfate (46 μg/ml) were added to a series of wells for 2 h. The concentration of formazan was then measured in the culture medium by photometry at 490 mm (the correlation between the photometric response and the number of cells per well was examined in preliminary experiments and found to be linear).
Statistical analyses and curve fittings.
All statistical analyses and curve fittings were done using Graph Pad Prism software (version 2.01; Graph Pad software Inc., San Diego, Calif.). Group comparisons were made by analysis of varience ANOVA, and, in the case of significant differences, by paired group comparisons using Student's t test.
RESULTS
Influence of the extracellular concentration of ciprofloxacin on its cellular accumulation.
In a first series of exploratory experiments, we noticed that the ability of cells to concentrate ciprofloxacin, rather than being impaired, was actually increased when the extracellular concentration was raised from clinically relevant concentrations (up to 10 mg/liter) to higher ones (typically above 50 mg/liter). Examining this apparently paradoxical property in detail with cells incubated for 2 h with the drug (Fig. 1), we observed that the apparent cellular-to-extracellular-concentration ratio of ciprofloxacin rose from ca. 6 to ca. 25 in a clearly sigmoidal fashion when the extracellular concentration was increased from about 8 to 250 mg/liter ca. 25 to ca. 800 μM). Half of this effect was obtained for an extracellular concentration of ca. 40 mg/liter (ca. 125 μM). Based on these findings, we explored the kinetics of accumulation and efflux of ciprofloxacin by using both a low and a high extracellular concentration (50 μM [17 mg/liter] and 200 μM [68 mg/liter], respectively). Figure 2 (left panel) shows that the accumulation was rapid in both cases, reaching equilibrium within 30 to 60 min. Cells incubated with the highest concentrations of ciprofloxacin showed a higher cellular-to-extracellular-concentration ratio at all times points. However, the relative rate constants of accumulation were similar in the two situations (see the insets; see also kinetic data in the figure legend). In contrast, cells that had accumulated a large amount of ciprofloxacin showed a slower efflux than did cells with a lower drug content. Figure 2 (right panel) shows that, indeed, the half-life of ciprofloxacin was about 4 min for cells loaded with a high extracellular concentration of ciprofloxacin (68 mg/liter; 200 μM) but only 1.7 min for cells loaded with a low concentration (17 mg/liter; 50 μM) (see the insets). This difference vanished at 8 min, after which efflux proceeded at the same fractional rate under both conditions. In view of the data presented in Fig. 1 and 2, all subsequent experiments were performed at a fixed ciprofloxacin extracellular concentration of 17 mg/liter (50 μM) to keep below the level at which concentration-dependent effects became clearly detectable while, at the same time, being able to assay cell-associated ciprofloxacin levels with sufficient accuracy.
FIG. 1.
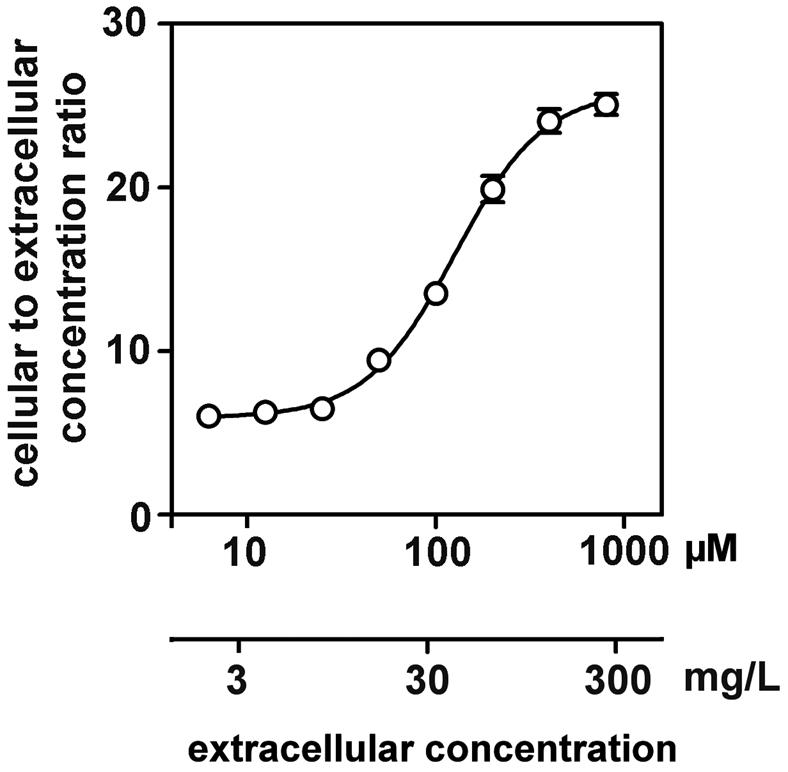
Influence of the extracellular concentration of ciprofloxacin on its accumulation at equilibrium (2 h of incubation) in J774 macrophages. Regression parameters (sigmoidal dose-response with variable slope): r2 = 0.999 Hill's slope = 1:845, 50% effective concentrain (EC50) = 126.4 ± 1.0 μM. For this experiment (and for all those described in Figures 2 to 6 and 8), all data are the mean of at least three independent measurements ± standard deviation [SD] (when not visible, the SD bar is smaller than the symbol).
FIG. 2.

Kinetics of accumulation (left) and efflux (right) of ciprofloxacin in J774 macrophages at two different extracellular drug concentrations (17 mg/liter [50 μM] [open circles] and 68 mg/liter [200 μM] [closed circles]). Regression parameters for influx (one-phase exponential association): for 50 μM, r2 = 0.932, k = 0.1103 ± 0.0265 min−1, ymax = 8.126; for 200 μM, r2 = 0.982, k = 0.0778 ± 0.0098 min−1, ymax = 18.45. Regression parameters for efflux (one-phase exponential decay): for 17 mg/liter [50 μM], r2 = 0.973, k = 0.4131 ± 0.0732 min−1; for 68 mg/liter [200 μM], r2 = 0.978, k = 0.1698 ± 0.0312 min−1). Insets: data presented as relative values (percentage of maximum; *, significant difference between cells incubated with 17 mg/liter [50 μM] and 68 mg/liter [200 μM] by paired t-test analysis with P < 0.05).
Influence of ATP depletion.
Cells were preincubated for 20 min with 5 mM NaN3 and 60 mM 2-d-deoxyglucose to deplete them of ATP (typical levels, 3.4 ± 0.2 versus 32.92 ± 1.50 nmol/mg of protein for controls [n = 3]). They were then challenged with 50 μM ciprofloxacin for up to 2 h in the continuing presence of NaN3 and 2-d-deoxyglucose (typical ATP levels at the end of the incubation period, 3.2 ± 0.1 versus 31.1 ± 3.2 nmol/mg of protein for controls [n = 6]). Figure 3 shows that ATP depletion caused a marked increase in ciprofloxacin accumulation which, qualitatively as well as quantitatively, was similar to that observed when comparing accumulation at low (17 mg/liter; 50 μM) and high (68 mg/liter; 200 μM) extracellular concentrations. Thus, we noted an increase in the net amount of drug accumulated but no apparent effect on the relative rate of intake when examined over the entire period of accumulation (0 to 120 min [see kinetic data in the caption of Fig. 3; see, however, the short-term kinetic data, for 0 to 5 min, in Fig. 6]). Ciprofloxacin efflux was then studied with cells depleted in ATP, loaded with ciprofloxacin in the presence of NaN3 and 2-d-deoxyglucose, and transferred to a drug-free medium in the continuing presence of NaN3 and 2-d-deoxyglucose (so as to maintain their state of ATP depletion). Figure 3 (right panel) shows that the rate of efflux of ciprofloxacin was very markedly decreased under these conditions compared to what was observed for control cells (no ATP depletion treatment). In parallel, we measured the efflux of ciprofloxacin from cells which were loaded with the drug while in a state of ATP depletion but which were then transferred to a drug-free and an NaN3 and 2-D-deoxyglucose-free medium (this allowed ATP levels to be restored to 25.5% ± 6.2% of control values within 15 min). Efflux proceeded at an intermediate rate between those for control cells and cells maintained in a state of ATP depletion.
FIG. 3.

Kinetics of accumulation (left) and efflux (right) of ciprofloxacin (extracellular concentration, 17 mg/liter [50 μM]) in J774 macrophages maintained under normal conditions (control) or in cells depleted in ATP (20-min preincubation with 2-d-deoxyglucose and NaN3 [ATP levels were reduced to less than 3% of the original content; influx was monitored in the continuing presence of the inhibitors; for efflux, cells were either transferred to a drug-free medium in the presence of the inhibitors or transferred to a drug-free medium without inhibitors [ATP resynthesis]). Regression parameters for influx (one-phase exponential association): control cells, r2 = 0.981, k = 0.0706 ± 0.0077 min−1; ymax = 7.08; ATP-depleted cells, r2 = 0.991, k = 0.0702 ± 0.055 min−1; ymax = 20.23. Regression parameters for efflux (one-phase exponential decay): control cells, r2 = 0.953, k = 0.4026 ± 0.0610 min−1; ATP-depleted cells, r2 = 0.993, k = 0.0646 ± 0.0017 min−1; ATP resynthesis, r2 = 0.988, k = 0.1414 ± 0.0059 min−1.
FIG. 6.
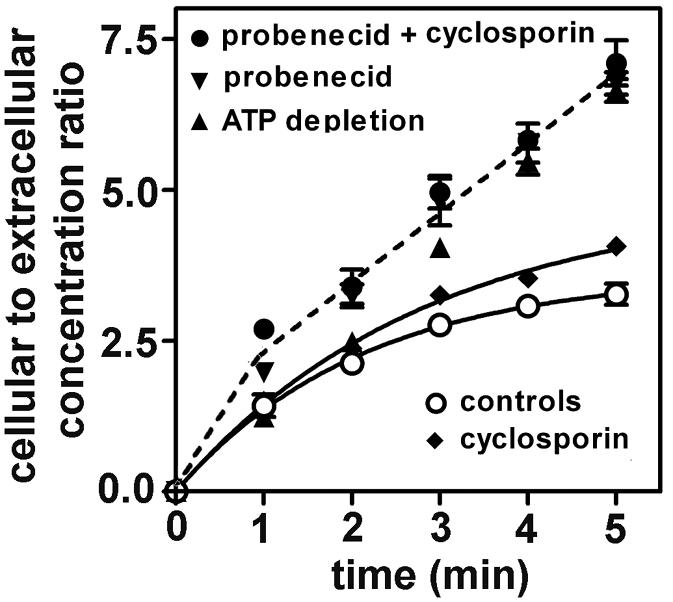
Short-term kinetics of the accumulation of ciprofloxacin (17 mg/liter; 50 μM) in control J774 macrophages in comparison with ATP-depleted J774 macrophages and with J774 macrophages coincubated with transporter inhibitors (5 mM probenecid, 100 μM cyclosporin A, and a combination of probenecid and cyclosporin A). For control cells and cells incubated with cyclosporin A alone, data could be fitted to a one-phase exponential association equation (r2 = 0.999 and 0.997). For all other conditions, fitting to either one-phase exponential decay or a simple linear equation were both considered statistically acceptable. Examination of the data, however, suggests a fast first phase of uptake (0 to 1 min) followed by a slower phase that is still linear with respect to time (1 to 5 min). Simple dotted lines illustrate these two successive linear processes, with no attempt to distinguish between the conditions tested (ATP depletion, probenecid, probenecid plus cyclosporin), since no significant difference could be noted between any of them.
Influence of glutathione depletion.
Cells were exposed for 24 h to 80 μM buthionine sulfoximine to obtain a stable reduction in glutathione cellular content (total sulfhydryl-containing constituents, 1.34 ± 0.13 versus 37.5 ± 5.4 nmol/mg of protein, expressed as glutathione, in treated versus control cells [n = 9]. The cells were then challenged with 50 μM ciprofloxacin for up to 2 h in the continuing presence of buthionine sulfoximine, in comparison with control cells. Cells depleted in glutathione showed a 47.58% ± 3.22% increase (n = 3) in ciprofloxacin cell content after 2 h (data not shown [this increase was observed at all time points during accumulation from 5 min to 2 h]).
Influence of pH and of the proton ionophore monensin.
In the next series of experiments, the accumulation of ciprofloxacin (17 mg/liter; 50 μM) in cells incubated with buffered media adjusted to specific pH values ranging from 6 to 8 was studied. The exact pH of each medium was measured before and after incubation and was found not to vary by more than 0.1 pH unit during the experiment. Efflux was thereafter studied by transferring cells to drug-free media of the same pH. Figure 4 (left panel) shows that an acidic medium (pH 6) caused an increase in the absolute amount of drug accumulated in comparison with cells incubated at neutral pH (pH 7). Conversely, incubation in an alkaline medium (pH 8) caused a lower accumulation of ciprofloxacin. Figure 4 (right panel) shows that the rate of ciprofloxacin efflux was inversely proportional to the acidity of the medium (but the decay remained of first order [see the legend to Fig. 4 for kinetic data]). Interestingly enough, cells transferred from an acidic to a neutral medium during efflux (for instance, at 8 min) reacted almost immediately to this change of condition. They indeed showed a rapid loss of ciprofloxacin, after which efflux proceeded at rate similar to what was observed in cells incubated in a neutral medium throughout (data not shown). Monensin, a natural carboxylic ionophore with antifungal properties, known to dissipate transmembrane gradients in higher eucaryotes (32), was then used here to further assess the importance of pH transmembrane gradients and intracellular pH on ciprofloxacin accumulation. Monensin did not affect ciprofloxacin accumulation (tested at pH 6, 7, and 8). As a control of an effective collapse of the transmembrane pH gradients with monensin, we checked that it entirely suppressed the accumulation of azithromycin, which accumulates in J774 macrophages by proton trapping as previously reported (52).
FIG. 4.

Kinetics of accumulation (left) and efflux (right) of ciprofloxacin (extracellular concentration, 17 mg/liter [50 μM]) in J774 macrophages maintained at three different pHs using bicarbonate-free medium buffered with 5 mM phosphate (the pH value was checked before and after the experiment). Regression parameters for influx (one-phase exponential association): pH 6, r2 = 0.995, k = 0.0648 ± 0.0043 min−1, ymax = 12.38 ± 0.27; pH 7, r2 = 0.993, k = 0.149 ± 0.0122 min−1, ymax = 8.06 ± 0.16; pH 8, r2 = 0.981, k = 0.1452 ± 0.0201 min−1 ymax = 5.63 ± 0.22. Regression parameters for efflux (one phase exponential decay): pH 6, r2 = 0.987, k = 0.0748 ± 0.0037 min−1; pH 7, r2 = 0.977, k = 0.1826 ± 0.0015 min−1); pH 8, r2 = 0.998, k = 0.3128 ± 0.0074 min−1.
Influence of probenecid and other efflux pump inhibitors.
Figure 5 (left panel) shows that probenecid (10 mM) added to cells simultaneously with ciprofloxacin (17 mg/liter; 50 μM) immediately caused a marked increase in the absolute amount of drug accumulated by cells in comparison to controls. Again, this effect was both qualitatively and quantitatively similar to what was observed with cells exposed to a high concentration of drug or with ATP-depleted cells. Probenecid-treated cells loaded with ciprofloxacin were then examined for ciprofloxacin efflux in the continuing presence of probenecid (10 mM), in comparison with cells in which ciprofloxacin accumulation and efflux were taking place in the absence of probenecid (controls). As shown in Fig. 5 right panel, probenecid also markedly reduced the rate of efflux of ciprofloxacin from probenecid-treated cells compared to controls. In parallel, we examined the efflux of ciprofloxacin from cells which were incubated with ciprofloxacin (17 mg/liter; 50 μM) in the absence of probenecid but which were then transferred to a ciprofloxacin-free medium containing 10 mM probenecid. Efflux was delayed almost exactly as in cells incubated in the presence of probenecid during both ciprofloxacin uptake and efflux (data not shown). This indicates that the influence of probenecid was immediate, directed toward efflux, and unrelated to the amount of ciprofloxacin accumulated by cells. In the next series of experiments, we examined the influence exerted by gemfibrozil, another nonspecific inhibitor of anionic transporters, and by MK571, a preferential MRP inhibitor. This was studied in comparison with two known P-glycoprotein (P-gp) modulators, namely, verapamil and cyclosporin A (both of which, however, are not specific for the P-gp) and with GF 120918 (a preferential inhibitor of P-gp). Table 1 shows the results obtained at a fixed concentration of these drugs. Azithromycin, a substrate of P-gp in J774 macrophages (46), was used for comparison purposes. It clearly appears that probenecid gemfibrozil, and MK571 enhanced ciprofloxacin accumulation to essentially similar extents (ca. 2.5- to 4-fold) whereas verapamil and GF 120918 had only minimal effects (see below for concentration-dependent experiments with these inhibitors). Cyclosporin A showed an intermediate behavior. In contrast, neither probenecid nor gemfibrozil affected azithromycin accumulation whereas verapamil, cyclosporin A, and GF 120918 markedly increased it.
FIG. 5.

Kinetics of accumulation (left) and efflux (right) of ciprofloxacin (extracellular concentration, 17 mg/liter [50 μM]) in J774 macrophages incubated in the presence or absence of 10 mM probenecid (the drugs were added simultaneously for the accumulation experiment). Regression parameters for influx (one-phase exponential association): control cells, r2 = 0.922; k = 0.1539 ± 0.0401 min−1; ymax = 6.94 ± 0.43; probenecid (10 mM), r2 = 0.988; k = 0.1103 ± 0.0117 min−1; ymax = 19.17 ± 0.55. Regression parameters for efflux (one-phase exponential decay): control cells, r2 = 0.998; k = 0.5414 ± 0.0224 min−1; probenecid (10 mM). r2 = 0.988; k = 0.0729 ± 0.0041 min−1 (k = 0.0631 ± 0.0072 for cells incubated in 10 mM probenecid for efflux only [data not shown]).
TABLE 1.
Influence of efflux pump modulators on the accumulation of ciprofloxacin and azithromycin by J774 macrophagesa
| Modulator | Concn | Increase in accumulation (% of controls)
|
|
|---|---|---|---|
| Ciprofloxacinb | Azithromycinc | ||
| Probenecid | 2.5 mM | 288 ± 4 | 101 ± 12 |
| Gemfibrozil | 200 μM | 308 ± 3 | 101 ± 12 |
| MK571 | 200 μM | 392 ± 5 | NDd |
| Verapamil | 100 μM | 136 ± 4 | 438 ± 45 |
| Cyclosporin A | 50 μM | 236 ± 5 | 353 ± 77e |
| GF 120918 | 1 μM | 116 ± 2 | 372 ± 61 |
Statistical analysis: for ciprofloxacin, all differences are significant; for azithromcyin, differences are significant for verapamil, cyclosporin A, and GF 120918 only (paired t test compared to controls).
17 mg/liter (50 μM); 2-h incubation.
5 mg/liter (6.8 μM); 3-h incubation (time to equilibrium).
ND, not determined.
20 μM.
Short-term kinetic studies.
To refine the analysis of the influence of ATP depletion and of the inhibitors on ciprofloxacin influx, we repeated the accumulation experiments, focusing on the first 5 min after addition of ciprofloxacin. As described above, the influence of ATP depletion was assessed with cells that had been preexposed to 2-d-deoxyglucose and NaN3 for 20 min to obtain a stable state of depletion before adding ciprofloxain. Conversely, the other inhibitors were added at the same time as ciprofloxacin to examine whether their effect would be immediate. As shown in Fig. 6, control cells and cells incubated with cyclosporin A showed curvilinear kinetics of accumulation right from the first minutes of the uptake (with slight but significantly faster accumulation for cyclosporin A-treated cells). In contrast, ATP-depleted cells as well as cells exposed to probenecid and cells exposed to probenecid plus cyclosporin A showed a faster accumulation, which was similar for all three situations. Moreover, after 1 min of contact, the kinetics remained almost perfectly linear for at least the subsequent 4 min, with no meaningful differences between the conditions tested.
Summary of the efflux kinetic data from wild-type cells.
Table 2 shows in a summary fashion the half-lives of ciprofloxacin that were recorded under the main conditions investigated in this study, which led to an increase in the cellular accumulation of the drug in wild-type cells. It clearly appears that all these conditions were associated with significant increases in half-lives, which systematically reached values three- to sixfold longer than under control conditions.
TABLE 2.
Half-lives of ciprofloxacin in J774 macrophages as measured during efflux studies under conditions that caused a 2.5- to 4-fold increase in drug accumulation at equilibriuma
| Condition | Half-life (min)b |
|---|---|
| Ciprofloxacin (17 mg/liter; 50 μM)c | 1.68 ± 0.30a |
| Ciprofloxacin (68 mg/liter; 200 μM)c | 4.08 ± 0.74b |
| Exposure to pH 6d | 9.26 ± 0.53c |
| ATP depletiond | 10.72 ± 0.24c |
| Addition of probenecid (10 mM)d | 9.51 ± 0.53c |
| Addition of MK571 (100 μM)d | 7.70 ± 1.19c |
Groups with the same letter are not different from one another but are significantly different from those with a different letter (paired t test analysis; P < 0.05)
Drug extracellular concentration during uptake only.
Ciprofloxacin extracellular concentration during uptake was 50 μM; indicated treatment applied during uptake and efflux.
Studies with probenecid-resistant cells.
Compared to wild cells, cells made resistant to the continuous presence of 3 mM probenecid were slightly less swollen (volume-to-protein ratio, 2:68 ± 0.27 [n = 6] versus 3.08 ± 0.16 [n = 21] μl/mg). They appeared larger and displayed conspicuous elongations extending far from the cell body, sometimes giving a pseudodendritic aspect to the cell (this aspect could be clearly evidenced with nonconfluent cells only). Under the electron microscope, probenecid-resistant cells appeared essentially normal except for an apparently larger content of mitochondria. The growth rate of probenecid-resistant cells was 30% lower than that of wild-type cells (based on formazan reduction; cells were, however, always compared at a same degree of confluency, i.e., after 2 to 3 days of culture for wild-type cells and 3 to 4 days for probenecid-resistant cells). Protein synthesis (measured by [3H]leucine incorporation 3 days after subculture for probenecid-resistant cells and 2 days after subculture for wild-type (cells) was slightly more intense in probenecid-resistant cells (38.46 ± 3.57 versus 28.51 ± 1.50 dpm of TCA-insoluble material per mg of protein [n = 3] after incubation of cells with [3H]leucine (incorporation: 75.2% ± 9.3% of available material in probenecid-resistant cells versus 62.1% ± 5.3% in wild-type cells). Other key parameters, such as phospholipid- or cholesterol-to-protein ratios and ATP content, were similar. As shown in Fig. 7, probenecid-resistant cells overexpressed the MRP1 transporter, as evidenced by Western blotting, with this protein being clearly displayed at the cell surface (as shown by confocal microscopy). Figure 8 (left panel) shows that the accumulation of ciprofloxacin (17 mg/liter; 50 μM) in probenecid-resistant cells was only marginally increased in comparison with that in wild-type cells. When probenecid-resistant cells were exposed to high concentrations of probenecid (up to 10 mM), gemfibrozil (up to 200 μM), or MK571 (up to 150 μM), the accumulation of ciprofloxacin (in 2 h) was increased to a maximum of 1.5- to 2-fold versus 3- to 4-fold for wild-type cells (data not shown). Wild-type and probenecid-resistant cells released ciprofloxacin at essentially a similar, high rate in the absence of probenecid. When efflux was monitored in the presence of a fixed concentration of probenecid (3 mM, i.e., the concentration at which the probenecid-resistant cells are maintained), wild-type cells showed a markedly reduced rate of efflux (as already illustrated for a higher concentration of probenecid in Fig. 5). In contrast, probenecid (3 mM) exerted much less effect on ciprofloxacin efflux in probenecid-resistant cells. Similar results were obtained when ciprofloxacin efflux from wild-type and probenecid-resistant cells was examined in the presence of gemfibrozil or MK571 (data not shown). The influence of verapamil, cyclosporin A, and GF 120918 was also examined in probenecid-resistant cells and found not to be significantly different from what was seen in wild-type cells (data not shown).
FIG. 7.

(Top) Western blots of lysates from probenecid-resistant and wild-type (control) J774 macrophages (15 μg of protein each), as revealed with an anti-human MRP1 antibody. The controls included Swiss 3T3 cells (which express only poorly constitutive MRP1) and Swiss 3T3 cells transfected with a multicopy plasmid containing the human MRP1 gene (pVD6-3 cells). Equal loading of the gels was checked by revealing actin (no difference detected). (Bottom) Confocal microscopy of probenecid-resistant and wild-type (control) J774 macrophages. Cells were immunostained with the same anti-human MRP1 antibody as for the Western blot analysis.
FIG. 8.

(Left) Kinetics of accumulation of ciprofloxacin (extracellular concentration, 17 mg/liter [50 μM]) in wild-type (control) J774 macrophages and in probenecid-resistant J774 macrophages. Regression parameters for influx (one-phase exponential association): wild cells, r2 = 0.901, k = 0.11162 ± 0.0335 min−1; probenecid-resistant cells, r2 = 0.934; k = 0.1414 ± 0.0340 min−1. (Right) Kinetics of efflux of ciprofloxacin from wild-type (control) and probenecid-resistant cells incubated for 2 h in the presence of ciprofloxacin (50 μM) and then transferred to a ciprofloxacin-free medium with or without probenecid (3 mM). Regression parameters for efflux (one-phase exponential decay): wild-type cells in the absence of probenecid, r2 = 0.992, k = 0.2926 ± 0.0145 min−1; wild-type cells in the presence of probenecid, r2 = 0.984, k = 0.0902 ± 0.0055 min−1; probenecid-resistant cells in the absence of probenecid, r2 = 0.990; k = 0.3178 ± 0.0183 min−1; probenecid resistant-cells in the presence of probenecid, r2 = 0.993; k = 0.1577 ± 0.0061 min−1.
DISCUSSION
Fluoroquinolones are potential substrates of a variety of drug transporters in eucaryotic cells (53). Transcellular transport of fluoroquinolones has been observed in kidney cells, intestinal cell lines, and human airway epithelial cells in vitro and in liver and leukaemia cells in vivo (54). Evidence for a role of fluoroquinolone transporters in macrophages, in relation to drug accumulation and efflux, has, however, remained indirect so far and is based primarily on the observation that organic anions, such as probenecid and gemfibrozil, significantly increase fluroquinolone accumulation in J774 macrophages (5, 40, 41). The present data confirm and significantly extend these findings in two respects. First, they provide direct evidence that probenecid and gemfibrozil impair ciprofloxacin efflux with no detectable change in its kinetics of influx. Second, they show that a similar effect on ciprofloxacin accumulation and efflux can be obtained with ATP depletion and with the selective MRP inhibitor MK571. We also show that cells exposed to a high concentration of ciprofloxacin accumulate the drug to a three- to fourfold-greater extent than those exposed to therapeutically meaningful concentrations. Glutathione deprivation and acidic pH also increase ciprofloxacin accumulation, and this effect is mediated by a corresponding change in efflux in the case of acidic pH. Our results suggest that J774 macrophages have an MRP transporter that is (i) sensitive to organic anions, (ii) dependent on energy and glutathione, (iii) inhibited by H+ and by ciprofloxacin itself. The fact that a high ciprofloxacin concentration during uptake causes a change in efflux in the early phase of the washout experiments only is probably because efflux is measured in the absence of drug in the extracellular milieu. As the intracellular drug concentration decreases, it probably falls rapidly below the level at which the transporter can be saturated.
Fluoroquinolones are zwitterionic and may therefore be recognized by both anion and cation transporters. Based on the Saier classification of transport in eucaryotic cells (43; M. H.Saier, Jr., http://www.biology.ucsd.edu/∼MSAIER/transport), anion transporters comprise (i) the OAT for “organic anion transporters”), which are members of the major facilitator superfamily (MFS); (ii) OATP and OATK, which are members of the OAT superfamily; and (iii) MRP, which are members of the ABC superfamily. OAT, OATP, and OATK are energized by ion gradients (most often H+) and act as symports, uniports, or antiports. MRP are energized by ATP hydrolysis and require glutathione as a cofactor. Cation transporters include (i) OCT (for “organic cation transporters”), which are, like OAT, members of the MFS; and; (ii) MDR (Multidrug resistance), also known as P-gp, which, like MRP, are members of the ABC superfamily (and therefore rely on ATP for activity) but do not require glutathione for optimal activity (10). If we analyze our results globally based on this classification, we may find the strong suggestion that we are dealing with an MRP transporter. Besides the characteristics mentioned above, we also observe an impairment of ciprofloxacin efflux by the preferential MRP inhibitor MK571 (16) and no marked effect exerted by cationic compounds such as verapamil and the preferential P-gp inhibitor GF 120918 (2, 42, 48). The intermediate effect observed with cyclosporin A may result from the fact that this compound not only acts on P-gp but also inhibits the MRP (27, 51). Impairment by acidic pH has been reported for MRP1 and may be related to the fact that the ATPase activity of MRP is itself adversely affected by acid pH (31). Since monensin, a drug which collapses transmembrane pH gradients (32), does not markedly influence ciprofloxacin accumulation, it is also reasonable to accept that pH gradients per se and intracellular pH are unimportant in this context, which globally excludes the transporters energized by H+ gradients.
The complete human ABC transporter subfamily (ABCC) has 12 identified members (ABCC1 to ABCC12), of which 9 belong to the multidrug resistance-like subgroup (MRP) (3, 39, 49). Based on recent reviews of the main properties of MRP transporters (3; http://nutrigene.4t.com/humanabc.htm), we may probably exclude MRP2, MRP3, and MRP4. MRP2 shows the same substrate specificity as MRP1 but is mostly localized in bile canaliculi in vivo and has been found in vitro in polarized cells only. MRP3 is restricted to intestine and kidney cells and is a poor transporter of glutathione conjugates (20). MRP4 is localized in specific tissues and is probably transporting phosphate conjugates only. The intervention of MRP5 can be excluded since it is not inhibited by MK571 (24) and transports substrates independently from glutathione (33). MRP6 is difficult to immediately exclude since it acts as a organic anion transporter, is inhibited by probenecid, and can be coamplified with MRP1 in cells that overexpress the latter transporter because of their proximity on the chromosome (23,25). However, MRP6 is expressed mainly in polarized cells (30, 44) and shows substrate specificities that are largely different from those of the other MRP. MRP7 has the lowest degree of relatedness to the other MRP transporters, and its RNA transcript is expressed at only very low levels in most tissues. Its capacity to transport anionic compounds is still unknown (22). MRP8, expressed in various adult human tissues including the liver, lungs, and kidneys, as well as in several fetal tissues, is a transporter of amphipathic anions. It is able to efflux cyclic AMP (cAMP) and cGMP and to function as a resistance factor for commonly employed purine and pyrimidine nucleotide analogues. MRP9 is an unusual truncated member of the ABC transporter superfamily that is highly expressed in breast cancer and in testes but is not expressed at detectable levels in essential normal tissues (1). Limiting the discussion to the evidence presented with wild-type J774 macrophages would make MRP1 a prime candidate for ciprofloxacin efflux. MRP1 indeed functions as a multispecific organic anion transporter, which transports drugs and other hydrophobic compounds in the presence of glutathione (http://nutrigene.4t.com/humanabc.htm). Yet, when this hypothesis was tested using the probenecid-resistant cells (which overexpress MRP1, as evidenced by both Western blot analysis and confocal microscopy), no marked change in the accumulation and efflux of ciprofloxacin was noted. The overexpressed MRP1 was nevertheless functional since probenecid, gemfibrozil, and MK571 are less effective inhibitors of ciprofloxacin efflux in these cells than in wild-type cells, which is as expected since anions are actively extruded in probenecid-resistant cells (4). The data must therefore be interpreted as demonstrating that MRP1 is not the actual ciprofloxacin transporter and indicate that additional research is needed in this area. Conversely, the data presented here unambiguously show that P-gp is not involved in ciprofloxacin efflux, even though J774 macrophages express a functional P-gp that acts on azithromycin (46). A similar conclusion was reached concerning the transport of ciprofloxacin in MDR1 (P-gp)-transfected MDCK-II epithelial cells (29). Interestingly enough, the closely related fluoroquinolone grepafloxacin was found to be a substrate of both P-gp and MRP2 in these cells and to compete for the ciprofloxacin-sensitive pathway. This suggests that apparently minor structural differences in fluoroquinolones may play a key role in transporter recognition (as they do with respect to antibacterial spectrum). It must, however, be stressed that the situation in polarized cells may be considerably more complex than in macrophages. It does not simply involve influx and efflux mechanism but also involves a succession of oriented transports in which several proteins may be implicated.
Although the nature of the ciprofloxacin transporter in J774 macrophages remains undefined, the data presented here may already have important biological and therapeutic significance. We show that the ciprofloxacin transporter causes a significant reduction of the intracellular concentration of this fluoroquinolone at therapeutically meaningful concentrations. If this takes place with macrophages in vivo, we may anticipate that it will reduce the accumulation of the antibiotic and thereby decrease its intracellular activity. The activity of fluoroquinolones is concentration dependent, including the activity toward intracellular organisms (7, 41), and suboptimal concentrations are associated with insufficient eradication of microorganisms and potentially rapid emergence of resistance (21). The design of specific inhibitors may therefore be of large interest in this context. It must also be emphasized that MRP transporters have a broad specificity and may be involved in the efflux of many anionic drugs, probably including other antibiotics like the β-lactams (26) and several anticancer drugs (28). It may therefore be interesting to monitor their overexpression following prolonged exposure and/or overuse of fluoroquinolones during antibacterial and anticancer therapy in critical situations.
Acknowledgments
We thank the following colleagues from the Université Catholique de Louvain: E. Balzi (present address: DG Research, European Commission, Brussels, Belgium) for critical discussions and guidance in the early steps of this work, V. D'Hondt for the kind gift of the pVD-6-3 cells, P. Buc Calderon for assistance in Western blot studies, and C. Seral for allowing us to use some of her unpublished data on azithromycin transport in macrophages. We also thank M. L. Den Boer (Department of Pediatric Hematology/Oncology, Academic Hospital, Vrije Universiteit, Amsterdam, The Netherlands) for advice on fixation procedures for confocal microscopy studies. F. Renoird, N. Aguilera, and M. C. Cambier provided dedicated technical assistance throughout this work.
J.-M.M. was the recipient of a fellowship of the Belgian Bourse Belge de la Vocation/Belgische Stichting Roeping and was thereafter supported by the Fonds Spécial de Recherches of the Université Catholique de Louvain. F.V.B. is Chercheur Qualifié of the Belgian Fonds National de la Recherche Scientifique. This work was supported by the Belgian Fonds de la Recherche Scientifique Médicale (grants 3.4.549.00 F, 3.4542.02 F, and 1.5.207.01 F) and by a grant-in-aid from Bayer AG, Leverkusen, Germany.
REFERENCES
- 1.Bera, T. K., C. Iavarone, V. Kumar, S. Lee, B. Lee, and I. Pastan. 2002. MRP9, an unusual truncated member of the ABC transporter superfamily, is highly expressed in breast cancer. Proc. Natl. Acad. Sci. USA 99:6997-7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boer, R., V. Gekeler, W. R. Ulrich, P. Zimmermann, W. Ise, A. Schodl, and S. Haas. 1996. Modulation of P-glycoprotein mediated drug accumulation in multidrug resistant CCRF VCR-1000 cells by chemosensitisers. Eur. J. Cancer 32A:857-861. [DOI] [PubMed] [Google Scholar]
- 3.Borst, P., R. Evers, M. Kool, and J. Wijnholds. 2000. A family of drug transporters: the multidrug resistance-associated proteins. J. Natl. Cancer Inst. 92:1295-1302. [DOI] [PubMed] [Google Scholar]
- 4.Cao, C., T. H. Steinberg, H. C. Neu, D. Cohen, S. B. Horwitz, S. Hickman, and S. C. Silverstein. 1993. Probenecid-resistant J774 cell expression of enhanced organic anion transport by a mechanism distinct from multidrug resistance. Infect. Agents Dis. 2:193-200. [PubMed] [Google Scholar]
- 5.Cao, C. X., S. C. Silverstein, H. C. Neu, and T. H. Steinberg. 1992. J774 macrophages secrete antibiotics via organic anion transporters. J. Infect. Dis. 165:322-328. [DOI] [PubMed] [Google Scholar]
- 6.Carlier, M. B., B. Scorneaux, A. Zenebergh, J. F. Desnottes, and P. M. Tulkens. 1990. Cellular, uptake, localization and activity of fluoroquinolones in uninfected and infected macrophages. J. Antimicrob. Chemother. 26(Suppl. B):27-39. [DOI] [PubMed] [Google Scholar]
- 7.Carryn, S., F. Van Bambeke, M. P. Mingeot-Leclercq, and P. M. Tulkens. 2002. Comparative intracellular (THP-1 macrophage) and extracellular activities of beta-lactams, azithromycin, gentamicin, and fluoroquinolones against Listeria monocytogenes at clinically relevant concentrations. Antimicrob. Agents Chemother. 46:2095-2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chapman, J. S., and N. H. Georgopapadakou. 1989. Fluorometric assay for fleroxacin uptake by bacterial cells. Antimicrob. Agents Chemother. 33:27-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cohn, V. H., and J. Lyle. 1966. A fluorometric assay for glutathione. Anal. Biochem. 14:434-440. [DOI] [PubMed] [Google Scholar]
- 10.Davey, M. W., R. M. Hargrave, and R. A. Davey. 1996. Comparison of drug accumulation in P-glycoprotein-expressing and MRP-expressing human leukaemia cells. Leuk. Res. 20:657-664. [DOI] [PubMed] [Google Scholar]
- 11.Den Boer, M. L., C. M. Zwaan, R. Pieters, K. M. Kazemier, M. M. Rottier, M. J. Flens, R. J. Scheper, and A. J. Veerman. 1997. Optimal immunocytochemical and flow cytometric detection of P-gp, MRP and LRP in childhood acute lymphoblastic leukemia. Leukemia 11:1078-1085. [DOI] [PubMed] [Google Scholar]
- 12.D'Hondt, V., M. Caruso, and A. Bank. 1997. Retrovirus-mediated gene transfer of the multidrug resistance-associated protein (MRP) cDNA protects cells from chemotherapeutic agents. Hum. Gene Ther. 8:1745-1751. [DOI] [PubMed] [Google Scholar]
- 13.Easmon, C. S., and J. P. Crane. 1985. Uptake of ciprofloxacin by macrophages. J. Clin. Pathol. 38:442-444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Easmon, C. S., J. P. Crane, and A. Blowers. 1986. Effect of ciprofloxacin on intracellular organisms: in-vitro and in- vivo studies. J. Antimicrob. Chemother. 18(Suppl. D):43-48. [DOI] [PubMed] [Google Scholar]
- 15.Flens, M. J., M. A. Izquierdo, G. L. Scheffer, J. M. Fritz, C. J. Meijer, R. J. Scheper, and G. J. Zaman. 1994. Immunochemical detection of the multidrug resistance-associated protein MRP in human multidrug-resistant tumor cells by monoclonal antibodies. Cancer Res. 54:4557-4563. [PubMed] [Google Scholar]
- 16.Gekeler, V., W. Ise, K. H. Sanders, W. R. Ulrich, and J. Beck. 1995. The leukotriene LTD4 receptor antagonist MK571 specifically modulates MRP associated multidrug resistance. Biochem. Biophys. Res. Commun. 208:345-352. [DOI] [PubMed] [Google Scholar]
- 17.Gollapudi, S., C. H. Kim, B. N. Tran, S. Sangha, and S. Gupta. 1997. Probenecid reverses multidrug resistance in multidrug resistance-associated protein-overexpressing HL60/AR and H69/AR cells but not in P-glycoprotein-overexpressing HL60/Tax and P388/ADR cells. Cancer Chemother. Pharmacol. 40:150-158. [DOI] [PubMed] [Google Scholar]
- 18.Gottesman, M. M., C. Cardarelli, S. Goldenberg, T. Licht, and I. Pastan. 1998. Selection and maintenance of multidrug-resistant cells. Methods Enzymol. 292:248-258. [DOI] [PubMed] [Google Scholar]
- 19.Hartwick, R. A. and P. R. Brown. 1975. The performance of microparticle chemically-bonded anion-exchange resins in the analysis of nucleotides. J. Chromatogr. 112:650-662. [DOI] [PubMed] [Google Scholar]
- 20.Hirohashi, T., H. Suzuki, and Y. Sugiyama. 1999. Characterization of the transport properties of cloned rat multidrug resistance-associated protein 3 (MRP3). J. Biol. Chem. 274:15181-15185. [DOI] [PubMed] [Google Scholar]
- 21.Hooper, D. C. 2000. Fluoroquinolones, p. 404-423. In G. L. Mandell, J. E. Bennett, and R. Dolin (ed.), Mandell, Douglas, and Bennett's principles and practice of infectious diseases. Churchill Livingstone, Philadelphia, Pa.
- 22.Hopper, E., M. G. Belinsky, H. Zeng, A. Tosolini, J. R. Testa, and G. D. Kruh. 2001. Analysis of the structure and expression pattern of MRP7 (ABCC10), a new member of the MRP subfamily. Cancer Lett. 162:181-191. [DOI] [PubMed] [Google Scholar]
- 23.Ilias, A., Z. Urban, T. L. Seidl, O. Le Saux, E. Sinko, C. D. Boyd, B. Sarkadi, and A. Varadi. 2002. Loss of ATP-dependent transport activity in pseudoxanthoma elasticum-associated mutants of human ABCC6 (MRP6). J. Biol. Chem. 277:16860-16867. [DOI] [PubMed] [Google Scholar]
- 24.Jedlitschky, G., B. Burchell, and D. Keppler. 2000. The multidrug resistance protein 5 functions as an ATP-dependent export pump for cyclic nucleotides. J. Biol. Chem. 275:30069-30074. [DOI] [PubMed] [Google Scholar]
- 25.Kool, M., M. van der Linden, M. de Haas, F. Baas, and P. Borst. 1999. Expression of human MRP6, a homologue of the multidrug resistance protein gene MRP1, in tissues and cancer cells. Cancer Res. 59:175-182. [PubMed] [Google Scholar]
- 26.Kusuhara, H., H. Suzuki, and Y. Sugiyama. 1998. The role of P-glycoprotein and canalicular multispecific organic anion transporter in the hepatobiliary excretion of drugs. J. Pharm. Sci. 87:1025-1040. [DOI] [PubMed] [Google Scholar]
- 27.Leier, I., G. Jedlitschky, U. Buchholz, S. P. Cole, R. G. Deeley, and D. Keppler. 1994. The MRP gene encodes an ATP-dependent export pump for leukotriene C4 and structurally related conjugates. J. Biol. Chem. 269:27807-27810. [PubMed] [Google Scholar]
- 28.Loe, D. W., R. G. Deeley, and S. P. Cole. 1996. Biology of the multidrug resistance-associated protein, MRP. Eur. J. Cancer 32A:945-957. [DOI] [PubMed] [Google Scholar]
- 29.Lowes, S., and N. L. Simmons. 2002. Multiple pathways for fluoroquinolone secretion by human intestinal epithelial (Caco-2) cells. Br. J. Pharmacol. 135:1263-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Madon, J., B. Hagenbuch, L. Landmann, P. J. Meier, and B. Stieger. 2000. Transport function and hepatocellular localization of mrp6 in rat liver. Mol. Pharmacol. 57:634-641. [DOI] [PubMed] [Google Scholar]
- 31.Mao, Q., E. M. Leslie, R. G. Deeley, and S. P. Cole. 1999. ATPase activity of purified and reconstituted multidrug resistance protein MRP1 from drug-selected H69AR cells. Biochim. Biophys. Acta 1461:69-82. [DOI] [PubMed] [Google Scholar]
- 32.Marnell, M. H., M. Stookey, and R. K. Draper. 1982. Monensin blocks the transport of diphtheria toxin to the cell cytoplasm. J. Cell Biol. 93:57-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McAleer, M. A., M. A. Breen, N. L. White, and N. Matthews. 1999. pABC11 (also known as MOAT-C and MRP5), a member of the ABC family of proteins, has anion transporter activity but does not confer multidrug resistance when overexpressed in human embryonic kidney 293 cells. J. Biol. Chem. 274:23541-23548. [DOI] [PubMed] [Google Scholar]
- 34.Michelet, C., J. L. Avril, C. Arvieux, C. Jacquelinet, N. Vu, and F. Cartier. 1997. Comparative activities of new fluoroquinolones, alone or in combination with amoxicillin, trimethoprim-sulfamethoxazole, or rifampin, against intracellular Listeria monocytogenes. Antimicrob. Agents Chemother. 41:60-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Michelet, C., J. L. Avril, F. Cartier, and P. Berche. 1994. Inhibition of intracellular growth of Listeria monocytogenes by antibiotics. Antimicrob. Agents Chemother. 38:438-446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nix, D. E., S. D. Goodwin, C. A. Peloquin, D. L. Rotella, and J. J. Schentag. 1991. Antibiotic tissue penetration and its relevance: models of tissue penetration and their meaning. Antimicrob. Agents Chemother. 35:1947-1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O'Donnell, M. E. 1993. Role of Na-K-Cl cotransport in vascular endothelial cell volume regulation. Am. J. Physiol. 264:C1316-C1326. [DOI] [PubMed] [Google Scholar]
- 38.Renard, C., H. J. Vanderhaeghe, P. J. Claes, A. Zenebergh, and P. M. Tulkens. 1987. Influence of conversion of penicillin G into a basic derivative on its accumulation and subcellular localization in cultured macrophages. Antimicrob. Agents Chemother. 31:410-416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Renes, J., E. G. de Vries, P. L. Jansen, and M. Muller. 2000. The (patho)physiological functions of the MRP family. Drug Resist. Update 3:289-302. [DOI] [PubMed] [Google Scholar]
- 40.Rispal, P., J. Grellet, C. Celerier, D. Breilh, M. Dorian, J. L. Pellegrin, M. C. Saux, and B. Leng. 1996. Comparative uptake of sparfloxacin and ciprofloxacin into human THP 1 monocytic cells. Arzneimittelforschung 46:316-319. [PubMed] [Google Scholar]
- 41.Rudin, D. E., P. X. Gao, C. X. Cao, H. C. Neu, and S. C. Silverstein. 1992. Gemfibrozil enhances the listeriacidal effects of fluoroquinolone antibiotics in J774 macrophages. J. Exp. Med. 176:1439-1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Safa, A. R. 1988. Photoaffinity labeling of the multidrug-resistance-related P-glycoprotein with photoactive analogs of verapamil. Proc. Natl. Acad. Sci. USA 85:7187-7191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Saier, M. H., Jr., and I. T. Paulsen. 2001. Phylogeny of multidrug transporters. Semin. Cell Dev. Biol. 12:205-213. [DOI] [PubMed] [Google Scholar]
- 44.Scheffer, G. L., X. Hu, A. C. Pijnenborg, J. Wijnholds, A. A. Bergen, and R. J. Scheper. 2002. MRP6 (ABCC6) detection in normal human tissues and tumors. Lab. Investig. 82:515-518. [DOI] [PubMed] [Google Scholar]
- 45.Seral, C., S. Carryn, P. M. Tulkens, and F. Van Bambeke. 2003. Influence of P-glycoprotein and MRP efflux pump inhibitors on the intracellular activity of azithromycin and ciprofloxacin in macrophages infected by Listeria monocytogenes or Staphylococcus aureus. J. Antimicrob. Chemother. 51:1167-1173. [DOI] [PubMed] [Google Scholar]
- 46.Seral, C., J. M. Michot, H. Chanteux, M. P. Mingeot-Leclercq, P. M. Tulkens, and F. Van Bambeke. 2003. Influence of P-glycoprotein inhibitors on accumulation of macrolides in J774 murine macrophages. Antimicrob. Agents Chemother. 47:1047-1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Steinberg, T. H., A. S. Newman, J. A. Swanson, and S. C. Silverstein. 1987. Macrophages possess probenecid-inhibitable organic anion transporters that remove fluorescent dyes from the cytoplasmic matrix. J. Cell Biol. 105:2695-2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tamai, I., and A. R. Safa. 1990. Competitive interaction of cyclosporins with the Vinca alkaloid-binding site of P-glycoprotein in multidrug-resistant cells. J. Biol. Chem. 265:16509-16513. [PubMed] [Google Scholar]
- 49.Tammur, J., C. Prades, I. Arnould, A. Rzhetsky, A. Hutchinson, M. Adachi, J. D. Schuetz, K. J. Swoboda, L. J. Ptacek, M. Rosier, M. Dean, and R. Allikmets. 2001. Two new genes from the human ATP-binding cassette transporter superfamily, ABCC11 and ABCC12, tandemly duplicated on chromosome 16q12. Gene 273:89-96. [DOI] [PubMed] [Google Scholar]
- 50.Tulkens, P. M. 1991. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 10:100-106. [DOI] [PubMed] [Google Scholar]
- 51.Twentyman, P. R. 1992. Cyclosporins as drug resistance modifiers. Biochem. Pharmacol. 43:109-117. [DOI] [PubMed] [Google Scholar]
- 52.Tyteca, D., P. Van Der Smissen, M. Mettlen, F. Van Bambeke, P. M. Tulkens, M. P. Mingeot-Leclercq, and P. J. Courtoy. 2002. Azithromycin, a lysosomotropic antibiotic, has distinct effects on fluid-phase and receptor-mediated endocytosis, but does not impair phagocytosis in J774 macrophages. Exp. Cell. Res. 281:86-100. [DOI] [PubMed] [Google Scholar]
- 53.Van Bambeke, F., E. Balzi, and P. M. Tulkens. 2000. Antibiotic efflux pumps. Biochem. Pharmacol. 60:457-470. [DOI] [PubMed] [Google Scholar]
- 54.Van Bambeke, F., J. M. Michot, and P. M. Tulkens. 2003. Antibiotic efflux pumps in eukaryotic cells: occurrence and impact on antibiotic cellular pharmacokinetics, pharmacodynamics and toxicodynamics. J. Antimicrob. Chemother. 51:1067-1077. [DOI] [PubMed] [Google Scholar]
- 55.Vilde, J. L., E. Dournon, and P. Rajagopalan. 1986. Inhibition of Legionella pneumophila multiplication within human macrophages by antimicrobial agents. Antimicrob. Agents Chemother. 30:743-748. [DOI] [PMC free article] [PubMed] [Google Scholar]