

Abstract
Airway remodeling, caused by inflammation and fibrosis, is a major component of chronic obstructive pulmonary disease (COPD) and currently has no effective treatment. Transforming growth factor–β (TGF-β) has been widely implicated in the pathogenesis of airway remodeling in COPD. TGF-β is expressed in a latent form that requires activation. The integrin αvβ8 (encoded by the itgb8 gene) is a receptor for latent TGF-β and is essential for its activation. Expression of integrin αvβ8 is increased in airway fibroblasts in COPD and thus is an attractive therapeutic target for the treatment of airway remodeling in COPD. We demonstrate that an engineered optimized antibody to human αvβ8 (B5) inhibited TGF-β activation in transgenic mice expressing only human and not mouse ITGB8. The B5 engineered antibody blocked fibroinflammatory responses induced by tobacco smoke, cytokines, and allergens by inhibiting TGF-β activation. To clarify the mechanism of action of B5, we used hydrodynamic, mutational, and electron microscopic methods to demonstrate that αvβ8 predominantly adopts a constitutively active, extended-closed headpiece conformation. Epitope mapping and functional characterization of B5 revealed an allosteric mechanism of action due to locking-in of a low-affinity αvβ8 conformation. Collectively, these data demonstrate a new model for integrin function and present a strategy to selectively target the TGF-β pathway to treat fibroinflammatory airway diseases.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is now the third leading cause of death in the United States (1). A major cause of airflow obstruction in COPD is airway narrowing caused by peribronchial chronic inflammation and fibrosis, known as airway remodeling, for which there is no effective treatments (2).
Airway remodeling severity is linked to cumulative cigarette smoke exposure and the frequency of sudden declines in lung function known as exacerbations (3). COPD exacerbations (~50%) are caused by viral infections (4). Such infections can directly cause increased transforming growth factor–β1 (TGF-β1) expression (5) and experimental airway remodeling accompanied by airway hyperresponsiveness (6). Exposing mice to the viral mimetic polyinosinic: polycytidylic acid [poly(I:C)] accelerates airway remodeling in response to cigarette smoke (7). Therefore, cigarette smoke combined with poly(I:C) models the self-amplifying cycle of cigarette smoke–induced injury, viral infection, and exacerbations that culminate in airway wall thickening and obstructive physiology in humans. TGF-β1 and interleukin-1β (IL-1β) drive this cycle and represent potential therapeutic targets in the airway remodeling process in COPD (8, 9). TGF-β is expressed in an inactive (latent) form and must be activated to have biologic activity. One protein that activates TGF-β in vivo is the integrin αvβ8 (encoded by the ITGB8 gene), which shows increased expression in COPD airways (10). Thus, selective inhibition of αvβ8-mediated TGF-β activation is a potential therapeutic strategy (11) that would bypass the toxicities of global TGF-β inhibition (12, 13).
Increased expression of αvβ8 is driven by IL-1β (10, 14). Increased IL-1β in COPD patient samples is linked to cigarette smoke and viruses (15, 16). Adenoviral (Ad) delivery of IL-1β leads to increased αvβ8-dependent TGF-β activation and airway remodeling that is blocked by conditional deletion of itgb8 in fibroblasts or by neutralizing pan–TGF-β antibodies (10). Intratracheal delivery of Ad-IL-1β initiates the αvβ8-dependent influx of lung dendritic cells (DCs), which increases adaptive T cell immunity [that is, CD4 T helper 1 (TH1) and TH17] and airway inflammation and fibrosis (10).
Lungs of intratracheal Ad-IL-1β–treated mice or IL-1β–stimulated mouse or human lung fibroblasts demonstrate αvβ8- and TGF-β–dependent increases in the potent DC chemokine CCL20, suggesting a proximal role in TGF-β–dependent airway remodeling (10). CCL20 and DCs are increased in COPD lung biospecimens (17). Thus, CCL20 is a physiologically relevant biomarker of αvβ8-mediated TGF-β activation, leading to DC accumulation (17).
We sought to understand the mechanism by which integrin αvβ8 activates TGF-β in fibroinflammatory airway disease to design a therapeutic strategy for its treatment. TGF-β is maintained in the inactive (latent) state by the noncovalent association with its propeptide, latency-associated peptide (18). The latency-associated peptides of TGF-β1 and TGF-β3 both contain RGD motifs (18), which bind to integrin αvβ8 with high affinity (19, 20).
The sentinel event in integrin function is ligand binding, widely thought to be facilitated by integrin “activation” (21). Mechanisms of integrin activation inferred using biochemical and structural data from the αvβ3, αIIbβ3, and α5β1 integrins support two distinct models of integrin activation: (i) a “switchblade-like” opening from a compact (bent) to extended conformation with an “open” headpiece, and (ii) a subtle headpiece opening occurring in a bent conformation (22–24). The former model addresses steric constraints imposed by the cell membrane because integrin extension increases access of large ligands of the extracellular matrix to the ligand-binding pocket (24). In either model, a closed headpiece conformation is thought to be inactive and of low affinity (22, 25, 26). How these models and assumptions apply to αvβ8 is not immediately obvious because of sequence differences between conformationally important regions of β8 compared with other β subunits (27, 28).
Integrin headpieces contain the ligand-binding pocket, located at the interface of the integrin β-subunit head (referred to as βI) and the α-subunit head domains (21). Interactions between the β-subunit βI domain α1 and α7 helices regulate integrin activation states and are influenced by ligand and metal ion occupancy (21, 25). Integrin βI domains contain three conserved metal binding sites except the βI domain of integrin β8, which only has two because it lacks two critical aspartate residues of the ADMIDAS cation binding site that allosterically couples the ligand-binding pocket to the rest of the integrin (24). As monitored by adhesion or ligand-binding assays of non-β8 integrins, Ca2+ and Mg2+ facilitate integrin low-affinity states, and Mn2+, high-affinity states (22). In the presence of Mn2+, integrins extend and open their headpieces (a process enhanced by RGD peptide) by a “swing-out” of the adjacent hybrid domain (24, 25, 29). A large body of work suggests that Mn2+ alters βI α1-α7 helix interactions, causing headpiece opening (24, 25, 29).
Here, we use hydrodynamic, electron microscopic, and mutational analyses to demonstrate that integrin αvβ8 predominantly adopts a constitutively active conformation with an extended-closed headpiece and thus does not fit current models of integrin activation. We affinity-matured an anti-human β8 monoclonal antibody (37E1) that binds to the α1 helix of the β8 βI domain to generate B5. B5 causes a β8 headpiece conformational change that efficiently inhibits TGF-β activation. The relevance of these findings for a therapeutic strategy is demonstrated using bacterial artificial chromosome (BAC) transgenic (Tg) mice expressing only human and not mouse itgb8. These mice were used to establish and extensively characterize a physiologically relevant airway remodeling system induced by a combination of cigarette smoke and poly(I:C). B5 blocked airway inflammation and fibrosis not only in cigarette smoke–induced airway disease but also in cytokine- and allergen-induced airway disease mouse models, suggesting that αvβ8-mediated TGF-β activation may be a new therapeutic target for treating airway remodeling diseases.
RESULTS
An optimized β8 antibody (B5) blocks TGF-β activation
B5 blocks IL-1β–induced airway remodeling
Intratracheal injection of Ad-IL-1β in mice induces airway disease that recapitulates key immunologic and pathologic features of airway remodeling in human COPD by increasing DCs that drive adaptive TH1 and TH1 immunity (10). We used intratracheal injection of Ad-IL-1β in BAC Tg mice expressing only human and not mouse itgb8 to test the efficacy of B5 (Fig. 1A). The expression of human ITGB8 in these BAC Tg mice is at similar levels and expression patterns as in human tissues (fig. S1), and rescues the developmental lethality of itgb8 knockout mice (fig. S2) (30–32). This shows that human β8 binds to murine latency-associated peptide (fig. S3).
Fig. 1. Optimized B5 antibody blocks TGF-β activation in vivo and intratracheal Ad-IL-1β–induced airway inflammation and fibrosis.

(A) Schematic of the creation of a mouse model of airway inflammation using adenovirally delivered IL-1β (Ad-IL-1β) administered intratracheally. (B) B5 blocks intratracheal Ad-IL-1β–induced pSMAD2/3, demonstrating that neutralization of αvβ8 inhibits TGF-β activation in vivo. Lung homogenates from mice treated with B5, compared with IgG2a isotype or wild-type (WT) non–Ad-IL-1β injected intratracheally, were evaluated by pSMAD2/3 enzyme-linked immunosorbent assay (ELISA). n = 4, *P = 0.03 by analysis of variance (ANOVA) and post-test for linear trend. (C to H) B5 (D and F) compared with isotype control (C and E) blocks inflammation of the airway wall (C, D, and G) and fibrosis (E, F, and H) induced by intratracheally administered Ad-IL-1β. Results expressed as area of inflammation or fibrosis per basement membrane (BM) length. Semiquantitative airway morphometry of standard hematoxylin and eosin (H&E)–stained (C and D) or trichrome-stained (E and F) sections. ***P < 0.0001, by ANOVA and Tukey’s post-test. Scale bar, 200 μm. (I to M) B5 blocks intratracheal Ad-IL-1β–induced inflammation in bronchoalveolar lavage. Total cells in bronchoalveolar lavage (I), macrophages (J), and neutrophils (K), as well as gene transcripts of ITGB8 (L) and col1a2 (M), were increased by intratracheal Ad-IL-1β, and this increase was inhibited by B5. n = 3, Ad-LacZ+isotype– or Ad-LacZ+B5–treated mice; n = 4, Ad-IL-1β+isotype–treated mice; or n = 6, Ad-IL-1β+B5–treated mice. *P < 0.05, ** P < 0.01, ***P < 0.001, by ANOVA and Tukey’s post-test.
B5 demonstrated a significant dose-dependent blockade of phosphorylation of the TGF-β signaling effector pSMAD2/3 from lung homogenates of BAC ITGB8 Tg mice treated by intratracheal injection of Ad-IL-1β, indicating successful blockade of TGF-β activation (P = 0.03, Fig. 1B). Decreased pSMAD2/3 correlated with dose-dependent decreases in CCL20 (fig. S4A), decreases in the total number of cells in bronchoalveolar lavage, and reduced numbers of neutrophils and macrophages (fig. S4, B and D). B5 effectively inhibited Ad-IL-1β–dependent inflammation of the airway wall (Fig. 1, C, D, and G), fibrosis (Fig. 1, E, F, and H), and increases in bronchoalveolar lavage total cells, macrophages, and neutrophils (Fig. 1, I to K). mRNA levels of ITGB8 and col1a2 were increased by intratracheal injection of Ad-IL-1β and were blocked by B5 (Fig. 1, L and M). B5 had no effect on airway histology of control animals (fig. S5). These results demonstrate that B5 had similar effects on airway remodeling in vivo, as previously shown with TGF-β neutralizing antibodies or conditional deletion of itgb8 on fibroblasts (10).
B5 blocks airway disease in a cigarette smoke exacerbation model of COPD
We established and characterized a mouse model of COPD that combined cigarette smoke with poly(I:C) treatment of BAC ITGB8 Tg mice (7) to test the efficacy of B5. Three weeks of whole-body cigarette smoke exposure alone caused minimal pathology, and 2 weeks of intranasal poly(I:C) alone caused vigorous neutrophilic inflammation but no significant airway remodeling (fig. S6). Three weeks of cigarette smoke together with intranasally administered poly(I:C) (Fig. 2A) caused increased airway remodeling; an increase in the number of total cells in bronchoalveolar lavage; an increase in macrophages, neutrophils, and lymphocytes; and increased expression of lung IL-1β, IL-17A, and CCL2 (fig. S6). This system therefore replicated key components of human COPD and COPD-relevant animal models (17). Over time, total inflammatory cells and macrophages increased steadily with a parallel increase in lung IL-1β and IL-17 (fig. S7). CCL2 and CCL20 expression peaked at 12 days after smoke exposure, similar to that observed with the intratracheal injection of Ad-IL-1β (fig. S7).
Fig. 2. B5 blocks airway inflammation and fibrosis induced by cigarette smoke and the viral mimetic poly(I:C).

(A) Schematic showing creation of the cigarette smoke intranasal (IN)–poly(I:C)–induced mouse model of airway remodeling. (B and C) Quantitative airway morphometry showing cigarette smoke–poly(I:C) (CS+PolyI:C)–induced inflammation of the airway wall (B) and wall thickening (C), and the effects of B5 compared to control IgG2a. (D to G) Photomicrographs of mouse lungs treated with cigarette smoke–poly(I:C) and control (D and F), and cigarette smoke–poly(I:C) with B5 (E and G); H&E (D and E) and trichrome (F and G). Scale bar, 75 μm. B5 blocks cigarette smoke–poly(I:C)–induced influx of neutrophils and lymphocytes. (H to K) Total cells from the bronchoalveolar lavage (BAL) (H), macrophages (I), neutrophils (J), and lymphocytes (K). (L to O) B5 blocks the expression of cigarette smoke–poly(I:C)–induced IL-1β (L), CCL2 (M), CCL20 (N), and IL-17 (O). ELISAs were performed on whole-lung lysates for expression of IL-1β, IL-17, and CCL2 or on bronchoalveolar lavage for expression of CCL20. (P) Analysis of pSMAD3 immunostaining. (Q to S) Lung phenotyping. (Q) Mean linear intercept; L(m), estimate of airspace enlargement. (R) Airway resistance with increasing acetylcholine (ach) concentrations (log2). (S) Concentration of acetylcholine as provocative challenge doubling baseline resistance (pc200); room air groups, n = 3; cigarette smoke–poly(I:C) groups, n = 4 to 5. *P < 0.05, **P < 0.01, ***P < 0.001, by ANOVA and Bonferroni’s post-test.
Combined cigarette smoke–poly(I:C) exposure (Fig. 2A) increased inflammation and fibrosis surrounding small airways of BAC ITGB8 Tg mice compared with control, untreated BAC ITGB8 Tg mice (P < 0.01, Fig. 2, B to D and F). These increases were inhibited by treatment (intraperitoneal) with the B5 antibody (P < 0.01; Fig. 2, B, C, E, and G). Inflammation (total bronchoalveolar lavage cell counts, neutrophils, and lymphocytes), inflammatory mediators (lung IL-1β, CCL2, CCL20, and IL-17), TGF-β signaling (pSMAD3), emphysema (airspace enlargement), and airway obstruction (airway resistance to acetylcholine challenge) were all increased by cigarette smoke–poly(I:C). B5 inhibited each of these increases (Fig. 2, H to S). These data demonstrate that B5 blocks airway remodeling, inflammation, inflammatory cytokine and chemokine production, airspace enlargement, and obstructive airway physiology in a model that mimics COPD exacerbation by cigarette smoke.
B5 blocks allergic airway inflammation
B5 was used in an ovalbumin sensitization and challenge model to determine mechanistic commonalities with IL-1β and cigarette smoke–poly(I:C) mouse models of airway disease (fig. S8A). Ovalbumin challenge increased inflammation surrounding small airways of BAC ITGB8 Tg mice (fig. S8, B and D), total bronchoalveolar lavage cell counts, eosinophils, IL-1β, CCL2, and CCL20, all of which were inhibited by B5 (fig. S8, C to I). These results are consistent with the ability of B5 to block a central pathway of αvβ8-dependent airway inflammation, as previously suggested using conditional deletion of itgb8 (10, 33).
The αvβ8 integrin has unique structural and functional features
Secreted integrin αvβ8 is constitutively active
Integrins are thought to transit between active and inactive states regulated by cation occupancy, with Mn2+ being the most strongly activating cation (34). As a first step to understand the interaction of αvβ8 with TGF-β, we asked whether Mn2+ also increases β8 affinity for latency-associated peptide. We used human 293 cells stably expressing the integrin β8 subunit, which binds well to latency-associated peptide in the presence of Ca2+ and Mg2+ (Fig. 3A). Mn2+ did not increase, but rather decreased, cell adhesion (Fig. 3A) and did not increase the already high level of binding of soluble truncated secreted αvβ8 to latency-associated peptide (Fig. 3B and fig. S3). In contrast, binding of soluble truncated αvβ3 to its ligands was markedly increased by Mn2+ (Fig. 3B), suggesting that αvβ8 is constitutively active, in contrast to αvβ3, which is not.
Fig. 3. Integrin αvβ8 is constitutively active in a high-affinity extended-closed conformation.

(A) Adhesion of β8-expressing 293 cells to latency-associated peptide (LAP) or bovine serum albumin (BSA; control), with Ca2+/Mg2+ or Mn2+, and reported as absorbance (A570). n = 3 experiments. (B) Binding of soluble αvβ8–alkaline phosphatase (AP) or αvβ3-AP fusion proteins to latency-associated peptide, or to the αvβ3 ligand fibronectin as a control, with Ca2+/Mg2+ (open bars) or Mn2+ (solid bars) reported as A405. n = 8 experiments. ***P < 0.001, by ANOVA and Tukey’s post-test. n.s., not significant. (C) Schematic of the domain structure of secreted integrins with or without a C-terminal clasp with locations of various domains. Clasped version has a 10–amino acid linker between the acid-base coil. (D) Size exclusion chromatography of clasped and unclasped αvβ8 secreted proteins. Clasped protein or unclasped protein in a solution containing Ca2+/Mg2+, unclasped protein with an RGD peptide in a solution containing Ca2+/Mg2+ or Mn2+. Particles too large to enter the medium are excluded and this volume is denoted as “void volume (V0)” as indicated. n = 3. (E) Negative staining electron microscopy of peak fractions shown in (D). Representative class averages showing extended-closed or bent conformations. Cartoon depicts domain structure. Below the micrographs are shown percentages of each subclass. Scale bar, 10 nm.
Integrin activation corresponds to global conformational rearrangements that can be monitored by size exclusion chromatography because receptor extension leads to axial asymmetry and a larger hydrodynamic radius than a compact (bent) form (21). The conformational heterogeneity of αvβ8 was investigated by comparing size exclusion chromatography profiles of secreted αvβ8 with and without a C-terminal clasp (Fig. 3C). The clasp mimics the spatial constraints of transmembrane domains that stabilize the bent conformation of other integrins (21). The size exclusion chromatography profile of clasped, secreted αvβ8 was identical to that of the nonclasped form of this integrin (Fig. 3D and Table 1). Elution peaks did not change in the presence of RGD peptide with or without Mn2+ (Fig. 3D and Table 1), suggesting that αvβ8, unlike αvβ3, predominantly adopts a single cation- and ligand-independent conformation.
Table 1.
Size exclusion chromatography of αv-integrin constructs.
| Condition | Elution volume (ml)* | Stokes radius (Å) |
|---|---|---|
| No Fab/ligand | ||
| αvβ8 (C-terminal clasp) | 10.51 ± 0.03 | 64.25 |
| αvβ8 (truncated) | 10.55 ± 0.02 | 63.87 |
| Plus ligand (RGD peptide) | ||
| αvβ8 (truncated) Ca2+ | 10.53 ± 0.12 | 63.98 |
| αvβ8 (truncated) Mn2+ | 10.49 ± 0.01 | 64.28 |
| Plus B5 Fab | ||
| αvβ8 (C-terminal clasp) | 10.0 ± 0.02 | 67.70† |
| αvβ8 (truncated) | 10.1 ± 0.03 | 67.04† |
| Plus clone 68 Fab | ||
| αvβ8 (C-terminal clasp) | 10.15 ± 0.02 | 66.78† |
| αvβ8 (truncated) | 10.18 ± 0.03 | 66.53† |
Mean ± SEM (n ≥ 3).
P < 0.01 by unpaired Student’s t test.
Negative staining electron microscopy was used to directly view conformations of αvβ8 in size exclusion chromatography fractions. Computational grouping of well-resolved particles into classes of nearly identical conformers (fig. S9) resulted in highly related classes in 20 of the clasped and 19 of 20 of the unclasped preparations (fig. S9). Both clasped and unclasped versions were almost entirely in an extended-closed state, similar to the αvβ3 integrin in Mn2+ without RGD peptide (21). Unclasped αvβ8 was more heterogeneous than clasped, and both contained a second most common form (6% of clasped, 39% of unclasped) that was extended but likely represented a side view or a conformational intermediate (Fig. 3E). The bent conformation (0% of clasped, 5% of unclasped) was a minor population that did not significantly affect the hydrodynamic profile (Table 1), which would be expected if the bent conformation was a major population (21). In addition, RGD peptide would be expected to induce integrin extension if a bent species was present (21); αvβ8 in the presence of RGD peptide in Ca2+/Mg2+ or Mn2+ remained extended-closed without headpiece opening (Fig. 3E). Together, these data demonstrate that the secreted form of αvβ8 is constitutively active in a thermodynamically favored, stable extended-closed conformation.
αvβ8 expressed on the cell surface is constitutively active
We next asked whether αvβ8 was constitutively and fully active on the cell surface. Other studies have used glycosylation modification of the head-hybrid domain interface to constitutively activate integrins on the cell surface (αIIbβ3, αvβ3, α5β1) (35, 36). The β8 integrin subunit contains a natural glycan at an almost identical location as one of these mutationally introduced glycans in the β1 integrin subunit (Fig. 4A) (35, 36). To further amplify the effects of activating glycans, we mutationally introduced a second glycosylation site at the head-hybrid domain interface (N294). At a homologous residue in the β3 (N303) or an adjacent residue in the β1 (N429 or N333) subunit, an artificial glycan induces extension, headpiece opening, and constitutive activation (Fig. 4B) (35, 36). Homology modeling of αvβ8 predicts that both the natural and artificial glycan “wedge” expressed by αvβ8 would have the same contact points on the adjacent hybrid domain as αvβ3 and would thus similarly favor the open headpiece conformation (Fig. 4B). Such swing-out of the hybrid domain increases the affinity of ligand binding by the αvβ3 (35), αIIbβ3 (35), and α5β1 (36) and thus would be predicted to also open the αvβ8 head domain (Fig. 4B).
Fig. 4. αvβ8 is constitutively active on the cell surface.

(A) Alignment of human β8 and β1 integrin subunits with positions of N-X-T consensus (underlined) and N-linked glycosylation sites in green. Native β8 glycosylation site (N414), adjacent to the mutant N429 glycosylation site in the β1 integrin subunit (49). This is modeled onto a space-filling rendering of homology-modeled (PyMOL V1.1r1) αvβ8 headpiece based on the αvβ3 crystal structure in a closed-conformation [Protein Data Bank (PDB) 3IJE] (25) with GlcNAc2Man7 glycan chain (brown) from human CD2 (1GYA) (49). Green, β-subunit head domain; black, β8-hybrid; blue, αv subunit. (B) Alignment of human β8 and β3 subunits with neo-β8 glycosylation site N294 (green) introduced by the N296T (red) mutation compared with the neo-β3 glycosylation site N303 (green) introduced by the N305T mutation (red) (35). Modeled rendering of the αvβ8 headpiece, as in (A). Glycans in position N414 (brown) and N294 (red) oriented laterally allowing the closed headpiece conformation. (C) Immunoprecipitation with β8 antibody of surface-labeled β8-HT1080 cells transfected with WT β8 (lanes 2 and 4) or N294 mutant (lanes 1 and 3), with or without peptide–N-glycosidase F (PNG) (nonreducing SDS-PAGE). *, a size increase of 3 kD in the neoglycosylated N294 mutant. (D) Histogram overlays of anti-β8–stained, stably transfected HT1080 pools of WT, N294 glycan mutants, or mock versus WT cells stained with secondary antibody only (PBS). (E and F) Cell adhesion (E) or TGF-β activation (F) assays using transfected sorted pools of HT1080 cells expressing equal surface concentrations of WT β8 or N294 glycan β8 compared with mock-transfected cells. n = 3; *P < 0.05, **P < 0.01, ***P < 0.001, by ANOVA and Tukey’s or Bonferroni’s post-test.
Pools of HT1080 cells transfected with the β8 glycan mutant or wild-type β8 subunit were surface-biotinylated and immunoprecipitated to reveal glycosylation status (Fig. 4C) and were sorted to equal levels of surface expression (Fig. 4D). Surface recognition by multiple domain-specific β8 antibodies (figs. S10 and S11) indicated proper folding of the glycan mutant. The glycan mutant β8 subunit migrated on SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gels with a ~3-kD larger molecular weight than that of the wild-type β8 subunit. The deglycosylating amidase, peptide–N-glycosidase F, reduced the size of the glycan mutant approximately to that of the deglycosylated wild-type β8 subunit (Fig. 4C). Cell adhesion assays (Fig. 4E) and TGF-β activation assays (Fig. 4F) revealed no functional differences between the glycan mutant and wild-type β8 subunits. These data demonstrate that forced opening of the hybrid domain does not further increase αvβ8 function, and are consistent with hydrodynamic and electron microscopy data, suggesting that the αvβ8 extended-closed conformation is constitutively (and maximally) active when on the cell surface.
B5 selectively targets αvβ8-mediated TGF-β activation
To develop a β8 antibody suitable for clinical use, we chose to optimize the neutralizing β8 antibody (clone 37E1) that inhibited TGF-β activation in coculture assays at high concentrations (20). We reasoned that if the effects of 37E1 were specific for inhibiting TGF-β activation at high concentrations, then it would be worth optimizing. Human fetal tracheal fibroblasts, a model of myofibroblasts [the cell type implicated in lung fibrosis (19)], were treated with 37E1 or 1D11, a pan–TGF-β isoform neutralizing antibody, to interrogate the spectrum of genes influenced by basal autocrine αvβ8-mediated TGF-β activation (tables S1 and S2). Two hundred fifty-two genes were significantly altered (B statistic >0) by TGF-β antibody treatment compared with control antibody treatment (table S1). The direction of change [increase versus decrease in response to β8 or TGF-β antibody (100 μg/ml)] was the same for 251 of these 252 genes (fig. S12), indicating virtually identical pathways affected by inhibiting αvβ8 or TGF-β. Thus, 37E1 does not produce off-target effects independent of TGF-β (P < 2 × 10−16, r2 = 0.88).
Gene array data encouraged engineering of an optimized version of the 37E1 antibody, B5, which improved the affinity to a range suitable for in vivo applications (figs. S13 and S14). When 37E1 and B5 were expressed as single-chain antibody fragments (scFv) on yeast, the apparent binding affinity improved from 117.51 to 15.42 nM, respectively. A second affinity-matured antibody, clone 68, was developed in parallel on the basis of hybridoma clone 11E8, which cross-competed with B5 for αvβ8 binding and therefore was directed against an overlapping epitope. Clone 68 had a higher affinity for αvβ8 than did B5 [as scFv, dissociation constant (Kd) = 1.42 nM] but did not significantly inhibit αvβ8-mediated TGF-β activation [inhibition as immunoglobulin G1 (IgG1) at 10 μg/ml = 0 ± 14%]. In contrast, B5 as an intact IgG (Kd of 0.54 nM) displayed a 36-fold increase in ability to block the binding of αvβ8-AP to latency-associated peptide relative to 37E1 (fig. S14), which was specific to αvβ8 because it did not inhibit the binding of αvβ6-AP (fig. S15). Optimized B5 markedly increased (~1000-fold) the ability to block TGF-β activation (P < 0.001, Fig. 5A) and IL-1β–induced CCL20 production by human lung fibroblasts, compared with 37E1 (P < 0.0001, Fig. 5B). These data demonstrate that the affinity-matured B5 derivative has significantly improved abilities to inhibit key TGF-β–dependent functional endpoints from relevant human cells.
Fig. 5. Affinity-optimized β8 antibody (B5) inhibits TGF-β–dependent chemokine expression by stimulated human lung fibroblasts.
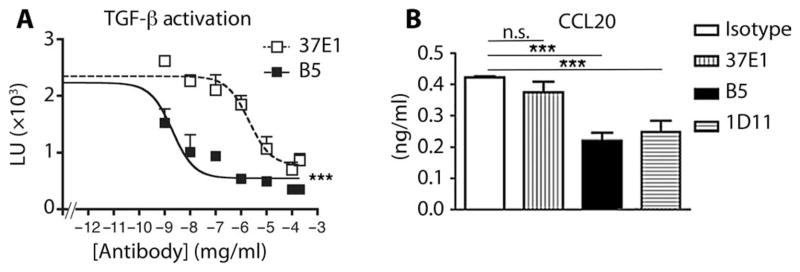
(A) Inhibition of TGF-β activation as measured using B5, an affinity-matured more potent inhibitor of αvβ8-mediated TGF-β activation than 37E1. Cocultures of β8-transfected HT1080 cells with transformed mink lung epithelial TGF-β reporter cells with varying concentrations (μg/ml) of B5 (filled squares) or 37E1 (open squares) reported by relative light units (LU; ×103). n = 5 experiments. (B) B5 is more potent than 37E1 in blocking TGF-β–dependent CCL20 secretion by IL-1β–stimulated human lung fibroblasts. ELISA for measuring CCL20 in culture supernatant from IL-1β–stimulated normal primary human lung fibroblasts treated with isotype control (open bar), 37E1 (vertical stripes), B5 (filled bar), or 1D11 (horizontal stripes) at 10 μg/ml. Non–IL-1β–treated fibroblast controls do not secrete detectable CCL20. n = 5 different patients. ***P < 0.001, by ANOVA and Tukey’s post-test. n.s., not significant.
B5 binds to the β8 head-hybrid interface and alters the head domain conformation
Hybridoma clone 37E1 recognized only human and not the mouse β8 subunit, suggesting that the 37E1 binding epitope depends on species-specific amino acid differences, reflecting its derivation from mice immunized with human αvβ8. Chimeric constructs swapping various regions between mouse and human β8 were expressed on 293 cells and used to identify a critical region (located between amino acids 133 and 138) of human β8 required for binding of B5, 37E1, and clone 68 antibodies (fig. S10). The blocking antibodies B5 and 37E1 and the nonblocking clone 68 recognized various combinations of the human species-specific residues R133, F137, and F138, which were located on the C-terminal α1 helix of the β8 βI domain (fig. S10 and Fig. 6A).
Fig. 6. αvβ8 is in a stable extended conformation with a closed headpiece; B5 is associated with inward bending of the β8 head-hybrid domain angle.

(A) Ribbon diagram (PyMOL V1.1r1) of the extended, closed structure of the β8 subunit generated by homology modeling (Modeller) (50) to αvβ3 (PDB 3IJE) (25). Modeled β8 (green) with the α1 and α7 helices (red) superimposed on αvβ3 (purple). Red spheres, atoms of the B5 epitope on the α1 helix (R133, F137, F138). Modeled RGD tripeptide (blue spheres) based on PDB 3ZDX (51) bound to the ligand-binding pocket in complex with the MIDAS Ca2+ cation (orange sphere). The distance from the edge of the ligand-binding pocket (A115) to R133 of the B5 epitope is 28 Å, indicated by dotted arrows. Head, hybrid, and Psi domains are indicated. The αv subunit and leg domains are not included. (B) Image analysis measuring the hybrid-head domain angles of clasped and unclasped αvβ8 with no Fab compared to size exclusion chromatography–purified αvβ8-B5 or αvβ8–clone 68 Fab complexes. n = 15, 24, 10, 15, 37, and 30 measurements from class averages of clasped alone (filled squares), clasped+B5 Fab (open upward triangles), clasped+clone 68 Fab (filled diamonds), unclasped alone (open downward triangles), unclasped+B5 Fab (closed circles), and unclasped+clone 68 Fab (open squares), respectively. **P < 0.01, ***P < 0.001, by ANOVA and Tukey’s post-test. (Insets) Representative electron microscopy class averages. Average head-hybrid domain angles shown below the micrographs. Scale bar, 10 nm. Cartoons show bound Fab with head (βI) and hybrid domain angles. (C) Rendered αvβ8 space filling model (PyMOL V1.1r1) of the closed headpiece structure of the αvβ8 subunit, as above, with docked prototype Fab (SG/19: PDB 3VI3, translucent gray with black outline) to the B5 epitope (R133, F137, F138) indicated by red spheres, approximating dimensions and orientation of B5 Fab with inward bending of the hybrid domain. Modeled β8 (green) with αv (light blue). RGD tripeptide (orange spheres) bound to ligand-binding pocket and angles of the closed head-hybrid domain (black lines). (D) Space-filling αvβ8 generated homology model, as above, except with a rendering of docked 68 Fab.
The mechanisms of action of integrin antibodies that function as allosteric activators and inhibitors have been successfully determined using size exclusion chromatography and electron microscopy of integrin–Fab fragment complexes (37). To determine the mechanism of action of B5, we asked whether B5 induced conformational changes of αvβ8 that could be detected by size exclusion chromatography and electron microscopy that were not seen with Fab from the non–function-blocking antibody clone 68, which binds to an overlapping epitope. The αvβ8–B5 Fab or αvβ8–clone 68 Fab complex eluted from the size exclusion chromatography column slightly earlier than αvβ8 alone, consistent with the binding of the Fabs, but did not reveal detectable conformational differences between αvβ8 bound by B5 or clone 68 (fig. S16 and Table 1).
Electron microscopy of peak size exclusion chromatography fractions of αvβ8-Fab complexes revealed that both B5 and clone 68 Fabs bound exclusively to the extended conformation at the β8 subunit head-hybrid junction oriented away from the ligand-binding pocket, agreeing well with epitope mapping data (Fig. 6A). Analysis of class averages of clasped and unclasped preparations revealed that nearly 100% of the well-resolved classes were extended with a closed headpiece with or without B5 and clone 68 Fab (Fig. 6B and fig. S17). Two-dimensional image analysis of class averages revealed that head-hybrid domain angles were reduced (~20°) when bound to B5 Fab, but were not significantly changed by clone 68 Fab (Fig. 6, B to D). The B5/clone 68 epitope was far (~28 Å) and oriented away from the ligand-binding pocket, making steric effects of B5 on the RGD ligand-binding site improbable (Fig. 6, A, C, and D). B5 Fab blocked the binding of secreted αvβ8 to latency-associated peptide nearly as well as the intact B5 IgG (at 40 μg/ml: B5 Fab 74 ± 10% versus B5 IgG 87 ± 1% inhibition), demonstrating that antibody avidity or the additional size of the intact IgG was not required for B5 function. Together, these data suggest that B5 is a noncompetitive allosteric inhibitor; subtle differences seen on electron microscopy between the orientations of the hybrid domain between the blocking B5 and nonblocking clone 68 Fabs reflect conformational changes in the head domain and ligand-binding pocket.
Homology modeling of αvβ8 to the closed headpiece conformations of αvβ3 (Fig. 4A) reveals a steric clash with a natural glycan (N414), highly conserved between species, absent from all other integrin β subunits, located on the hybrid domain close to the vertex of the head-hybrid domain angle, where the interdomain distances are predicted to be closest (35). This site faces the α5-β5 loop at the bottom of the βI domain, and when a glycan is computationally docked on this site, several glycan conformations sterically clash with the βI head, and thus predicted to open the head-hybrid angle (Fig. 4B), clearly not seen on any electron microscopy projections (Fig. 3, D and E). Thus, the natural (N414) (Fig. 4A) and the mutant (N294) glycans (Fig. 4B) of β8 are almost certainly laterally displaced in the closed headpiece conformation of αvβ8, with or without inward hybrid bending by ~20°, as seen with B5. Together, these data suggest that the closed conformation of αvβ8 is highly thermodynamically favored.
B5 is a noncompetitive allosteric inhibitor that induces a low-affinity ligand binding state of αvβ8
We sought to determine whether B5 directly competes for latency-associated peptide because allosteric antibody inhibitors directed to the C-terminal α1 helix of other integrins noncompetitively inhibit ligand binding (36, 38). Latency-associated peptide decreased B5 binding to β8-expressing HT1080 cells by 29% (Fig. 7A). To determine whether this decreased binding was due to competitive or noncompetitive inhibition, we used a linear regression model of a solid-phase ligand-binding assay (Fig. 7B). Plots revealed a noncompetitive mode of inhibition by B5 (Fig. 7B), suggesting that B5 binds to a low-affinity conformation of αvβ8 and acts as a noncompetitive allosteric inhibitor.
Fig. 7. B5 is a noncompetitive allosteric inhibitor that induces a low-affinity state.

(A) Latency-associated peptide decreases B5 binding to αvβ8-expressing HT1080 cells. β8-expressing HT1080 cells stained with B5 (2 μg/ml) with increasing concentrations of latency-associated peptide (LAP) reported as mean fluorescence intensity (MFI) (n = 6). (B) Lineweaver-Burk plots of solid-phase binding assays of αvβ8-AP binding to latency-associated peptide with two different concentrations of B5 (squares), or RGD peptide (circles) as a competitive inhibitor control, or no inhibitor (triangles-dotted line). B5 plots show similar x intercepts as uninhibited receptor but different slopes and y intercepts consistent with noncompetitive inhibition. RGD plots intersect above the x axis with the uninhibited receptor consistent with a competitive mode of inhibition. Representative of two experiments with similar results. (C to E) B5 induces low-affinity binding sufficient to mediate cell adhesion, but insufficient to support TGF-β activation. (C) β8-expressing 293 cells adhered to latency-associated peptide with saturating concentrations of RGD peptides and B5 (filled squares, solid line) or isotype control (open squares, hashed line). Assays performed as in Fig. 3A. n = 3. **P < 0.01 by unpaired Student’s t test. (D) B5 induces a low-affinity state maximally inhibiting the binding of soluble αvβ8 to latency-associated peptide by 92% in the presence of RGE peptide (filled inverted triangles, hashed lines); remaining binding blocked completely by RGD peptide (filled squares, solid line). Boxed magnified area of the highest B5 and RGD/E concentrations shows small amount of residual binding remaining with B5 and RGE peptide completely blocked by B5+RGD. Isotype with RGD peptide (open squares, solid line) or RGE peptide (open inverted triangles, hashed line). *P = 0.039 by nonlinear regression and F test of the bottom of each data set. (E) B5 blocks TGF-β activation by ~70%, and the addition of RGD peptide completely blocks remaining activation. β8-transfected 293 cells with RGD peptide and saturating concentrations of B5 (filled squares, solid line) or isotype control (open squares, hashed lines) at the indicated concentrations. Mock-transfected 293 cells with RGD peptide and saturating concentrations of B5 (filled diamonds, solid line) or isotype control (open triangles, hashed lines). n = 4. ***P < 0.001, by ANOVA and Tukey’s post-test of the highest concentration of RGD peptide and antibodies.
Allosteric inhibitors reduce but do not completely eliminate ligand binding (36). Indeed, B5 (or 37E1) maximally blocked receptor binding, TGF-β activation, or TGF-β–dependent gene expression by 60 to 90% (Figs. 1B, 4F, and 7, C to E, and figs. S12, S14, and S15). We hypothesized that induction of a low-affinity state that changes the shape of the ligand-binding pocket of αvβ8 would increase the ability of a linear RGD peptide to compete for latency-associated peptide binding. An RGD peptide based on the latency-associated peptide of the TGF-β 1 RGD binding loop normally blocks αvβ8–latency-associated peptide interactions at high concentrations, because RGD peptides must be constrained into conformations that mimic the natural ligands from which they were derived to achieve high affinity for integrins (39). Saturating concentrations of B5 had no significant effect on cell adhesion, but allowed RGD peptide to block adhesion completely at concentrations where RGD peptide alone had minimal effect, suggesting that B5 induced a low-affinity state where binding of a linear RGD peptide could more easily compete with latency-associated peptide (Fig. 7C). B5 maximally blocked soluble αvβ8 receptor binding or TGF-β activation by 92 or 68%, respectively, and the remaining binding or activation could be blocked completely by high concentrations of RGD peptide (Fig. 7, D and E). Together, these data demonstrate that B5 allosterically induces a low-affinity state that has minimal impact on cell adhesion, but markedly inhibits soluble receptor binding and TGF-β activation.
DISCUSSION
This study addresses the hypothesis that selective targeting of TGF-β activation with an β8 antibody can treat airway remodeling. Airway remodeling in severe asthma and COPD is refractory to current therapies. Here, we present development of an antibody, B5, that selectively blocks αvβ8-mediated TGF-β activation, airway inflammation, and fibrosis in vivo. We use mouse models that phenocopy many of the relevant features of human airway disease to illuminate the mechanistic connections between TGF-β and the pathologic, cellular, and biochemical endpoints that are altered in human COPD specimens.
Therapeutic effects of B5 in disease models
We have recently characterized Ad-IL-1β and chronic ovalbumin airway remodeling mouse models and found that they mimic key pathologic, chemokine, and cytokine features of human airway remodeling in both COPD and asthma (10). Here, extensive characterization demonstrates the validity of the combined cigarette smoke–poly(I:C) airway remodeling system because proinflammatory and profibrogenic cytokines that are increased in human disease biospecimens [that is, IL-1β (16), CCL2 (40), CCL20 (17), and IL-17 (41)] were also increased in mice exposed to cigarette smoke–poly(I:C). These increases were not observed in mice treated only with cigarette smoke, and were greatly amplified by the concomitant exposure to the viral mimetic poly(I:C). These data add to the evidence that viral infection plays an important role as a cofactor in airway remodeling. B5 treatment decreased airway inflammation and inflammatory mediator production in mice exposed to Ad-IL-1β, ovalbumin, and cigarette smoke–poly(I:C), as well as chemokine production by stimulated human lung fibroblasts, suggesting that B5 inhibits a central and common fibro-inflammatory mechanism involving lung fibroblasts.
These data support a general model of the pathologic function of αvβ8 in TGF-β activation in the airway, whereby active IL-1β and TGF-β together increase fibroblast chemokine expression, DC recruitment, and priming of pathologic adaptive immunity, which culminates in obstructive airway physiology. This mechanistic cascade assumes that DC behavior is indirectly influenced by fibroblasts. However, αvβ8-mediated TGF-β activation directly by DCs has also been shown to increase priming of α/β CD4+ IL-17–producing cells in asthma models (33), suggesting that B5 also has the capacity to directly influence DC function (10, 33). Whatever the case, the function of αvβ8 by the relatively restricted cell types that support αvβ8-mediated TGF-β activation shares in the common alterations of proinflammatory cytokines. In particular, IL-17A provides a common mechanistic basis for the therapeutic effects of B5 in a variety of injury and fibroinflammation models in the lung and other organs where αvβ8 is expressed (that is, kidney and liver).
However, current mouse models do not replicate the full spectrum of human COPD (that is, progressive fibrotic airway wall thickening and physiologic fixed airway obstruction). These differences reflect fundamental biologic and anatomic differences between humans and mice. For instance, mice lack subsegmental and respiratory bronchioles, which are the major sites of fixed airway obstruction in humans (42). Despite these shortcomings, the inhibiting effects of B5 on cigarette smoke–poly(I:C)–induced airspace enlargement and airway hyperresponsiveness in mice suggest that B5 may delay disease progression or prevent the exacerbation of COPD symptoms. The prevention of airspace enlargement by B5 is notable because loss of function of the TGF-β–activating integrin αvβ6 causes spontaneous airspace enlargement (43), demonstrating a fundamental difference in the biology of αvβ8 compared to αvβ6.
The role of integrin conformation in αvβ8 function
Mechanism-of-action studies performed on the process of therapeutic development of B5 reveal that αvβ8 is in a constitutively active extended-closed conformation. This conclusion is reinforced by (i) lack of evidence that αvβ8 assumes either a bent or an extended-open conformation by size exclusion chromatography and negative electron microscopy staining; (ii) αvβ8 has a naturally occurring glycan wedge (N414) in a nearly identical location to a mutationally introduced glycan that constitutively activates and opens the α5β1 integrin headpiece but does not cause headpiece opening of αvβ8; (iii) introduction of a second glycan (N294) that constitutively activates the β3 integrins but does not further increase the function of αvβ8. These data reinforce predictions of an extended constitutively active αvβ8 conformation based on domain-swapping of the “knee” region of the β8 subunit (44) and add to the debate about the relative importance of “switchblade” and bent integrin activation models (22, 25, 26) by demonstrating that the extended-closed conformation can be stable and functional.
The constitutive activity of αvβ8 raises questions about how αvβ8-mediated TGF-β activation is regulated. Current evidence suggests that metalloproteolytic cleavage of latent TGF-β bound to the cell surface is involved in the activation mechanism and thus could indirectly modulate αvβ8 function (20). Hence, αvβ8 could position latency-associated peptide for metalloproteases to cleave the latency-associated peptide and release mature TGF-β from the cell surface (20). Evidence would argue against mechanical forces induced from the integrin to latent TGF-β as causing αvβ8-mediated TGF-β activation. Such mechanical forces require dynamic integrin conformational changes or tension transduced from the cytoskeleton to integrin cytoplasmic domains to cell- or matrix-tethered latent TGF-β. These dynamic conformational changes are not seen with αvβ8 because it adopts mainly a single conformation, and the β8 cytoplasmic domain is not required for cell adhesion and TGF-β activation (20).
Conformational changes induced by B5
The B5 epitope located at the head-hybrid junction would be predicted to be altered by hybrid domain swing-out, as illustrated by the α5β1 glycan (N429) wedge mutant (36). B5 retains its ability to bind to and inhibit the function of the β8 glycan mutant, suggesting that the β8 hybrid domain does not swing-out even with two glycans occupying the vertex of the head-hybrid domain angle. Negative electron microscopy staining is consistent with a stabilizing effect of B5 on an exaggerated closed-head conformation with a ~20° inward swing of the hybrid domain. This conformation was not well represented in negative electron microscopy stains when bound to the non–function-blocking clone 68 Fab, suggesting that it is not thermodynamically favored and thus represents a rare low-affinity conformer.
Regulation of αvβ8 affinity for binding to latent TGF-β
Integrin hybrid domain motions are coupled to changes in the ligand-binding pocket. Exactly how the inward hybrid domain motion seen with the B5 Fab is coupled to changes in the ligand-binding pocket remains to be determined, but some predictions can be made from the extensive structural literature involving other integrins. In particular, two of the homologous residues comprising the B5 epitope (R133 and F137) on the α1 helix of βI domains of other integrins are involved in allosteric effects on ligand binding and affect hybrid domain motions, but none cause inward bending of the hybrid domain (36, 37).
Positional changes of the α1 helix affect the neighboring α7 helix, which is directly coupled to the hybrid domain. Such coordinated changes would be required for inward bending of the hybrid domain. The lack of conservation of the β8 ADMIDAS domain indicates that such coordinated positional changes are different for β8 compared with other integrins. In all other integrin β-subunits, the ADMIDAS cation links the top of the α1 helix to the cation of the MIDAS domain, which forms the edge of the ligand-binding pocket that coordinates the aspartate of RGD (29). We speculate that binding of B5 to the C terminus of the α1 helix affects the positioning of the top of the β8 α1 helix, which is relatively nonconstrained by the lack of the ADMIDAS, changing the shape of the ligand-binding pocket into a low-affinity state. Such allosteric effects are consistent with the observed attenuated latency-associated peptide binding to the αvβ8-B5 complex and the improved ability of linear RGD peptides to inhibit the residual binding to this complex.
Separation of TGF-β activation and cell adhesion functions of αvβ8
Therapeutics targeting αvβ8-mediated TGF-β activation but not cell adhesion likely have improved safety profiles compared to therapies that inhibit both of these functions. For example, genetic deficiency of αvβ8 is associated with a paradoxical increase in TGF-β activation in the kidney, presumably due to unmasking of αvβ8-independent pathways activated by TGF-β (45). Our results suggest that allosteric inhibition of αvβ8 by B5 produces highly specific targeting of the TGF-β pathway only in the cells expressing αvβ8 with minimal non–TGF-β–specific effects, providing a safer therapy than ligand-mimetic αvβ8 inhibitors. This expectation is borne out by the observation that B5 does not appear to cause toxicities in treated mice.
Mechanistically, αvβ8-mediated TGF-β activation differs from αvβ8-mediated cell adhesion to latency-associated peptide in that adhesion was not affected by affinity changes induced by B5. Cell adhesion requires integrin clustering, which effectively increases receptor avidity (46). However, clustering is apparently not required for αvβ8-mediated TGF-β activation, because B5 blocks TGF-β activation under conditions where it does not block adhesion. This suggests that the TGF-β–activating function of αvβ8 requires a high-affinity state, whereas cell adhesion requires only a low-affinity state. This property of B5 could explain why the transcriptomes of fibroblasts treated with B5 or anti–TGF-β are almost identical, with no integrin adhesion–specific effects.
In summary, the constitutively active integrin αvβ8 can be targeted by an allosteric inhibitor to prevent experimental airway disease. B5 has now been fully humanized and is currently in preclinical development.
MATERIALS AND METHODS
Study design
We sought to develop a therapeutic strategy to selectively target the TGF-β pathway to treat airway remodeling in COPD. To accomplish this, studies were designed in five parts: (i) to determine the active conformation of the TGF-β activating integrin αvβ8; (ii) to develop a therapeutic neutralizing antibody designed to the active conformation; (iii) to test the mechanism of action of the therapeutic antibody; (iv) to develop airway disease models that are relevant to human pathophysiology; (v) to use the therapeutic antibody for efficacy and mechanism-of-action studies in mice designed to regulate human αvβ8 under the control of the human promoter. Sample sizes were predetermined for cell culture, biochemistry, and murine experiments based on large expected effect sizes (based on pilot studies d > 0.8) requiring an n = 3 to 6 (α = 0.5, β = 0.8). For the ovalbumin experiment, effect sizes were only moderate, requiring n = 12. Data collection was terminated when these sample sizes were met, with the exception of secreted receptor data for which unanticipated interassay variation necessitated larger sample sizes (n = 8). For studies using primary human cells, a minimum of three (gene array) and a maximum of five normal donors (CCL20 ELISA) were predetermined on the basis of practical considerations of maintaining simultaneously multiple primary cultures. No formal outlier analysis was performed.
Production of BAC ITGB8 Tg mice
Tg mice expressing human ITGB8 were generated to test the efficacy of B5 in disease models. The human BAC successfully rescued the lethal phenotype associated with deficiency of mouse itgb8. Genetic germline deletion of itgb8 is lethal in mice by postnatal day 0 in inbred strains and lethal within 3 months in mixed or outbred strains (30, 32). Mice die of complications of developmental defects in vasculogenesis or neonatal hemorrhage/neurogenesis (30, 32). DC-specific deletion (CD11c-cre) results in mice that develop autoimmune colitis and die by 6 months (31). A human chromosome BAC (RP11-431K20) containing the entire 80-kb ITGB8 gene and 70 and 30 kb of 5′ and 3′ flanking regions, respectively, was purified (Nucleobond, Clontech), determined to be intact by pulse-field electrophoresis, microinjected into pronuclei of FVB/N mice zygotes in microinjection buffer [10 mM tris, 0.1 mM EDTA (pH 7.4)], and surgically transferred to oviducts of pseudopregnant females. Twenty-eight pups survived and tail tip DNA was screened using BAC end primers, 5′ BAC end pair (forward 5′-CCTGTGTAACTACCACCAC-3′; reverse 5′-CTCACTTGACAA-TCTAGTCCTC-3′), and 3′ BAC end pair (forward 5′-CAACCAATC-AGTAGCACCC-3′; reverse 5′-CTCAGTGAGATCTGTTATGAAC-3′). Four founders (lines A to D) had completely integrated copies of the full-length transgene and were crossed to C57B/6 itgb8+/- mice. Lines B to D successfully transmitted the transgene to progeny to generate BAC; itgb8+/- mice on a mixed FVB/C57B/6 background. Sibling matings generated mice heterozygous or homozygous for the BAC and homozygous for the itgb8 knockout allele.
Antibody dosing
Initial dosing for B5 was based on dose-response curves generated with the B-line of mice where 7 mg/kg outperformed lower doses after IT-Ad-Il-1β provocation (Fig. 1B and fig. S4). To replicate these findings in an independent line of BAC Tg mice, C-line mice were dosed with B5 or isotype control (7 mg/kg), and the experiment was repeated (Fig. 1, I to K). The timing of the first antibody administration was based on the observation that the maximal inflammation in the Ad-IL-1β model occurs at 7 days after intratracheal injection (10). Thus, administration of B5 was initiated at 7 days after intratracheal injection. In the cigarette smoke–poly(I:C) model, B5 or isotype antibody administration was begun 1 week after cigarette smoke exposure, at a point when significant lung neutrophilia was observed. For the ovalbumin experiment, antibody dosing was initiated 1 day before antigen challenge.
Cigarette smoke and poly(I:C) exposure
B-line mice were exposed using a whole-body cigarette smoke exposure system (Teague Enterprises) within a barrier facility. Mice are acclimated using increasing smoke exposures for 5 days starting at a total suspended particulates (TSP) of 40 mg/m3 for 1 hour, and increasing incrementally to final smoke exposures of 100 TSP using 3R4F cigarettes. Full-dose exposures were begun in week 1 with 5 hours of continuous exposure, with rest on weekends. In week 2, intranasal doses of poly(I:C) (Invivogen, 50 μg per dose) were given on days 9 and 12, and again in week 3 on days 15 and 18.
Electron microscopy
Samples were applied to glow-discharged carbon-coated copper grids, washed, stained with 0.75% (w/v) uranylformate, and aspirated to dryness (47). Images were taken on a Tecnai T12 electron microscope (FEI Company) equipped with a LaB6 filament and operating at a 120-kV acceleration voltage. Micrographs were recorded using a 4K charge-coupled device camera (UltraScan 4000, Gatan Inc.), where one pixel equals 2.21 Å on the specimen. Particles were selected manually. Individual particles were windowed out from two binned images and were subjected to six cycles of multireference alignment and K-means classification using SPIDER to generate well-resolved class averages (48). The number of class-averages to capture the sample heterogeneity was adjusted between 20 and 50. Manual selection was used to assign obviously related extended conformations into subclasses: extended-closed, extended-intermediate, and bent. Bent conformations were assigned when the headpiece-tailpiece angle was less than 90°. The head-hybrid angles were determined from class averages where the head and hybrid axis was oriented to allow for definitive angle assignment [ImageJ 1.42q, National Institutes of Health (NIH)]. Images where the assignment was ambiguous were not included in the analysis. Individual angles were measured from class averages where the head and hybrid domains were well-resolved (ImageJ).
Statistical analysis
For microarray analysis, the data set was normalized using Lowess normalization. No background subtraction was performed, and median feature pixel intensity was used as the raw signal before normalization. A one-way ANOVA linear model was fit to each comparison of interest to estimate log2 fold change and calculated B statistic. All procedures were carried out using functions in the R package limma in Bioconductor. To regress the log ratios of (anti-β8/control) against the log ratios of (anti–TGF-β/control) among differentially expressed genes, we used log2 (anti-β8/control) = a + b * log2 (anti–TGF-β/control) to obtain intercept and slope estimates. To identify differentially expressed genes, a B statistic of B ≥ 0 was used. All other data are reported as means ± SEM. Comparisons between two different groups were determined using Student’s t test. One-way ANOVA was used for multiple comparisons, and Tukey’s or Bonferroni’s post hoc test was used to test for statistical significance, as indicated. Significance was defined as P < 0.05. All statistical analyses were performed using the software package Prism 4.0b (GraphPad Software).
Supplementary Material
Acknowledgments
We thank J. Munger (New York University) for TMLC reporter cells, H. Weiner (Harvard Medical School) for the murine TGF-β1–expressing P3U1 cells, X. Huang [University of California, San Francisco (UCSF) Sandler Airway Physiology Core], and S. Farr-Jones for editorial assistance.
Funding: This work was supported by NIH HL113032, HL063993, HL090662, NS044155, UCTRDRP, UCSF Academic Senate, UCOP POC award, Sponsored Research agreement from MedImmune, LLC (S.L.N.), UCSF Liver Center (P30DK026743, to S.L.N. and J.L.B.), UCSF PBBR (Y.C.), and Nina Ireland Lung Disease Program (P.W.).
Footnotes
Author contributions: S.M. performed all in vivo experiments and helped to establish the BAC Tg mouse line. J.L., W.W., and J.D.M. helped to conceive and perform affinity maturation and antibody engineering and to create and prepare recombinant proteins including integrins and IgGs. R.I.S. performed size exclusion chromatography experiments, developed ELISAs, and conceived electron microscopy experiments. S.W. and Y.C. performed and interpreted electron microscopy experiments. L.M., P.T., J.C., and R.H. performed optimization of IgG, affinity measurements, and oversight of IgG production. C.G. and A.C. provided structural and computational expertise. T.B., S.C., R.M., and K.J.M. performed secreted binding assays and TGF-β activation assays, and established and characterized the BAC Tg line. R.C., H.Y., A.G., M.H., H.K., and O.J.B. performed extensive characterization of BAC Tg mice. L.F.R. provided itgb8+/− mice. A.H., D.J., and P.W. provided fresh human lung tissue. J.A., A.J.B., and D.J.E. performed microarray experiments. J.L.B. helped to produce original hybridomas, and S.L.N. conceived and oversaw the entire project, and wrote and prepared the manuscript.
Data and materials availability: All reagents are property of the UC Regents and can be obtained under a material transfer agreement.
www.sciencetranslationalmedicine.org/cgi/content/full/6/241/241ra79/DC1
Materials and Methods
Fig. S1. ITGB8 BAC Tg mice express αvβ8 at similar expression levels and tissue distribution to humans.
Fig. S2. ITGB8 BAC transgene rescues early lethality of mouse itgb8 deficiency.
Fig. S3. Secreted human αvβ8 integrin–placental AP fusion proteins bind to murine latency-associated peptide.
Fig. S4. Dose response of B5 antibody treatment of intratracheal Ad-IL-1β–injected B-line BAC ITGB8 Tg mice.
Fig. S5. Antibody treatment with B5 does not have any effect on lung morphology.
Fig. S6. Cigarette smoke and poly(I:C) synergistically produce airway disease that resembles COPD in humans.
Fig. S7. Effects of combined exposure of cigarette smoke and poly(I:C) on inflammation and inflammatory mediators.
Fig. S8. B5 antibody treats allergic airway inflammation.
Fig. S9. Electron microscopy of integrin αvβ8.
Fig. S10. β8 antibody epitope mapping.
Fig. S11. Non–function-blocking antibodies binding to the Psi, hybrid, or epidermal growth factor (EGF) 1–2 domains.
Fig. S12. Genome-wide comparison of the effects of β8 and TGF-β neutralizing antibodies on human fibroblast gene expression.
Fig. S13. VH and VL sequences of 37E1 and B5.
Fig. S14. B5 improves the ability of 37E1 to inhibit the binding of soluble αvβ8 to latency-associated peptide.
Fig. S15. B5 specifically blocks binding of αvβ8, and not αvβ6, to latency-associated peptide.
Fig. S16. Gel filtration of clasped or unclasped αvβ8 in complex with B5 Fab.
Fig. S17. Electron microscopy of integrin αvβ8.
Table S1. Fibroblast differentially expressed gene array data.
Table S2. Autocrine TGF-β activation mediated by αvβ8 in human fetal tracheal fibroblasts. References (52–62)
Competing interests: Some authors are listed on the following U.S. patents: “Integrin αvβ8 neutralizing antibody” and “Antibodies that bind integrin αvβ8” (Nos. 61/305,749 and 61/428,814, respectively, to J.L., J.D.M., J.L.B., and S.L.N.), and IgG vectors (U.S. Patent Application No. 61/305,749 to J.D.M.). Some of this work was funded by a sponsored research agreement from MedImmune, LLC. J.L., J.D.M., J.L.B., and S.L.N. have received royalty payments from the Regents of the University of California for β8 neutralizing antibodies used in this publication. The other authors declare no competing interests.
REFERENCES AND NOTES
- 1.Centers for Disease Control and Prevention (CDC) Chronic obstructive pulmonary disease among adults—United States, 2011. MMWR Morb Mortal Wkly Rep. 2012;61:938–943. [PubMed] [Google Scholar]
- 2.Postma DS, Timens W. Remodeling in asthma and chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2006;3:434–439. doi: 10.1513/pats.200601-006AW. [DOI] [PubMed] [Google Scholar]
- 3.Han MK, Kazerooni EA, Lynch DA, Liu LX, Murray S, Curtis JL, Criner GJ, Kim V, Bowler RP, Hanania NA, Anzueto AR, Make BJ, Hokanson JE, Crapo JD, Silverman EK, Martinez FJ, Washko GR COPDGene Investigators. Chronic obstructive pulmonary disease exacerbations in the COPDGene study: Associated radiologic phenotypes. Radiology. 2011;261:274–282. doi: 10.1148/radiol.11110173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wilkinson TM, Donaldson GC, Johnston SL, Openshaw PJ, Wedzicha JA. Respiratory syncytial virus, airway inflammation, and FEV1 decline in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2006;173:871–876. doi: 10.1164/rccm.200509-1489OC. [DOI] [PubMed] [Google Scholar]
- 5.Uhl EW, Castleman WL, Sorkness RL, Busse WW, Lemanske RF, Jr, McAllister PK. Parainfluenza virus-induced persistence of airway inflammation, fibrosis, and dysfunction associated with TGF-beta 1 expression in brown Norway rats. Am J Respir Crit Care Med. 1996;154:1834–1842. doi: 10.1164/ajrccm.154.6.8970378. [DOI] [PubMed] [Google Scholar]
- 6.Sterk PJ. Virus-induced airway hyperresponsiveness in man. Eur Respir J. 1993;6:894–902. [PubMed] [Google Scholar]
- 7.Kang MJ, Lee CG, Lee JY, Dela Cruz CS, Chen ZJ, Enelow R, Elias JA. Cigarette smoke selectively enhances viral PAMP– and virus-induced pulmonary innate immune and remodeling responses in mice. J Clin Invest. 2008;118:2771–2784. doi: 10.1172/JCI32709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Churg A, Zhou S, Wang X, Wang R, Wright JL. The role of interleukin-1β in murine cigarette smoke-induced emphysema and small airway remodeling. Am J Respir Cell Mol Biol. 2009;40:482–490. doi: 10.1165/rcmb.2008-0038OC. [DOI] [PubMed] [Google Scholar]
- 9.Podowski M, Calvi C, Metzger S, Misono K, Poonyagariyagorn H, Lopez-Mercado A, Ku T, Lauer T, McGrath-Morrow S, Berger A, Cheadle C, Tuder R, Dietz HC, Mitzner W, Wise R, Neptune E. Angiotensin receptor blockade attenuates cigarette smoke–induced lung injury and rescues lung architecture in mice. J Clin Invest. 2012;122:229–240. doi: 10.1172/JCI46215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamura H, Cambier S, Somanath S, Barker T, Minagawa S, Markovics J, Goodsell A, Publicover J, Reichardt L, Jablons D, Wolters P, Hill A, Marks JD, Lou J, Pittet JF, Gauldie J, Baron JL, Nishimura SL. Mouse and human lung fibroblasts regulate dendritic cell trafficking, airway inflammation, and fibrosis through integrin αvβ8–mediated activation of TGF-β. J Clin Invest. 2011;121:2863–2875. doi: 10.1172/JCI45589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nishimura SL. Integrin-mediated transforming growth factor-β activation, a potential therapeutic target in fibrogenic disorders. Am J Pathol. 2009;175:1362–1370. doi: 10.2353/ajpath.2009.090393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vitsky A, Waire J, Pawliuk R, Bond A, Matthews D, Lacasse E, Hawes ML, Nelson C, Richards S, Piepenhagen PA, Garman RD, Andrews L, Thurberg BL, Lonning S, Ledbetter S, Ruzek MC. Homeostatic role of transforming growth factor-β in the oral cavity and esophagus of mice and its expression by mast cells in these tissues. Am J Pathol. 2009;174:2137–2149. doi: 10.2353/ajpath.2009.080723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anderton MJ, Mellor HR, Bell A, Sadler C, Pass M, Powell S, Steele SJ, Roberts RR, Heier A. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol. 2011;39:916–924. doi: 10.1177/0192623311416259. [DOI] [PubMed] [Google Scholar]
- 14.Markovics JA, Araya J, Cambier S, Somanath S, Gline S, Jablons D, Hill A, Wolters PJ, Nishimura SL. Interleukin-1β induces increased transcriptional activation of the transforming growth factor-β-activating integrin subunit β8 through altering chromatin architecture. J Biol Chem. 2011;286:36864–36874. doi: 10.1074/jbc.M111.276790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eltom S, Stevenson CS, Rastrick J, Dale N, Raemdonck K, Wong S, Catley MC, Belvisi MG, Birrell MA. P2X7 receptor and caspase 1 activation are central to airway inflammation observed after exposure to tobacco smoke. PLOS One. 2011;6:e24097. doi: 10.1371/journal.pone.0024097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sapey E, Ahmad A, Bayley D, Newbold P, Snell N, Rugman P, Stockley RA. Imbalances between interleukin-1 and tumor necrosis factor agonists and antagonists in stable COPD. J Clin Immunol. 2009;29:508–516. doi: 10.1007/s10875-009-9286-8. [DOI] [PubMed] [Google Scholar]
- 17.Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, Joos GF, Brusselle GG. Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2007;175:998–1005. doi: 10.1164/rccm.200608-1113OC. [DOI] [PubMed] [Google Scholar]
- 18.Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature. 2011;474:343–349. doi: 10.1038/nature10152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Araya J, Cambier S, Morris A, Finkbeiner W, Nishimura SL. Integrin-mediated transforming growth factor-β activation regulates homeostasis of the pulmonary epithelial-mesenchymal trophic unit. Am J Pathol. 2006;169:405–415. doi: 10.2353/ajpath.2006.060049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mu D, Cambier S, Fjellbirkeland L, Baron JL, Munger JS, Kawakatsu H, Sheppard D, Broaddus VC, Nishimura SL. The integrin αvβ8 mediates epithelial homeostasis through MT1-MMP–dependent activation of TGF-β1. J Cell Biol. 2002;157:493–507. doi: 10.1083/jcb.200109100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Takagi J, Petre BM, Walz T, Springer TA. Global conformational rearrangements in integrin extracellular domains in outside-in and inside-out signaling. Cell. 2002;110:599–511. doi: 10.1016/s0092-8674(02)00935-2. [DOI] [PubMed] [Google Scholar]
- 22.Dong X, Mi LZ, Zhu J, Wang W, Hu P, Luo BH, Springer TA. aVb3 integrin crystal structures and their functional implications. Biochemistry. 2012;51:8814–8828. doi: 10.1021/bi300734n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ye F, Hu G, Taylor D, Ratnikov B, Bobkov AA, McLean MA, Sligar SG, Taylor KA, Ginsberg MH. Recreation of the terminal events in physiological integrin activation. J Cell Biol. 2010;188:157–173. doi: 10.1083/jcb.200908045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Askari JA, Buckley PA, Mould AP, Humphries MJ. Linking integrin conformation to function. J Cell Sci. 2009;122:165–170. doi: 10.1242/jcs.018556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong JP, Mahalingham B, Alonso JL, Borrelli LA, Rui X, Anand S, Hyman BT, Rysiok T, Müller-Pompalla D, Goodman SL, Arnaout MA. Crystal structure of the complete integrin αVβ3 ectodomain plus an α/β transmembrane fragment. J Cell Biol. 2009;186:589–600. doi: 10.1083/jcb.200905085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schürpf T, Springer TA. Regulation of integrin affinity on cell surfaces. EMBO J. 2011;30:4712–4727. doi: 10.1038/emboj.2011.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moyle M, Napier MA, McLean JW. Cloning and expression of a divergent integrin subunit β8. J Biol Chem. 1991;266:19650–19658. [PubMed] [Google Scholar]
- 28.Nishimura SL, Sheppard D, Pytela R. Integrin αvβ8. Interaction with vitronectin and functional divergence of the β8 cytoplasmic domain. J Biol Chem. 1994;269:28708–28715. [PubMed] [Google Scholar]
- 29.Xiong JP, Stehle T, Zhang R, Joachimiak A, Frech M, Goodman SL, Arnaout MA. Crystal structure of the extracellular segment of integrin αVβ3 in complex with an Arg-Gly-Asp ligand. Science. 2002;296:151–155. doi: 10.1126/science.1069040. [DOI] [PubMed] [Google Scholar]
- 30.Aluwihare P, Mu Z, Zhao Z, Yu D, Weinreb PH, Horan GS, Violette SM, Munger JS. Mice that lack activity of αvβ6- and αvβ8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci. 2009;122:227–232. doi: 10.1242/jcs.035246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Travis MA, Reizis B, Melton AC, Masteller E, Tang Q, Proctor JM, Wang Y, Bernstein X, Huang X, Reichardt LF, Bluestone JA, Sheppard D. Loss of integrin αvβ8 on dendritic cells causes autoimmunity and colitis in mice. Nature. 2007;449:361–365. doi: 10.1038/nature06110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhu J, Motejlek K, Wang D, Zang K, Schmidt A, Reichardt LF. β8 integrins are required for vascular morphogenesis in mouse embryos. Development. 2002;129:2891–2903. doi: 10.1242/dev.129.12.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, Wang Y, Bernstein X, Li JT, Atabai K, Huang X, Sheppard D. IL-17A produced by αβ T cells drives airway hyperresponsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med. 2012;18:547–554. doi: 10.1038/nm.2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Campbell ID, Humphries MJ. Integrin structure, activation, and interactions. Cold Spring Harb Perspect Biol. 2011;3:a004994. doi: 10.1101/cshperspect.a004994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luo BH, Springer TA, Takagi J. Stabilizing the open conformation of the integrin headpiece with a glycan wedge increases affinity for ligand. Proc Natl Acad Sci USA. 2003;100:2403–2408. doi: 10.1073/pnas.0438060100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nagae M, Re S, Mihara E, Nogi T, Sugita Y, Takagi J. Crystal structure of α5β1 integrin ectodomain: Atomic details of the fibronectin receptor. J Cell Biol. 2012;197:131–140. doi: 10.1083/jcb.201111077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen X, Xie C, Nishida N, Li Z, Walz T, Springer TA. Requirement of open headpiece conformation for activation of leukocyte integrin αXβ2. Proc Natl Acad Sci USA. 2010;107:14727–14732. doi: 10.1073/pnas.1008663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mould AP, Akiyama SK, Humphries MJ. The inhibitory anti-β1 integrin monoclonal antibody 13 recognizes an epitope that is attenuated by ligand occupancy. Evidence for allosteric inhibition of integrin function. J Biol Chem. 1996;271:20365–20374. doi: 10.1074/jbc.271.34.20365. [DOI] [PubMed] [Google Scholar]
- 39.Tung JS, Jakubowski JA, Heath WF, Utterback BG, Herron DK. Correlation of molecular shape with GPIIb-IIIa receptor antagonist activity in RGD peptides. Receptor. 1993;3:343–356. [PubMed] [Google Scholar]
- 40.Liu SF, Chin CH, Wang CC, Lin MC. Correlation between serum biomarkers and BODE index in patients with stable COPD. Respirology. 2009;14:999–1004. doi: 10.1111/j.1440-1843.2009.01608.x. [DOI] [PubMed] [Google Scholar]
- 41.Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, McCormick M, Woods J, May R, Sleeman MA, Anderson IK, Brightling CE. Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest. 2010;138:1140–1147. doi: 10.1378/chest.09-3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vanoirbeek JA, Rinaldi M, De Vooght V, Haenen S, Bobic S, Gayan-Ramirez G, Hoet PH, Verbeken E, Decramer M, Nemery B, Janssens W. Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol. 2010;42:96–104. doi: 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 43.Morris DG, Huang X, Kaminski N, Wang Y, Shapiro SD, Dolganov G, Glick A, Sheppard D. Loss of integrin αvβ6-mediated TGF-β activation causes Mmp12-dependent emphysema. Nature. 2003;422:169–173. doi: 10.1038/nature01413. [DOI] [PubMed] [Google Scholar]
- 44.Smagghe BJ, Huang PS, Ban YE, Baker D, Springer TA. Modulation of integrin activation by an entropic spring in the β-knee. J Biol Chem. 2010;285:32954–32966. doi: 10.1074/jbc.M110.145177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Khan S, Lakhe-Reddy S, McCarty JH, Sorenson CM, Sheibani N, Reichardt LF, Kim JH, Wang B, Sedor JR, Schelling JR. Mesangial cell integrin αvβ8 provides glomerular endothelial cell cytoprotection by sequestering TGF-β and regulating PECAM-1. Am J Pathol. 2011;178:609–620. doi: 10.1016/j.ajpath.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bunch TA. Integrin αIIbβ3 activation in Chinese hamster ovary cells and platelets increases clustering rather than affinity. J Biol Chem. 2010;285:1841–1849. doi: 10.1074/jbc.M109.057349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ohi M, Li Y, Cheng Y, Walz T. Negative staining and image classification—Powerful tools in modern electron microscopy. Biol Proced Online. 2004;6:23–34. doi: 10.1251/bpo70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 49.Luo BH, Strokovich K, Walz T, Springer TA, Takagi J. Allosteric β1 integrin antibodies that stabilize the low affinity state by preventing the swing-out of the hybrid domain. J Biol Chem. 2004;279:27466–27471. doi: 10.1074/jbc.M404354200. [DOI] [PubMed] [Google Scholar]
- 50.Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993;234:779–815. doi: 10.1006/jmbi.1993.1626. [DOI] [PubMed] [Google Scholar]
- 51.Zhu J, Zhu J, Springer TA. Complete integrin headpiece opening in eight steps. J Cell Biol. 2013;201:1053–1068. doi: 10.1083/jcb.201212037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Finkbeiner WE. In: The Lung: Scientific Foundations. Crystal RG, editor. Lippincott-Raven Publishers; Philadelphia, PA: 1997. pp. 415–433. [Google Scholar]
- 53.Oida T, Weiner HL. TGF-β induces surface latency associated peptide expression on murine CD4 T cells independent of Foxp3 induction. PLOS One. 2010;5:e15523. doi: 10.1371/journal.pone.0015523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Razai A, Garcia-Rodriguez C, Lou J, Geren IN, Forsyth CM, Robles Y, Tsai R, Smith TJ, Smith LA, Siegel RW, Feldhaus M, Marks JD. Molecular evolution of antibody affinity for sensitive detection of botulinum neurotoxin type A. J Mol Biol. 2005;351:158–169. doi: 10.1016/j.jmb.2005.06.003. [DOI] [PubMed] [Google Scholar]
- 55.Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, Barzcak A, Xiao Y, Erle DJ, Nishimura SL. Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest. 2007;117:3551–3562. doi: 10.1172/JCI32526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Munger JS, Harpel JG, Giancotti FG, Rifkin DB. Interactions between growth factors and integrins: Latent forms of transforming growth factor-β are ligands for the integrin αvβ1. Mol Biol Cell. 1998;9:2627–2638. doi: 10.1091/mbc.9.9.2627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kehoe JW, Velappan N, Walbolt M, Rasmussen J, King D, Lou J, Knopp K, Pavlik P, Marks JD, Bertozzi CR, Bradbury AR. Using phage display to select antibodies recognizing post-translational modifications independently of sequence context. Mol Cell Proteomics. 2006;5:2350–2363. doi: 10.1074/mcp.M600314-MCP200. [DOI] [PubMed] [Google Scholar]
- 58.Ganter MT, Roux J, Miyazawa B, Howard M, Frank JA, Su G, Sheppard D, Violette SM, Weinreb PH, Horan GS, Matthay MA, Pittet JF. Interleukin-1β causes acute lung injury via αvβ5 and αvβ6 integrin–dependent mechanisms. Circ Res. 2008;102:804–812. doi: 10.1161/CIRCRESAHA.107.161067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kolb M, Margetts PJ, Anthony DC, Pitossi F, Gauldie J. Transient expression of IL-1β induces acute lung injury and chronic repair leading to pulmonary fibrosis. J Clin Invest. 2001;107:1529–1536. doi: 10.1172/JCI12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shalaby KH, Gold LG, Schuessler TF, Martin JG, Robichaud A. Combined forced oscillation and forced expiration measurements in mice for the assessment of airway hyper-responsiveness. Respir Res. 2010;11:82. doi: 10.1186/1465-9921-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, Paré PD. The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med. 2004;350:2645–2653. doi: 10.1056/NEJMoa032158. [DOI] [PubMed] [Google Scholar]
- 62.Gline SE, Cambier S, Govaerts C, Nishimura SL. A 50-Å separation of the integrin αvβ3 extracellular domain C termini reveals an intermediate activation state. J Biol Chem. 2004;279:54567–54572. doi: 10.1074/jbc.M406582200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.