

Abstract
The co-occurrence of both Alzheimer disease (AD) pathology and vascular brain injury (VBI) is very common, especially amongst the oldest of old. In neuropathologic studies, the prevalence of AD, VBI, and mixed AD/VBI lesions ranks ahead of Lewy bodies and hippocampal sclerosis. In the modern era of structural magnetic resonance imaging (MRI) and amyloid positron emission tomography (PET) imaging, this review examines 1) the prevalence of mixed AD and VBI pathology, 2) the significance of these pathologies for cognitive impairment (AD and vascular cognitive impairment (VCI)), and 3) the diagnosis and treatment of mixed AD/VCI. Although epidemiologic studies report that vascular risk factors for arteriosclerosis increase the risk of incident AD, both autopsy and amyloid PET studies indicate that AD and VBI contribute additively, but independently, to the risk of dementia. The literature confirms the malignancy of AD and highlights the adverse effects of microinfarcts on cognitive function. For the clinical diagnosis of mixed AD/VCI, the presence of AD can be recognized by neuropsychological profile, structural imaging, cerebrospinal fluid biomarkers, and glucose PET and amyloid PET imaging. The diagnosis of VBI, however, still hinges predominantly on the structural MRI findings. Severe amnesia and atrophy of the hippocampus are characteristic of early AD, whereas the cognitive profile for VCI is highly variable and dependent on size and location of VBI. The cognitive profile of mixed AD/VBI is dominated by AD. With the notable exception of microinfarcts (which elude in vivo detection), infarcts, hemorrhages, and white matter hyperintensities on structural MRI currently represent the best markers for the presence VBI. Better markers that reflect the health and reactivity of intracerebral blood vessels are needed. For prevention and treatment, the type of underlying cerebrovascular disease (for example, arteriosclerosis or cerebral amyloid angiopathy) should be considered. It is likely that reduction of vascular risk factors for arteriosclerosis can significantly reduce vascular contributions to mixed dementia.
Introduction
Traditionally, Alzheimer disease (AD) and vascular dementia are recognized as the two most prevalent forms of dementia in late life. A combination of AD and vascular pathologies (so-called mixed dementia) is usually registered as a close third, moving up to first or second in rank in community-based studies of the oldest of old. Conceptualization and diagnosis of these entities has been evolving from clinical-pathological phenotypes, which could not be resolved until autopsy, to new research diagnostic criteria that incorporate molecular biomarkers (for example, amyloid-beta (Aβ) and phosphorylated tau), and in vivo structural, functional, and perfusion imaging. Neuropsychological features (for example, pattern and severity of impairment across principle cognitive domains) remain relevant to diagnosis and clinical care. Innovations are underway in computerized neuropsychological assessment (for example, computerized assessment with continuous measures, such as the National Institutes of Health tool box) and rethinking cognitive impairment in terms of functional neural networks. In this review, we consider mainly how recent advances in biomarkers and imaging have altered our conceptualization and diagnosis of mixed AD and vascular pathologies.
Medically speaking, the approach to mixed AD and vascular pathologies should be tied to prevention and treatment. During the past 30 years, emphasis has moved from dementia to mild cognitive impairment to preclinical disease, in the hopes that prevention and treatment can be instituted earlier in the disease course. AD is currently conceptualized as a combined amyloidopathy and tau-related neurodegeneration. Mainstream strategies to prevent and treat AD target these misfolded proteins.
The model for vascular cognitive impairment (VCI) adopted here posits that vascular risk factors (VRFs) lead to cerebrovascular disease (CVD), which leads to vascular brain injury (VBI), which leads to VCI (Figure 1). It is paramount to maintain focus on the type of CVD (the cause), as well as on the resulting VBI and VCI. For example, the prevention and treatment of cerebral amyloid angiopathy (CAA), which is intrinsically associated with AD, is likely to differ fundamentally from the prevention and treatment of atherosclerosis. It may pay off to pay some attention to terminology, rather than to lump all vascular dementias into the category of VCI.
Figure 1.
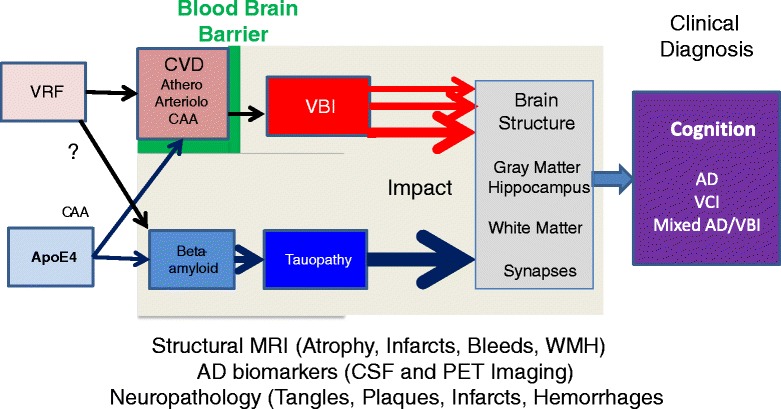
Mixed Alzheimer disease/vascular brain injury. Additive parallel or interactive pathways? AD, Alzheimer disease; ApoE, apolipoprotein E; CAA, cerebral amyloid angiopathy; CSF, cerebrospinal fluid; CVD, cerebrovascular disease; MRI, magnetic resonance imaging; PET, positron emission tomography; VBI, vascular brain injury; VCI, vascular cognitive impairment; VRF, vascular risk factor; WMH, white matter hyperintensity.
In this review of clinical and imaging features of mixed AD/VCI, we have chosen nuanced terminology to distinguish levels of vascular factors: mixed AD/VRFs, mixed AD/CVD, mixed AD/VBI, and mixed AD/VCI. We use the term VCI when clinical criteria are used to define groups, VBI when study groups are defined by infarcts/hemorrhages noted by imaging/pathology, and CVD to refer to specific disorders of blood vessels (for example, atherosclerosis or amyloid angiopathy). Finally, we use the term VRFs to refer to traditional risk factors for atherosclerosis (for example, hypertension, diabetes mellitus, and hyperlipidemia) and not for amyloid angiopathy (for example, apolipoprotein E (ApoE) ε4 is mentioned distinctly and not included under the label VRF in this review).
The diagnosis of VCI is reflected in recent clinical criteria [1], which draw heavily on evidence of infarcts, white matter hyperintensities (WMHs) and microbleeds (MBs) using structural magnetic resonance imaging (MRI). Multiple autopsy studies have shown that microinfarcts (one type of VBI) are major risk factors for VCI; however, microinfarcts fall below the resolution of 1.5 and 3 T MRI. This is one of several gaps in in vivo detection that we must close going forward.
For the diagnosis of AD in this review, we adopt research-level criteria for high likelihood of preclinical, mild cognitive impairment and dementia due to AD based on biomarker evidence [2-4]. This requires evidence of 1) amyloid deposition by autopsy, low cerebrospinal fluid (CSF) Aβ, or positive amyloid positron emission tomography (PET) and 2) neurodegeneration by autopsy, high CSF ptau, and AD pattern of atrophy on structural imaging. While amyloid and tau biomarkers are not required in clinical practice for the diagnosis of AD, this review is intentionally more selective.
In attempting to define clinical and imaging features of entities that are defined by clinical and imaging phenotype, we must be aware of possible circular reasoning. For example, in seeking to characterize neuropsychological features, we have selected study groups based on mixed AD/VBI, not mixed AD/VCI (unless the diagnosis of VCI was reached without knowledge of the neuropsychological profile).
Diagnostic classifications such as AD, VCI and mixed AD/VCI represent an oversimplified dichotomous framework, necessary for clinical practice. With the expanding repertoire of neuroimaging modalities, it makes sense in the long term to adopt a multi-factorial model that plugs in continuous measures of AD and VBI as independent variables and looks to continuous measures of various cognitive domains as the outcome. Indeed, many of the studies reviewed below used multi-factorial models where imaging features are correlated with cognitive performance.
We are now at the infancy of testing whether these cognitive outcomes are mediated by injury to corresponding cognitive networks (using functional MRI and diffuse tensor imaging) and how these systems change over time. We look forward to visualizing how AD pathology and VBI impact neural networks and how treatment might protect, sustain, and repair them.
Prevalence of mixed AD/VCI or mixed AD/VBI in prospective longitudinal autopsy studies
Over four decades ago, the high prevalence of AD and vascular pathology in older patients (mean age = 76.4 years) was brought to light by Tomlinson, Blessed, and Roth [5,6]. Previously, AD had been considered a relatively rare cause of early onset dementia, while arteriosclerosis was widely held to be the most common cause of late onset dementia. Among 50 cases with dementia living in a mental hospital, widespread plaques and tangles (that is, AD changes) were the predominant pathological lesions in 50%, while cerebral softening (that is, territorial infarcts) due to atherosclerosis accounted for 12 to 17%, and mixed AD and VBI pathologies were found in 8 to 18%. At this juncture, the pendulum of clinical diagnosis and investigation swung dramatically from arteriosclerotic dementia to AD.
In 1997, three decades later, the Nun Study heightened interest in mixed dementia, by showing additive effects of AD and VBI on risk of dementia [7]. The Nun Study introduced a generation of prospective, longitudinal, clinic-to-autopsy cohort studies which have systematically addressed clinical-pathological correlations in dementia (Tables 1 and 2). We review some of these studies from the United States, United Kingdom, and Japan. The prevalence of mixed AD/VBI among cases with dementia ranges from 14 to 44%.
Table 1.
Community-based, autopsy studies are required to estimate the prevalence and incidence of mixed Alzheimer disease/vascular brain injury
| Autopsy study | N | Mean age (years) | Diagnosis of AD | Diagnosis of VBI or VCI | Prevalence of mixed | Interaction between VBI and AD on risk of dementia | Neuropsychology |
|---|---|---|---|---|---|---|---|
| Nun Study [7] | 102 | 87 (76-100) | Khachaturian plaque criteria | Number of infarcts > or <1.5 cm | 39% (24/61) of dementia cases were mixed | Number of tangles and number of lacunes exert independent additive effect on MMSE and likelihood of dementia | MMSE |
| CERAD | |||||||
| ROS [8,9] | 550 | 87 | Mean number of neurofibrillary tangles, neuritic plaques, and diffuse plaques in five lobes | Number of macroscopic and microscopic infarcts | 28% of dementia cases were mixed | AD and VBI pathology have additive effect on odds of dementia | |
| MAP [8,9] | 425 | 87 | Mean number of neurofibrillary tangles, neuritic plaques, and diffuse plaques in five lobes | Number of macroscopic and microscopic infarcts | 44% of dementia cases were mixed | ||
| BLSA [10] | 179 | 87.6 ± 7.1 | CERAD | Macroscopic infarcts | Hemispheral infarcts alone or with AD account for 35% of dementia cases | In subjects with intermediate AD pathology scores, a single macroscopic hemispheral infarct was sufficient to cause dementia | Blessed Memory Information Concentration Test |
| Braak and Braak stage | Microscopic infarcts | ||||||
| BLSA [11] | 200 | Atherosclerosis (0-3) of coronary, aorta, and intracranial vessels | 45% have remote infarct | 68% of cases have atherosclerosis, which increased the odds of dementia independent of AD pathology or cerebral infarcts | |||
| 175 complete autopsies, including heart and aorta | |||||||
| MRC CFAS [12] | N = 456 | 87 (SD = 7): range 66 to 100 (63% ≥85) | CERAD scale (0-3) | Regional infarcts (>1 cm) | Association between AD pathology and cognitive status goes down with age | MMSE | |
| 243 dementia | Diffuse plaques, neuritic plaques, tangles, atrophy | AGECAT | |||||
| 183 without dementia | Small vessel disease: lacunes, microinfarcts, white matter change | ||||||
| Association between atrophy and age continues to go up | |||||||
| 30 unknown | |||||||
| MRC CFAS [13] | Self-reported vascular risk factors | Vascular risk factors were not associated with an increased burden of AD pathology at death in old age | |||||
| 26% of non dementia cases had CVA; 43% of dementia cases had CVA | |||||||
| CC75 + C [14] | 224 | 91 | CERAD | 22% of 113 dementia cases | |||
| Hisayama [15] | N = 469 | CERAD | NINDS-AIREN | 4.7% of dementia cases were mixed | |||
| 275 incident dementia cases | NIA-Reagan | ||||||
| (164 autopsies) | |||||||
| HAAS [16] | N = 443 | 86 ± 5.2 | Mean number of neurofibrillary tangles and neuritic plaques over 20 fields in 4 lobes | High correlations noted between MBIs and lacunar infarcts (Spearman r = 0.45, P < 0.0001) | 14.2% of dementia cases were mixed | No correlation between AD and microvascular lesions | CASI |
| (72-90+) | |||||||
| HAAS [17] | N = 436 | Number of infarcts > or <1.0 cm, microinfarcts | MBI found in 72% of demented and 61% of non-demented | ||||
| 144 with dementia | |||||||
| 292 without dementia | MBI and AD exert independent additive effects on cognition |
AD, Alzheimer disease; AGECAT, Automated Geriatric Examination for Computer Assisted Taxonomy; BLSA, Baltimore Longitudinal Aging Study; CASI, Cognitive Abilities Screening Instrument; CC75 + C, Cambridge City Over-75 s Cohort; CERAD, Consortium to Establish a Registry for Alzheimer Disease; CFAS, Cognitive Function and Ageing Study; CVA, cerebrovascular accident; HAAS, Honolulu Asia Aging Study; MAP, Rush Memory and Aging Project; MBI, microscopic brain infarct; MMSE, Mini-Mental State Exam; MRC, Medical Research Council; NIA, National Institute on Aging; NINDS-AIREN, National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences; ROS, Rush Religious Order Study; SD, standard deviation; VBI, vascular brain injury; VCI, vascular cognitive impairment.
Table 2.
Subcortical Ischemic Vascular Dementia (SIVD) Neuropathology study: imaging and clinical correlations
| Autopsy study | N/pathological diagnosis | Mean age (years) | Diagnosis of AD | Diagnosis of VBI | Prevalence of mixed | Neuropsychology | Interaction between VBI and AD on risk of dementia |
|---|---|---|---|---|---|---|---|
| Subcortical Ischemic Vascular Dementia (SIVD) study [18] | N = 61 | 78 (at time of neuropsych) | CERAD | CVD-PS | Linear composite measures | AD patient scores were lower than EXEC by nearly a standard deviation on average. VBI patients were rather equally impaired on EXEC,MEM and NVMEM | |
| AD = 23 | Braak and Braak ≥ IV | Infarct score ≥20 | |||||
| VBI = 11 | GLOB | ||||||
| Mixed = 9 | MEM | ||||||
| NSP = 19 | EXEC | ||||||
| SIVD study [19] | N = 79 | 82.8 ± 7.0 | NINDS-ADRDA criteria | ADDTC criteria | Clin Dx = 28% | Positive likelihood ratios: AD = 6.4 > VCI = 3.7 > mixed = 2.3 | |
| AD = 34 | At time of death | ||||||
| VBI = 15 | CERAD | CVD-PS | Path Dx = 11% | Cognitive status (CN, CIND, Dem) | AD pathology and hippocampal sclerosis have relatively greater effect on cognition than VBI | ||
| Mix = 9 | Braak and Braak ≥ IV | Infarct score ≥20 | |||||
| NSP = 21 | |||||||
| SIVD study [20] | N = 93 | 84 | Lacunes associated with WMH | ||||
| Normal = 12 | AD, atherosclerosis, and VBI contribute independently to cortical gray matter atrophy | ||||||
| AD = 46 | AD and hippocampal sclerosis are associated with hippocampal atrophy | ||||||
| VBI = 14 | |||||||
| Mixed = 9 | |||||||
| NSP = 12 | |||||||
| SIVD study [21] | N = 163 | 84 | Cerebral atherosclerosis was positively associated with microinfarcts (OR, 2.3; 95% CI, 1.2-4.4) and cystic infarcts (OR, 2.0; 95% CI, 1.0-4.2) but not AD pathology | ||||
| Normal = 23 | |||||||
| AD = 81 | Cerebral amyloid angiopathy was inversely associated with lacunar infarcts (OR, 0.6; 95% CI, 0.41-1.1), but positively associated with Braak and Braak stage (OR, 1.5; 95% CI, 1.1-2.1) and Consortium to Establish a Registry for Alzheimer Disease plaque score (OR, 1.5; 95% CI, 1.1-2.2) | ||||||
| VBI = 21 | |||||||
| Mixed = 15 | |||||||
| NSP = 23 | |||||||
| SIVD study (Zheng et al., manuscript submitted for publication) | N = 116 | 84 | Pathological measures of AD, atherosclerosis, and VBI contribute independently to MRI cortical gray matter atrophy and cognitive impairment. Path analyses show that the adverse effects of atherosclerosis on cognition are largely mediated through subcortical infarcts to brain atrophy, while the effects of AD on cognition work equally through cortical atrophy as well as through a yet unmeasured direct pathway | ||||
| Normal = 12 | |||||||
| AD = 53 | |||||||
| VBI = 18 | |||||||
| Mixed = 9 | |||||||
| NSP = 24 | |||||||
AD, Alzheimer disease; ADDTC, Alzheimer’s Disease Diagnostic and Treatment Centers; CERAD, Consortium to Establish a Registry for Alzheimer Disease; CI, confidence interval; CIND, Cognitive impairment not meeting criteria for dementia; Clin Dx, clinical diagnosis; CN, cognitively normal; CVD-PS, cerebrovascular disease-parenchymal score; Dem, dementia; EXEC, executive score; GLOB, global cognition score; MEM, memory score; NINDS-ADRDA, National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences; NSP, non-significant pathology; NVMEM, non-verbal memory; OR, odds ratio; Path Dx, pathological diagnosis; VBI, vascular brain injury; WMH, white matter hyperintensity.
The Rush Religious Orders Study (ROS) and the Rush Memory and Aging Project (MAP) constitute parallel, but independent, longitudinal clinical-pathological studies. Cases of clinically diagnosed probable AD often revealed a combination of AD plus other pathologies, especially VBI [8,22]. In the combined ROS and MAP autopsy samples (n = 804), mixed AD/VBI lesions were found in 16% of cases between ages 65 and 89 years, and grew to 28% after age 90 years [23]. Among cases with dementia, the prevalence of mixed AD/VCI was higher in the community-based MAP cohort (44%) than in the religiously defined ROS cohort (28%) [8]. The high prevalence of mixed AD/VBI and AD/VCI, especially in the oldest of old, underscores the importance of reducing VRFs as a public health priority.
In the Baltimore Longitudinal Aging Study (BLSA; n = 200), AD pathology alone accounted for 50% of the dementia seen in this cohort; hemispheral infarcts alone (VBI) or in conjunction with AD pathology (AD/VBI) accounted for 35%. In subjects with intermediate AD pathology scores, a single macroscopic hemispheral infarct was sufficient to cause dementia [10]. Atherosclerosis, brain infarcts, and AD pathology all contributed independently to risk of dementia [11]. Atherosclerosis scores were not correlated with AD pathology.
In the population-based Medical Research Council Cognitive Function and Ageing Study (CFAS; n = 456), the association between AD pathology (neuritic plaques and tangles) and cognitive status went down with age, while the association between atrophy and age continued to go up [12]. Self-reported VRFs were associated with infarcts, but not AD pathology [13]. Specifically, hypertension and heart attack were associated with microinfarcts in both dementia and non-dementia cases. In the Cambridge City over-75 s cohort, 22% of 118 dementia cases were classified as mixed AD/VBI [14].
In the population-based Hisayama Study, among 275 cases of incident dementia followed to autopsy, 45% were classified as AD, 30% as VCI, 5% as mixed AD/VCI and 4% as dementia with Lewy bodies [15]. The incidences of AD, combined dementia and other types of dementia rose with increasing age, particularly after the age of 85 years; this tendency was not observed for VCI or dementia with Lewy bodies. In an earlier autopsy study (n = 135), diabetes mellitus and insulin resistance were associated with neuritic plaques, but not neurofibrillary tangles [24].
In the Honolulu Asia Aging Study (HAAS) of older Japanese American men (n = 443 autopsies), microvascular lesions were the predominant lesion in 33% of dementia cases, AD the predominant lesion in 18.6% and mixed lesions (most often AD and VBI) in 14.2% [16]. The frequency of AD pathology and brain atrophy increased steadily after age 72 years (12 to 35% and 25 to 63%, respectively), whereas the frequency of microvascular lesions remained fairly constant at around 30% across the older age spectrum. Lewy bodies and hippocampal sclerosis jumped up in frequency to 15% and 10%, respectively, after age 80 years. The ratio of VBI to AD has been relatively higher in the HAAS cohort compared with other cohorts, raising the question of possible ethnic differences in gene-environment-brain interactions.
Based on the above community and cohort studies of prevalence, we conclude that mixed AD/VBI ranks in the top three most prevalent pathologies (with AD and VCI), well ahead of dementia with Lewy bodies and hippocampal sclerosis. Several studies have noted that the prevalence of AD pathology and atrophy continue to grow with advancing age, while the prevalence of infarcts remains more constant across the younger, middle and oldest of old. The prevalence of VBI compared with AD is higher in studies of Japanese (Hisayama and HAAS) compared with studies of predominantly Caucasians in the United States and United Kingdom (ROS, MAP, BLSA, CFAS, Cambridge City over-75 s cohort). However, among infarcts of various sizes, regardless of ethnicity, microinfarcts show the strongest correlation with cognitive impairment (HAAS, ROS, CFAS, BLSA). Consistently, autopsy studies have demonstrated that VBI and AD exert independent and additive effects on the risk of dementia.
Weighting the relative effects of VBI, AD, and other pathological lesions on cognition
Beyond knowing the relative prevalence of pathological lesions, it is essential to weight their importance for cognitive impairment. The two variables (prevalence and clinical relevance) are not necessarily correlated. In the combined Rush ROS and MAP (n = 856) [25], subjects with normal cognition at enrollment who were followed longitudinally (mean 7.5 years) had a high prevalence of neuropathological findings at autopsy (mean age 88 years): 99% evinced plaques or tangles; 36% had at least one gross infarct; 28% had at least one microinfarct; and 11% had neocortical Lewy bodies.
The relative weighting of these pathological lesions on longitudinal cognitive decline was also studied: 22% of the rate of decline was explained by global AD pathology, 6% by amyloid plaques, 34% by tangles, 2% by macroscopic infarcts, and 8% by neocortical Lewy bodies. When analyzed in conjunction, all pathological indices explained 41% of the total variation in cognitive performance. In these two studies combined, the rank order significance of pathologic lesions for cognitive decline was: tangles > Lewy bodies > amyloid plaques > macroscopic infarcts.
The prevalence and impact of microinfarcts was examined in the ROS (n = 425 autopsies) [26]. Microinfarcts were present in 36.5% of persons with dementia and 25.3% in persons without dementia. The presence of microinfarcts, especially in multiple cortical locations, increased the odds of dementia (odds ratio, 1.77; 95% confidence interval, 1.07 to 2.92) and lowered average global cognition (estimate, -0.287; standard error (SE), 0.113; P = 0.012). Microinfarcts were associated (in order of effect size) with lower perceptual speed (estimate, -0.400; SE, 0.117; P < 0.001), semantic memory (estimate, -0.391; SE, 0.130; P = 0.003), and episodic memory (estimate, -0.279; SE, 0.138; P = 0.044). These associations were not modified by the presence of macroscopic infarcts or AD pathology, suggesting that the effects of microinfarcts were independent. Of relevance, 58 of 129 (45%) people with microinfarcts did not have macroscopic infarcts, reminding us that we cannot rely on MRI to exclude VBI.
In the HAAS, 65% of autopsied cases harbored microscopic brain infarcts (MBIs), which contributed significantly and independently to brain atrophy and cognitive impairment, particularly before dementia was clinically evident [17]. Scalars were developed to represent the severity of five different pathologic lesions (AD, MBI, hippocampal sclerosis, Lewy bodies, and atrophy). Spearman rank correlations (r) between these scalars and the last Cognitive Abilities Screening Instrument score were all significant, as follows (in rank order): atrophy, r = -0.453; AD lesions, r = -0.299; hippocampal sclerosis, r = -0.200; MBI, r = -0.195; and Lewy bodies, r = -0.158 [27]. The five types of pathology explained 40% of the variance in the last Cognitive Abilities Screening Instrument score [17].
In the Subcortical Ischemic Vascular Dementia study, a cohort was recruited from university-affiliated memory clinics, enriched for people who had lacunes and WMHs on MRI (Table 2). The correlation between level of cognitive impairment was far stronger with AD pathology and hippocampal sclerosis than VBI [19]. The profile of cognitive impairment for neuropathologically defined mixed AD/subcortical vascular dementia (SVD) resembled that seen in AD cases (memory scores were lower than executive scores by nearly one standard deviation) rather than SVD (where all cognitive domains were impaired more or less equally) [18]. These finding suggest that, in general, when SVD is combined with AD, the effects of AD on severity and profile of cognitive impairment overwhelm those contributed by SVD.
Analyses of these longitudinal, clinical, neuropathological, and imaging studies have moved our understanding from prevalence to clinical relevance of various pathological hallmarks. These data confirm the malignancy of AD pathology and highlight the importance of microinfarcts as one form of VBI. They demonstrate the usefulness of multivariate, continuous approaches to understanding brain-behavior relationships. At the same time, they indicate the limitations of current neuroimaging and neuropathological measures to model and predict cognitive decline.
Pathological correlates of structural MRI in mixed AD/VBI
The advent of structural imaging computerized tomography (CT) scans in the 1970s and MRI scans in the 1980s) revolutionized our ability to visualize regional atrophy and large and small infarcts as well as WMHs and MBs [28]. A few longitudinal studies have attempted to validate structural MRI measures (for example, atrophy, WMHs) in reference to neuropathologic lesions and cognitive impairment. Pointedly, however, MRI scans at 1.5 and 3 T are not able to visualize microinfarcts, a form of VBI with demonstrated relevance to cognitive impairment. Recently, structural MRI at 7 T has revealed larger microinfarcts (for example, 0.7 mm diameter), whereas average microinfarcts (0.3 mm diameter) still fall below the detection threshold [29].
A variety of pathological changes in the brain parenchymal and vasculature have been ascribed to WMHs in late life (reviewed in [30]). Smooth, periventricular rims and punctuate lesions appear to have little clinical relevance. Irregular and confluent WMHs are correlated with a host of parenchymal changes (for example, variable loss of myelin and axons, and microglial and inflammatory changes) as well as a spectrum of vascular pathologies (for example, arteriolosclerosis, amyloid angiopathy, dilated perivascular spaces) [30]. Deep WMH lesions are thought to result from chronic hypoperfusion and hypoxia in terminal vascular beds fed by long, penetrating arterioles [31] and/or from breakdown of the blood-brain barrier and activation of matrix metalloproteinases [32,33]. In the Oregon Brain Aging Study, arteriolosclerosis (as opposed to atherosclerosis) was found to be the strongest correlate of WMHs [34].
In patients with AD, loss of fractional anisotropy, and increased mean and radial diffusivity are seen in white matter association tracts, especially the corpus callosum and cingulate and uncinate fasciculi [35,36]. These changes may be seen in the absence of WMHs, and most likely reflect secondary (Wallerian) degeneration from loss of cortical neurons/axons, but to some degree there is also a component of primary white matter degeneration [37]. The co-occurrence of WMHs in AD is commonly associated with the ApoE ε4 genotype, CAA, or arteriolosclerosis [30,34]. Thus, for the most part, WMHs in late life can be viewed as a marker for white matter change related to vasculopathy.
In the Subcortical Ischemic Vascular Dementia study, volumes of cortical gray matter, WMHs, hippocampus, and lacunes were obtained from structural MRI and correlated with pathology (Table 2). WMHs were straightforwardly associated with pathologic measures of vascular white matter injury; discrete lesions >3 mm and brighter than CSF correlated well with lacunar infarcts. On the other hand, neuropathology correlations for hippocampal volume and cortical gray matter were more complex. Hippocampal sclerosis and AD pathology explained 33% of the variance in hippocampal volume. A combination of AD pathology, arteriosclerosis, and subcortical VBI explained 25% of the variance in cortical gray matter [20].
To improve the clinical diagnosis of mixed AD/VBI, these findings suggest the requirement for a multi-variable and multi-modality algorithm. Structural MRI measures alone have limited sensitivity and specificity. WMHs, infarcts, and hemorrhages on MRI may be taken as suitable markers for VBI (minus microinfarcts).
Hippocampal volume is a suitable marker for AD (but could also mean hippocampal sclerosis). MRI atrophy, at least as a global measure, cannot be relied upon to differentiate a neurodegenerative versus vascular etiology. Several measures of amyloid and tau pathology (for example, amyloid and tau PET or CSF biomarkers) have been validated and would improve diagnostic specificity for AD.
Differentiation of mixed AD/VBI in the era of amyloid PET imaging
Prior to the arrival of amyloid PET imaging [38] and the validation of CSF biomarkers [39], autopsy was essential to disclose the neuropathological hallmarks of AD (namely plaques and tangles). Hence our decision to begin a review of mixed AD/VCI with neuropathologically defined AD (Tables 1 and 2). During the past few years, however, the detection of early AD changes beginning in preclinical stages has been improved by amyloid PET ligands (for example, Pittsburgh Imaging Compound B (PiB), florbetapir, flutemetamole, and so on) [40,41].
A team of investigators at Samsung Medical Center, Seoul, Korea, separated patients with severe WMHs (evidence of SVD) into two groups based on positive or negative retention of amyloid on PiB PET scans (Table 3). Mixed AD/SVD (31% of 45 patients) performed worse on delayed recall, had fewer lacunar infarcts and had greater hippocampal atrophy than pure SVD [42]. The group went on to test the ability of MRI shape analysis to discriminate mixed AD/SVD from pure SVD [43]. Among 68 patients with SVD defined by severe WMHs, 23 (33.8%) patients were defined as mixed AD/SVD based on positive PiB binding. With use of hippocampal shape analysis alone, mixed SVD could be distinguished from pure SVD with 95.7% sensitivity and 68.9% specificity. With use of amygdalar shape, the discrimination accuracy was 87.0% sensitivity and 68.9% specificity. The two groups could also be distinguished based on the shape of the hippocampus and amygdala.
Table 3.
Studies of mixed Alzheimer disease/vascular brain injury with Alzheimer disease defined by amyloid PET imaging
| Study | N | Age (years) | AD = PiB+ | VBI | Mixed AD/VBI | Mixed AD/VBI findings |
|---|---|---|---|---|---|---|
| Samsung [42] | N = 45 SVD severe WMH |
74.2 ± 7.6 | Pure SVD | Mixed AD/SVD | Mixed AD/SVD performed worse on delayed recall, had fewer lacunar infarcts and had greater hippocampal atrophy than pure SVD. | |
| N = 31 (69%) PiB- | N = 14 (31%) PiB+ | |||||
| Samsung [43] | N = 68 SVD | Pure SVD | Mixed AD/SVD | Differentiation of mixed AD/SVD from pure SVD: | ||
| N = 45 (66.2%) | N = 23 (33.8%) | Hippocampal shape analysis: 95.7% sensitivity; 68.9% specificity | ||||
| PiB- | PiB+ | |||||
| Amygdalar shape analysis: 87.0% sensitivity; 68.9% | ||||||
| Aging Brain Study [44] | N = 54 | 79 | PiB+ | VBI: WMHs and infarcts | No relationship between VBI (infarcts or WMHs) and PiB | |
| VBI is associated with impairment in EXEC | ||||||
| N = 33 PiB- | N = 27 VBI- | No relationship between PiB and cognition | ||||
| N = 21 PiB+ | N = 27 VBI+ | |||||
| Aging Brain Study [45] | N = 61 | 79 | VBI: Infarcts | Infarction, particularly in cortical and subcortical gray matter, was associated with lower cognitive performance in all domains (P < 0.05 for all comparisons) | ||
| N = 32 PiB- | N = 27 infarct- | |||||
| N = 29 PiB+ | N = 34 infarct+ | |||||
| Aging Brain Study [46] | N = 43 | 78.9 (6.7) | FCRP accounted for 16% of the variance in PiB index (P < 0.008) and the positive association remained significant controlling for age and sex, and apolipoprotein E genotype | |||
| Aging Brain Study [47] | N = 74 | 79 | Higher LDL-C and lower HDL-C levels were both associated with a higher PiB index, independent of apolipoprotein E genotype | |||
| 33 NCI | ||||||
| 38 MCI | ||||||
| 3 CDR1 | ||||||
| Aging Brain Study [48] | N = 67 | 79 | A relationship between Aβ and memory was mediated by cortical thickness. | |||
| 35 NCI | The relationship between Aβ and cortical thickness was eliminated after controlling for FCRP, except in PiB+ subjects (n = 22), where Aβ remained associated with thinner cortex in precuneus and occipital lobe | |||||
| 31 MCI | ||||||
| 1 CDR1 | Vascular risk and Aβ both contribute to cortical thickness | |||||
Aβ, amyloid-beta; AD, Alzheimer disease; CDR, Clinical Dementia Rating; EXEC, executive score; FCRP, Framingham Coronary Risk Profile; HDL-C, high density lipoprotein cholesterol; LDL-C, low density lipoprotein-cholesterol; MCI, mild cognitive impairment; NCI, no cognitive impairment; PET, positron emission tomography; PiB, Pittsburgh Imaging Compound B; SVD, subcortical vascular dementia; VBI, vascular brain injury; WMH, white matter hyperintensity.
In the Aging Brain Study [44,45], a longitudinal cohort enriched for vascular disease, no associations were found between measures of VBI on MRI and amyloid retention on PiB PET (Table 3). Participants with infarcts showed lower executive functioning (P = 0.001). Subcortical infarcts were inversely associated with declines in executive and memory function, whereas cortical infarcts were mainly associated with decline in executive function. Global PiB retention was associated with diminished non-verbal memory. Within this spectrum of normal aging to mild dementia, VBI and Aβ aggregation appeared to be independent processes with VBI having a greater impact on cognition than PiB retention. These studies illustrate how amyloid PET imaging will greatly improve our ability to recognize mixed AD/VCI cases of dementia.
Differentiation of mixed AD/VBI with CSF biomarkers
CSF biomarkers for Aβ42 and tau also provide molecular evidence for AD, but the findings are more difficult to interpret. In the Amsterdam Dementia Cohort, the presence of both MBs and WMHs was associated with lower CSF levels of Aβ42, while lacunar infarcts were associated with higher Aβ42 and lower tau (Table 4) [49]. The investigators concluded that the data supported a direct relationship between SVD and AD pathology. However, if type of SVD is considered (that is, atherosclerosis versus CAA), the direct relationship between MBs and WMHs and AD biomarkers might reflect underlying CAA and AD, driven in common by ApoE ε4 genotype.
Table 4.
Studies of mixed Alzheimer disease/vascular brain injury with Alzheimer disease defined by cerebrospinal fluid Aβ and phosphorylated tau
| Study | N | AD = | VCI/VBI | VBI and AD correlations |
|---|---|---|---|---|
| ADNI [50] | N = 819 | NINDS-ADRDA | VRFs | VRFs were not associated with AD biomarkers |
| 229 NCI | WMH | Increased time-varying WMHs were associated with faster decline in executive function and lower FDG uptake in NCI | ||
| 397 CI | ||||
| 193 AD | ||||
| Amsterdam Dementia Cohort [49] | N = 914 | NINDS-ADRDA | VCI by NINDS-AIREN criteria | The presence of both MBs and WMHs was associated with lower CSF levels of Aβ42, indicating a direct relationship between SVD and AD pathology (note: could SVD be CAA?) |
| 337 NCI | ||||
| 547 AD | ||||
| 30 VCI | The presence of lacunes was associated with higher Aβ42 in vascular dementia (standardized beta = 0.17, P = 0.07) and lower tau in AD (standardized beta = -0.07, P = 0.05) but there were no effects for Aβ42 or phosphorylated tau181 in AD (note: could SVD with lacunes be arteriolosclerosis?) |
Aβ, amyloid-beta; AD, Alzheimer disease; ADNI, Alzheimer’s Disease Neuroimaging Initiative; CAA, cerebral amyloid angiopathy; CI, cognitively impaired; CSF, cerebrospinal fluid; FDG, [18 F]fluorodeoxyglucose; MB, microbleed; NCI, no cognitive impairment; NINDS-ADRDA, National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences; NINDS-AIREN, National Institute of Neurological Disorders and Stroke-Association Internationale pour la Recherche et l’Enseignement en Neurosciences; SVD, subcortical vascular dementia; VBI, vascular brain injury; VCI, vascular cognitive impairment; VRF, vascular risk factor; WMH, white matter hyperintensity.
In the Alzheimer Disease Neuroimaging Initiative, VRFs were not associated with AD biomarkers (that is, CSF amyloid, [18 F]fluorodeoxyglucose (FDG) PET uptake or MRI hippocampal atrophy). In normal controls, progressive increases in WMH over time were associated with greater decline in executive function and lower FDG PET uptake (Table 4) [50]. One should keep in mind that the Alzheimer Disease Neuroimaging Initiative is focused on AD not CVD. The Framingham Coronary Risk Profile scores for the cohort were relatively low, approximately 18% across normal cognition, mild cognitive impairment and AD groups. Nonetheless, no interactions were noted between VRFs and AD biomarkers.
Other possible pathophysiological interactions between VRFs, CVD, and AD pathology
Converging evidence from epidemiologic [51-53], neuropathologic, amyloid PET, and CSF biomarker studies show that VBI and AD exert additive adverse effects on cognitive health (Figure 1). Do VRFs and CVD merely increase the co-occurrence of two separate processes (that is, AD and silent/symptomatic VBI), which shifts the syndromal diagnosis of dementia and AD earlier (reviewed in [9,54])? Or do VRFs and CVD potentiate AD-specific pathophysiological pathways, such as amyloidopathy and tauopathy? Several mechanisms have been postulated by which Aβ may be cleared in the brain: 1) enzymatic degradation (for example, neprilysin, insulin degrading enzyme) by microglia and astrocytes; 2) active transport from brain to blood through an endothelial lipoprotein receptor-related protein-mediated process [55-57]; and 3) passive transport through a perivascular ‘lymphatic-like’ drainage system of the brain [58,59]. Recently, associations between the accumulation of Aβ on PiB PET scans with serum cholesterol and aortic arterial stiffness have been described [46,47,60]. However, direct associations between Aβ clearance and levels of brain cholesterol or cerebral arteriolar/venous stiffness in human beings have yet to be shown. Thus, interactions at the pathophysiological level between VRFs/CVD and AD pathology, while plausible, are still unresolved.
Type of cerebrovascular disease matters for prevention and treatment
VCI and VBI refer to phenotypes rather than treatable etiologies. It should be apparent that clinical diagnosis must go further to identify the underlying type of CVD. While there are many possible types of CVD, the three major types are atherosclerosis, arteriolosclerosis, and CAA. Risk factors for atherosclerosis and arteriolosclerosis overlap considerably and include hypertension, diabetes mellitus, and hyperlipidemia, which are easily measured and effectively treated with medications and lifestyle modifications.
The location of some, but not all, structural imaging features may help to distinguish underlying arteriolosclerosis from CAA. WMHs are associated with both hypertensive arteriosclerosis and severe CAA, with evidence of a more posterior location favoring CAA [61,62]. Lacunar infarcts are more likely to be associated with arteriolosclerosis than with CAA [21]. The regional distribution of MBs may help to differentiate SVD and CAA; the occurrence of MBs in deep gray nuclei suggests SVD, while MBs in lobar or cortical regions suggest CAA [63,64]. Though promising, the sensitivity and specificity of categorizing strictly lobar as CAA, strictly deep as hypertensive vasculopathy, or both lobar and deep as coexisting CAA and hypertensive vasculopathy has not yet been studied [64].
Evidence-based treatment recommendations for VCI have been recently reviewed [1]. Also, based on comprehensive reviews of the literature, and assuming a causal relationship and intervention at an appropriate age to reduce the prevalence of each of seven risk factors by 10% per decade, it has been estimated that the prevalence of AD/dementia could be reduced by 8.3% worldwide by 2050 [53]. This would translate to 1.1 to 3 million AD/dementia cases worldwide and 184,000 to 492,000 cases in the USA [65]. These risk factors include five vascular risk factors (that is, midlife hypertension, diabetes mellitus, mid-life hyperlipidemia, smoking, sedentary life style), as well as depression and low educational attainment. Arguably the epidemiological diagnosis of AD may well include cases with subclinical VBI or mixed AD/VBI. Regardless of etiological label, assuming internal consistency in the definitions of risk factors and the diagnosis of dementia subtype, the epidemiological data present a valid and worthy public health objective, namely a 10% reduction in VRFs for 10 years for an 8% reduction in dementia.
The major risk factor for CAA is the ApoE ε4 allele, which can be determined readily by genotyping, though is not yet recommended by best practice guidelines. Strategies to reduce risks related to the ApoE ε4 genotype have been explored in cultured neurons [66] and in animal models of AD [67]. Bexaratone reduces amyloid plaques in animals, but no clinical trials have been conducted in humans.
Conclusion
The co-occurrence of AD and VBI in elderly persons is very common. There is now a large body of evidence showing that AD and VBI lead in an additive and independent fashion to cognitive dysfunction. In AD, there is a characteristic pattern of tau-related, neurofibrillary neurodegeneration spreading from the medial temporal lobe to other multi-modal association areas and a corresponding pattern of memory loss spreading to other cognitive domains. By contrast, there is tremendous variation in the neuropsychological profile associated with VBI. In the SVD subtype of VCI, executive dysfunction often equals or may exceed memory impairment, but depending on location all varieties of cognitive impairment may ensue.
WMHs, small and large infarcts and hemorrhages are visible on structural MRI and CT imaging and currently serve as the most reliable marker for VBI. However, microinfarcts, which have been most strongly related to cognitive impairment in neuropathology studies, continue to elude clinical detection. The validation of amyloid PET imaging adds considerable specificity for the diagnosis of AD, beyond the long recognized atrophy of the hippocampus. The addition of tau PET imaging is expected shortly. Global measures of atrophy on MRI are important markers of overall brain injury, but cannot be used reliably to determine etiology.
Longitudinal studies with repeat neuropsychological testing support multivariate approaches to model the effects of various types of pathology on dementia risk and cognitive decline. Once comparable measures of AD and VBI pathology are available from in vivo neuroimaging studies, we can anticipate that one day dichotomous classifications will be replaced by more sophisticated modeling. Still, the best models available today predict less than half of the variance in cognitive performance.
For prevention and treatment, it is important to bear in mind the type of CVD underlying VBI and VCI, as well as to consider that subclinical CVD and VBI may contribute additively to cognitive impairment in patients with AD. Epidemiological data suggest that attention to ‘life’s simple seven’, referring to seven health factors and lifestyle behaviors identified by the American Heart Association that include being physically active, eating foods low in cholesterol and saturated fats, monitoring high blood pressure and blood sugar, maintaining a healthy weight, controlling cholesterol, and avoiding tobacco smoking, can significantly reduce the risk of dementia.
Note
This article is part of a series on Cerebral multi-morbidity of the aging brain edited by Johannes Attems and Julie Schneider. Other articles in the series can be found at http://alzres.com/series/cerebral_multimorbidity.
Acknowledgements
This work was supported by the National Institute on Aging: 1P01AG12435, 1P50AG05142.
Abbreviations
- Aβ
Amyloid-beta
- AD
Alzheimer disease
- ApoE
Apolipoprotein E
- BLSA
Baltimore Longitudinal Aging Study
- CAA
Cerebral amyloid angiopathy
- CFAS
Cognitive Function and Ageing Study
- CSF
Cerebrospinal fluid
- CT
Computerized tomography
- CVD
Cerebrovascular disease
- FDG
[18 F]fluorodeoxyglucose
- HAAS
Honolulu Asia Aging Study
- MAP
Memory and Aging Project
- MB
Microbleed
- MBI
Microscopic brain infarct
- MRI
Magnetic resonance imaging
- PET
Positron emission tomography
- PiB
Pittsburgh Imaging Compound B
- ROS
Religious Orders Study
- SE
Standard error
- SVD
subcortical vascular dementia
- VBI
Vascular brain injury
- VCI
Vascular cognitive impairment
- VRF
Vascular risk factor
- WMH
White matter hyperintensity
Footnotes
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Helena C Chui, Email: chui@usc.edu.
Liliana Ramirez-Gomez, Email: liliana.ramirezgomez@med.usc.edu.
References
- 1.Gorelick PB, Scuteri A, Black SE, Decarli C, Greenberg SM, Iadecola C, et al. Vascular contributions to cognitive impairment and dementia: a statement for healthcare professionals from the American Heart Association/American Stroke Association. Stroke. 2011;42:2672–713. doi: 10.1161/STR.0b013e3182299496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sperling RA, Aisen PS, Beckett LA, Bennett DA, Craft S, Fagan AM, et al. Toward defining the preclinical stages of Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:280–92. doi: 10.1016/j.jalz.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Albert MS, DeKosky ST, Dickson D, Dubois B, Feldman HH, Fox NC, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:270–9. doi: 10.1016/j.jalz.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Jr, Kawas CH, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7:263–9. doi: 10.1016/j.jalz.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tomlinson BE, Blessed G, Roth M. Observations on the brains of non-demented old people. J Neurol Sci. 1968;7:331–56. doi: 10.1016/0022-510X(68)90154-8. [DOI] [PubMed] [Google Scholar]
- 6.Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. J Neurol Sci. 1970;11:205–42. doi: 10.1016/0022-510X(70)90063-8. [DOI] [PubMed] [Google Scholar]
- 7.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease: The Nun Study. JAMA. 1997;277:813–7. doi: 10.1001/jama.1997.03540340047031. [DOI] [PubMed] [Google Scholar]
- 8.Schneider JA, Aggarwal NT, Barnes L, Boyle P, Bennett DA. The neuropathology of older persons with and without dementia from community versus clinic cohorts. J Alzheimers Dis. 2009;18:691–701. doi: 10.3233/JAD-2009-1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bennett DA, Wilson RS, Arvanitakis Z, Boyle PA, de Toledo-Morrell L, Schneider JA. Selected findings from the Religious Orders Study and Rush Memory and Aging Project. J Alzheimers Dis. 2013;33:S397–403. doi: 10.3233/JAD-2012-129007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Troncoso JC, Zonderman AB, Resnick SM, Crain B, Pletnikova O, O’Brien RJ. Effect of infarcts on dementia in the Baltimore longitudinal study of aging. Ann Neurol. 2008;64:168–76. doi: 10.1002/ana.21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dolan H, Crain B, Troncoso J, Resnick SM, Zonderman AB, Obrien RJ. Atherosclerosis, dementia, and Alzheimer disease in the Baltimore Longitudinal Study of Aging cohort. Ann Neurol. 2010;68:231–40. doi: 10.1002/ana.22055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Savva GM, Wharton SB, Ince PG, Forster G, Matthews FE, Brayne C. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302–9. doi: 10.1056/NEJMoa0806142. [DOI] [PubMed] [Google Scholar]
- 13.Richardson K, Stephan BC, Ince PG, Brayne C, Matthews FE, Esiri MM. The neuropathology of vascular disease in the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Curr Alzheimer Res. 2012;9:687–96. doi: 10.2174/156720512801322654. [DOI] [PubMed] [Google Scholar]
- 14.Brayne C, Richardson K, Matthews FE, Fleming J, Hunter S, Xuereb JH, et al. Neuropathological correlates of dementia in over-80-year-old brain donors from the population-based Cambridge city over-75 s cohort (CC75C) study. J Alzheimers Dis. 2009;18:645–58. doi: 10.3233/JAD-2009-1182. [DOI] [PubMed] [Google Scholar]
- 15.Matsui Y, Tanizaki Y, Arima H, Yonemoto K, Doi Y, Ninomiya T, et al. Incidence and survival of dementia in a general population of Japanese elderly: the Hisayama study. J Neurol Neurosurg Psychiatry. 2009;80:366–70. doi: 10.1136/jnnp.2008.155481. [DOI] [PubMed] [Google Scholar]
- 16.White L. Brain lesions at autopsy in older Japanese-American men as related to cognitive impairment and dementia in the final years of life: a summary report from the Honolulu-Asia aging study. J Alzheimers Dis. 2009;18:713–25. doi: 10.3233/JAD-2009-1178. [DOI] [PubMed] [Google Scholar]
- 17.Launer LJ, Hughes TM, White LR. Microinfarcts, brain atrophy, and cognitive function: the Honolulu Asia Aging Study Autopsy Study. Ann Neurol. 2011;70:774–80. doi: 10.1002/ana.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reed BR, Mungas DM, Kramer JH, Ellis W, Vinters HV, Zarow C, et al. Profiles of neuropsychological impairment in autopsy-defined Alzheimer’s disease and cerebrovascular disease. Brain. 2007;130:731–9. doi: 10.1093/brain/awl385. [DOI] [PubMed] [Google Scholar]
- 19.Chui HC, Zarow C, Mack WJ, Ellis WG, Zheng L, Jagust WJ, et al. Cognitive impact of subcortical vascular and Alzheimer’s disease pathology. Ann Neurol. 2006;60:677–87. doi: 10.1002/ana.21009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jagust WJ, Zheng L, Harvey DJ, Mack WJ, Vinters HV, Weiner MW, et al. Neuropathological basis of magnetic resonance images in aging and dementia. Ann Neurol. 2008;63:72–80. doi: 10.1002/ana.21296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng L, Vinters HV, Mack WJ, Zarow C, Ellis WG, Chui HC. Cerebral atherosclerosis is associated with cystic infarcts and microinfarcts but not Alzheimer pathologic changes. Stroke. 2013;44:2835–41. doi: 10.1161/STROKEAHA.113.001945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community-dwelling older persons. Neurology. 2007;69:2197–204. doi: 10.1212/01.wnl.0000271090.28148.24. [DOI] [PubMed] [Google Scholar]
- 23.James BD, Bennett DA, Boyle PA, Leurgans S, Schneider JA. Dementia from Alzheimer disease and mixed pathologies in the oldest old. JAMA. 2012;307:1798–800. doi: 10.1001/jama.2012.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsuzaki T, Sasaki K, Tanizaki Y, Hata J, Fujimi K, Matsui Y, et al. Insulin resistance is associated with the pathology of Alzheimer disease: the Hisayama study. Neurology. 2010;75:764–70. doi: 10.1212/WNL.0b013e3181eee25f. [DOI] [PubMed] [Google Scholar]
- 25.Boyle PA, Wilson RS, Yu L, Barr AM, Honer WG, Schneider JA, et al. Much of late life cognitive decline is not due to common neurodegenerative pathologies. Ann Neurol. 2013;74:478–89. doi: 10.1002/ana.23964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arvanitakis Z, Leurgans SE, Barnes LL, Bennett DA, Schneider JA. Microinfarct pathology, dementia, and cognitive systems. Stroke. 2011;42:722–7. doi: 10.1161/STROKEAHA.110.595082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.White L, Small BJ, Petrovitch H, Ross GW, Masaki K, Abbott RD, et al. Recent clinical-pathologic research on the causes of dementia in late life: update from the Honolulu-Asia Aging Study. J Geriatr Psychiatry Neurol. 2005;18:224–7. doi: 10.1177/0891988705281872. [DOI] [PubMed] [Google Scholar]
- 28.Patel B, Markus HS. Magnetic resonance imaging in cerebral small vessel disease and its use as a surrogate disease marker. Int J Stroke. 2011;6:47–59. doi: 10.1111/j.1747-4949.2010.00552.x. [DOI] [PubMed] [Google Scholar]
- 29.van Veluw SJ, Zwanenburg JJ, Engelen-Lee J, Spliet WG, Hendrikse J, Luijten PR, et al. In vivo detection of cerebral cortical microinfarcts with high-resolution 7 T MRI. J Cereb Blood Flow Metab. 2013;33:322–9. doi: 10.1038/jcbfm.2012.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gouw AA, Seewann A, van der Flier WM, Barkhof F, Rozemuller AM, Scheltens P, et al. Heterogeneity of small vessel disease: a systematic review of MRI and histopathology correlations. J Neurol Neurosurg Psychiatry. 2011;82:126–35. doi: 10.1136/jnnp.2009.204685. [DOI] [PubMed] [Google Scholar]
- 31.Pantoni L, Garcia JH. Pathogenesis of leukoaraiosis: a review. Stroke. 1997;28:652–9. doi: 10.1161/01.STR.28.3.652. [DOI] [PubMed] [Google Scholar]
- 32.Taheri S, Gasparovic C, Shah NJ, Rosenberg GA. Quantitative measurement of blood-brain barrier permeability in human using dynamic contrast-enhanced MRI with fast T1 mapping. Magn Reson Med. 2011;65:1036–42. doi: 10.1002/mrm.22686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenberg GA, Bjerke M, Wallin A. Multimodal markers of inflammation in the subcortical ischemic vascular disease type of vascular cognitive impairment. Stroke. 2014;45:1531–8. doi: 10.1161/STROKEAHA.113.004534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Erten-Lyons D, Woltjer R, Kaye J, Mattek N, Dodge HH, Green S, et al. Neuropathologic basis of white matter hyperintensity accumulation with advanced age. Neurology. 2013;81:977–83. doi: 10.1212/WNL.0b013e3182a43e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Alves GS, O’Dwyer L, Jurcoane A, Oertel-Knöchel V, Knöchel C, Prvulovic D, et al. Different patterns of white matter degeneration using multiple diffusion indices and volumetric data in mild cognitive impairment and Alzheimer patients. PLoS One. 2012;7:e52859. doi: 10.1371/journal.pone.0052859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Amlien IK, Fjell AM. Diffusion tensor imaging of white matter degeneration in Alzheimer’s disease and mild cognitive impairment. Neuroscience. 2014;276C:206–15. doi: 10.1016/j.neuroscience.2014.02.017. [DOI] [PubMed] [Google Scholar]
- 37.Bozzali M, Falini A, Franceschi M, Cercignani M, Zuffi M, Scotti G, et al. White matter damage in Alzheimer’s disease assessed in vivo using diffusion tensor magnetic resonance imaging. J Neurol Neurosurg Psychiatry. 2002;72:742–6. doi: 10.1136/jnnp.72.6.742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 39.Jack CR, Jr, Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kantarci K. Molecular Imaging of Alzheimer Disease Pathology. AJNR Am J Neuroradiol. 2014;35:S12–7. doi: 10.3174/ajnr.A3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Landau SM, Thomas BA, Thurfjell L, Schmidt M, Margolin R, Mintun M, et al. Amyloid PET imaging in Alzheimer’s disease: a comparison of three radiotracers. Eur J Nucl Med Mol Imaging. 2014;41:1398–407. doi: 10.1007/s00259-014-2753-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee JH, Kim SH, Kim GH, Seo SW, Park HK, Oh SJ, et al. Identification of pure subcortical vascular dementia using 11C-Pittsburgh compound B. Neurology. 2011;77:18–25. doi: 10.1212/WNL.0b013e318221acee. [DOI] [PubMed] [Google Scholar]
- 43.Kim HJ, Kim J, Cho H, Ye BS, Yoon CW, Noh Y, et al. Individual subject classification of mixed dementia from pure subcortical vascular dementia based on subcortical shape analysis. PLoS One. 2013;8:e75602. doi: 10.1371/journal.pone.0075602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marchant NL, Reed BR, DeCarli CS, Madison CM, Weiner MW, Chui HC, et al. Cerebrovascular disease, beta-amyloid, and cognition in aging. Neurobiol Aging. 2012;33:1006 e1025-1036. doi: 10.1016/j.neurobiolaging.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Marchant NL, Reed BR, Sanossian N, Madison CM, Kriger S, Dhada R, et al. The aging brain and cognition: contribution of vascular injury and abeta to mild cognitive dysfunction. JAMA Neurol. 2013;70:488–95. doi: 10.1001/2013.jamaneurol.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reed BR, Marchant NL, Jagust WJ, DeCarli CC, Mack W, Chui HC. Coronary risk correlates with cerebral amyloid deposition. Neurobiol Aging. 2012;33:1979–87. doi: 10.1016/j.neurobiolaging.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71:195–200. doi: 10.1001/jamaneurol.2013.5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villeneuve S, Reed BR, Wirth M, Haase CM, Madison CM, Ayakta N, et al. Cortical thickness mediates the effect of beta-amyloid on episodic memory. Neurology. 2014;82:761–7. doi: 10.1212/WNL.0000000000000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kester MI, Goos JD, Teunissen CE, Benedictus MR, Bouwman FH, Wattjes MP, et al. Associations between cerebral small-vessel disease and Alzheimer disease pathology as measured by cerebrospinal fluid biomarkers. JAMA Neurol. 2014;71:855–62. doi: 10.1001/jamaneurol.2014.754. [DOI] [PubMed] [Google Scholar]
- 50.Lo RY, Jagust WJ, Alzheimer’s Disease Neuroimaging I Vascular burden and Alzheimer disease pathologic progression. Neurology. 2012;79:1349–55. doi: 10.1212/WNL.0b013e31826c1b9d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Luchsinger JA, Reitz C, Honig LS, Tang MX, Shea S, Mayeux R. Aggregation of vascular risk factors and risk of incident Alzheimer disease. Neurology. 2005;65:545–51. doi: 10.1212/01.wnl.0000172914.08967.dc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meng XF, Yu JT, Wang HF, Tan MS, Wang C, Tan CC, et al. Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014;42:1295–310. doi: 10.3233/JAD-140954. [DOI] [PubMed] [Google Scholar]
- 53.Norton S, Matthews FE, Barnes DE, Yaffe K, Brayne C. Potential for primary prevention of Alzheimer’s disease: an analysis of population-based data. Lancet Neurol. 2014;13:788–94. doi: 10.1016/S1474-4422(14)70136-X. [DOI] [PubMed] [Google Scholar]
- 54.Chui HC, Zheng L, Reed BR, Vinters HV, Mack WJ. Vascular risk factors and Alzheimer’s disease: are these risk factors for plaques and tangles or for concomitant vascular pathology that increases the likelihood of dementia? An evidence-based review. Alzheimers Res Ther. 2012;3:36. doi: 10.1186/alzrt98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holtzman DM, Herz J, Bu G. Apolipoprotein E and apolipoprotein E receptors: normal biology and roles in Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2:a006312. doi: 10.1101/cshperspect.a006312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zlokovic BV. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat Rev Neurosci. 2011;12:723–38. doi: 10.1038/nrn3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zlokovic BV. Cerebrovascular effects of apolipoprotein E: implications for Alzheimer disease. JAMA Neurol. 2013;70:440–4. doi: 10.1001/jamaneurol.2013.2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 59.Xie L, Kang H, Xu Q, Chen MJ, Liao Y, Thiyagarajan M, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hughes TM, Kuller LH, Barinas-Mitchell EJ, Mackey RH, McDade EM, Klunk WE, et al. Pulse wave velocity is associated with beta-amyloid deposition in the brains of very elderly adults. Neurology. 2013;81:1711–8. doi: 10.1212/01.wnl.0000435301.64776.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, et al. Cerebral amyloid angiopathy burden associated with leukoaraiosis: a positron emission tomography/magnetic resonance imaging study. Ann Neurol. 2013;73:529–36. doi: 10.1002/ana.23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Thanprasertsuk S, Martinez-Ramirez S, Pontes-Neto OM, Ni J, Ayres A, Reed A, et al. Posterior white matter disease distribution as a predictor of amyloid angiopathy. Neurology. 2014;83:794–800. doi: 10.1212/WNL.0000000000000732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Park JH, Seo SW, Kim C, Kim GH, Noh HJ, Kim ST, et al. Pathogenesis of cerebral microbleeds: in vivo imaging of amyloid and subcortical ischemic small vessel disease in 226 individuals with cognitive impairment. Ann Neurol. 2013;73:584–93. doi: 10.1002/ana.23845. [DOI] [PubMed] [Google Scholar]
- 64.Martinez-Ramirez S, Greenberg SM, Viswanathan A. Cerebral microbleeds: overview and implications in cognitive impairment. Alzheimers Res Ther. 2014;6:33. doi: 10.1186/alzrt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barnes DE, Yaffe K. The projected effect of risk factor reduction on Alzheimer’s disease prevalence. Lancet Neurol. 2011;10:819–28. doi: 10.1016/S1474-4422(11)70072-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen HK, Liu Z, Meyer-Franke A, Brodbeck J, Miranda RD, McGuire JG, et al. Small molecule structure correctors abolish detrimental effects of apolipoprotein E4 in cultured neurons. J Biol Chem. 2012;287:5253–66. doi: 10.1074/jbc.M111.276162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cramer PE, Cirrito JR, Wesson DW, Lee CY, Karlo JC, Zinn AE, et al. ApoE-directed therapeutics rapidly clear beta-amyloid and reverse deficits in AD mouse models. Science. 2012;335:1503–6. doi: 10.1126/science.1217697. [DOI] [PMC free article] [PubMed] [Google Scholar]