

A novel signaling pathway consisting of Gai, PLC, PKCβ, PKD, SSH2, and cofilin is crucial for GPCR-mediated chemotaxis in neutrophils. This pathway regulates depolymerization of the actin network that drives the directional migration of neutrophils.
Abstract
Chemotaxis requires precisely coordinated polymerization and depolymerization of the actin cytoskeleton at leading fronts of migrating cells. However, GPCR activation-controlled F-actin depolymerization remains largely elusive. Here, we reveal a novel signaling pathway, including Gαi, PLC, PKCβ, protein kinase D (PKD), and SSH2, in control of cofilin phosphorylation and actin cytoskeletal reorganization, which is essential for neutrophil chemotaxis. We show that PKD is essential for neutrophil chemotaxis and that GPCR-mediated PKD activation depends on PLC/PKC signaling. More importantly, we discover that GPCR activation recruits/activates PLCγ2 in a PI3K-dependent manner. We further verify that PKCβ specifically interacts with PKD1 and is required for chemotaxis. Finally, we identify slingshot 2 (SSH2), a phosphatase of cofilin (actin depolymerization factor), as a target of PKD1 that regulates cofilin phosphorylation and remodeling of the actin cytoskeleton during neutrophil chemotaxis.
INTRODUCTION
Chemotaxis, a chemoattractant gradient–directed cell migration, plays critical roles in many physiological processes, such as neuron patterning, immune response, angiogenesis, and metastasis of cancer cells. Eukaryotic cells detect many chemoattractants by G protein–coupled receptors (GPCRs). Activation of chemokine GPCRs promotes the dissociation of heterotrimeric G proteins into Gα and Gβγ subunits, which, in turn, activate downstream signal transduction pathways that ultimately regulate the spatiotemporal organization of the actin cytoskeleton (Jin et al., 2008). In fast-migrating amoeba-like cells such as human neutrophils, the actin network forms branched filaments leading to sheet-like protrusions at the leading edge of chemotaxing cells. Chemokine GPCRs control multiple signaling pathways to activate the Rho family of small GTPases (cdc42 and Rac1/2) and promote the growth of “new” F-actin filaments by removing capping proteins and stimulating the Arp2/3 complexes that initiate the formation of new actin branches from existing ones (Li et al., 2000, 2003, 2005; Servant et al., 2000; Welch et al., 2002; Sun et al., 2007; Gan et al., 2012). Whereas the front of the actin sheet continuously extends by forming a “new” dendritic actin network, the “old” F-actin meshwork at the rear of the leading edge depolymerizes F-actin to provide actin monomers for rapid and continuous assembly of the F-actin network in the leading front of chemotaxing cells. Compared to the extensive studies that have been done on the signaling pathway and molecular mechanism of actin polymerization, little is known about how chemokine GPCRs control the depolymerization of actin cytoskeleton in neutrophils.
Cofilin is one well-known F-actin depolymerization factor (ADF; Mizuno, 2013). The activity of cofilin is regulated mainly through phosphorylation and dephosphorylation: phosphorylation at Ser‑3 by LIM kinases and testicular protein kinases (TESKs) inhibits its actin-binding, -severing, and -depolymerizing activities (Arber et al., 1998; Yang et al., 1998; Toshima et al., 2001a, b), and dephosphorylation at Ser‑3 by slingshot proteins (SSHs) and chronophin reactivates cofilin (Niwa et al., 2002; Ohta et al., 2003; Gohla et al., 2005). In neutrophils, chemoattractants induce rapid dephosphorylation/activation of cofilin (van Rheenen et al., 2009). A PLCβ/phosphoinositide 3-kinase γ (PI3Kγ)‑GSK3 signaling pathway was recently discovered to regulate cofilin phosphatase slingshot 2 (SSH2), which in turn regulates cofilin dephosphorylation (Tang et al., 2011). In spite of this progress, we still do not fully understand the signal transduction pathways that link a chemokine GPCR to the phosphorylation/dephosphorylation of cofilin in neutrophils.
Phospholipase C (PLC) activation is a key event in response to a wide range of chemoattractants and generates two important second messengers: diacylglycerol (DAG) and inositol-3,4,5-triphosphate (IP3; Suh et al., 2008). The role of PLCs in neutrophil chemotaxis has been studied (Jiang et al., 1997; Li et al., 2000; Tang et al., 2011). An early study reported that PLCβ2/3 activation is essential for Ca2+ signaling and oxidative burst in neutrophils, whereas murine neutrophils deficient in both PLCβ2/3 were still able to chemotax (Li et al., 2000). It thus appeared that PLC signaling is not essential for neutrophil chemotaxis. However, a later report indicated that PLCβ plays a role upstream of the GSK3 signaling pathway in regulating SSH2 activity in neutrophil chemotaxis (Tang et al., 2011). Although PLCγ2 is highly expressed in neutrophils (Suh et al., 2008), its role in neutrophil chemotaxis is not known. Therefore, the role of PLC signaling in neutrophil chemotaxis requires further investigation.
The protein kinase D (PKD) family of serine/threonine kinases plays critical roles in many physiological processes, including cell growth, protein trafficking, and lymphocyte biology (Wang, 2006). A study showed that PKD1 phosphorylates SSH1 and regulates cofilin to mediate F-actin depolymerization in slow-moving breast cancer cells (Eiseler et al., 2009). PKD isoforms are also highly expressed in immune cells, including neutrophils (Balasubramanian et al., 2002). However, it is not clear whether PKD activation plays a role in neutrophil chemotaxis and whether or how PKD is controlled by chemoattractant GPCR signaling.
In the present study, we show that PKDs are required for neutrophil chemotaxis. We find that chemoattractant GPCR controls spatiotemporal activation of PKDs and that GPCR-mediated PKD activation requires PLC/PKC signaling. GPCR activation recruits PLCγ2 to the leading edge of neutrophil cells and activates PLCγ2 in a PI3K-dependent manner. We show that PLC signaling is required for chemoattractant GPCR-mediated activation of PKC and PKD and also important for chemoattractant-induced PKD activation in neutrophils. Inhibitors that block PLC, PKC, or PKD abolish neutrophil chemotaxis, indicating that the signaling of PLC, PKC, and PKD is required for neutrophil chemotaxis. Furthermore, we show that PKCβ specifically interacts with PKD and is essential for neutrophil chemotaxis. Finally, we find that PKD1 interacts with SSH2 to regulate cofilin activity. Integrating this new information, we identify a novel signaling pathway consisting of a chemoattractant GPCR/Gαi protein, PLC, PKCβ, and PKDs that regulates SSH2/cofilin activity in neutrophil chemotaxis. We propose that this pathway transduces signals from the chemoattractant GPCR to promote F-actin polymerization and thus coordinates the directional assembly of the actin cytoskeleton that drives neutrophil chemotaxis.
RESULTS
PKDs contribute to GPCR-controlled chemotaxis of leukocytes
To examine the role of PKDs in leukocyte chemotaxis, we determined the expression profile of PKDs (PKD1, PKD2, and PKD3) in a panel of leukocyte cell lines (Figure 1A). We found that human leukocyte cell line HL60 expressed all three PKD isoforms (Figure 1A and Supplemental Figure S1), and other cell lines expressed one or more isoforms. HL60 is a well-established cell line for leukocyte chemotaxis assay (Millius and Weiner, 2009). Using the EZ-TAXI Scan assay (Supplemental Figure S2), we showed that HL60 cells displayed robust chemotaxis in a 100 nM f-Met-Leu-Phe (fMLP) gradient (Figure 1B). We also found that chemotaxis of HL60 cells was severely impaired by either of two PKD inhibitors (CID755673 or Gö 6976; Gschwendt et al., 1996; Sharlow et al., 2008; Figure 1, B and C). Treatment of PKD inhibitors did not block fMLP-induced Ca2+ responses in HL60 cells (Supplemental Figure S3 and Supplemental Figure S3 Supplemental Movies 1–6), indicating that PKD inhibitors did not abolish chemoattractant-induced GPCR signaling in general.
FIGURE 1:

The PKD plays essential roles in neutrophil chemotaxis. (A) Expression profile of PKD family members in variant cell lines. (B) Scheme showing the chemotaxing paths of HL60 cells in variant conditions monitored by EZ-TaxiScan. (C) Quantification of chemotaxis parameters as total travel path, speed, and directionality using DYIS software. (D) Establishment of a stable pkd1-knockdown clones (pkd1kd) using shRNA virus particles. (E) Decreased expression of PKD1 is correlated with a decreased chemotaxis capability of Raw 264.7 cells. (F, G) Dose-dependent inhibitory effect of PKD1 inhibitors (CID755673 and Gö 6976) on Raw 264.7 cell chemotaxis. SDF1α 100 nM was used for the Transwell chemotaxis assay presented.
We next examined the role of PKDs in the chemotaxis of the murine leukemia cell line Raw 264.7, which expresses predominantly PKD1 but little of the other isoforms (Figure 1A and Supplemental Figure S1). We established a stable PKD1-knockdown clone (pkd1kd) using short hairpin RNA interference (shRNAi) technology (Figure 1D). Knocking down PKD1 expression did not affect the expression profile of either PKD isoforms or the chemokine receptor CXCR4 in pkd1kd cells (Supplemental Figure S4). Control Raw264.7 cells displayed a clear dose-dependent chemotaxis in response to a gradient of CXCR4 ligand SDF1α (stromal cell–derived factor-1α), using a Transwell assay (Supplemental Figure S5). Decreased expression of PKD1 in pkd1kd cells resulted in reduced cell migration in SDF1α gradient (Figure 1E). We also found that treatment with PKD inhibitor Gö 6976 or CID755673 significantly impaired chemotaxis of Raw 264.7 cells (Figure 1, F and G). However, Raw 264.7 cells, unlike HL60 cells, did not display robust chemotaxis behavior in the EZ-TAXI Scan assay, which provides detailed measurements of multiple chemotaxis parameters, including total travel path, speed, and directionality. Therefore, we chose HL60 cells to further study the role of PKDs in chemotaxis.
Chemoattractants trigger dynamic membrane translocation and activation of PKDs
To study the function of PKDs in leukocyte chemotaxis, we ectopically expressed PKD1–green fluorescent protein (GFP), PKD2–cyan fluorescent protein (CFP), or PKD3-GFP in HL60 cells (Figure 2A) and examined their cellular localization in response to chemoattractant stimuli. We found that all three PKDs mainly localized to the cytosol in resting cells and transiently translocated to the plasma membrane in response to a uniformly applied fMLP stimulus and then localized at the leading edge of polarized HL60 cells (Figure 2, B and C, and Figure 2 Supplemental Movies 1–4). We also observed similar membrane-targeting dynamics upon uniform stimulation with SDF1α (Figure S6, A and B, and Figure S6A Supplemental Movies 1–4), indicating that the activation of chemoattractant Gαi-GPCRs generally controls the membrane translocation of PKDs. A previous study showed that activation of a Gαq-coupled GPCR induced PKD1 phosphorylation, a critical marker of PKD1 activation (Jacamo et al., 2008). We found that fMLP stimulation also triggered phosphorylation of PKD1 (Figure 2D). In either an fMLP or an SDF1α gradient, chemotaxing cells actively recruited all three PKD proteins to the leading edge (Figure 2E, Figure 2 Supplemental Movies 5–8, Supplemental Figure S6C, and Figure S6 Supplemental Movies 5–8), suggesting that chemoattractant GPCRs control the spatial localization and activation of PKD in chemotaxing neutrophils.
FIGURE 2:
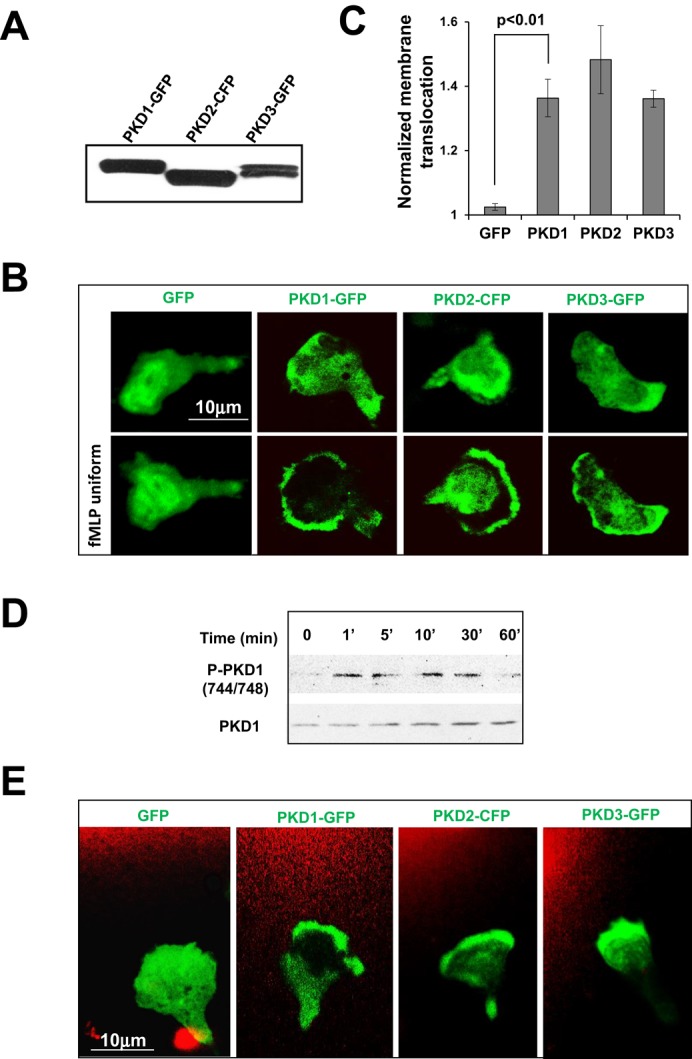
Chemoattractant-induced GPCR signaling activates PKD. (A) Transient expression of GFP- or CFP-tagged PKD1 (PKD1-GFP), PKD2 (PKD2-CFP), or PKD3 (PKD3-GFP) (green) in HL60 cells. (B) Montage shows the membrane translocation of PKD members (green) upon uniform application of 1 μM fMLP. Also see Figure 2 Supplemental Movies 1–4 (fMLP was mixed with Alexa 594 and consequently appears red in the videos to indicate homogeneously applied uniform stimulation). (C) Quantitative measurement of normalized membrane translocation of PKD members in response to 1 μM fMLP stimulation. (D) Activation profile of PKD1 upon fMLP stimulation. (E) Leading edge localization of PKD members (green) in 1 μM fMLP gradient. Also see Figure 2 Supplemental Movies 5–8. Alexa 594 (red) was mixed with 1 μM fMLP to visualize the gradient. Scale bar, 10 μm.
C1a domain of PKD1 is required for its membrane translocation
To understand how chemoattractant GPCR signaling controls PKD function, we determined which domain of PKD1 is essential for fMLP-induced membrane translocation. For this, we compared four GFP-tagged PKD1 constructs: wild type (PKD1-GFP); PKD1-C1a(P157G) (C1a-GFP; proline at position 157 mutated to glycine in the C1a domain); PKD1-C1b(P281G) (C1b-GFP; proline at position 281 mutated to glycine in C1b domain); and kinase-inactive mutant PKD1(K612W)-GFP (Figure 3A). These constructs were expressed in HL60 cells and assessed for their abilities to translocate to the membrane in response to fMLP stimulation (Figure 3, B– D, and Figure 3 Supplemental Movies 1–6). Membrane translocation of PKD1(K612W)-GFP was not affected upon fMLP stimulation. Consistent with this observation, the PKD1 inhibitor did not block membrane translocation of WT PKD1-GFP, indicating that PKD kinase activity is not essential for membrane targeting. Of interest, the DAG low-affinity mutant C1a-GFP, but not C1b-GFP, failed to translocate to the membrane, indicating that C1a is required for the binding of DAG and the subsequent translocation of PKD1 to the membrane. This result is consistent with a previous report showing that the C1a domain of PKD binds to DAG analogues with 48-fold-higher affinity than the C1b domain and is mainly responsible for the binding of DAG in intact cells (Chen et al., 2008). Our data suggest that the C1a domain and its binding ability to DAG are required for the membrane translocation of PKD1.
FIGURE 3:

Molecular mechanism of PKD1 membrane targeting upon chemoattractant stimulation. (A) Scheme showing the domain composition of GFP-tagged mutants and wild-type (WT) PKD1 (more explanation of the mutant constructs). (B) Protein levels of the mutant GFP-PKD1 in HL60 cells, as detected using anti-PKD1 antibodies. (C) Membrane translocation of GFP alone or GFP-tagged WT PKD1 and its mutants (green) upon uniformly applied 1 μM fMLP (red) stimulation (Figure 3 Supplemental Movies 1–6). fMLP 5 μM was mixed with Alexa 594 (red) to indicate homogeneously applied uniform stimulation. Scale bar, 10 μm. (D) Quantitative measurements of normalized membrane translocation of WT PKD1 or its mutant upon uniform 1 μM fMLP stimulation.
PKD1 is activated through the PLC signaling pathway
Our results suggest that the binding of the C1a domain and DAG is essential for the chemoattractant GPCR-induced membrane targeting of PKD1. It is well known that the activation of chemoattractant GPCR induces the dissociation of heterotrimeric proteins into Gα and Gβγ subunits, which, in turn, activate PLC to generate DAG on the plasma membrane (Li et al., 2000). We therefore reasoned that chemoattractant GPCR-induced PLC activation is essential for regulating the spatiotemporal activity of PKD. To test this idea, we examined the effect of the PLC inhibitor U73122 (Smith et al., 1996) on the cellular localization and phosphorylation of PKD1 in response to fMLP stimulation (Figure 4 and Figure 4 Supplemental Movies 1–12). We first monitored fMLP-induced DAG production using a fluorescence probe for DAG production (GFP-tagged DAG receptor [DAGR-GFP]) and subsequently examined the effect of the PLC inhibitor U73122 (Figure 4A and Figure 4 Supplemental Movies 1–3). fMLP stimulation triggered membrane translocation of DAGR-GFP, consistent with chemoattractant GPCR-mediated PLC activation. The PLC inhibitor U73122 completely blocked fMLP-induced membrane translocation of DAGR-GFP (Figure 4A and Figure 4 Supplemental Movies 1–3) and its localization to the leading edge in chemotaxing cells (Figure 4B and Figure 4 Supplemental Movies 4-6) at 1 μM but not at a lower concentration (100 nM). U73122 at a concentration of 1 μM also abolished fMLP-induced PKD1 membrane translocation (Figure 4, C and D, and Figure 4 Supplemental Movies 7–9), its leading-edge localization (Figure 4E and Figure 4 Supplemental Movies 10–12), and fMLP-triggered PKD1 phosphorylation (Figure 4, F and G), suggesting that chemoattractant GPCR-triggered and PLC-dependent DAG production is required for PKD1 activation. When PLC activity was blocked, neither DAGR-GFP nor PKD1-GFP localized to the leading front of chemotaxing cells, indicating that PLC activation is required for the chemoattractant gradient–induced spatial localization of DAG and PKD1 in chemotaxing cells.
FIGURE 4:

PLC activation is required for PKD1 membrane targeting and activation. (A) Transient activation of PLC and dose-dependent inhibition of PLC activation by PLC inhibitor U73122 upon uniformly applied fMLP stimulation (Figure 4 Supplemental Movies 1–3). (B) Montage showing the enriched PLC activation (green) and dose-dependent inhibitory effect of U73122 on leading edge activation of PLC in chemotaxing cells in 1 μM fMLP gradient (red; Figure 4 Supplemental Movies 4–6). In A and B, HL60 cells expressed YFP-tagged DAG receptor (DAGR-GFP). In B and E, 1 μM fMLP was mixed with Alexa 594 (red) to indicate fMLP stimulation. Scale bar, 10 μm. (C) Montage showing that PLC inhibitor U73122 blocked PKD1 membrane translocation upon uniform 1 μM fMLP stimulation in a dose-dependent inhibition manner (Figure 4 Supplemental Movies 7–9). (D) Normalized membrane translocation. (E) Montage showing blockage of the leading edge localization of PKD1-GFP by the PLC inhibitor U73122 (Figure 4 Supplemental Movies 10–12). (F) Western blot shows the activation profile of PKD1 and the inhibitory effects of PLC inhibitor (U73122). (G) Quantitative measurement of PKD1 activation with or without the treatment of PLC inhibitor. Data were obtained from three independent experiments.
PLC activation is required for chemotaxis
Neutrophils express multiple PLC isoforms, including PLCβ2, ‑β3, and ‑γ2 (Li et al., 2000; Suh et al., 2008). We found that treatment with PLC inhibitor U73122, which blocks the activation of all PLC isoforms, resulted in clear chemotaxis defects in both HL60 and Raw 264.7cells (Figure 5, A and B, and Supplemental Figure S7). However, no significant chemotaxis defect was observed in murine neutrophils lacking both PLCβ2 and PLCβ3 (Li et al., 2000), suggesting that other PLC isoform(s) besides PLCβ2 and PLCβ3 may play a role in human neutrophil chemotaxis. PLCγ2 is highly expressed in human neutrophils (Suh et al., 2008). To test the function of PLCγ2 in neutrophil chemotaxis, we expressed GFP-tagged PLCγ2 in HL60 cells and examined its cellular localization upon fMLP stimulation (Figure 5, C and D, and Figure 5 Supplemental Movies 1–3). We found that uniform fMLP stimulation induced clear membrane translocation of PLCγ2-GFP. Treatment with Gαi inhibitor pertussis toxin abolished fMLP-induced membrane translocation of PLCγ2-GFP, indicating that chemoattractant GPCR-triggered Gαi signaling is required for membrane recruitment of PLCγ2. More important, PI3K inhibitor LY294002 blocked membrane translocation of PLCγ2-GFP, indicating that PIP3 binding by the PH domain of PLCγ2 is required for its membrane targeting. This result is consistent with the finding that PLCγ1, an isoform of PLCγ2, is recruited to the membrane through PIP3 binding of its PH domain and consequently activated (Falasca et al., 1998). We also found that PLCγ2-GFP localized to the leading edge of chemotaxing cells (Figure 5E and Figure 5 Supplemental Movie 4). Our results indicate that chemoattractant GPCR/Gαi signaling spatially controls activation of not only PLCβ2/3 but also PLCγ2, to produce DAGs as an intracellular cue to control the temporal-spatial activation of its downstream effectors, such as PKCs and PKDs, in chemotaxing cells.
FIGURE 5:

PLC activation is required for chemotaxis. (A) Montage showing the traveling path of chemotaxing cells under different concentration of PLC inhibitor U73122 monitored using EZ-TaxiScan chemotaxis assay. (B) Quantitative measurements of chemotaxis index. (C) Gαi signaling and PI3K dependence of PLCγ2-GFP (green) membrane translocation in response to chemoattractant fMLP stimulation (red; Figure 5 Supplemental Movies 1–3). Cells were treated with the indicated inhibitors for 30 min before the experiments. (D) Normalized quantitative measurement of PLCγ2-GFP membrane translocation. (E) Leading edge localization of PLCγ2-GFP (green) in chemotaxing cells in 1 μM fMLP (red) gradient (Figure 5 Supplemental Movie 4).
PKCβ activation is required for chemotaxis
DAG is the physiological activator of PKCs, and some PKC isoforms can activate PKDs (Wang, 2006). We found that the PKC inhibitor Gö 6983 significantly inhibited phosphorylation of PKD1 (Figure 6, A and B), suggesting that PKCs play a role in chemoattractant GPCR-mediated PKD activation. Human neutrophils express five PKC isoforms: PKC‑α, ‑βI, ‑βII, ‑δ, and ‑ζ (Bertram and Ley, 2011). To identify the PKC isoform that activates PKD, we carried out coimmunoprecipitation experiments using PKD1 as bait (Figure 6C). Using an anti–phospho-PKC (pan) antibody that recognizes all PKC isoforms, we detected only one band, at ∼75 kDa, indicating that either PKCα or β, both of which are ∼75 kDa, interacts with PKD1. Next we used Western blotting analysis with anti-PKCα or -β–specific antibodies and confirmed that PKCβ was the major PKC isoform that interacted with PKD1. Because PKCβI and PKCβII display overall sequence and domain similarity and identical membrane translocation behavior upon fMLP stimulation (Supplemental Figure S8 and Figure S8 Supplemental Movies 1–3), we reported only on the dynamics of GFP-tagged PKCβII upon fMLP stimulation. In a fMLP gradient, PKCβII-GFP localized to the leading edge of chemotaxing cells (Figure 6D and Figure 6 Supplemental Movie 1). Uniform fMLP stimulation induced robust membrane translocation of PKCβII-GFP (Figure 6, E and F, and Figure 6 Supplemental Movies 2–4). We also found that inhibiting PLC activation blocked PKCβII membrane translocation, consistent with the notion that PKC is recruited and activated at the membrane via PLC-generated DAG (Wang, 2006). We then examined the role of PKCβII in neutrophil chemotaxis (Figure 6, G and H, and Supplemental Figure S9). Abrogation of PKCβII activity by either treatment of a PKC inhibitor (Gö 6983) or overexpression of a dominant-negative PKCβII mutant (PKCβII-DN) or a constitutively active PKCβII mutant (PKCβII-CA) impaired chemotaxis of HL60 cells. Taken together, our data suggest that PLC-mediated PKCβ activation is required for neutrophil chemotaxis.
FIGURE 6:

PLC-dependent PKCβ activation is required for chemotaxis. (A) Gö 6983 (PKC inhibitor) inhibits PKD1 phosphorylation. (B) Quantitative measurements of PKD1 phosphorylation profile with or without PKC inhibitor treatment from three independent experiments. (C) Identification of PKCβ as the major PKC isoform interacting with PKD1 by immunoprecipitation using PKD1-GFP as bait. Western blot detection of PKD and PKC isoforms with anti-GFP, anti–phospho-PKC(pan) (βII-Ser-660), and anti-PKCα. (D) Leading edge localization of PLCβII-GFP (green) in the chemotaxing cells in response to 1 μM fMLP gradient (red; Figure 6 Supplemental Movie 1). (E) Montage showing PLC-dependent activation and membrane translocation of PKCβII upon 1 μM fMLP stimulation. GFP-tagged PKCβII (green) was transiently expressed in HL60 cells. Cells were treated with PLC inhibitor U63122 at indicated concentration for 30 min before experiments. Alexa 594 (red) was mixed with 1 μM fMLP to indicate uniform application of fMLP or gradient stimulations in D and E; also see Figure 6 Supplemental Movies 1-4. Scale bar, 10 μm. (F) Quantitative measurement of PKCβII membrane translocation with or without PLC inhibitor treatment. (G) Montage showing the traveling path of chemotaxing cells either treated with PKC inhibitor Gö 6983 or transiently overexpressing a dominant-negative (PKCβII-DN) or constitutively active mutant (PKCβII-CA) of PKCβII, as monitored by EZ-TaxiScan chemotaxis assay. (H) Quantitative measurements of chemotaxis indexes as total path, speed, and directionality.
PKD1 interacts with SSH2 to regulate cofilin activity
F-actin depolymerization factor ADF/cofilin is a key regulator of cell migration (Mizuno, 2013). The activity of cofilin is mainly regulated by phosphorylation and dephosphorylation events. Cofilin is dephosphorylated and consequently activated by the phosphatase slingshot protein family (SSHs). SSH1 has been identified as the substrate of PKD1 that regulates cofilin activation during breast cancer cell migration (Eiseler et al., 2009). Here we first determined which slingshot protein interacts with PKD1 by immunoprecipitation, using PKD1-GFP as bait. We found that SSH2, but not SSH1, interacts with PKD1, consistent with the observation that SSH2 is the dominant isoform of slingshot proteins expressed in neutrophils (Tang et al., 2011). As previously reported (Eiseler et al., 2009), we found that GPCR-mediated PKD1 activation increased its interaction with SSH2 (Figure 7, A and B). PKD1 activation also increased the interaction between SSH2 and 14‑3‑3, consistent with the findings that activation of PKD1 facilitates its interaction with SSH1 and also SSH1/14‑3‑3 interaction (Eiseler et al., 2009). Hence PKD1 activation results in decreased phosphatase activity of SSH2 toward cofilin. Thus we next examined the effect of PKD inhibitors on cofilin phosphorylation and found that PKD1 inhibitors caused a decreased level of cofilin phosphorylation in cells treated with PKD1 inhibitors (Figure 7C). We found that PKD inhibitors did not change the level of actin amount in the cells (Figure 7D) but changed the ratio of F-actin to global actin with or without activation (Figure 7, E and F). These results together indicate that PKD1 interacts with SSH2 to regulate cofilin activity in the cells.
FIGURE 7:
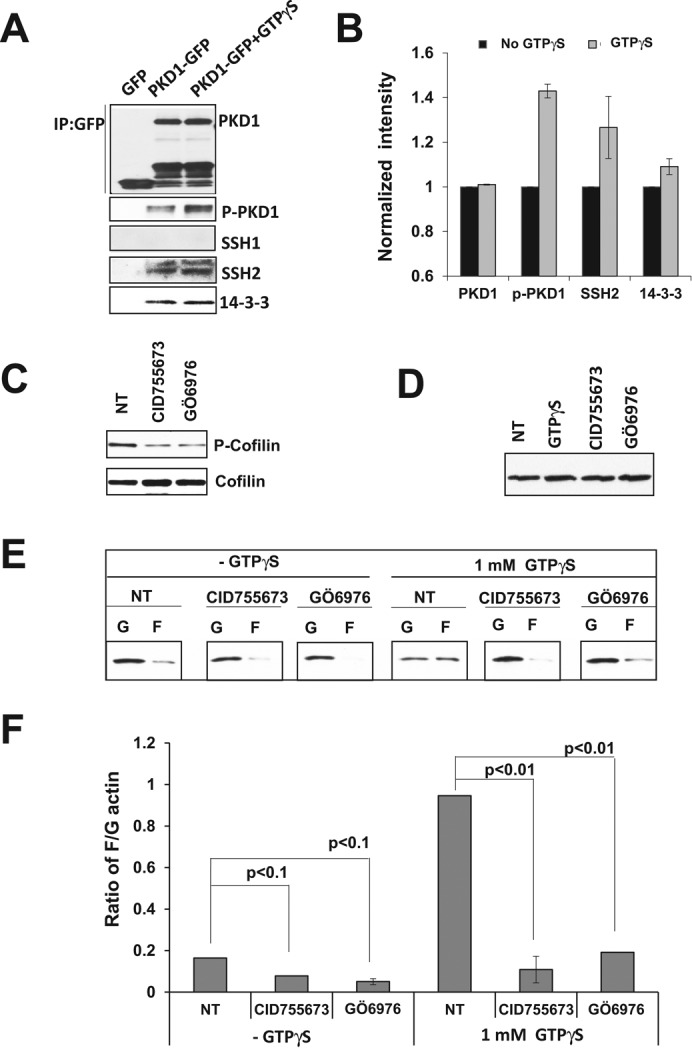
PKD1 interacts with SSH2 to regulate cofilin activity. (A) GPCR activation enhances the interaction between PKD1 and SSH2. Raw 264.7 cells transiently expressed GFP only or GFP-tagged PKD1 protein. Immunoprecipitation was carried out with anti–GFP-agarose beads. (B) Quantitative measurement of the activation and interaction of indicated molecules. Intensity of PKD1 without GTPγS treatment was normalized by 1. (C) Treatment of PKD inhibitor increased cofilin activity in the Raw 264.7 cells. (D) Treatment of PKD inhibitors does not affect the actin level in the Raw 264.7 cells. (E) In vivo assay to determine the ratio of filamental (F) and globular (G) actin amounts with or without PKD inhibitor treatments. (F) Quantification of F/G actin ratio in the Raw 264.7 cells.
PLC/PKC/PKD1 axis is required for peripheral neutrophil chemotaxis
To understand whether a similar signaling pathway that regulates chemotaxis in the foregoing cell lines also acts in polymorphonuclear neutrophils (PMNs), we determined the inhibitory effects of PLC, PKC, and PKD inhibitors on PMN chemotaxis by Transwell assay (Figure 8). We found that all three groups of PLC, PKC, and PKD inhibitors significantly blocked the chemotaxis of PMN toward SDF1α (p < 0.001), indicating a general function of the PLC/PKC/PKD signaling pathway in neutrophil-like cell lines and PMN chemotaxis.
FIGURE 8:
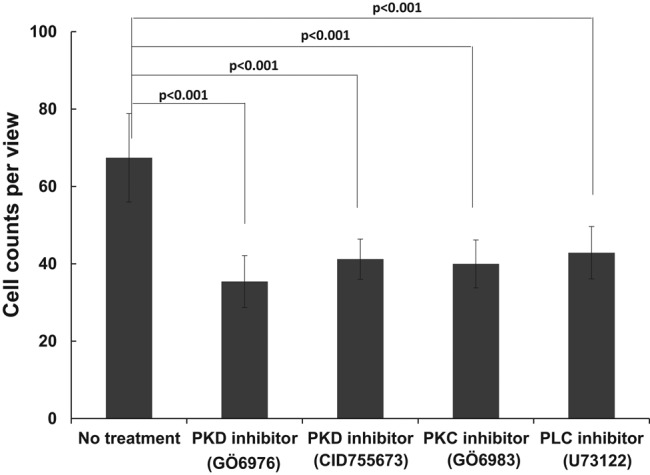
PLC/PKC/PKD signaling pathway plays an essential role in chemotaxis of PMNs. Inhibitory effect of PLC (1 μM U73122), PKC (100 nM Gö 6983), and PKD (1 μM CID755673 and 100 nM Gö 6976) inhibitors on PMN chemotaxis determined by Transwell assay. Transwell chemotaxis assays were performed with 100 nM SDF-1α gradient. Data were obtained from three independent experiments.
DISCUSSION
In the present study, we revealed a novel signaling pathway in which the PKD family serves as the direct downstream target of a PLC/PKCβ signaling axis to regulate SSH2 activity and control cofilin activity in neutrophil chemotaxis (Figure 9).
FIGURE 9:
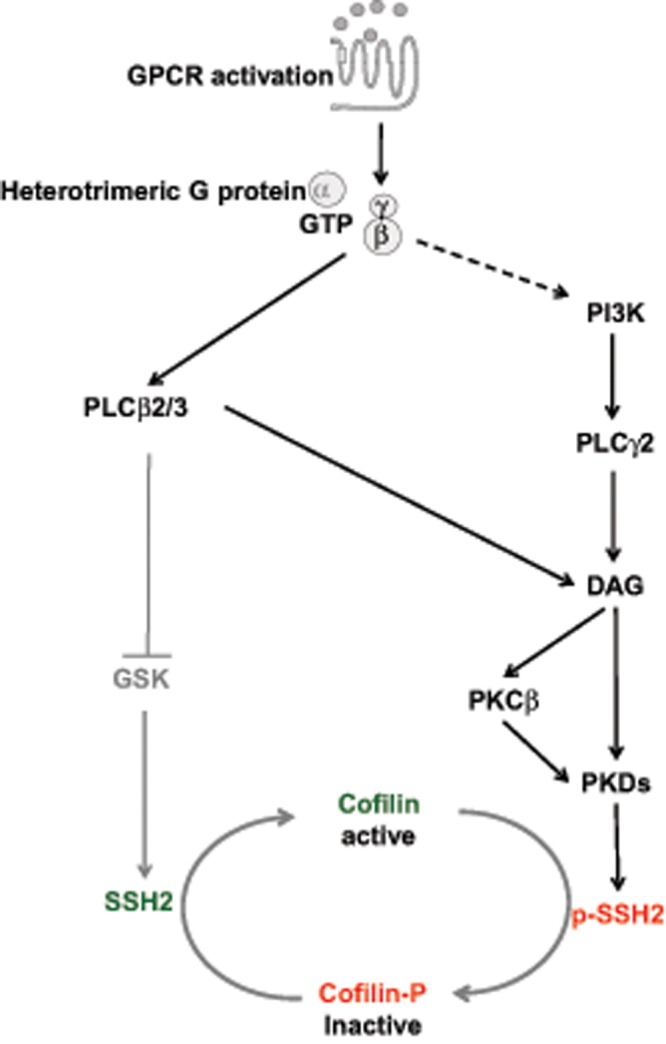
GPCR-mediated PLC/PKCβ/PKD/SSH2 signaling pathway to regulate cofilin activity. The Gβ-dependent PLCβ2/3 and PI3K-dependent PLCγ2 activation of PLC family members upon chemokine stimulation functions as the upstream activator of the PKC/PKD signaling pathway to regulate SSH2/cofilin dynamics.
PLC signaling is required for neutrophil chemotaxis
We demonstrated the essential role of PLC signaling in neutrophil chemotaxis. There are 13 phosphatidylinositide-specific PLCs, divided into six subgroups: ‑β, ‑γ, ‑δ, ‑e, ‑ξ, and ‑η (Suh et al., 2008). Chemoattractants activate PLCβ2, ‑β3, and ‑e (Jiang et al., 1994; Li et al., 2000; Kelley et al., 2006). Neutrophils express three PLC isoforms, namely PLCβ2, ‑β3, and ‑γ2 (Suh et al., 2008). Deficiency of both PLCβ2 and β3 did not affect chemotaxis of murine neutrophils (Li et al., 2000). This result leads to the assumption that PLCβ2/3 signaling might not play an essential role in neutrophil chemotaxis. However, in this study, we first revealed that PLC signaling is essential for neutrophil chemotaxis (Figure 5, A and B). Furthermore, we discovered that chemokine stimulation triggers dynamic membrane translocation of PLCγ2, a highly expressed PLC isoform in neutrophils. Membrane targeting of PLCγ2 depends on Gαi signaling, more specifically PI3K activation, indicating that PIP3 binding by the PH domain of PLCγ2 serves as the major determinant of its membrane targeting (Figure 5C). This result is consistent with the finding that activation of PLCγ1 is a consequence of its PH domain binding to PIP3 produced on the membrane by multiple signaling pathways (Falasca et al., 1998). Although PLCe is also activated by GPCR signaling (Kelley et al., 2006), peripheral blood leukocytes express little PLCe (Suh et al., 2008), indicating that it might not be a major player in leukocyte chemotaxis. In conclusion, PLCγ2, in addition to PLCβ2/β3, can be activated by chemokine GPCR activation, and PLC signaling is required for neutrophil chemotaxis.
Chemoattractant-mediated PLC/PKCβ/PKD signaling is essential for neutrophil chemotaxis
In addition to the essential role of PLC signaling, of greater importance, our study revealed the connection between PLC/PKCβ/PKD signaling and the regulation of cofilin activity in neutrophil chemotaxis. Several signaling pathways activate PLCs to generate DAGs, physiological activators of PKCs (Wang, 2006). We also observed that, in neutrophils, fMLP stimulation induces both DAG production (Figure 4A) and membrane translocation of PKCβII-GFP (Figure 6E). Consistent with the previous observation that PKC activation requires PLCβ in murine neutrophils (Tang et al., 2011), we found that a PLC inhibitor abolished the membrane translocation of PKCβII, indicating that PLC activation acts upstream of PKCβ.
Chemoattractants triggered membrane translocation of all three PKD isoforms (Figure 2). Specifically, the DAG-binding domain (C1a) is required for membrane translocation of PKD1 (Figure 3), indicating that PLC activation is necessary for PKD1 membrane translocation. Moreover, PLC inhibitor abrogated fMLP-induced membrane translocation and phosphorylation (activation) of PKD1 (Figure 4, E– G) at a concentration that completely inhibited DAG production (Figure 4A) and membrane translocation of PKCβII-GFP (Figure 6, E and F). PKC is a physiological activator of PKDs (Wang, 2006). PKC inhibitors also impaired PKD1 phosphorylation/activation (Figure 6, A and B). We further identified PKCβ, which specifically interacts with PKD1 and plays an essential role in neutrophil chemotaxis (Figure 6, G and H). In an fMLP gradient, PLCγ2-GFP (Figure 5E), PKCβII-GFP (Figure 6D), and PKD1-GFP (Figure 4E) are all enriched at the leading front of neutrophils. PLC inhibitors abolished the localization of PLC, PKC, or PKD to the leading front of a cell in a chemoattractant gradient. All of these results suggest that chemoattractant GPCRs activate PLC to generate DAGs as an intracellular cue that guides activation of PKCβ and PKD in chemotaxing neutrophils.
Regulation of cofilin activity in neutrophil chemotaxis
The coordinated polymerization and depolymerization of F-actin–based cytoskeleton in chemotaxing cells require temporal and spatial regulation of cofilin activity. The most important and best-studied mechanism of cofilin activity regulation is through phosphorylation: LIMK and TESK phosphorylate and deactivate cofilin; slingshot proteins and chronophin dephosphorylate and reactivate cofilin (Mizuno, 2013). In neutrophils, chemoattractants mediate the rapid dephosphorylation of cofilin (van Rheenen et al., 2007). The chemoattractant-mediated PLCβ/PI3Kγ/GSK3 signaling pathway has been found to increase SSH2 activity to regulate dephosphorylation of cofilin (Tang et al., 2011). It is not clear whether the chemoattractant activates LIMK or TESKs to phosphorylate/deactivate cofilin in neutrophils. Recent studies identify PKDs as the kinase that phosphorylates SSH1 at multiple Ser residues to inhibit its cofilin phosphatase activity (Barisic et al., 2011; Eiseler et al., 2009; Nagel et al., 2010). In the present study, we showed that PKD1 interacts with SSH2—the major isoform of SSH protein in neutrophils—to regulate cofilin phosphorylation (Figure 7, A and B). More important, the inhibition of PKD1 resulted in decreased phosphorylation level of cofilin (Figure 7C) and significantly altered F-actin dynamics upon GPCR activation (Figure 7, E and F). Although all three PKD proteins have also been reported to regulate LIMK activity by direct phosphorylation and activation of upstream kinase p21–activated kinase 4 (PAK4) in order to indirectly regulate cofilin activity (Spratley et al., 2011), we did not detect the interaction between PAK4 and PKD1. Instead, chemoattractant-induced PKD1 activation increased the interaction between SSH2 and 14‑3‑3 (Figure 7, A and B). This result is consistent with the finding that binding of 14‑3‑3 to phosphorylated SSH1 alters SSH1 subcellular localization and F-actin binding capability, consequently decreasing its cofilin-phosphatase activity (Eiseler et al., 2009). These results together indicate that chemoattractant-mediated PKD activation regulates SSH2 activity to control cofilin activity and reorganization of F-actin cytoskeleton upon chemoattractant stimulation in neutrophils.
The other important mechanism of cofilin regulation is through PIP2. PIP2 binds to dephosphorylated cofilin to inhibit the actin-severing activity of cofilin (van Rheenen et al., 2007). In carcinoma cells, epidermal growth factor–induced rapid loss of PIP2 through PLCγ2 activation results in a release of membrane-bound active cofilin (van Rheenen et al., 2007). In neutrophils, chemoattractant triggers PLCβ2/3 activation (Li et al., 2000). In the present study, we showed that the chemoattractant fMLP also induces PLCγ2 activation (Figure 5). However, it is not clear whether a rapid loss of PIP2 on the membrane by PLCβ/γ activation in neutrophils also releases cofilin. If so, the released dephosphorylated cofilin might serve as a fast-activation loop of cofilin in neutrophils (van Rheenen et al., 2009). Future study may reveal how these pathways function together to facilitate the temporal and spatial coordination of actin polymerization and depolymerization processes during chemotaxis.
MATERIALS AND METHODS
Cell lines, cell culture, and differentiation
All the cell lines were obtained from the American Type Culture Collection (ATCC) and cultured according to the supplied protocol. IC21 and U937 cells were maintained in RPMI 1640 growth medium (Gibco, Grand Island, NY) supplemented with 10% (vol/vol) fetal bovine serum (FBS; Invitorgen, Grand Island, NY), 0.1 mM sodium pyruvate, and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES; Gibco). HL60 cells were maintained in RPMI 1640 medium supplemented with 10% (vol/vol) FBS, 0.1 mM sodium pyruvate, and 25 mM HEPES. Raw 264.7 cells were maintained in DMEM medium supplemented with 10% (vol/vol) FBS. The cells were incubated at 37°C in a humidified 5% CO2 atmosphere. HL60 cells were passaged in RPMI 1640 medium supplemented with 10% FBS and 25 mM HEPES. HL60 cells were differentiated in their culture medium containing 1.3% dimethyl sulfoxide (DMSO) for 5 d before the experiment.
Purification of polymorphonuclear neutrophils
Whole blood (150 ml) was drawn from healthy volunteers at the Blood Bank of the National Institutes of Health. Coagulation was prevented by heparin. The majority of the red blood cells were removed by dextran (0.2 g/l; GE Healthcare, Pittsburgh, PA) sedimentation (30 min, room temperature). Upper phases containing white blood cells were collected and washed twice in phosphate-buffered saline (PBS). Cells were resuspended in PBS and layered on top of a five-step Percoll gradient (65, 70, 75, 80, and 85%; Sigma-Aldrich) in 15-ml conical tubes. After centrifugation (800 × g, 20 min, room temperature), the 70/75/80% Percoll layers containing granulocytes were collected and washed twice in PBS. The collected neutrophils were resuspended in PBS, the cell concentration was determined, and the cells were kept at room temperature until use. Cell viability was determined by trypan blue dye extrusion, and the results were >98% viable neutrophils. The purity of the preparations was determined by Wright–Giemsa staining, and the yield was >95% neutrophil granulocytes.
Establishment of stable PKD1-knockdown cells
On day 1, 2.5 × 105 cells were seeded in a six-well plate. On day 2, the culture medium in the six-well plate was replaced by 1 ml of infection cocktail (complete culture medium containing 12 μg/ml Polybrene). We added 10 μl of virus particle encoding to the six-well plate. After 6 h, the infection cocktail of each well was replaced by complete culture medium without Polybrene. On day 3, 10 μg/ml puromycin was added to select positively virus-infected cells. Culture media were changed every 2–3 d until the infected cells proliferated into enough cells to allow for the detection of endogenous PKD1 expression and the other experiments indicated.
Reagents and antibodies
SDF-1α and CSF-1 were purchased from R&D Systems (Minneapolis, MN) and Pepro Tech (Rocky Hill, NJ), respectively. Gö 6976, Gö 6983, and U73122 were purchased from Calbiochem (Billerica, MA). CID755673 was purchased from Tocris (United Kingdom). fMLP and DMSO were purchased from Sigma-Aldrich (St. Louis, MO). Antibodies anti–phospho-PKD/PKCmu(S744/748), anti–phospho-PKD1(Ser-916), and anti-PKD/PKCμ were from Cell Signaling (Danvers, MA). Anti-SSH2 antibody was from Novus Biologicals. Anti–14‑3‑3 antibody was from Santa Cruz Biotechnology. Anti-GFP antibody was from BD Bioscience (San Jose, CA). Horseradish peroxidase (HRP)–conjugated anti-actin was from Santa Cruz Biotechnology (Dallas, TX). HRP-conjugated anti-mouse or anti-rabbit immunoglobulin G was obtained from Jackson ImmunoResearch (United Kingdom). All tissue culture reagents were purchased from Invitrogen.
Plasmids and transfection of cells
The plasmids encoding cDNA of PKCβI–yellow fluorescent protein (YFP), PKCβII-YFP, dominant-negative PKCβII-YFP (DN-PKCβII-YFP), constitutive active PKCβII-YFP (CA- PKCβII-YFP), and DAGR-YFP were purchased from Addgene (www.addgene.org/). The plasmid encoding PLCγ2-GFP was from Origene. The procedure used to transfect HL60 cells mostly followed the manufacturer's instructions. Briefly, 2 × 106 cells were centrifuged at 100 × g for 10 min and resuspended in 100 μl of nucleofection solution V at room temperature. A total of 6 μg of plasmid DNA encoding the desired protein was used for a single transfection reaction using T-019 on the Amaxa Nucleofector II (Lonza).
Chemotaxis assays
Transfilter chemotaxis assay.
Cells were collected and resuspended in RPMI 1640 medium containing 1% BSA and 25 μM HEPES at a cell density of 107 cells/ml. The desired concentration of SDF-1α (R&D Systems) or CSF-1 (PeproTech) containing RPMI medium was applied to the lower part of the transfilter cassette. A total of 50 μl of cells was added to the top part of the transfilter assay cassette and incubated at 37°C for 90 min. Nontransfiltered cells were wiped away with a cotton applicator. The membrane along with the transfiltered cells was washed with phosphate buffer (PB), pH 7.2, and fixed, and the cells were counted. Each experiment was performed in three independent wells.
TaxiScan chemotaxis assay.
HL60 cells were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated FBS and 25 mM HEPES at 37°C with 5% CO2. Cells were differentiated according to published methods (Millius and Weiner, 2009). Briefly, cells under passage 10 were incubated with 1.3% DMSO at 1.5 × 105 cells/ml for 6 d. Differentiated cells were incubated with U73122 at different concentrations for 1 h at 37°C before being loaded onto fibronectin-coated, 4‑μm EZ-TaxiScan chambers. Cells were allowed to migrate toward 100 nM fMLP or no chemoattractant control for 30 min at 37°C. Pictures were captured at 15‑s intervals. The image data were analyzed with DYIS software. Each treatment group contained ∼15 different cells, and data are shown as mean and SEM. The bar graphs were plotted with Excel (Microsoft).
PKD1 activation profile
About 3 × 106 cells were seeded and starved in six-well plates with RPMI medium with 25 mM HEPES for 3 h. Cells were collected and resuspended at 2 × 106 cell/ml and then stimulated with 1 μM fMLP, 100 nM CSF-1α, or 100 ng/ml SDF-1α for 0, 1, 5, 10, 30, and 60 min. The cells were then centrifuged and lysed with 100 μl lysis buffer containing 25 mM Tris, 150 mM NaCl, 1 mM EDTA, 1 mM NaVO3, 10 μM NaF, proteinase inhibitor, and 1% Triton X for 30 min on ice. Cell lysates were then centrifuged at 100,000 × g for 30 min, and 90 μl of the supernatant was mixed with 75 μl of SDS loading buffer for further detection of the protein by Western blot.
Calcium response
HL60 cells were treated with Fluo-4 (Invitrogen) at a final concentration of 100 ng/ml and incubated in a 37°C culturing hood for 30 min. To remove unstained Fluo-4, the cells were washed with RPMI 1640 medium without FBS twice and then subjected to experiments.
Confocal imaging and data analysis
To facilitate live-cell imaging, one- or four-well glass chambers (NUNC, Germany) were coated with 0.2% freshly prepared gelatin in PBS at 37°C for 30 min of incubation. The chambers were then washed with room temperature RPMI 1640 medium three times before the cells were seeded. Confocal images were collected using a Zeiss 510 META NLO microscope (Carl Zeiss, Thornwood, NY) with Zeiss Plan-Apochromat 63×/1.4 numerical aperture oil objective. Images were exported with Zeiss 510 META software and further analyzed with Photoshop (Adobe). The intensity change of whole cells or cytoplasm was measured and exported with Zeiss 510 META software. For quantitative measurements of the membrane translocation of indicated protein, we first measured the intensity change of the cytoplasm in response to uniformly applied stimuli over time. To obtain the relative intensity change of each individual cell during the time lapse, we divided its intensity at given time (It) by its intensity at time 0 (I0); consequently, relative intensity of any cells at time 0 became 1. Then we divided the normalized intensity at time 0 (I0) by the normalized intensity at given time (It) to convert the normalized intensity change of the cytoplasm to membrane translocation. Finally, we calculated and present mean ± SD of peak membrane translocation from more than five independent cells.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health Intramural Fund from the National Institute of Allergy and Infectious Diseases, National Institutes of Health. Q. Jane Wang was supported in part by National Institutes of Health grant R01CA142580. We acknowledge Thomas Leto and Blazs Rada, Laboratory of Immunogenetics, National Institute of Allergy and Infectious Diseases, National Institutes of Health, for their help with the PMN experiments. We also thank Joseph Brzostowski, Laboratory of Immunogenetics Imaging Facility, for his help.
Abbreviations used:
- DAG
diacylglycerol
- GPCRs
G protein–coupled receptors
- LIMKs
LIM kinases
- PKC
protein kinases C
- PKD
protein kinases D
- PLC
phospholipase C
- SSHs
slingshot proteins
- TESKs
testicular protein kinases.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-05-0982) on January 7, 2015.
REFERENCES
- Arber S, Barbayannis FA, Hanser H, Schneider C, Stanyon CA, Bernard O, Caroni P. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393:805–809. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- Balasubramanian N, Advani SH, Zingde SM. Protein kinase C isoforms in normal and leukemic neutrophils: altered levels in leukemic neutrophils and changes during myeloid maturation in chronic myeloid leukemia. Leuk Res. 2002;26:67–81. doi: 10.1016/s0145-2126(01)00098-4. [DOI] [PubMed] [Google Scholar]
- Barisic S, Nagel AC, Franz-Wachtel M, Macek B, Preiss A, Link G, Maier D, Hausser A. Phosphorylation of Ser 402 impedes phosphatase activity of slingshot 1. EMBO Rep. 2011;12:527–533. doi: 10.1038/embor.2011.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertram A, Ley K. Protein kinase C isoforms in neutrophil adhesion and activation. Arch Immunol Ther Exp (Warsz) 2011;59:79–87. doi: 10.1007/s00005-011-0112-7. [DOI] [PubMed] [Google Scholar]
- Chen J, Deng F, Li J, Wang QJ. Selective binding of phorbol esters and diacylglycerol by individual C1 domains of the PKD family. Biochem J. 2008;411:333–342. doi: 10.1042/BJ20071334. [DOI] [PubMed] [Google Scholar]
- Eiseler T, Doppler H, Yan IK, Kitatani K, Mizuno K, Storz P. Protein kinase D1 regulates cofilin-mediated F-actin reorganization and cell motility through slingshot. Nat Cell Biol. 2009;11:545–556. doi: 10.1038/ncb1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. EMBO J. 1998;17:414–422. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan X, Wang J, Wang C, Sommer E, Kozasa T, Srinivasula S, Alessi D, Offermanns S, Simon MI, Wu D. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat Cell Biol. 2012;14:686–696. doi: 10.1038/ncb2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gohla A, Birkenfeld J, Bokoch GM. Chronophin, a novel HAD-type serine protein phosphatase, regulates cofilin-dependent actin dynamics. Nat Cell Biol. 2005;7:21–29. doi: 10.1038/ncb1201. [DOI] [PubMed] [Google Scholar]
- Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- Jacamo R, Sinnett-Smith J, Rey O, Waldron RT, Rozengurt E. Sequential protein kinase C (PKC)-dependent and PKC-independent protein kinase D catalytic activation via Gq-coupled receptors: differential regulation of activation loop Ser(744) and Ser(748) phosphorylation. J Biol Chem. 2008;283:12877–12887. doi: 10.1074/jbc.M800442200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Kuang Y, Wu Y, Xie W, Simon MI, Wu D. Roles of phospholipase C beta2 in chemoattractant-elicited responses. Proc Natl Acad Sci USA. 1997;94:7971–7975. doi: 10.1073/pnas.94.15.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Wu D, Simon MI. Activation of phospholipase C beta 4 by heterotrimeric GTP-binding proteins. J Biol Chem. 1994;269:7593–7596. [PubMed] [Google Scholar]
- Jin T, Xu X, Hereld D. Chemotaxis, chemokine receptors and human disease. Cytokine. 2008;44:1–8. doi: 10.1016/j.cyto.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley GG, Kaproth-Joslin KA, Reks SE, Smrcka AV, Wojcikiewicz RJ. G-protein-coupled receptor agonists activate endogenous phospholipase Cepsilon and phospholipase Cbeta3 in a temporally distinct manner. J Biol Chem. 2006;281:2639–2648. doi: 10.1074/jbc.M507681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L, et al. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005;7:399–404. doi: 10.1038/ncb1236. [DOI] [PubMed] [Google Scholar]
- Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, et al. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell. 2003;114:215–227. doi: 10.1016/s0092-8674(03)00559-2. [DOI] [PubMed] [Google Scholar]
- Li Z, Jiang H, Xie W, Zhang Z, Smrcka AV, Wu D. Roles of PLC-beta2 and -beta3 and PI3Kgamma in chemoattractant-mediated signal transduction. Science. 2000;287:1046–1049. doi: 10.1126/science.287.5455.1046. [DOI] [PubMed] [Google Scholar]
- Millius A, Weiner OD. Chemotaxis in neutrophil-like HL-60 cells. Methods Mol Biol. 2009;571:167–177. doi: 10.1007/978-1-60761-198-1_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25:457–469. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- Nagel AC, Schmid J, Auer JS, Preiss A, Maier D. Constitutively active protein kinase D acts as negative regulator of the Slingshot-phosphatase in Drosophila. Hereditas. 2010;147:237–242. doi: 10.1111/j.1601-5223.2010.02200.x. [DOI] [PubMed] [Google Scholar]
- Niwa R, Nagata-Ohashi K, Takeichi M, Mizuno K, Uemura T. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108:233–246. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
- Ohta Y, Kousaka K, Nagata-Ohashi K, Ohashi K, Muramoto A, Shima Y, Niwa R, Uemura T, Mizuno K. Differential activities, subcellular distribution and tissue expression patterns of three members of Slingshot family phosphatases that dephosphorylate cofilin. Genes Cells. 2003;8:811–824. doi: 10.1046/j.1365-2443.2003.00678.x. [DOI] [PubMed] [Google Scholar]
- Servant G, Weiner OD, Herzmark P, Balla T, Sedat JW, Bourne HR. Polarization of chemoattractant receptor signaling during neutrophil chemotaxis. Science. 2000;287:1037–1040. doi: 10.1126/science.287.5455.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, Bravo-Altamirano K, Wipf P, Lazo JS, Wang QJ. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–33526. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith RJ, Justen JM, McNab AR, Rosenbloom CL, Steele AN, Detmers PA, Anderson DC, Manning AM. U-73122: a potent inhibitor of human polymorphonuclear neutrophil adhesion on biological surfaces and adhesion-related effector functions. J Pharmacol Exp Ther. 1996;278:320–329. [PubMed] [Google Scholar]
- Spratley SJ, Bastea LI, Doppler H, Mizuno K, Storz P. Protein kinase D regulates cofilin activity through p21-activated kinase 4. J Biol Chem. 2011;286:34254–34261. doi: 10.1074/jbc.M111.259424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh PG, Park JI, Manzoli L, Cocco L, Peak JC, Katan M, Fukami K, Kataoka T, Yun S, Ryu SH. Multiple roles of phosphoinositide-specific phospholipase C isozymes. BMB Rep. 2008;41:415–434. doi: 10.5483/bmbrep.2008.41.6.415. [DOI] [PubMed] [Google Scholar]
- Sun CX, Magalhaes MA, Glogauer M. Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol. 2007;179:239–245. doi: 10.1083/jcb.200705122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang W, Zhang Y, Xu W, Harden TK, Sondek J, Sun L, Li L, Wu D. A PLCbeta/PI3Kgamma-GSK3 signaling pathway regulates cofilin phosphatase slingshot2 and neutrophil polarization and chemotaxis. Dev Cell. 2011;21:1038–1050. doi: 10.1016/j.devcel.2011.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Amano T, Yang N, Narumiya S, Mizuno K. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001a;12:1131–1145. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toshima J, Toshima JY, Takeuchi K, Mori R, Mizuno K. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J Biol Chem. 2001b;276:31449–31458. doi: 10.1074/jbc.M102988200. [DOI] [PubMed] [Google Scholar]
- van Rheenen J, Condeelis J, Glogauer M. A common cofilin activity cycle in invasive tumor cells and inflammatory cells. J Cell Sci. 2009;122:305–311. doi: 10.1242/jcs.031146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rheenen J, Song X, van Roosmalen W, Cammer M, Chen X, Desmarais V, Yip SC, Backer JM, Eddy RJ, Condeelis JS. EGF-induced PIP2 hydrolysis releases and activates cofilin locally in carcinoma cells. J Cell Biol. 2007;179:1247–1259. doi: 10.1083/jcb.200706206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang QJ. PKD at the crossroads of DAG and PKC signaling. Trends Pharmacol Sci. 2006;27:317–323. doi: 10.1016/j.tips.2006.04.003. [DOI] [PubMed] [Google Scholar]
- Welch HC, Coadwell WJ, Ellson CD, Ferguson GJ, Andrews SR, Erdjument-Bromage H, Tempst P, Hawkins PT, Stephens LR. P-Rex1, a PtdIns(3,4,5)P3- and Gbetagamma-regulated guanine-nucleotide exchange factor for Rac. Cell. 2002;108:809–821. doi: 10.1016/s0092-8674(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Yang N, Higuchi O, Ohashi K, Nagata K, Wada A, Kangawa K, Nishida E, Mizuno K. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393:809–812. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.