

A novel mechanism is described for the agrin-mediated focal delivery of acetylcholine receptors (AChRs) to the postsynaptic membrane of the neuromuscular junction. Microtubule capture mediated by CLASP2 and its interaction partner, LL5β, and an intact subsynaptic actin cytoskeleton are both required for focal AChR transport to the synaptic membrane.
Abstract
A hallmark of the neuromuscular junction (NMJ) is the high density of acetylcholine receptors (AChRs) in the postsynaptic muscle membrane. The postsynaptic apparatus of the NMJ is organized by agrin secreted from motor neurons. The mechanisms that underlie the focal delivery of AChRs to the adult NMJ are not yet understood in detail. We previously showed that microtubule (MT) capture by the plus end–tracking protein CLASP2 regulates AChR density at agrin-induced AChR clusters in cultured myotubes via PI3 kinase acting through GSK3β. Here we show that knockdown of the CLASP2-interaction partner LL5β by RNAi and forced expression of a CLASP2 fragment blocking the CLASP2/LL5β interaction inhibit microtubule capture. The same treatments impair focal vesicle delivery to the clusters. Consistent with these findings, knockdown of LL5β at the NMJ in vivo reduces the density and insertion of AChRs into the postsynaptic membrane. MT capture and focal vesicle delivery to agrin-induced AChR clusters are also inhibited by microtubule- and actin-depolymerizing drugs, invoking both cytoskeletal systems in MT capture and in the fusion of AChR vesicles with the cluster membrane. Combined our data identify a transport system, organized by agrin through PI3 kinase, GSK3β, CLASP2, and LL5β, for precise delivery of AChR vesicles from the subsynaptic nuclei to the overlying synaptic membrane.
INTRODUCTION
The density of neurotransmitter receptors accumulated in the postsynaptic membrane of synapses is determined by the balance between receptor insertion and removal. Although numerous factors involved in receptor density regulation, including molecular signals and the pattern of impulse traffic across the synapse, have been identified, the cell biological mechanisms underlying these processes are poorly understood. The neuromuscular junction (NMJ)—the synapse between motor neurons and skeletal muscle fibers—is particularly well suited for their examination because the differentiation of the postsynaptic muscle membrane is organized by one major factor secreted by the motor nerve terminal, agrin (McMahan et al., 1992; Cohen et al., 1997; Jones et al., 1997; Wu et al., 2010), which acts through its receptor/effector LRP4/MuSK in the muscle fiber membrane (Kim et al., 2008; Zhang et al., 2008). As a consequence, synaptic membranes comparable in many respects to the postsynaptic muscle membrane of the NMJ in vivo and amenable to molecular and cell biological manipulation, can be easily generated by culturing myotubes on patches of agrin attached to the culture substrate.
One factor contributing to synaptic AChR accumulation in fetal muscle is the trapping and anchoring of constitutively expressed AChRs diffusing in the surface membrane; the AChRs are trapped via the agrin-induced subsynaptic apparatus, which is anchored to a synapse-specific, cortical actin cytoskeleton (Hall et al., 1981; Dai et al., 2000; Geng et al., 2008; Chen et al., 2014). In the adult, however, in which extrasynaptic AChR levels are very low, a transport system for focal delivery to the synaptic membrane is needed. Indeed, sorting of post-Golgi vesicles containing AChRs polarized to the postsynaptic membrane has been observed in Torpedo electric organ, a homologue of skeletal muscle (Camus et al., 1998). Recently we showed that agrin stimulates the capture of microtubules (MTs) at agrin-induced AChR clusters in cultured myotubes through regulation of the MT plus end–tracking protein (+TIP) CLASP2, a process required for MT-dependent AChR transport to the cluster membrane. Similar to CLASP2-mediated MT capture at the leading edge of migrating cells (Akhmanova et al., 2001; Watanabe et al., 2009), MT capture in myotubes occurs through inhibition of GSK3β-dependent phosphorylation of strategically located serine residues in CLASP2 (Basu et al., 2014), which increases CLASP2 binding to MT plus ends (Kumar et al., 2009) and thus enables their capture through interaction with the cluster membrane. Of interest, a known interaction partner of CLASP2 in migrating cells, the phosphatidylinositol-3,4,5-triphosphate (PIP3)–binding protein LL5β (Lansbergen et al., 2006), is also present at the NMJ (Kishi et al., 2005), where it may be involved in the transition of the AChR cluster topology from plaque to pretzel (Smith and Slater, 1983) that occurs during normal NMJ maturation (Proszynski et al., 2009; Proszynski and Sanes, 2013).
Here we show that LL5β is recruited by agrin to the postsynaptic membrane through its PH domain. LL5β thus enriched preferentially captures CLASP2-decorated MT plus ends at the synapse, thus providing for focal transport of AChR vesicles. Accordingly, knockdown of LL5β in vivo reduces AChR insertion and AChR density at the NMJ in adult mice. Unexpectedly, MTs also require filamentous actin for focused capture, revealing a role for the subsynaptic actin cytoskeleton in AChR clustering not only directly through AChR anchoring, but also indirectly through its involvement in MT capture and AChR delivery.
RESULTS
LL5β is recruited to agrin-induced clusters
Focal, CLASP2-mediated MT capture at agrin-induced AChR clusters in cultured primary myotubes requires a CLASP2-interaction partner at the cluster membrane. A prime candidate to mediate agrin/CLASP2-dependent MT capture is LL5β: first, it binds CLASP2 in fibroblasts (Lansbergen et al., 2006), and second, it is elevated at the postsynaptic membrane of the NMJ, where it is involved in AChR clustering (Kishi et al., 2005). However, its mode of action at the NMJ is unknown. To see whether LL5β affects AChR clustering through capturing of CLASP2-decorated MTs that in turn deliver AChRs to the cluster membrane, we first examined the localization of LL5β and CLASP2 in relation to agrin-induced AChR clusters. Primary myotubes were cultured on a laminin substrate that was focally impregnated with agrin in small patches (20–60 μm in diameter). Where they contacted agrin deposits, myotubes formed large clusters of AChRs. LL5β stained with a specific antibody (monoclonal antibody 1H12 from Kishi et al., 2005) was colocalized with the agrin-induced AChR clusters (Figure 1A). Within some of these, the levels of LL5β over the cluster area were uneven (Figure 1A, arrowheads), but levels were always greater than those outside the cluster; moreover, within clusters, LL5β levels correlated with those of (force expressed) green fluorescent protein (GFP)–CLASP2 (Figure 1, B and C). Thus CLASP2 and LL5β colocalize at discrete regions of the synaptic cluster, consistent with a role for LL5β in CLASP2-mediated MT capture.
FIGURE 1:

LL5β colocalizes with CLASP2 at agrin-induced AChR clusters. (A) LL5β is enriched at agrin-induced AChR clusters. Primary myotubes were cultured on focal agrin deposits on a laminin substrate, and AChR clusters were stained with α-BTX–Alexa 488 (green) and for LL5β (red). Arrowheads mark uneven distribution of LL5β. Scale bar, 5 μm. (B) Regions of elevated LL5β (red) inside AChR clusters (blue) are enriched for CLASP2 (green, marked with arrowheads), consistent with an LL5β-CLASP2 interaction and CLASP2-dependent MT capture at synaptic membranes by LL5β. At 48 h postinfection, myotubes expressing adenovirus-delivered GFP-CLASP2 were stained for AChRs (blue), endogenous LL5β (red), and GFP with antibody. Bottom, magnification of boxed area at top. Scale bar, 10 μm (top), 5 μm (bottom). (C) Quantification of LL5β and CLASP2 inside AChR clusters. Bar graph shows that CLASP2 load/area increases with LL5β levels within the myotube region indicated on x-axis. Means ± SE; **p < 0.01; *p < 0.05; n = 6 myotubes with >10 comets/cell.
Interaction between CLASP2 and LL5β is required for MT capture at agrin-induced clusters
If LL5β is involved in CLASP2-dependent MT capture at AChR clusters, knockdown of LL5β by RNA interference (RNAi) should abolish the enrichment of MTs normally observed at the clusters. The efficiency of LL5β knockdown by RNAi was confirmed by Western blotting of cultured myotube lysates (Figure 2A). Similarly, the accumulation of LL5β at AChR clusters was also abolished (Supplemental Figure S1). Because individual myotubes expressing short hairpin RNAs (shRNAs) could be identified by coexpressed red fluorescent protein (RFP; see Materials and Methods), the effect of LL5β knockdown on MT capture could be assessed by immunofluorescence staining of MT plus end densities inside and outside AChR clusters. MT plus ends, visualized by staining for EB3 (Figure 2B), were about twice as dense at agrin-induced AChR clusters in myotubes expressing a scrambled shRNA (shScrambled) than in myotubes expressing the shLL5β that effectively knocked down endogenous levels of LL5β protein (Figure 2, B and D). These data show that LL5β is required for MT capture at agrin-induced AChR clusters.
FIGURE 2:

Knockdown of LL5β expression and overexpression of CLASP2-C abolish capture of MT plus ends at agrin-induced AChR clusters. (A) Western blot of LL5β isolated from cultured primary myotubes expressing shLL5β and shScrambled (ctrl). Myotubes were infected with lentiviral constructs encoding shLL5β (hairpin no. 2, see Materials and Methods) or shScrambled for control. Two days later, LL5β expression levels were analyzed. (B) MT plus end densities visualized by staining for EB3 are reduced inside AChR clusters upon LL5β knockdown with shLL5β. Two days after infection with shLL5β or with shScrambled (control), respectively, myotubes were stained for AChRs with α-BTX–Alexa 488 (green) and EB3 (red), and densities of EB3-decorated comets inside vs. outside AChR clusters were compared. Boxes 1 (inside cluster) and 2 (outside cluster) are enlarged on the right. Scale bars, 10 μm; enlarged, 2.5 μm. Similar data were obtained with hairpin no. 4 (see Supplemental Methods). (C) Forced expression of GFP-CLASP2-C (green) abolishes increase in EB3 (red) comet density at agrin-induced AChR clusters compared with noncluster region, indicating impairment of MT capture. Boxes are enlarged at the bottom. Scale bar, 10 μm; enlarged, 2.5 μm. (D) Density of EB3 comets inside relative to outside AChR clusters in scrambled control vs. LL5β-knocked-down myotubes and in GFP control vs. GFP-CLASP2-C–transfected myotubes. Means ± SE. N = 12 ctrl, 12 shLL5β, 6 GFP-CLASP2-C, and 7 GFP alone. *p < 0.05. (E) Clustering of prelabeled AChRs by agrin is not affected by LL5β knockdown, indicating that LL5β does not act by trapping AChRs. Representative images of primary myotubes infected with control and shLL5β lentivirus and stained with α-BTX–Alexa 488, followed by addition of 10 nM mini-agrin for 18 h. Arrowheads point to examples of AChR clusters. Scale bar, 50 μm. (F) Quantification of cells from E. Number of clusters per myotube was quantified (left); N = 167 clusters for control and 138 for shLL5β knockdown cells. Quantification of relative AChR cluster density (middle) and area (right); N = 65 clusters in control and 74 clusters in shLL5β-knockdown cells. All bars depict means ± SE.
Because in HeLa cells, CLASP2 interacts with cortical LL5β to attach distal MT ends to the cell cortex (Lansbergen et al., 2006), overexpression of a CLASP2 fragment that binds to LL5β but does not bind to MT plus ends would be expected to abolish MT capture at agrin-induced AChR clusters. Such a fragment, CLASP2-C, comprising amino acids 1254–1338, was identified biochemically (Lansbergen et al., 2006); when overexpressed, CLASP2-C should saturate the LL5β domain interacting with CLASP2 without binding to MT plus ends (Kumar et al., 2009), thus abolishing MT capture at the cluster membrane. We examined, therefore, whether overexpressing GFP-CLASP2-C abolished MT capture and AChR clustering at agrin-induced AChR clusters. Indeed, unlike in myotubes expressing GFP alone, in myotubes induced to express GFP-CLASP2-C, the increase in EB3 comet density normally seen inside versus outside AChR clusters was abolished (Figure 2, C and D) and AChR cluster size declined (Supplemental Figure S2). Combined these experiments indicate that LL5β promotes AChR clustering by mediating MT capturing at agrin-induced AChR clusters through its interaction with MT plus end–bound CLASP2.
Clustering of prelabeled surface AChRs is not affected by LL5β knockdown
LL5β knockdown impairs clustering of AChRs in myotubes grown on laminin alone (Kishi et al., 2005), consistent with the present data. However, AChRs in those experiments were stained after clusters had formed, leaving open the possibility that LL5β affects AChR clustering through trapping rather than through delivery or that spontaneous and agrin-induced AChR clustering are differentially affected by LL5β. Therefore we tested whether LL5β affected AChR trapping. To this end, we cultured myotubes on agrin-free substrate. At 48 h after LL5β knockdown, surface AChRs were pulse labeled with α-bungarotoxin (α-BTX)–Alexa 488, and soluble chicken mini-agrin cN257C21B8 (Lin et al., 2008) was added to the culture medium at 10 nM for 18 h. AChR clusters were then compared with those in control myotubes infected with shScrambled. In two independent experiments, the AChR clusters were similar in LL5β-knockdown and control myotubes with respect to cluster number per myotube, AChR density, and cluster size (Figure 2, E and F). These experiments indicate that LL5β affects agrin-induced AChR clustering by a mechanism other than AChR trapping or metabolic stabilization.
Knockdown of LL5β by RNAi reduces AChR density and insertion at NMJs in vivo
LL5β may be recruited to the synaptic membrane via its PH domain (Kishi et al., 2005), which is known to bind to PIP3 (Paranavitane et al., 2003). Given the local activation of phosphoinositide 3-kinase (PI3K) at the cluster membrane by agrin (Schmidt et al., 2012), we first examined whether LL5β is recruited to synaptic membrane in vivo through mutations in its PH domain critical for binding to PIP3. To this end, we electroporated constructs coding for wild-type (WT) LL5β-GFP and a mutant form (Mut) of LL5β-GFP, in which two point mutations in its PH domain (K1162A/R1163A) abolished LL5β binding to PIP3 (Paranavitane et al., 2003), into soleus muscle; 2–3 wk later, the intensities of GFP fluorescence at the synaptic relative to extrasynaptic membrane were about five times lower in fibers expressing MutLL5β-GFP than in fibers expressing WT-LL5β-GFP (Supplemental Figure S3A). This is in agreement with data by Kishi et al. (2005), who found impaired synaptic localization of a LL5β mutant in which the entire PH domain was deleted. Thus LL5β is localized to the synaptic membrane depending on its PH domain and on agrin-induced PI3-K activity at the clusters.
Abolition of CLASP2-dependent MT capture at NMJs in vivo results in lowering of synaptic AChR density, likely through abolition of MT-mediated AChR transport to the synaptic membrane (Schmidt et al., 2012). If LL5β is involved in AChR delivery at the NMJ through CLASP2-mediated MT capturing, its knockdown should reduce synaptic AChR density as does Clasp2 deletion (Schmidt et al., 2012). We thus examined whether lowering the expression level of LL5β by RNAi affects synaptic AChR density. To this end, we electroporated sternomastoid muscles of wild-type mice with constructs expressing shLL5β or a scrambled shRNA (shScrambled) control together with Histone-2B-monomeric RFP serving to mark nuclei of successfully electroporated fibers (Supplemental Figure S3B). Eleven days after electroporation, AChRs on the surface of muscle cells were labeled to saturation in vivo with fluorescent α-BTX–Alexa 488, and the fluorescence intensities of labeled AChRs in fibers containing mRFP positive nuclei were compared in knockdown and control fibers. In LL5β-knockdown muscles, AChR fluorescence at NMJs was significantly lower (∼30%) than in control (scrambled shRNA) synapses (Supplemental Figure S3, C and D). Our data indicate, therefore, that LL5β recruited to the synaptic membrane via its PH domain contributes to AChR density at the NMJ in vivo. Given its function to mediate CLASP2-dependent MT capture (Figure 2), which in turn promotes AChR delivery to AChR clusters (Schmidt et al., 2012), we predict that LL5β acts by promoting AChR delivery to the synaptic membrane.
To test this prediction at NMJs in vivo, we stained sternomastoid muscles electroporated to express shLL5β and imaged them as summarized in Figure 3A. Specifically, 4 d postelectroporation, synapses were stained to saturation with α-BTX–Alexa 488 and then immediately imaged. Reimaging them again 6 d later revealed the amount of AChR removal. As shown in Figure 3, B and C, the AChR removal in LL5β-knockdown synapses was slightly increased compared with nonelectroporated, neighboring synapses in the same muscles. To estimate the rate at which new AChRs were inserted into the synaptic membrane, we resaturated the synapses with α-BTX–Alexa 488 and reimaged. Electroporated synapses showed only ∼60% recovery of the initial intensity, as compared with close to 100% recovery for nonelectroporated synapses (Figure 3C). Therefore knockdown of LL5β reduces the rate of insertion of acetylcholine receptors in vivo. Of importance, on the time scale used in these experiments (10–11 d total), neither the electroporation procedure itself nor the LL5β knockdown affected gross endplate morphology as judged by the absence of NMJ fragmentation (Figure 3 and Supplemental Figure S3C).
FIGURE 3:

Knockdown of LL5β expression levels in muscle fibers reduces the stability and insertion rate of AChRs at the neuromuscular junction in vivo. (A–C) Stability and insertion rate of synaptic AChRs are reduced at NMJs upon knockdown of LL5β. Histone2B-RFP–expressing pLKO.1-shLL5β or pLKO.1-shScrambled was electroporated into sternomastoid muscles. (A) Schematic depiction of the experimental protocol. (B) Examples of synapses imaged (in pseudocolor) and nuclei marked by RFP in their subsynaptic nuclei used for identification of successfully electroporated fibers. Scale bar, 10 μm. (C) Bar graphs summarizing the effect of LL5β knockdown on AChR stability and insertion. Numbers in columns give numbers of synapses analyzed. Bars indicate mean fluorescence intensities (± SE) relative to that at t = 0. ***p <0.0001; **p < 0.005; two sided t test. (D–F) The stability and insertion rate of both recycled and newly inserted AChRs at NMJs are reduced upon knockdown of LL5β. Electroporation of constructs was as in A). (D) Schematic depiction of the experimental protocol. (E) Examples of synapses in pseudocolor. Scale bar, 10 μm. (F) Graphs summarizing the effect of LL5β knockdown on stability and insertion of recycled and newly inserted AChRs. All other indications are as in B and C. Numbers on columns give numbers of synapses analyzed. Bars depict means ± SE. ***p <0.0001; two sided t test.
On their removal from the synapse, a fraction of the internalized AChRs becomes reinserted into the synaptic membrane (recycled AChRs; Bruneau et al., 2005). To examine whether LL5β knockdown differentially affects the rate of insertion of newly made and recycled AChRs, we stained and imaged AChRs as outlined in Figure 3D. Briefly, to estimate the initial AChR density, we first imaged 7-d-postelectroporation synapses immediately after labeling with biotin-conjugated α-BTX, followed by saturating streptavidin–Alexa 488. Three days later, the rate of removal was calculated by imaging existing AChRs without further labeling. Then a fresh treatment of streptavidin–Alexa 488 was used to estimate the amount of recycled AChRs, and finally, the fraction of newly inserted AChRs was calculated by relabeling with biotin–α-BTX and streptavidin–Alexa 488 (see also Materials and Methods). Quantitative analysis of fluorescence recovery showed that the insertion rates of recycled and newly made AChRs were affected similarly (Figure 3, E and F), consistent with similar insertion routes for the two types of AChRs.
LL5β and actin are required for MT-dependent AChR delivery to agrin-induced AChR clusters in cultured myotubes
To investigate the mechanisms of AChR delivery in more detail, we developed an assay to monitor, in cultured myotubes, the dynamics of AChR insertion at agrin-induced clusters. To this end, we induced myotubes to express GFP-tagged AChRs (GFP-AChRs), using an adenoviral vector (Gensler et al., 2001); together with constitutively expressed AChR subunits, GFP–AChR-γ will form GFP-tagged AChRs that cluster at agrin patches; 2 d after GFP–AChR-γ infection, upon photobleaching the GFP-AChR clusters, we observed fluorescence recovery after photobleaching (FRAP), sampling fluorescence every minute by total internal reflection fluorescence microscopy (TIRFM; Figure 4, A and B). Recovery of GFP fluorescence upon bleaching was ∼80% complete in control cells by 40 min, when observation was stopped due to monitor bleaching (Figure 4B; see also Materials and Methods). The contribution to recovery by diffusion of GFP-AChRs from the nonbleached into the bleached region of the cluster was minor, since 5–10 min after bleaching, no gradient of GFP fluorescence from the periphery toward the center of the bleached region could be resolved (Figure 4, C and D). Moreover, fusions of vesicles containing GFP-AChRs (see later discussion of Figure 6) were slightly more frequent near the periphery of the bleached region than in its central region, probably due to vesicles originating from outside the bleached zone. Thus the recovery into the bleached region can be largely attributed to the insertion of new, unbleached GFP-AChRs.
FIGURE 4:

LL5β and actin are required for MT-dependent AChR delivery to agrin-induced AChR clusters in cultured myotubes. (A–D) Primary myotubes grown on laminin substrates with local deposits of neural agrin were infected with adenovirus expressing the AChR–γ-GFP subunit, and GFP-AChR clusters were visualized by TIRF microscopy. Photobleaching was begun 90 min after addition of drugs and carried out in their presence. (A) Examples of GFP-AChR clusters in control and nocodazole- (10 μM, >90 min) and CytoD-treated myotubes (2 μM, >90 min) before, immediately at, and 20 min after bleaching. Images were acquired every minute for 45 min after bleaching, when monitor bleaching began to interfere. Scale bars, 10 μm. (B) Fluorescence recovery is reduced in myotubes upon LL5β knockdown upon treatment with nocodazole or with CytoD. Note that suppression of fluorescence recovery is almost identical for all treatments. Values of mean fluorescence ± SE as a function of time. Clusters containing bleached regions were imaged every minute for a total of 45 min; background fluorescence outside cells was subtracted; the fluorescence values for each time point were processed to correct for monitor bleaching, and residual fluorescence at t = 0 was normalized to zero as described in Materials and Methods. Means ± SE from eight untreated, six nocodazole-treated, six CytoD treated, and seven shLL5β-treated cells. (C) Scheme used for testing whether fluorescence recovery was due to diffusion of GFP-AChRs from unbleached to the bleached regions. To this end, total fluorescence intensity per area in an annulus (Ir) at the periphery of the bleached region was compared, during recovery, with that in a circle (Ic) at its center. Their radii were as follows: entire FRAP region, 6 μm; inner circle, 2.3 μm; inner edge of annulus, 4.0 μm (resulting in a distance between the outer edge of the circle and the inner edge of the annulus of 1.7 μm); and outer edge of annulus, 4.7 μm (resulting in width of the annulus of 0.70 μm). (D) Graph of the ratio Ic/Ir as a function of time. A value of 1, reached within the first 10 min after bleaching, indicates no gradient from the periphery to the center and, thus, negligible diffusion, consistent with FRAP due to insertion of new GFP-AChRs. Means ± SE from N = 5 control cells. (E) CytoD treatment used for experiments in A and B is effective in breaking down actin cables, as revealed by phalloidin staining. Primary myotubes cultured on agrin substrate were stained with α-BTX–Alexa 488 (green) and phalloidin–Alexa 594 (red) 90 min after CytoD treatment. Scale bar, 10 μm.
FIGURE 6:

Analysis of AChR vesicle delivery to agrin-induced AChR clusters in myotubes expressing GFP–AChR-γ subunit. (A–F) GFP-AChR clusters were bleached and fluorescence recovery observed at 3 frames/s for 30 s in TIRFM. (A) AChR cluster before, immediately after, and 30 s after bleaching. Scale bar. 10 μm. (B) Example of a single frame containing a vesicle fusion event (red box) and its intensity profile during the fusion process as calculated using the SOS plug-in (see Materials and Methods). Scale bar, 2.5 μm. (C) Enlarged red square, showing the complete sequence of frames for the same vesicle as in B during the entire fusion process. (D) Double-fusion event occurring at the same site as in B at a later time point during the acquisition and its corresponding intensity profile. (E) Snapshot of a schematic three-dimensional projection showing the time lapse of the cell in A to illustrate the appearance of double-fusion events at the same position. The bleached region was cropped, individual events were marked as green circles, and double events corresponding to events 8 and 15 were marked as red circles using the MTrackJ plug-in. The time lapse was then projected in the Vaa3D software. The z-axis corresponds to the time scale. (F) GFP fluorescence intensities of all 15 fusion events in the cell shown in A. Bars denote mean ± SE of intensities sampled over the entire duration of the fusion event at 3 frames/s (see the example shown in B). The open triangles denote the number of frames used in the calculation of each such event intensity and SE. Note the uniform intensities of events. The fusions 8 and 13 each can be interpreted as events arising from two consecutive fusions at the same site partly overlapping in time, thus accounting for the higher SE.
MTs contributed significantly to the recovery of GFP fluorescence. Their contribution was revealed by pretreating myotubes with nocodazole (10 μM, >90 min), a drug that disrupts MTs. In the presence of nocodazole, FRAP of GFP-AChRs was significantly reduced. Of importance, and consistent with LL5β knockdown abolishing MT capturing described earlier (Figure 2), knocking down LL5β suppressed AChR delivery to agrin-induced AChR clusters to a degree similar to that by nocodazole (Figure 4B). Finally, FRAP of GFP-AChRs was reduced to a similar degree after myotubes had been pretreated with ZSTK474 (unpublished data), a specific inhibitor of PI3K, which blocks MT capture through inhibition of CLASP2 binding to MT plus ends (Schmidt et al., 2012; Basu et al., 2014). These experiments suggest that LL5β affects AChR delivery by mediating CLASP2-dependent MT capture, consistent with the decrease in MT plus end density upon LL5β knockdown and overexpression of CLASP2-C shown earlier (Figure 2, B–D).
Synaptic targeting of AChRs is believed to require a synapse-specific actin cytoskeleton that traps AChRs diffusing in the surface membrane (Dai et al., 2000; Geng et al., 2009). However, indirect evidence also suggests the involvement of actin in active AChR transport mechanisms to the cluster membrane (Lee et al., 2009). To see whether a role of actin in AChR delivery could be resolved in our FRAP assay, we treated myotubes with cytochalasin D (CytoD; 2 μM, >90 min), a drug that disrupts actin filaments by inhibiting polymerization of actin monomers (Figure 4E). Indeed, CytoD suppressed FRAP of GFP-AChRs and, thus, AChR delivery to the clusters, down to the levels found after nocodazole treatment or LL5β knockdown (Figure 4B).
An intact actin cytoskeleton is required for MT capture at AChR clusters
Capture of dynamic MTs at AChR clusters can be visualized in living cultured myotubes by observing MT plus ends decorated with GFP-tagged CLIP170 in the TIRF microscope (Schmidt et al., 2012). To examine whether actin was involved in AChR delivery at the level of MT capture in living myotubes, we pretreated myotubes derived from Gfp-Clip-170 knock-in mice (Akhmanova et al., 2005) with CytoD (Figure 5). The dynamics of GFP-tagged MT plus ends was then analyzed from kymographs (Figure 5, A–C) generated from time-lapse movies. As previously observed in untreated control myotubes, the mobility of MTs inside the clusters was significantly smaller than outside, indicating MT capture preferentially at the cluster membrane (Schmidt et al., 2012). This difference was reduced upon treatment with CytoD, that is, the majority of MT plus ends was mobile inside the clusters (Figure 5C). In contrast, outside AChR clusters, MT dynamics appeared unaffected by CytoD treatment (compare red columns in Figure 5C). The average speeds of GFP-CLIP170 plus ends were also unchanged, with GFP-CLIP170 having an average speed of 0.163 ± 0. 0.078 μm/s in control cells and 0.147 ± 0.069 μm/s in CytoD-treated cells (mean ± SD from 30 tracks each), in quantitative agreement with previous measurements (Schmidt et al., 2012). This indicates that the behavior of MT plus ends near the membrane is greatly altered inside but remains unchanged outside clusters. We also observed that the total density of plus ends at the cluster relative to outside was slightly decreased upon CytoD treatment (unpublished data), thereby pointing to a role of synaptic actin in guiding plus ends to AChR clusters.
FIGURE 5:

The actin cytoskeleton is required for MT capture at AChR clusters. (A–C) CytoD inhibits MT plus end capture at agrin-induced AChR cluster in live myotubes. Myotubes derived from GFP–CLIP170-KI mutant mice were cultured on agrin patches, and dynamics of GFP-labeled MT plus ends was recorded in TIRFM at 1 frame/s for 180 s with and without CytoD treatment. (A) First frames (left) and projection of all 180 frames superimposed in control (top) and CytoD-treated myotubes (bottom). The outlines of AChR clusters are shown by solid black lines. Note higher number of stable comets (dots in projection) in clusters in control than in CytoD-treated myotubes. Scale bar, 5 μm. (B) Examples of kymographs generated from stable and moving MT plus ends; kymographs were generated from time-lapse recordings as illustrated in A. (C) Graph summarizing the percentages of stable MT plus ends inside and outside AChR clusters. Note the lowered percentage of stable MT plus ends inside AChR clusters in CytoD-treated myotubes, whereas percentages outside clusters remained unchanged. Owing to differing MT plus end abundance between myotubes, percentage values of stable comets inside relative to outside AChR clusters were calculated for each myotube, and percentages were averaged. Bars give means ± SE (N = 4 cells each for control and CytoD-treated myotubes). (D, E) CytoD inhibits MT plus end capture at agrin-induced AChR clusters, as visualized in fixed myotubes by density of EB3-stained MT plus ends. (D) Myotubes were cultured on agrin patches, fixed after 90 min of CytoD treatment, and stained with α-BTX–Alexa 488 (AChRs, green) and an antibody to EB3 (red). Right, boxes 1 (inside) and 2 (outside clusters) enlarged to visualize EB3-positive plus end densities. Scale bar, 7.5 μm; boxes, 2.5 μm. (E) Bars summarizing densities of EB3-stained MT plus ends inside relative to outside clusters. Means ± SE; N = 18 cells for control, 19 cells for CytoD-treated myotubes; *p < 0.05.
A similar result was obtained when the density of comets visualized by EB3 staining in fixed cells was used as a readout of MT capture. Specifically, in control myotubes, EB3 comet density observed by confocal laser scanning microscopy (CLSM) was increased inside compared with outside clusters, and it was largely abolished in CytoD-treated myotubes (Figure 5, D and E). These data indicate that MT capture at AChR clusters requires an intact actin cytoskeleton.
Finally, we tested whether the intact actin cytoskeleton at the cluster may be required for the MT capture process itself or for stabilizing the signaling cascade linking agrin to MT capture (Schmidt et al., 2012). The latter possibility was excluded by our findings that CytoD treatment did not affect the agrin-induced phosphorylation (= inactivation) of GSK3β (Supplemental Figure S4A) or the recruitment of LL5β to the agrin patch (Supplemental Figure S4B).
Analysis of AChR delivery at the level of single vesicles
Close inspection of the bleached region at higher acquisition rates showed that soon after bleaching, spots of GFP-AChR fluorescence ∼200 nm in diameter appeared, and they disappeared within a few seconds (Figure 6, A–C, and Supplementary Movies S1 and S2). Such events observed in TIRFM can be interpreted as the arrival of vesicles filled with GFP-tagged AChRs into the TIRFM plane of view, followed by fusion of the vesicle with the cell membrane. Indeed, we observed internal AChR vesicles along dynamic MTs by costaining intracellular AChRs with tyrosinated tubulin (Supplemental Figure S5). To estimate the AChR insertion rate, we counted the occurrence of fusion events in the bleached region within the first 30 s after bleaching at an acquisition rate of 3 frames/s using automated tracking (see Materials and Methods). In accordance with previously observed docking times for vesicle fusion events (Schmoranzer and Simon, 2003; Toonen et al., 2006; Grigoriev et al., 2007), we considered only discrete spots of fluorescence that persisted for >1 s in our analysis, as events that lasted <1 s most likely represent untethered vesicles moving in and out of the TIRF plane. With these criteria, we observed in control myotubes between 15 and 20 fusion events per 30 s in a bleached region 7 μm in diameter. For further characterization, we examined the spatial distribution of the fusion events over the bleached area, their durations, and their fluorescence intensities.
Fusion events were not randomly distributed over the bleached region. For example, in the fusions shown in Figure 6, two of 15 events occurred at the same site (Figure 6, B and D, and F, arrowheads) at different times after bleaching. The appearance of these events is shown schematically in Figure 6E as a three-dimensional visualization of the time lapse for the cell shown, in which the two double events are indicated in red and the rest are depicted in green. Given the area of a single event of ∼0.32 μm2 (diameter, 0.2 μm) and the total area of the bleached region of 38 μm2 (diameter, 7 μm), the probability for two fusions occurring randomly at one and the same site is calculated to be 0.32/38 = 8 × 10−3, and for two more fusions coinciding at another site, it is 6.4 × 10−5, that is, much lower than actually observed. Similar findings were obtained in a total of 12 cells analyzed, where the total number of fusions was 178, 30 of which were double events. These data suggest that there are preferred sites of vesicle delivery within the agrin-controlled AChR clusters.
Of interest, the mean fluorescence intensities of each fluorescent spot appearing in time (those from the representative cell shown in Figure 6A are plotted in Figure 6F) were quite similar, except for some for which fluorescence was about twice as high (Figure 6F, events 8 and 13). These results support our conclusion that the fluorescent spots are indeed vesicles filled with similar amounts of AChR molecules. Events with twice the fluorescence intensity can be interpreted as two fusions at the same site overlapping in time.
Finally, the events appeared to occur with slight preference at the periphery of the bleached region (unpublished results), perhaps reflecting unbleached GFP-AChRs moving in from the nonbleached region of the myotube (see also Figure 4, B and D).
Agrin-induced AChR vesicle transport to the synapse depends on LL5β and F-actin integrity
In accordance with our interpretation that suppression of recovery of “global” GFP-AChR fluorescence was due to impairment of AChR delivery by interfering with MT capture, the frequency of fusion events within the 30-s observation period was strongly reduced after LL5β knockdown (Figure 7A) and even more after CytoD treatment (Figure 7D). As expected for a function in vesicle trafficking rather than in loading of receptors into vesicles, the fluorescence intensity was not affected by either treatment (Figure 7, C and F). However, LL5β knockdown and CytoD appeared to interfere with different aspects of the fusion process. First, the fusion frequency was reduced more strongly by CytoD than by LL5β knockdown (compare Figure 7, A and D); second, in the LL5β-knockdown myotubes, although the frequency of fusion events was reduced, the fraction of double-fusion events in all fusions resolved (103 fusions in 14 clusters examined) remained unchanged (0.16 in LL5β knockdown; 0.17 in controls); and third, unlike LL5β knockdown, which had no effect on the duration of the fusion events (Figure 7B), this parameter was reduced ∼25% by interfering with the normal function of the actin cytoskeleton (Figure 7E).
FIGURE 7:

LL5β knockdown and CytoD treatment differentially affect frequency, duration, and GFP fluorescence intensity of AChR vesicle fusions. All methods and parameters as defined in Figure 6. Bars give means ± SE. (A–C) LL5β-knockdown myotubes. Note the reduction of fusion numbers during 30-s observation period upon LL5β knockdown without changes in duration and intensity of fusion events. The relative number of double fusions at the same sites was not affected (see text). N = 12 cells for control and 14 for cells for LL5β-knockdown myotubes. Means ± SE. *p < 0.05. (D–F) CytoD-treated myotubes. Note stronger reduction of fusion numbers during 30-s observation period upon CytoD treatment, with significant reduction in duration but not intensity of fusion events. Means ± SE. N = 14 cells for control and 14 cells for CytoD-treated myotubes. **p < 0.001, ***p < 0.0001.
Finally, pretreating myotubes with the PI3K inhibitor ZSTK474 reduced the frequency of fusion events to 25% of that in controls without an effect on event duration or fluorescence intensity (unpublished data); this is in full agreement with the demonstrated role of PI3K in LL5β recruitment and GSK3β inactivation (Schmidt et al., 2012). These data combined indicate that whereas LL5β is required for MT-dependent vesicle delivery to the synaptic membrane, it is apparently not required for membrane fusion itself. Instead, the failure of LL5β knockdown to affect the duration and intensity of fusion events suggests that fusion occurs normally in LL5β-depleted cells. However, an intact actin cytoskeleton appears to be required for several aspects of focal AChR delivery, including MT capture and vesicle fusion with the membrane.
DISCUSSION
The present work demonstrates a new role for LL5β in the maintenance of the NMJ. Specifically, LL5β in the synaptic membrane enables the MT-dependent focal trafficking of AChR vesicles to the synapse through capture of CLASP2-decorated MT plus ends.
LL5β is enriched at the synaptic membrane by elevated expression of the LL5β gene from the subsynaptic nuclei (Kishi et al., 2005) and the specific recruitment of its protein product to the synaptic membrane. Recruitment is via its PH domain to PIP3 (Paranavitane et al., 2003) generated by agrin at the synapse through focal activation of PI3K (Schmidt et al., 2012) and/or to synaptic filamin (Bloch and Hall, 1983; Shadiack and Nitkin, 1991). Through its synaptic localization by agrin, LL5β directs the capture of CLASP2-decorated MT plus ends to the synaptic membrane. The captured MTs mediate AChR transport to the cluster membrane in cultured myotubes and to the NMJ in vivo. That the CLASP2-dependent MT capture is controlled by LL5β is indicated by the findings that in myotubes deprived of LL5β by RNAi or expressing CLASP2-C (the CLASP2 domain interacting with LL5β), the capture of MT plus ends at AChR clusters is abolished. The involvement of MTs captured through CLASP2/LL5β-interaction in AChR delivery is reflected by the reduction in the recovery of GFP-AChR fluorescence upon photobleaching, which was quantitatively similar upon LL5β knockdown, treatment of myotubes with the MT-depolymerizing drug nocodazole, and with the PI3K inhibitor ZSTK474; consistent with our model, both nocodazole and ZSTK474 also abolish CLASP2-dependent MT capture and focal delivery of AChR vesicles. Finally, the physiological relevance of this process for the maintenance of synaptic AChR density in vivo is demonstrated by the reduced density and insertion of new and recycled AChRs at NMJs in mice upon LL5β knockdown and in Clasp2−/− mice (Schmidt et al., 2012).
AChR delivery to clusters was not continuous but appeared to occur in discrete steps, which we interpret as the focal fusion of GFP-AChR–containing vesicles with the cluster membrane. A small proportion of these discrete events also showed lateral movement within the bleached region; however, most of them seemed to appear, persist in the TIRF plane, and then disappear in time. We propose therefore that whereas the laterally moving vesicles might represent movement along MT tracks into the membrane, the majority of fusions observed come from MT tracks directly underneath the plasma membrane. Some of the characteristics of the fusion events in response to LL5β knockdown are suggestive of specific mechanisms involved. First, individual fusions occurred at preferred sites, consistent with preferred sites of MT capture, which may be explained by the speckled distribution of LL5β within the cluster (see, e.g., Figure 1). Second, with abolished MT capture upon LL5β knockdown, only the frequency, but neither the duration of the fusion events nor their intensity, was reduced, suggesting, within the limits of the resolution of our experiments, that LL5β is not involved in the fusion process itself. The more robust reduction by LL5β knockdown in FRAP of GFP-AChR fluorescence (∼2.5-fold; Figure 4B) than in MT capture (∼2-fold; Figure 2D) cannot be interpreted to suggest an involvement of LL5β in the fusion process because CLSM, by probing MT plus ends more deeply into the cell than TIRFM (used to assess GFP-AChR insertion), likely included MTs that did not reach the cell cortex and thus were not captured; as a consequence, when probed by CLSM, the densities of effectively captured MTs both at clusters and in noncluster regions will be overestimated, leading to underestimation of their ratio (e.g., Figures 2D and 5E). Third, MT capture and AChR delivery depend on the integrity of the actin cytoskeleton; again, their suppression by the actin- depolymerizing drug CytoD was quantitatively similar to that after nocodazole treatment and LL5β knockdown. However, unlike LL5β knockdown, CytoD treatment affected not only the fusion frequency, but also the fusion process itself; this is indicated by the shortening of its duration.
The striking effect of CytoD treatment to dramatically reduce dynamic MT capture implies some interaction between MTs and actin filaments in directing MTs to or in the MT capture process at the cluster membrane itself, likely involving the subsynaptic actin cytoskeleton. We did observe a slight decrease in the delivery of plus ends to the synaptic cluster (albeit not significant) but not outside it. Taking this together with our observation that average vesicle fusion times were significantly reduced upon CytoD treatment, we conclude that synapse-specific actin regulates the guidance and capture of MTs, as well as vesicle fusion into the synaptic membrane.
Considering the importance of MT capture for vesicular AChR delivery, the amount of MTs captured at AChR clusters appears to be very low. Indeed, whereas capture of GFP-CLIP170–decorated MT plus ends is readily resolved by TIRFM at agrin-induced AChR clusters in living MTs, immunofluorescence staining of MTs with antibodies in fixed cells to α-tubulin or to detyrosinated tubulin did not reveal MT accumulations at AChR clusters (unpublished results). The question remains, therefore, of how dynamic MTs contribute significantly to AChR delivery. In CNS neurons, dynamic MTs transiently entering dendritic spines to influence actin dynamics (Jaworski et al., 2009) may also be involved in the transport of cargo toward synaptic sites (Penzes et al., 2009); such cargo may be transferred from MT transporters to actin motor proteins. Indeed, myosin Va, which has been reported to mediate AChR recycling at the NMJ (Roder et al., 2008), can track MT plus ends by hitchhiking on EB1 in melanocytes (Wu et al., 2005). Further, F-actin itself can regulate the entry of MTs into dendritic spines through the actin- and MT-binding partner drebrin (Merriam et al., 2013), a process believed to play a role in the trafficking of synaptic proteins, although the molecular cargo believed to be transported by spine MTs remains unidentified.
Recently LL5β has been implicated, in, besides mediation of CLASP2-dependent MT capture and AChR delivery, another aspect of NMJ differentiation: it has been suggested that the postnatal transformation of the AChR cluster from an immature, plaque-like to a mature, pretzel-shaped topology is regulated through corralling AChRs, involving podosomes rich in LL5β and several of its associated proteins (Kishi et al., 2005; Proszynski et al., 2009; Proszynski and Sanes, 2013). In our experiments with clusters induced by spots of agrin on the substrate, we did not see the complementary staining of LL5β foci interspersed among AChR microclusters, as is seen in laminin-induced clusters, perhaps because the high concentration of agrin in our agrin spots overrides this muscle-autonomous process.
In summary, combined with our previous findings (Schmidt et al., 2012), the present experiments strongly suggest that agrin promotes the delivery of AChRs (and possibly other synapse-specific molecules) to the synaptic muscle membrane via the following mechanisms: 1) local inactivation, through activation of PI3K, of GSK3β maintaining its substrate CLASP2 in a dephosphorylated state near the synaptic membrane, 2) binding of dephosphorylated CLASP2 to MT plus ends, 3) recruitment of LL5β to the synaptic membrane through activation of PI3-K, and 4) local interaction of MT plus end–bound CLASP2 with synaptic LL5β, thus promoting MT capture at the synapse. In support of this model, the reduction in synaptic AChR density at NMJs in vivo upon deletion of Clasp2 (Schmidt et al., 2012) is similar to that upon knockdown of LL5β found here.
MATERIALS AND METHODS
Animals
For electroporation, C57Bl6 mice were anesthetized with ketamine (87 mg/kg body weight) and xylazine (13 mg/kg body weight). Postoperative analgesia was by four injections of buprenorphine at 12-h intervals. Mice were killed with CO2. Animal handling was approved by the Cantonal Veterinary Office of Basel-Stadt. Generation of the Gfp-Clip170 knock-in mice has been described previously (Akhmanova et al., 2005).
Electroporation of adult mouse muscle
A 40-μg amount of DNA (LL5β-GFP or Mut-LL5β-GFP) in 10 μl of 0.9% NaCl was injected with a 27-gauge Hamilton syringe into the soleus muscle of anesthetized 8- to 10-wk-old C57BL76 mice. After suturing of the skin, eight pulses (20 ms, 1 Hz, 200 V/cm) were applied to the leg using an ECM 830 square wave electroporation system (Harvard Apparatus, Holliston, MA).
For the sternomastoideus, electroporation was done as described in Bruneau and Akaaboune (2010). Briefly, the sternomastoid muscle was surgically exposed, and 7 μl of shRNA (shLL5β or shScrambled; 4 mg/ml) was layered over the muscle surface. Gold electrodes were placed parallel to the muscle fibers on either side of the muscle, and eight monopolar square-wave pulses were applied perpendicular to the long axis of the muscle. After electroporation, the mouse was sutured and allowed to recover in a heated recovery chamber.
In vivo labeling, live-animal imaging, and quantitative fluorescence measurements
Histone2B-RFP expressing pLKO.1-shLL5β or pLKO.1-shScrambled (described in the Constructs section of Supplemental Methods) was electroporated in mouse sternomastoid muscle as described and analyzed 2–3 wk later. To determine AChR density at the synapse, the sternomastoid muscle was labeled for 1 h with 5 μg/ml α-BTX–Alexa 594 (a dose that has been demonstrated to be sufficient to saturate all receptors; Bruneau et al., 2005), and superficial synapses were imaged. Only fibers in which synaptic nuclei were marked by Histone2B-RFP, indicative of either shLL5β or shScrambled expression, were analyzed.
To determine the removal rate of the different AChR pools (preexisting and recycled AChRs), the sternomastoid muscle of anesthetized mice was exposed to α-BTX–biotin (1 μg/ml, 10 min), followed by a single saturating dose of streptavidin–Alexa 488, so that synaptic transmission remained functional (Akaaboune et al., 2002), and synapses were imaged. Three days later, preexisting receptors from the same synapses were imaged, following which, recycled receptors that had lost the streptavidin–Alexa 488 in the process of recycling (Bruneau et al., 2005) and were reinserted into the synaptic membrane were labeled with streptavidin–Alexa 488 and reimaged. Finally, newly added receptors were labeled with a saturating dose of BTX-biotin/streptavidin-Alexa 488 and imaged. All controls for the specificity of the biotin-streptavidin dissociation have been worked out previously (Bruneau et al., 2005). Live-animal imaging and quantitative fluorescence measurements were performed as described (Turney et al., 1996; Akaaboune et al., 1999; Martinez-Pena y Valenzuela et al., 2005).
Analysis of LL5β recruitment to synaptic muscle membranes in vivo
A 40-μg amount of pGFP-LL5β or pGFP-LL5β-Mut (Paranavitane et al., 2003) was electroporated in mouse soleus muscle as described. At 10–14 d later, electroporated muscles were dissected, AChRs stained with 1 μg/ml α-BTX–Alexa 594 for 1 h at room temperature, fixed in 4% paraformaldehyde for 2 h, placed in 30% sucrose/phosphate-buffered saline overnight, and frozen. Muscles were then embedded and sectioned at 12-μm thickness in a cryostat (CM 1950; Leica Biosystems, Nussloch, Germany). Sections were mounted in Citifluor and imaged using an ACS APO 63×/1.3 numerical aperture (NA) objective on an SPE confocal microscope (DMI 4000B; Leica Microsystems, Mannheim, Germany).
For the measurement of synaptic GFP intensity, the α-BTX–stained synapse of GFP-positive fibers was defined as a region of interest (ROI). Using ImageJ (National Institutes of Health, Bethesda, MD), we measured green fluorescence in this synaptic ROI, moved the ROI to the extrasynaptic region of the same fiber profile, and measured the extrasynaptic green fluorescence . Because the level of GFP expression varies across fibers within an individual muscle, the ratio of green fluorescence at the synapse versus extrasynaptic regions was calculated individually for every fiber.
Preparation of primary muscle cultures
Preparation of primary myotube cultures from mouse neonatal leg muscle has been described (Schmidt et al., 2012). Myoblasts were cultured on laminin-coated dishes focally impregnated with agrin. For the preparation of the dishes, COS-1 cells transfected with a plasmid coding for full-length chicken agrin (Jones et al., 1996) were seeded at a density of (7–20) × 103 cells per 30-mm laminin-coated culture dish. Myoblasts were then seeded as described (Schmidt et al., 2011). Subsequent differentiation was in DMEM, 5% horse serum, and 1% antibiotic/antimycotic solution. Note that throughout this article, agrin was applied attached to the culture substrate rather than in solution to mimic the in vivo situation, unless otherwise mentioned.
For gene delivery in vitro using conventional transfection, myoblasts were transfected at confluency immediately before switching to fusion medium using FuGENE6 (Roche Diagnostics, Basel, Switzerland). For adenoviral and lentiviral transduction, cells were infected after formation of myotubes.
Immunofluorescence
Staining of +TIPs and other proteins in cultured myotubes was performed as described (Schmidt et al., 2012) after fixation in −80°C methanol. For staining intracellular AChRs, primary myotubes cultured on agrin substrate were first treated with a saturating dose of α-BTX–Alexa 488 (8 μg/ml, 4 h). After frequent washes in prewarmed differentiation medium for 30 min, cells were fixed in −80°C methanol as described. Cells were then incubated in Tyr-tubulin antibody overnight at 4°C and subsequently treated with secondary antibody for 45 min. α-BTX–Alexa 647 (0.3 μg/ml) was added during the last 10 min of incubation, followed by washing and mounting in Prolong Gold (Invitrogen, Life Technologies, Carlsbad, CA).
Confocal laser scanning microscopy
Confocal microscopy was performed as described (Schmidt et al., 2012). Myotubes were imaged with an SPE confocal scanning laser microscope (DMI 4000B; Leica) at a resolution of 1024 × 1024 pixels using an HCX PL APO 100× objective (NA 1.46) or an ACS APO 63× objective (NA 1.30). Image stacks were acquired with a step size of 300 nm. For comparison of different samples, the same laser settings and gains were applied.
Structured illumination microscopy
Fixed samples were imaged using the ELYRA PS.1 structured illumination microscope (Carl Zeiss AG, Jena, Germany). Images were acquired using a 63×/1.40 oil plan Apochromat objective and an Andor iXon DU 885 electron-multiplying charge-coupled device (EMCCD) camera (Oxford Instruments, Belfast, UK). Stacks of images of cultured myotubes were acquired at a step size of 0.110 μm. Image processing was performed using the Zeiss Zen software. For colocalization experiments, precise pixel alignment between different acquisition channels was ensured by correcting potential pixel shifts via the channel alignment function within the Zen software (Zeiss). Maximum-intensity projections and volume rendering was performed using Fiji (Schindelin et al., 2012).
Total internal reflection microscopy
The dynamics of the insertion of AChRs into and their stability in agrin-induced clusters were examined in primary mouse myotubes by TIRFM. Myotubes derived from C57Bl/6 mice were stained with α-BTX–Alexa 594 for 30 min and then washed with prewarmed DMEM for 1 h before imaging. For viral infections, myotubes were infected with the indicated viral supernatants 2 d before imaging. Cells were imaged at 37°C in Krebs Ringer's solution (140 mM NaCl, 5 mM KCl, 1 mM Mg2+, 2 mM Ca2+, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 1 mM NaHPO4, and 5.5 mM glucose) at pH 7.4. Images were acquired either on an inverted Nikon TI Eclipse microscope (Chiyoda, Tokyo, Japan), fitted with a 100× Apo TIRF objective from Nikon (NA 1.49), using an EMCCD camera (Evolve; Photometrics, Tucson, AZ) with 491- and 561-nm laser lines (20 mW) and a filterblock from Chroma (Chroma Technology, Bellows Falls, VT) and the Lambda 10-3 shutter (Sutter Instrument Company, Novato, CA), or on a TIRF Nikon Eclipse Ti-E inverted microscope with Nikon CFI APO TIRF 100× (1.49 NA) oil objective equipped with a Nikon perfect focus system and QuantEM 512SC EMCCD cameras (Photometrics) and with dichroic filter 565dcxr (Chroma) and a HQ530/30m emission filter (Chroma). The iLAS2 FRAP module (Roper Scientific, SAS, Evry Cedex, France) integrated into MetaMorph (Molecular Devices, Sunnyvale, CA) was used during photobleaching. Focus was maintained throughout imaging with the help of a perfect focus system.
Fluorescence recovery after photobleaching
AChR insertion was assessed from the recovery of GFP fluorescence in clusters of GFP-AChRs upon photobleaching a circled region of 6-μm radius with 95% laser power for 1100 ms. After photobleaching, fluorescence in the bleached and surrounding nonbleached regions of the cluster was monitored every minute for 45 min under identical acquisition settings and exposure times of 300 ms. The raw fluorescence data were then processed as follows: for every cluster at every time point, a circle of similar size as the bleached region was defined in the adjacent nonbleached region (red dotted in Figure 4A) and just outside the cell, and their mean fluorescence intensities were determined using ImageJ (Schneider et al., 2012). For every time point, background fluorescence outside myotubes was subtracted from those of the bleached and nonbleached regions, respectively. To correct for bleaching due to TIRFM sampling (monitor bleaching), the fluorescence within the bleached region relative to that of immediately adjacent, unbleached region was calculated. This ratio was then normalized to a scale between 0 (= fluorescence intensity [FI] immediately after bleaching, FI(0)) and 1 (= FI before bleaching), according to recovery, R(t) = 1 − [1 − F(t)]/[1 − F(0)]; this procedure sets recovery to 0, independently of completeness of bleaching (which was normally between 70 and 80%). A major contribution to fluorescence recovery by diffusion of GFP-AChRs from nonbleached regions could not be resolved (Figure 4, B and C).
Visualization of GFP-AChR vesicle fusions with the cluster membrane
Vesicles could be resolved in the bleached region when the bleaching time was reduced to 585 ms to minimize bleaching in the Golgi underlying the bleached region, and image acquisition was performed continuously every 300 ms (exposure time, 300 ms). Analysis of vesicle delivery upon CytoD treatment was performed using the same settings but a different microscope from the one used for LL5β knockdown (see Total internal reflection microscopy).
+TIP comet dynamics
Dynamics of GFP-CLIP170 comets was measured in primary myotubes derived from Gfp-Clip170 knock-in mice. Agrin-induced AChR clusters were stained with α-BTX–Alexa 594 (1 μg/ml, 30 min) and, after washing with prewarmed DME cells, were incubated at 37°C for 1 h before imaging. For TIRF imaging of GFP-CLIP170, images were acquired at 1 frame/s for 160–180 s with an exposure of 1000 ms. Dynamics of CLIP170 inside and outside agrin-induced AChR clusters was analyzed using ImageJ as described in Schmidt et al. (2012).
Quantitative analysis of vesicle fusion events
Single-particle tracking was performed using a custom-written plug-in for ImageJ. The plug-in first applied a detection algorithm based on a weighted least-squares Gaussian fitting of point-spread-functions (Thompson et al., 2002) within manually outlined regions of interest (FRAP regions). To efficiently localize particles of interest in the noisy images and deal with spurious detections, the images were first preprocessed using the multiscale isotropic undecimated wavelet transform (Olivo-Marin, 2002). The local maxima in the resulting images were taken as the candidate positions. To discriminate between true particles and noise, a two-dimensional Gaussian fitting algorithm (Thompson et al., 2002) was applied to the original images at these maxima, which yielded for each candidate position its subpixel coordinates (x, y), amplitude I, and size, expressed as the SD σ of the Gaussian fit. All positions for which I > Imin and  were then accepted as true particle positions. Typical values used for the parameters were
were then accepted as true particle positions. Typical values used for the parameters were 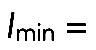 30–40 (for 8-bit images),
30–40 (for 8-bit images), 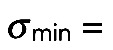 40–50 nm, and
40–50 nm, and 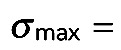 150–200 nm. Tracks were subsequently created for the obtained detections using a nearest-neighbor (based on the Euclidean distance) linking approach and visualized using the MTrackJ plug-in of ImageJ (Meijering et al., 2012). In other words, each trajectory consisted of detections with the interframe displacement not larger than 120 nm. Further, only trajectories >3 frames were used for the analysis. Three-dimensional projections of the bleached region were generated in Vaa3D (Peng et al., 2010).
150–200 nm. Tracks were subsequently created for the obtained detections using a nearest-neighbor (based on the Euclidean distance) linking approach and visualized using the MTrackJ plug-in of ImageJ (Meijering et al., 2012). In other words, each trajectory consisted of detections with the interframe displacement not larger than 120 nm. Further, only trajectories >3 frames were used for the analysis. Three-dimensional projections of the bleached region were generated in Vaa3D (Peng et al., 2010).
Quantitative analysis of +TIP comet density in fixed cells
Immunostaining in cultured myotubes was performed as described previously (Schmidt et al., 2012). The AChR cluster was defined as an ROI, and total number of EB3 comets was counted using ImageJ. A similar ROI was selected outside the cluster for each cell, and EB3 comets were counted within this as well. The number of comets per unit area was then calculated inside and outside the clusters to obtain EB3 comet density.
Quantitative analysis of AChR cluster size
To examine the effect of LL5β knockdown on clustering of surface AChRs, primary myotubes were infected with the indicated lentiviral constructs 24 h after switching to differentiation medium, and surface AChRs were stained with 1 μg/ml α-BTX–Alexa 488 at 36 h later, followed by frequent washing (every 5 min for 1 h at 37°C). Then 10 nM soluble chicken mini-agrin was added to the cultures for 18 h, following which, cultures were fixed with 2% paraformaldehyde for 10 min and imaged on an inverted Nikon Ti Eclipse microscope using a 40× objective (Nikon Plan Fluor/1.30 NA). To analyze AChR area and density in CLASP2-C–transfected myotubes, primary myoblasts grown on agrin patches were transfected with appropriate constructs and switched to differentiation medium the next day. At 48 h later, myotubes were stained with 1 μg/ml α-BTX–Alexa 594, fixed in 2% paraformaldehyde, and imaged as described. Quantification was done in ImageJ, as described in Basu et al. (2014).
Supplementary Material
Acknowledgments
We thank M. Ruegg (Biozentrum, University of Basel, Basel, Switzerland) for critical reading of the manuscript, J. R. Sanes (Department of Molecular and Cellular Biology, Harvard University, Cambridge, MA) for anti-LL5β antibodies, V. Witzemann (Max Planck Institute for Medical Research, Heidelberg, Germany) for the GFP–AChR-γ expression construct, O. Pertz (Department of Biomedicine, University of Basel, Basel, Switzerland) for assistance with TIRF microscopy, and J. Slotman and W. van Cappellen (Optical Imaging Center, Erasmus MC, Rotterdam, Netherlands) for their assistance with structured illumination microscopy. This work was supported by the Swiss National Science Foundation, the Swiss Foundation for Research on Muscle Diseases, the Netherlands Organization for Scientific Research (ZonMW Grant 91208002 to N.G.), and National Institutes of Health Grant NS-047332.
Abbreviations used:
- AChR
acetycholine receptor
- BTX
bungarotoxin
- CLSM
confocal laser scanning microscopy
- GFP
green fluorescent protein
- LRP4
low-density lipoprotein receptor–related protein 4
- MT
microtubule
- MuSK
muscle-specific kinase
- NMJ
neuromuscular junction
- RFP
red fluorescent protein
- +TIP
plus-end tracking protein
- TIRFM
total internal reflection microscopy.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E14-06-1158) on January 14, 2015.
S.B. performed experiments, analyzed and interpreted data, and edited the manuscript; S.S., I.M.P.V., and M.A. performed experiments and data analysis; I.S. developed software for data analysis; K.M. supervised and assisted in microscopy; N.G. drafted and edited the manuscript and interpreted the data; and H.R.B. conceived the study, interpreted the data, and drafted and edited the manuscript.
REFERENCES
- Akaaboune M, Culican SM, Turney SG, Lichtman JW. Rapid and reversible effects of activity on acetylcholine receptor density at the neuromuscular junction in vivo. Science. 1999;286:503–507. doi: 10.1126/science.286.5439.503. [DOI] [PubMed] [Google Scholar]
- Akaaboune M, Grady RM, Turney S, Sanes JR, Lichtman JW. Neurotransmitter receptor dynamics studied in vivo by reversible photo-unbinding of fluorescent ligands. Neuron. 2002;34:865–876. doi: 10.1016/s0896-6273(02)00739-0. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Hoogenraad CC, Drabek K, Stepanova T, Dortland B, Verkerk T, Vermeulen W, Burgering BM, De Zeeuw CI, Grosveld F, et al. CLASPs Are CLIP-115 and -170 associating proteins involved in the regional regulation of microtubule dynamics in motile fibroblasts. Cell. 2001;104:923–935. doi: 10.1016/s0092-8674(01)00288-4. [DOI] [PubMed] [Google Scholar]
- Akhmanova A, Mausset-Bonnefont AL, van Cappellen W, Keijzer N, Hoogenraad CC, Stepanova T, Drabek K, van der Wees J, Mommaas M, Onderwater J, et al. The microtubule plus-end-tracking protein CLIP-170 associates with the spermatid manchette and is essential for spermatogenesis. Genes Dev. 2005;19:2501–2515. doi: 10.1101/gad.344505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Sladecek S, Pemble H, Wittmann T, Slotman JA, van Cappellen W, Brenner HR, Galjart N. Acetylcholine receptor (AChR) clustering is regulated both by glycogen synthase kinase 3beta (GSK3beta)-dependent phosphorylation and the level of CLIP-associated protein 2 (CLASP2) mediating the capture of microtubule plus-ends. J Biol Chem. 2014;289:30857–30867. doi: 10.1074/jbc.M114.589457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch RJ, Hall ZW. Cytoskeletal components of the vertebrate neuromuscular junction: vinculin, alpha-actinin, and filamin. J Cell Biol. 1983;97:217–223. doi: 10.1083/jcb.97.1.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau EG, Akaaboune M. Dynamics of the rapsyn scaffolding protein at the neuromuscular junction of live mice. J Neurosci. 2010;30:614–619. doi: 10.1523/JNEUROSCI.4595-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau E, Sutter D, Hume RI, Akaaboune M. Identification of nicotinic acetylcholine receptor recycling and its role in maintaining receptor density at the neuromuscular junction in vivo. J Neurosci. 2005;25:9949–9959. doi: 10.1523/JNEUROSCI.3169-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camus G, Jasmin BJ, Cartaud J. Polarized sorting of nicotinic acetylcholine receptors to the postsynaptic membrane in Torpedo electrocyte. Eur J Neurosci. 1998;10:839–852. doi: 10.1046/j.1460-9568.1998.00091.x. [DOI] [PubMed] [Google Scholar]
- Chen Y, Ip FC, Shi L, Zhang Z, Tang H, Ng YP, Ye WC, Fu AK, Ip NY. Coronin 6 regulates acetylcholine receptor clustering through modulating receptor anchorage to actin cytoskeleton. J Neurosci. 2014;34:2413–2421. doi: 10.1523/JNEUROSCI.3226-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen I, Rimer M, Lomo T, McMahan UJ. Agrin-induced postsynaptic-like apparatus in skeletal muscle fibers in vivo. Mol Cell Neurosci. 1997;9:237–253. doi: 10.1006/mcne.1997.0623. [DOI] [PubMed] [Google Scholar]
- Dai Z, Luo X, Xie H, Peng HB. The actin-driven movement and formation of acetylcholine receptor clusters. J Cell Biol. 2000;150:1321–1334. doi: 10.1083/jcb.150.6.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng L, Qian YK, Madhavan R, Peng HB. Transmembrane mechanisms in the assembly of the postsynaptic apparatus at the neuromuscular junction. Chem Biol Interact. 2008;175:108–112. doi: 10.1016/j.cbi.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Geng L, Zhang HL, Peng HB. The formation of acetylcholine receptor clusters visualized with quantum dots. BMC Neurosci. 2009;10:80. doi: 10.1186/1471-2202-10-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gensler S, Sander A, Korngreen A, Traina G, Giese G, Witzemann V. Assembly and clustering of acetylcholine receptors containing GFP-tagged epsilon or gamma subunits: selective targeting to the neuromuscular junction in vivo. Eur J Biochem. 2001;268:2209–2217. doi: 10.1046/j.1432-1327.2001.02093.x. [DOI] [PubMed] [Google Scholar]
- Grigoriev I, Splinter D, Keijzer N, Wulf PS, Demmers J, Ohtsuka T, Modesti M, Maly IV, Grosveld F, Hoogenraad CC, et al. Rab6 regulates transport and targeting of exocytotic carriers. Dev Cell. 2007;13:305–314. doi: 10.1016/j.devcel.2007.06.010. [DOI] [PubMed] [Google Scholar]
- Hall ZW, Lubit BW, Schwartz JH. Cytoplasmic actin in postsynaptic structures at the neuromuscular junction. J Cell Biol. 1981;90:789–792. doi: 10.1083/jcb.90.3.789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworski J, Kapitein LC, Gouveia SM, Dortland BR, Wulf PS, Grigoriev I, Camera P, Spangler SA, Di Stefano P, Demmers J, et al. Dynamic microtubules regulate dendritic spine morphology and synaptic plasticity. Neuron. 2009;61:85–100. doi: 10.1016/j.neuron.2008.11.013. [DOI] [PubMed] [Google Scholar]
- Jones G, Herczeg A, Ruegg MA, Lichtsteiner M, Kroger S, Brenner HR. Substrate-bound agrin induces expression of acetylcholine receptor epsilon-subunit gene in cultured mammalian muscle cells. Proc Natl Acad Sci USA. 1996;93:5985–5990. doi: 10.1073/pnas.93.12.5985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones G, Meier T, Lichtsteiner M, Witzemann V, Sakmann B, Brenner HR. Induction by agrin of ectopic and functional postsynaptic-like membrane in innervated muscle. Proc Natl Acad Sci USA. 1997;94:2654–2659. doi: 10.1073/pnas.94.6.2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 Is a receptor for agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishi M, Kummer TT, Eglen SJ, Sanes JR. LL5β. J Cell Biol. 2005;169:355–366. doi: 10.1083/jcb.200411012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P, Lyle KS, Gierke S, Matov A, Danuser G, Wittmann T. GSK3β phosphorylation modulates CLASP–microtubule association and lamella microtubule attachment. J Cell Biol. 2009;184:895–908. doi: 10.1083/jcb.200901042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansbergen G, Grigoriev I, Mimori-Kiyosue Y, Ohtsuka T, Higa S, Kitajima I, Demmers J, Galjart N, Houtsmuller AB, Grosveld F, et al. CLASPs attach microtubule plus ends to the cell cortex through a complex with LL5β. Dev Cell. 2006;11:21–132. doi: 10.1016/j.devcel.2006.05.012. [DOI] [PubMed] [Google Scholar]
- Lee CW, Han J, Bamburg JR, Han L, Lynn R, Zheng JQ. Regulation of acetylcholine receptor clustering by ADF/cofilin-directed vesicular trafficking. Nat Neurosci. 2009;12:848–856. doi: 10.1038/nn.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, Landmann L, Ruegg MA, Brenner HR. The role of nerve-versus muscle-derived factors in mammalian neuromuscular junction formation. J Neurosci. 2008;28:3333–3340. doi: 10.1523/JNEUROSCI.5590-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Pena y Valenzuela I, Hume RI, Krejci E, Akaaboune M. In vivo regulation of acetylcholinesterase insertion at the neuromuscular junction. J Biol Chem. 2005;280:31801–31808. doi: 10.1074/jbc.M502874200. [DOI] [PubMed] [Google Scholar]
- McMahan UJ, Horton SE, Werle MJ, Honig LS, Kroger S, Ruegg MA, Escher G. Agrin isoforms and their role in synaptogenesis. Curr Opin Cell Biol. 1992;4:869–874. doi: 10.1016/0955-0674(92)90113-q. [DOI] [PubMed] [Google Scholar]
- Meijering E, Dzyubachyk O, Smal I. Methods for cell and particle tracking. Methods Enzymol. 2012;504:183–200. doi: 10.1016/B978-0-12-391857-4.00009-4. [DOI] [PubMed] [Google Scholar]
- Merriam EB, Millette M, Lumbard DC, Saengsawang W, Fothergill T, Hu X, Ferhat L, Dent EW. Synaptic regulation of microtubule dynamics in dendritic spines by calcium, F-actin, and drebrin. J Neurosci. 2013;33:16471–16482. doi: 10.1523/JNEUROSCI.0661-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivo-Marin JC. Extraction of spots in biological images using multiscale products. Pattern Recognition. 2002;35:1989–1996. [Google Scholar]
- Paranavitane V, Coadwell WJ, Eguinoa A, Hawkins PT, Stephens L. LL5beta is a phosphatidylinositol (3,4,5)-trisphosphate sensor that can bind the cytoskeletal adaptor, gamma-filamin. J Biol Chem. 2003;278:1328–1335. doi: 10.1074/jbc.M208352200. [DOI] [PubMed] [Google Scholar]
- Peng H, Ruan Z, Long F, Simpson JH, Myers EW. V3D enables real-time 3D visualization and quantitative analysis of large-scale biological image data sets. Nat Biotechnol. 2010;28:348–353. doi: 10.1038/nbt.1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penzes P, Srivastava DP, Woolfrey KM. Not just actin? A role for dynamic microtubules in dendritic spines. Neuron. 2009;61:3–5. doi: 10.1016/j.neuron.2008.12.018. [DOI] [PubMed] [Google Scholar]
- Proszynski TJ, Gingras J, Valdez G, Krzewski K, Sanes JR. Podosomes are present in a postsynaptic apparatus and participate in its maturation. Proc Natl Acad Sci USA. 2009;106:18373–18378. doi: 10.1073/pnas.0910391106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proszynski TJ, Sanes JR. Amotl2 interacts with LL5beta, localizes to podosomes and regulates postsynaptic differentiation in muscle. J Cell Sci. 2013;126:2225–2235. doi: 10.1242/jcs.121327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roder IV, Petersen Y, Choi KR, Witzemann V, Hammer JA, 3rd, Rudolf R. Role of Myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo. PLoS One. 2008;3:e3871. doi: 10.1371/journal.pone.0003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. doi: 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N, Akaaboune M, Gajendran N, Martinez-Pena y Valenzuela I, Wakefield S, Thurnheer R, Brenner HR. Neuregulin/ErbB regulate neuromuscular junction development by phosphorylation of alpha-dystrobrevin. J Cell Biol. 2011;195:1171–1184. doi: 10.1083/jcb.201107083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt N, Basu S, Sladecek S, Gatti S, van Haren J, Treves S, Pielage J, Galjart N, Brenner HR. Agrin regulates CLASP2-mediated capture of microtubules at the neuromuscular junction synaptic membrane. J Cell Biol. 2012;198:421–437. doi: 10.1083/jcb.201111130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmoranzer J, Simon SM. Role of microtubules in fusion of post-Golgi vesicles to the plasma membrane. Mol Biol Cell. 2003;14:1558–1569. doi: 10.1091/mbc.E02-08-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shadiack AM, Nitkin RM. Agrin induces alpha-actinin, filamin, and vinculin to co-localize with AChR clusters on cultured chick myotubes. J Neurobiol. 1991;22:617–628. doi: 10.1002/neu.480220607. [DOI] [PubMed] [Google Scholar]
- Smith MA, Slater CR. Spatial distribution of acetylcholine receptors at developing chick neuromuscular junctions. J Neurocytol. 1983;12:993–1005. doi: 10.1007/BF01153346. [DOI] [PubMed] [Google Scholar]
- Thompson RE, Larson DR, Webb WW. Precise nanometer localization analysis for individual fluorescent probes. Biophys J. 2002;82:2775–2783. doi: 10.1016/S0006-3495(02)75618-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toonen RF, Kochubey O, de Wit H, Gulyas-Kovacs A, Konijnenburg B, Sorensen JB, Klingauf J, Verhage M. Dissecting docking and tethering of secretory vesicles at the target membrane. EMBO J. 2006;25:3725–3737. doi: 10.1038/sj.emboj.7601256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turney SG, Culican SM, Lichtman JW. A quantitative fluorescence-imaging technique for studying acetylcholine receptor turnover at neuromuscular junctions in living animals. J Neurosci Methods. 1996;64:199–208. doi: 10.1016/0165-0270(95)00135-2. [DOI] [PubMed] [Google Scholar]
- Watanabe T, Noritake J, Kakeno M, Matsui T, Harada T, Wang S, Itoh N, Sato K, Matsuzawa K, Iwamatsu A, et al. Phosphorylation of CLASP2 by GSK-3β regulates its interaction with IQGAP1, EB1 and microtubules. J Cell Sci. 2009;122:2969–2979. doi: 10.1242/jcs.046649. [DOI] [PubMed] [Google Scholar]
- Wu H, Xiong WC, Mei L. To build a synapse: signaling pathways in neuromuscular junction assembly. Development. 2010;137:1017–1033. doi: 10.1242/dev.038711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XS, Tsan GL, Hammer JA., 3rd Melanophilin and myosin Va track the microtubule plus end on EB1. J Cell Biol. 2005;171:201–207. doi: 10.1083/jcb.200503028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008;60:285–297. doi: 10.1016/j.neuron.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.