

Abstract
Males and females of many animal species differ in their sex-chromosome karyotype, and this creates imbalances between X-chromosome and autosomal gene products that require compensation. Although distinct molecular mechanisms have evolved in three highly studied systems, they all achieve coordinate regulation of an entire chromosome by differential RNA-polymerase occupancy at X-linked genes. High-throughput genome-wide methods have been pivotal in driving the latest progress in the field. Here we review the emerging models for dosage compensation in mammals, flies and nematodes, with a focus on mechanisms affecting RNA polymerase II activity on the X chromosome.
In many animal species, the chromosomal basis for sex has led to specific evolutionary pressures on the chromosomes containing sex-determination genes. It is commonly thought that heterologous sex chromosomes evolved as a consequence of impaired meiotic recombination near sex-determining genes, which led to the gradual degeneration of the nonrecombining Y chromosome1. In mammals and the fruitfly Drosophila melanogaster, one of the two sexes remained associated with a homologous pair of sex chromosomes (XX females), whereas the other sex acquired a heterologous pair (XY males). The X chromosome remained gene rich, with many essential, non–sex-specific functions, whereas the Y chromosome became relatively gene poor and dedicated to male-specific sex determination and/or reproduction. In the nematode Caenorhabditis elegans, males bear a single copy of chromosome X (X0 karyotype), and hermaphrodites have two X chromosomes (XX) and display a female body plan but also have the ability to make sperm and to self-fertilize. In each of these organisms, sex-chromosome evolution led to a copy-number difference between the sexes and an imbalance in gene expression between X chromosomes and autosomes. Compensation for this dosage difference is thought to be important, especially for gene products belonging to multimeric complexes, for which an imbalance in the stoichiometry of components can be detrimental.
In mammals, fruitflies and nematodes, there is evidence for the evolution of increased gene expression from the X chromosome in response to the unfavorable X-to-autosome balance in XY or X0 males (Fig. 1a). In fruitflies, the upregulation of X-linked gene expression is primarily male specific and equalizes transcription between the sexes as well as between the X chromosome and autosomes2. This regulatory mechanism can be measured by comparing males and females or by comparing wild-type males and dosage-compensation mutants. However, in mammals and nematodes, and to a lesser extent in fruitflies3,4, it is thought that a general increase in transcriptional output from X chromosomes evolved that was not limited to males. Molecular evidence to support the evolutionary theory comes from the observation that average gene expression on the X chromosome in both mammals and nematodes is higher than average autosomal gene expression5–7. Although the evidence has sparked some debate8,9 (as reviewed in ref. 10), analyses from several studies suggest that increased output is confirmed when silent genes (i.e., those not targeted by dosage compensation) are excluded from the analysis11–15.
Figure 1.

Comparison of three modes of dosage compensation. (a) Dosage compensation is required to balance gene expression between sexes and between autosomes and sex chromosomes. In mammals (top), one of the two X chromosomes is inactivated in females (blue color for silencing) to balance expression, but X-chromosome upregulation is also required in both males and females to balance expression from diploid autosomes (red color for upregulation). In fruitflies (middle), the two-fold upregulation of the male X chromosome is sufficient to balance expression between sexes and relative to autosomes. In nematode hermaphrodites (XX) (bottom), downregulation of both copies of the X chromosome is superimposed upon upregulation of X chromosomes in both sexes, to balance autosomal expression levels. (b) Distinct complexes (DCC) are involved in dosage compensation in different species, but similarities have been proposed in the targeting and spreading of the various DCCs along the target chromosomes. The models propose that binding occurs at distinct nucleation sites (top) and spreads to additional target sites (bottom), which may be distant in the linear sequence of the chromosome, in addition to local spreading (dashed arrows) of the deposited epigenetic marks around primary target sites. This scheme is meant to highlight common principles, although of course there are species-specific differences, as detailed in the main text.
The multistep model for sex-chromosome evolution is attractive because it can explain how dosage compensation arose in hermaphrodite nematodes and female mammals. Eventually, as a consequence of X chromosomes with increased transcriptional activity, the selective pressure would switch to females, where having two X chromosomes would cause repressive mechanisms to evolve to maintain a balance with autosomes. In nematodes, this occurs by repression of gene expression by ~50% from both copies of the X chromosome (reviewed in ref. 16), whereas in mammals, this is achieved by random inactivation of one of the two female X chromosomes and subsequent clonal inheritance of the inactive state (Xi; reviewed in ref. 17) (Fig. 1a).
Despite the ultimate differences in molecular mechanisms, models for targeting each dosage-compensation complex (DCC) onto the X chromosome have converged recently (Fig. 1b). In fruitflies, nematodes and mammals, the regulators of dosage compensation are thought to bind initially to a few hundred nucleation sites (with specific sequence motifs identified in fruitflies and nematodes) and then to spread in cis on the target X chromosome(s)18–20. These models provide a framework to explain how coordinate regulation of a single chromosome can occur, in part, in the absence of recognizable sequence elements at each defined binding site.
Dissecting the molecular basis for transcriptional control in dosage compensation has been difficult. In fruitflies and nematodes, a two-fold change in mRNA output occurs over an entire chromosome. This small average variation is difficult to measure accurately at the level of individual loci. Furthermore, the selective advantage for precise two-fold regulation is very likely to vary from gene to gene. In contrast, X inactivation in mammals is solved in an ‘all or none’ fashion rather than by two-fold regulation, but it presents another experimental difficulty: that the two distinctly regulated but homologous chromosomes reside in the same nucleus and thus can be distinguished only if there are sequence differences (for example, single-nucleotide polymorphisms).
Moving from gene-specific to genome-wide analysis methods has been instrumental in driving recent progress in understanding the control of gene expression occurring in dosage compensation. In this Review, we focus on current models for the transcriptional-regulatory mechanisms responsible for dosage compensation in mammals, fruit-flies and nematodes. We specifically examine the results of genome-wide analyses that have driven recent progress in the field and also highlight challenges in analysis and interpretation of high-throughput-data.
Key advances enabled by sequencing-based methods
Recent technological advances in high-throughput sequencing have allowed development of a number of new assays for genome-wide measurement of chromatin modifications and transcriptional dynamics, and the applications of these assays have resulted in important advances in the study of dosage compensation. These methods are summarized in Box 1. Chromatin immunoprecipitation followed by sequencing (ChIP-seq) has replaced hybridization on microarrays (ChIP-chip) as the most widely adopted method to study not only transcription factors but also histone marks and other chromatin-associated proteins involved in epigenetic regulation21,22. These techniques have been used in the study of dosage compensation for mapping histone marks associated with silencing20,23–27 or increased expression28–30 and for studying transcriptional dynamics by mapping the distribution of both global RNA polymerase II (Pol II)31,32 and Pol II with specific modifications that indicate active engagement in elongation33. ChIP-based genome-wide techniques have also been crucial for mapping the distribution of the DCC itself. However, in this review, we focus on the activity of the DCC rather than on its targeting and spreading mechanisms.
BOX 1. Transcription analysis methods.
RNA Pol II transcription comprises several phases: promoter recruitment, initiation, 5′ proximal pausing, pausing release, elongation and termination. Genome-wide methods can provide information about specific phases.
ChIP-seq of Pol II
This is used to examine the distribution of Pol II over transcribed genes. DNA and associated proteins are cross-linked, DNA is fragmented (typically through sonication), and immunoprecipitation (IP) of Pol II and associated DNA fragments is performed with specific antibodies. The DNA fragments are then sequenced, and their different distributions in IP and control samples allow identification of Pol II-enriched regions22. This method covers all stages of transcription but does not allow discrimination between them, although antibodies specifically targeting Pol II isoforms phosphorylated on residues Ser2 or Ser5 can be used to examine Pol II enriched in the elongation or initiation stage of transcription, respectively.
GRO-seq
This is used to profile the locations of engaged Pol II along actively transcribed genes34. Transcription is extended in vitro in the presence of Br-UTP nucleotide, with sarkosyl detergent used to release paused Pol II. The in vitro–transcribed RNAs are purified by their Br-UTP labeling and sequenced. A variation of this protocol without the sarkosyl treatment allows profiling of actively elongating RNA Pol II, without the contribution from paused Pol II63. GRO-cap is another variant using additional enzymatic steps to enrich for RNAs with a 5′ cap5.
Nascent-seq
This is based on isolation of nuclei and subsequent extraction of RNA Pol II and nascent transcripts, by virtue of their stable association36. Two nascent-seq variants using direct single-molecule sequencing have been adopted to measure transcriptional dynamics during dosage compensation86. Net-seq also involves deep sequencing of nascent transcripts but has not been used in dosage-compensation studies so far87.
Short 5′ RNAs
Short RNAs attached to paused polymerase at 5′ ends are isolated by means of their characteristics (size <100 nt, nuclear localization and 5′ cap) and sequenced from both ends to map both TSSs and Pol II pausing sites37. Figure adapted from ref. 64.
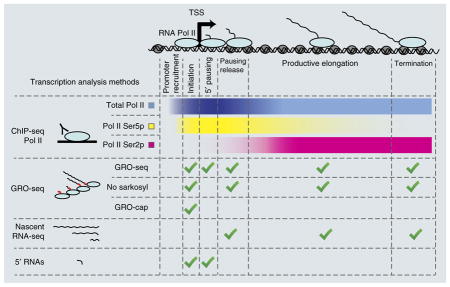
A careful study of transcriptional dynamics requires probing distinct stages of a complex process. A large variety of genome-wide techniques have been developed to gather information about specific steps of transcription (Box 1). Among them, several techniques for studying nascent transcripts have been useful in the study of dosage compensation. For example, global run-on sequencing (GRO-seq)34 measures the position of engaged Pol II along genes and has been used to study Pol II pausing and elongation in fruitfly dosage compensation35. GRO-seq and its variant GRO-cap have allowed precise mapping of nematode transcription start sites (TSSs), which was important for the analysis of dosage compensation5. Nascent RNA sequencing (nascent-seq) is an alternative method based on purification of active Pol II and associated nascent transcripts on the chromatin template36. The sequencing of 5′ short RNAs can serve as a proxy for analysis of Pol II pausing at 5′ ends of genes37.
In recent years, several methods for studying chromosome conformation were developed (reviewed in ref. 38; Box 2). These methods have been used to study regulatory interactions between distant chromatin regions; in studies of dosage compensation, they have provided insights into chromosome-X compartmentalization and its potential role in the spreading of the DCC and related chromatin marks39,40. In particular, a potential relationship between the three-dimensional (3D) folding of chromatin and DCC spreading has been investigated in mammals41,42 by use of Hi-C data43. More insights are expected in the future from the use of chromosome conformation analyses, because a number of open questions remain about the role of global chromosome condensation versus local control in gene silencing, both in mammals and in nematodes.
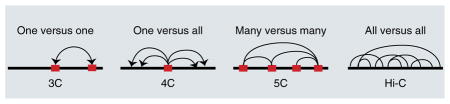
BOX 2. Chromosome conformation–analysis methods.
Dramatic chromosome rearrangements are associated with X-chromosome silencing mechanisms, and the spreading of dosage-compensation complexes has been related to chromosome conformation in mammals. Chromosome conformation capture (3C) was proposed more than 10 years ago as a method to measure colocalization of distant chromatin regions88. 3C has been used to identify chromatin loops that bring enhancers and promoters into proximity. A number of more-high-throughput variants have been derived over the years (for example, 4C, 5C and Hi-C) and are sometimes collectively referred to as ‘C’ technologies38. These techniques have been used in the study of dosage compensation, especially in mammals, in which X inactivation and transcriptional silencing are associated with global rearrangements and condensation of chromosome X. All of these techniques are based on (i) stabilization of DNA-protein interactions by chemical cross-linking of chromatin; (ii) digestion of DNA with a restriction enzyme; (iii) ligation of sticky ends in highly dilute conditions, thus joining only DNA fragments belonging to the same cross-linked complex (originally located near each other in the 3D nuclear space); (iv) reversal of cross-linking. The C technologies differ in the methods used to analyze ligated DNA fragments to identify distant chromatin portions that were interacting in proximity. The original 3C method uses a pair of specific primers to assess the interaction between two predefined regions (one versus one)88; 4C uses a fixed primer for a genome-wide analysis of all fragments ligated to a specific target region (bait) detected by either hybridization on microarrays (4C-chip) or deep sequencing (4C-seq) (one versus all)89,90; 5C uses a panel of predesigned primers to extensively assess all possible pairwise interactions within a user-defined set of target regions, and microarrays or sequencing have both been used to measure the interaction frequencies (many versus many)91,92; Hi-C and its variants make use of very deep sequencing to measure interactions between any possible pair of genomic regions at a lower resolution (all versus all)43. Red boxes in the cartoon indicate target regions of specific user-designed primers.
Along with the widespread adoption of genome-wide experimental techniques came the challenges in analyzing the data10,44–46 and interpreting the results47. In fact, in several instances, controversies in the field have revolved around specific analysis issues, such as proper selection of the list of specific genes for downstream analysis8,13. In other cases, the lack of a clear normalization strategy might result in ambiguous interpretation of results. Most normalization procedures used for genome-wide expression data rely on the assumption that expression of most of the genes does not change; however, when a large number of genes are potentially regulated as in dosage compensation, this assumption may be violated. For instance, it is difficult to discriminate between upregulation of the X chromosome and downregulation of the autosomes—the latter has been the basis for the ‘inverse effect’ hypothesis in Drosophila, in which the MSL complex balances X-chromosome and autosomal expression by sequestering an activator (MOF) away from autosomes rather than by upregulating the X chromosome48,49. Although the bulk of the molecular evidence and a normalization based on absolute signal support the ‘X upregulation’ model50–52, this controversy persists. Moreover, many analyses involve selection of appropriate sets of genes and suitable analytical methods, and the final results can depend on the various parameters. For example, measuring the magnitude of dosage compensation may depend on how ‘expressed’ genes are defined; analysis of elongation effects may depend on whether short genes are removed or not. (Box 3 discusses some of the considerations in drawing ‘metagene’ profiles.) It is therefore important to confirm any genome-wide observations with alternative analysis parameters or algorithms and independent data sets whenever possible, by using an ever-growing body of data in public repositories. In addition, testing of proposed hypotheses by biochemical or molecular genetic experiments will always be critical for establishing mechanistic insights.
BOX 3. Computational considerations in creating metagene profiles.
Transcriptomic or epigenetic data are often summarized in metagene profiles that show the average distribution around genes. Accurate visualization of data with appropriate parameters is important for an unbiased view of the molecular mechanism under study.
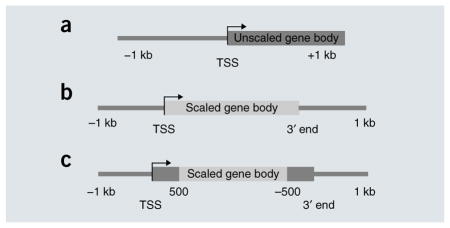
Absolute versus scaled coordinates
The simplest view is an average profile around the TSS or 3′ end, with absolute distances as coordinates (for example, a and Fig. 2, top). To summarize the profile across the gene body, genes must be rescaled to have the same length (Fig. 2, middle and bottom). When the whole region between the 5′ end and 3′ end is rescaled (b), some artifactual features may emerge, owing to averaging genes at different scales. To ameliorate this problem, another option is to preserve absolute distances near the ends (c); in this case, sharp transitions may arise at the interface between the scaled and unscaled portions, and they should be interpreted with caution. When appropriate, a heat map showing each gene separately should be plotted to ensure that the metagene profile summarizes the data correctly.
Gene length
Filtering by length is often necessary to avoid extreme stretching or compression with rescaling of genomic coordinates. Alternatively, multiple profiles representing distinct ranges of gene lengths can be used.
Distance to neighboring genes
Metagenes usually show flanking regions beyond the 5′ and 3′ ends. Filtering of neighboring loci may be required, because signal associated with neighbors may confound the profile.
Expression threshold
When analyzing transcriptional dynamics, it is crucial to focus on ‘active’ genes, which must be defined carefully; otherwise, the pattern could be confounded by random background fluctuations in silent genes.
Normalization
Ideally, a control sample should be obtained for empirical assessment of technical biases (for example, GC content, copy-number variation and read mappability biases). If such a sample is not available, biases should be estimated separately with more sophisticated computational approaches. Additional normalization might be required for comparison of different samples.
Variability
Outliers can sometimes affect the metagene profiles significantly. Profiles of individual genes should be viewed (for example, in a heat map) to assess variability, and robust statistics and confidence intervals should be used when appropriate.
Models for increased expression of the male X chromosome in Drosophila
In male fruitflies, the expression of chromosome X is upregulated approximately two-fold, to balance the expression between males (XY) and females (XX) as well as between autosomes and sex chromosomes. This occurs primarily through the male-specific expression and function of the MSL complex, which is composed of five proteins (MSL1, MSL2, MSL3, MOF and MLE) and two noncoding RNAs (roX1 or roX2). Many studies have focused on testing a model in which the MSL complex first targets chromatin-entry sites (also called high-affinity sites) and then spreads along the X chromosome to bind most active genes30,53,54. This process results in acetylation of K16 of histone H4 (to form H4K16ac) in the bodies of X-linked genes by the MOF histone acetyltransferase55.
Despite a general agreement in the field as to where and how the MSL complex targets the single male X chromosome, two alternative models have been proposed for how the complex causes increased transcription of X-linked genes. The key difference in the models lies in the stage of the transcriptional cycle that is the primary target of the complex: the recruitment and initiation steps or the elongation process. The ‘initiation-based’ model proposes that increased transcription is induced by enhanced recruitment of Pol II and thus by increased initiation of transcription at promoters of active X-linked genes. According to this scenario, Pol II density over transcribed genes is expected to increase by the same magnitude throughout their length. A recent report of ChIP-seq data for male and female salivary-gland cells in fruitflies supported this model31, but it was later disputed because of a methodological error that distorted the fold difference between results for male and female cells44–46.
The ‘elongation-based’ model proposes that the upregulation of X-chromosome genes is based on facilitation of RNA Pol II movement as it traverses gene bodies after the initiation step. This model was first proposed more than a decade ago55, on the basis of the observed increase in H4 K16 acetylation within the bodies of X-linked genes and the rationale that this model would still allow each gene to retain its individual 5′-initiation–based regulation while undergoing increased expression. The elongation-based model is also consistent with the MSL binding pattern and H4K16ac distribution, which show enrichment within the bodies of X-linked genes28,29,56,57. H4K16ac is known to interfere with the formation of more-compact chromatin and to reduce self-association of nucleosome particles58,59. Moreover, the capacity of the MSL complex to reduce negative supercoiling has been proposed as an additional mechanism favoring elongation60,61. Larschan et al.35 interpreted GRO-seq data in male S2 cells to be supportive of the elongation model because the 5′ initiation and pausing peaks of Pol II were very similar for the X chromosome and autosomal genes, whereas the Pol II density within gene bodies was clearly different (Fig. 2). Although the selection of genes used to form the metagene profiles in this study has been questioned31 and chromosome-arm biases have been noted (particularly for chromosome 4)62, the initial GRO-seq analyses have been corroborated by results from a recent independent data set63,64. In addition, independent genetic studies demonstrated that a decreased abundance of SPT5, a known elongation factor, compromised the efficiency of dosage compensation65. Furthermore, recent results from a study using a variation of nascent-seq36 provided independent confirmation that postinitiation differences in X-linked versus autosomal genes are detected specifically in male and not in female cell lines64. Reanalysis of independent public data sets for GRO-seq63 and ChIP-seq of elongating Pol II33 and short 5′-end RNAs37 also support a ‘jump-start and gain’ model64, in which a combination of enhanced pausing release and facilitated elongation are involved in increased transcription of X-linked genes in males. Reconciliation of this model with the recently reported ~1.2-fold increase in ChIP-seq Pol II density on X-linked genes, with no additional increase from the 5′ end to the 3′ end of the gene body, remains an important issue31,45,46. Overall, current evidence supports a mechanism for MSL-dependent dosage compensation in which local, gene-by-gene regulation occurs via increased H4K16ac within bodies of active genes to directly facilitate transcription by RNA Pol II.
Figure 2.

Metagene visualization of dosage compensation. Dosage-compensation mechanisms have a small effect in magnitude (approximately two-fold) but are observed over a large number of genes (entire chromosomes). For these characteristics, genome-wide experimental methods are particularly useful, because visualizing the average effect over many genes is a commonly adopted solution. Metagene profiles are average summaries of a quantitative score derived from genomic data, plotted against relative coordinates over gene loci. Metagene profiles can be plotted relative to a single position (top), for example, around the transcription start site (TSS), or relative to the gene body (middle and bottom), with scaling of the distance between the TSS and 3′ end for multiple genes (Box 3). In mammals (top), analysis of Pol II occupancy in the active (Xa) and the inactive (Xi) shows the lack of Pol II recruitment at the TSS of genes on the inactive X25. In male fruitflies (middle), an average relative increase in elongating Pol II density on X versus autosomes has been reported, and this effect is lost after RNA-interference (RNAi) knockdown of key components of the dosage-compensation complex (MSL)35. In hermaphrodite nematodes (bottom), mutants for the sdc-2 component of the dosage-compensation complex shows approximately double density in Pol II recruited to X-linked genes5.
Models for reduced X-chromosome expression in hermaphrodite C. elegans
In nematodes, the DCC is a specialized condensin complex containing dedicated subunits (DPY-27) and subunits shared with condensin I (DPY-26, DPY-28, MIX-1 and CAPG-1). Condensin I changes the shape of meiotic chromosomes66 and affects mitotic chromosome segregation67. Additional components of the DCC not found in other condensin complexes are dosage-compensation regulators (SDC-1, SDC-2, SDC-3, DPY-21 and DPY-30)16. The model for the loading of the DCC onto the X chromosome18,68–71 (Fig. 1b) features nucleation sites named ‘recruitment elements on X’ (rex), which carry a consensus DNA sequence motif and can function autonomously, and direct spreading to ‘dependent on X’ sites (dox), which are sequence independent, tend to be in promoters of active genes and are not autonomous32,70.
Recently, a potential connection between DCC function and a chromosome-wide histone modification was discovered: hermaphrodite X chromosomes are enriched for monomethylated H4 K20 (H4K20me1), and this enrichment is dependent on the DCC23,26,27,72. H4 K20 mono-methylation is catalyzed by the SET-1 protein, and set-1 mutants show increased expression of selected X-linked genes relative to autosomal genes, although this has been tested on only a few genes23. Increased H4 K20 monomethylation by SET-1 is unlikely to be the sole driver of dosage compensation, although the assessment of phenotype is complicated by the close relationship between H4 K20 monomethylation and di- and trimethylation catalyzed by a different enzyme, SET-4. Evidence suggests that X-linked H4K20me1 enrichment is due to selective inhibition of SET-4 on X chromosomes rather than to increased SET-1 activity23. Hermaphrodite X chromosomes are also relatively depleted for H4K16ac26,27 and the H2A.Z variant histone73,74, but how these chromatin components might be related to DCC function is not clear.
Given the targeting of the DCC near promoters, it was initially assumed that repression might be the direct action of the complex on adjacent genes. However, only a fraction of promoters appear to be bound, and gene expression analysis in wild type versus dosage-compensation mutants fails to support a correlation between binding and repression. Indeed, 43% of dosage-compensated genes lack DCC binding, whereas 61% of non–dosage-compensated genes have DCC bound70. The data suggest that a number of genes are escaping dosage compensation on the X chromosome, although the exact numbers are dependent on the filtering criteria for defining compensated versus noncompensated genes. For example, Jans et al.70 reported 290 non-compensated and 374 compensated genes according to their specific filtering criteria. The discrepancies between DCC binding and dosage compensation suggest that regulation in nematodes is unlikely to occur through strictly local control but might instead occur through selective sensitivity to long-distance regulation.
One important caveat to these findings was the complication that many TSSs were not precisely annotated in nematodes. This was due to the high incidence of trans splicing, in which the sequences of the mature 5′ ends of many transcripts did not reveal their authentic TSSs. The precise identification of TSSs has been addressed in three recent publications3,74,75. Among these, Kruesi et al.3 focused on dosage compensation, by using a combination of GRO-seq and its variant GRO-cap, to precisely map capped nascent transcription, i.e., transcription before the trans-splicing event occurred. This refinement of TSS mapping was critical to the conclusion that reduction of 5′ recruitment of Pol II is the basis for dosage compensation in nematodes. The key results showed that 5′ recruitment and Pol II density along X-linked genes was uniformly increased when DCC function was compromised (Fig. 2). An interesting additional finding was that 5′ Pol II pausing is not a prevalent step in transcription in nematodes, in contrast to both fruitflies and mammals. Instead, the typical active gene profile by GRO-seq analysis shows the greatest Pol II accumulation at the 3′ ends of genes.
After considering the refinements in TSS mapping, DCC-binding sites are even more precisely linked with promoters, yet these binding sites still show poor correlation with the magnitude of repression. Therefore, any current model for dosage compensation in nematodes must invoke regulation at a distance. The chromosome-wide enrichment in H4K20me1 along with depletion of H4K16ac and H2A.Z suggests that chromosome-wide epigenetic mechanisms may be involved in DCC-mediated repression. Given that the DCC appears to be a specialized version of condensin, the mitotic chromosome compaction machinery, it seems reasonable to propose that some step in chromosome compaction is at play, and this is strengthened by the documented link with H4K20me1, known to be enriched in mitotic chromosomes in mammals76,77. However, to date, no physical evidence for compaction of the hermaphrodite X chromosomes has been documented. Among the proposed hypotheses for regulating distant genes, there is a possible role of the DCC in reorganizing chromatin and forming long loops69. Genome-wide data on chromosome conformation will be very helpful for addressing this model in the future.
Models for gene silencing in X inactivation
In mammals, one of the two X chromosomes is inactivated in females (Xi versus Xa). In contrast to the fruitfly and nematode systems discussed so far, in this case the regulatory factor that is best understood is a long noncoding RNA, Xist. The site of Xist transcription, the X-inactivation center, is essential for the establishment of Xi. The identities of associated proteins remain mostly unknown, because saturating genetic screens are less feasible in mammalian systems, and biochemical purification of a complex starting with an RNA component is technically challenging in any system. However, a few genome-wide techniques have been developed to map the locations of noncoding RNAs that interact with chromatin, including capture hybridization analysis of RNA targets with deep sequencing (CHART-seq)41,75, chromatin isolation by RNA purification with deep sequencing (ChIRP-seq)76 and RNA antisense purification and deep sequencing (RAP-seq)42. The key proteins implicated so far are members of the Polycomb repressive complex 2 (PRC2) and to a lesser extent PRC1 (ref. 77). The EZH2 histone-methyltransferase component of PRC2 can interact directly with Xist RNA78,79, and initial targeting is postulated to involve nucleation sites and spreading on one of the two X chromosomes20,41,42,80, thus leading to the enrichment of trimethylated histone H3 K27 (H3K27me3) specifically on Xi (Fig. 1b). Mapping of H3K27me3 by ChIP20,24,25 and Xist RNA by CHART-seq41 provides evidence that active genes on Xi are initially the preferential targets of these factors, whereas a study using RAP-seq reports early Xist localization on inactive genes, at the periphery of active gene-dense regions42. Additional increases in H3 K9 methylation, histone hypoacetylation, H4 K20 monomethylation, macroH2A and promoter-DNA methylation culminate in a relative condensation of the Xi during interphase, to form the Barr body.
Still, there are many unknowns, especially with regard to the molecular mechanism of gene silencing. Although Polycomb-associated silencing of gene expression is a well-studied phenomenon, it is still relatively unclear how this is achieved at the molecular level. Does it occur through local compaction of chromatin structure or through a specific silencing property of the H3K27me3 mark on promoters? The H3K27me3 mark can cover large regions of the genome around target genes, with especially high enrichment over the promoters and TSSs. Indeed, recent ChIP-seq analysis of Pol II occupancy on the Xi versus Xa in trophoblast cells demonstrated that, at least in imprinted X inactivation, Pol II is prevented from binding to promoters of genes on Xi (Fig. 2)25. Similar trends have been reported in somatic cells of females with skewed X inactivation12,14. In addition, H3K27me3 enrichment is seen at the TSSs of X-inactivated genes, along with H4K20me1 and increased DNase I hypersensitivity. Considering these data and observations on genes escaping X inactivation, Calabrese et al.25 favor a model in which X inactivation occurs through local modifications of specific regulatory elements, perhaps via gene-by-gene repression, rather than by a 3D global-inaccessibility model that originates from cytological observations of the Barr body. However, the use of mouse trophoblastic cells, in which X inactivation is nonrandom (with preferential inactivation of paternal X), leaves open questions related to potential differences with random X inactivation in somatic cells.
A gene-by-gene model is also consistent with previous observations of chromosome conformation showing that two genes escaping inactivation interact with each other, thus presumably escaping condensation in the Barr body40. But chromatin folding data for additional loci would be desirable to draw a firm conclusion regarding this point. Whether Xist RNA itself might have a direct role in silencing, other than recruiting PRC2, is not known. Furthermore, the potential gene-specific or global roles of all of the other chromatin changes enriched in Xi in response to Xist RNA targeting (for example, H4K20me1, macroH2A, deacetylated nucleosomes, H3K9me2 and H3K9me3 and methylated promoter DNA) remain to be determined (Fig. 3). For example, Nozawa et al.81 recently found that HBiX1 binds both H3K9me3 and H3K27me3 domains through a PRC2-independent pathway and is required for Xi compaction. Therefore, the connection between the exclusion of RNA pol II from Xi genes and the local or global enrichment of repressive factors on Xi remains to be understood.
Figure 3.

Modulation of Pol II transcription within the context of distinct dosage-compensation models. The three mechanisms of transcription regulation (upregulation, downregulation and X inactivation, left) culminate in changes in chromatin composition, histone modifications and Pol II occupancy on the respective X chromosomes (right). A central question is whether these changes are mediated through gene-specific or chromosome-wide, higher-order mechanisms (depicted within the nucleus at the center of the diagram). In fruitflies, dosage compensation probably occurs via gene-by-gene upregulation of an entire chromosome, in which increased levels of H4K16ac within gene bodies directly facilitates pausing release and elongation by RNA Pol II. In nematodes, dosage compensation is achieved by a reduction of Pol II recruitment to the promoters of X-linked genes, driven by a chromosome-restructuring condensin complex. In addition, specific histone modifications (such as increased H4K20me1 and decreased H4K16ac) may also contribute to transcriptional downregulation on the X chromosome. Most importantly, the discrepancies between DCC targeting and dosage compensation suggest that in nematodes compensation occurs through selective sensitivity to chromosome-wide, long-distance regulation. In mammals, X inactivation (Xi) is probably accomplished by exclusion of RNA Pol II from Xi genes. In general, X inactivation is associated with global changes in chromatin accessibility and an increase of inactive chromatin marks such as H3K27me3, H4K20me1, H3K9me2 and H3K9me3, macroH2A and DNA methylation, as well as a decrease of active chromatin marks, such as H3K4me2 and H3K4me3 and histone acetylation. The question of whether local or global enrichment of the repressive factors is an initial event driving RNA Pol II exclusion from Xi genes remains unanswered. Black nucleosomes indicate inactive chromatin.
Challenges in distinguishing local and global mechanisms
Substantial challenges remain in understanding the mechanistic details of transcriptional control in dosage compensation. In fruit-flies, we favor a model for upregulation of the male X chromosome by facilitated pausing release and elongation. However, owing to limitations in current technologies, we are unable to distinguish between increased processivity (i.e., decreased premature termination) or increased recycling or positive feedback to 5′ Pol II complexes82–84. Furthermore, although a strong case can be made for local control of genes in the fruitfly, as the MSL complex directly binds its regulated targets, changes in 3D structure of the male X chromosome may also contribute to global control85.
In nematodes, in which the repression of X chromosomes in hermaphrodites is controlled at the recruitment level, gene expression data suggest that this is not a local control mechanism deployed at the level of individual promoters but rather is a consequence of long-distance or even chromosome-wide organization. Therefore, a direct experimental approach to dissect a functional role for higher-order structure is clearly needed, to clarify the role of chromosome- wide chromatin compaction in leading to a more challenging environment for Pol II recruitment.
Among the latest genomic techniques, the study of 3D organization has gained interesting insights into dosage compensation in mammalian models41,42. However, it is still unclear whether spatial compartmentalization of Xi in the form of the Barr body should be considered the cause or the consequence of the coordinated inactivation of the entire chromosome. Similarly to the other systems, in mammals there are alternative models proposing either local or global regulation of Pol II access.
Despite the mechanistic differences, remarkable similarities have been observed among specific characteristics of dosage-compensation mechanisms in mammals, fruitflies and nematodes, such as the targeting and spreading patterns of key regulators. In addition, the remaining open questions in these three systems revolve around the relative importance of local versus chromosome-wide mechanisms for the control of dosage compensation. We look forward to the development of new genome-wide experimental methods as a means to further extend the current understanding of molecular mechanisms involved in dosage compensation.
Acknowledgments
We are grateful to G. Csankovszki, J.T. Lee, T.R. Magnuson, B.J. Meyer, B. Alver and D. Day for critical reading of the manuscript. Our research on dosage compensation is supported by the US National Institutes of Health (GM45744 to M.I.K.).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Charlesworth D, Charlesworth B, Marais G. Steps in the evolution of sex chromosomes. Heredity (Edinb) 2005;95:118–128. doi: 10.1038/sj.hdy.6800697. [DOI] [PubMed] [Google Scholar]
- 2.Lucchesi JC, Kelly WG, Panning B. Chromatin remodeling in dosage compensation. Annu Rev Genet. 2005;39:615–651. doi: 10.1146/annurev.genet.39.073003.094210. [DOI] [PubMed] [Google Scholar]
- 3.Zhang Y, et al. Expression in aneuploid Drosophila S2 cells. PLoS Biol. 2010;8:e1000320. doi: 10.1371/journal.pbio.1000320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang Y, Oliver B. An evolutionary consequence of dosage compensation on Drosophila melanogaster female X-chromatin structure? BMC Genomics. 2010;11:6. doi: 10.1186/1471-2164-11-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kruesi WS, Core LJ, Waters CT, Lis JT, Meyer BJ. Condensin controls recruitment of RNA polymerase II to achieve nematode X-chromosome dosage compensation. Elife. 2013;2:e00808. doi: 10.7554/eLife.00808. Reports genome-wide analyses demonstrating repression of RNA-polymerase recruitment to X-linked genes as the molecular mechanism for C. elegans dosage compensation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gupta V, et al. Global analysis of X-chromosome dosage compensation. J Biol. 2006;5:3. doi: 10.1186/jbiol30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nguyen DK, Disteche CM. Dosage compensation of the active X chromosome in mammals. Nat Genet. 2006;38:47–53. doi: 10.1038/ng1705. Refs. 6 and 7 present initial genomic analyses supporting a generalized increase in X-linked gene expression as an evolutionary precursor to repressive mechanisms in C. elegans and to mammalian dosage compensation. [DOI] [PubMed] [Google Scholar]
- 8.Xiong Y, et al. RNA sequencing shows no dosage compensation of the active X-chromosome. Nat Genet. 2010;42:1043–1047. doi: 10.1038/ng.711. [DOI] [PubMed] [Google Scholar]
- 9.Lin F, Xing K, Zhang J, He X. Expression reduction in mammalian X chromosome evolution refutes Ohno’s hypothesis of dosage compensation. Proc Natl Acad Sci USA. 2012;109:11752–11757. doi: 10.1073/pnas.1201816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Birchler JA. Claims and counterclaims of X-chromosome compensation. Nat Struct Mol Biol. 2012;19:3–5. doi: 10.1038/nsmb.2218. [DOI] [PubMed] [Google Scholar]
- 11.Deng X, et al. Evidence for compensatory upregulation of expressed X-linked genes in mammals, Caenorhabditis elegans and Drosophila melanogaster. Nat Genet. 2011;43:1179–1185. doi: 10.1038/ng.948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deng X, et al. Mammalian X upregulation is associated with enhanced transcription initiation, RNA half-life, and MOF-mediated H4K16 acetylation. Dev Cell. 2013;25:55–68. doi: 10.1016/j.devcel.2013.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kharchenko PV, Xi R, Park PJ. Evidence for dosage compensation between the X chromosome and autosomes in mammals. Nat Genet. 2011;43:1167–1169. doi: 10.1038/ng.991. [DOI] [PubMed] [Google Scholar]
- 14.Yildirim E, Sadreyev RI, Pinter SF, Lee JT. X-chromosome hyperactivation in mammals via nonlinear relationships between chromatin states and transcription. Nat Struct Mol Biol. 2012;19:56–61. doi: 10.1038/nsmb.2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lin H, et al. Relative overexpression of X-linked genes in mouse embryonic stem cells is consistent with Ohno’s hypothesis. Nat Genet. 2011;43:1169–1170. doi: 10.1038/ng.992. author reply 1171–1172. [DOI] [PubMed] [Google Scholar]
- 16.Meyer BJ. Targeting X chromosomes for repression. Curr Opin Genet Dev. 2010;20:179–189. doi: 10.1016/j.gde.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schulz EG, Heard E. Role and control of X chromosome dosage in mammalian development. Curr Opin Genet Dev. 2013;23:109–115. doi: 10.1016/j.gde.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 18.Csankovszki G, McDonel P, Meyer BJ. Recruitment and spreading of the C. elegans dosage compensation complex along X chromosomes. Science. 2004;303:1182–1185. doi: 10.1126/science.1092938. [DOI] [PubMed] [Google Scholar]
- 19.Kelley RL, et al. Epigenetic spreading of the Drosophila dosage compensation complex from roX RNA genes into flanking chromatin. Cell. 1999;98:513–522. doi: 10.1016/s0092-8674(00)81979-0. [DOI] [PubMed] [Google Scholar]
- 20.Pinter SF, et al. Spreading of X chromosome inactivation via a hierarchy of defined Polycomb stations. Genome Res. 2012;22:1864–1876. doi: 10.1101/gr.133751.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park PJ. ChIP-seq: advantages and challenges of a maturing technology. Nat Rev Genet. 2009;10:669–680. doi: 10.1038/nrg2641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vielle A, et al. H4K20me1 contributes to downregulation of X-linked genes for C. elegans dosage compensation. PLoS Genet. 2012;8:e1002933. doi: 10.1371/journal.pgen.1002933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marks H, et al. High-resolution analysis of epigenetic changes associated with X inactivation. Genome Res. 2009;19:1361–1373. doi: 10.1101/gr.092643.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Calabrese JM, et al. Site-specific silencing of regulatory elements as a mechanism of X inactivation. Cell. 2012;151:951–963. doi: 10.1016/j.cell.2012.10.037. Describes chromosome-wide differences in RNA Pol II and H3K27me3 occupancy on Xa and Xi genes during imprinted X inactivation in mammalian trophoblastic cells. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wells MB, Snyder MJ, Custer LM, Csankovszki G. Caenorhabditis elegans dosage compensation regulates histone H4 chromatin state on X chromosomes. Mol Cell Biol. 2012;32:1710–1719. doi: 10.1128/MCB.06546-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu T, et al. Broad chromosomal domains of histone modification patterns in C. elegans. Genome Res. 2011;21:227–236. doi: 10.1101/gr.115519.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kind J, et al. Genome-wide analysis reveals MOF as a key regulator of dosage compensation and gene expression in Drosophila. Cell. 2008;133:813–828. doi: 10.1016/j.cell.2008.04.036. [DOI] [PubMed] [Google Scholar]
- 29.Gelbart ME, Larschan E, Peng S, Park PJ, Kuroda MI. Drosophila MSL complex globally acetylates H4K16 on the male X chromosome for dosage compensation. Nat Struct Mol Biol. 2009;16:825–832. doi: 10.1038/nsmb.1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Larschan E, et al. MSL complex is attracted to genes marked by H3K36 trimethylation using a sequence-independent mechanism. Mol Cell. 2007;28:121–133. doi: 10.1016/j.molcel.2007.08.011. [DOI] [PubMed] [Google Scholar]
- 31.Conrad T, Cavalli FM, Vaquerizas JM, Luscombe NM, Akhtar A. Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters. Science. 2012;337:742–746. doi: 10.1126/science.1221428. Provides evidence in favor of improved RNA-polymerase promoter recruitment as the molecular mechanism underlying dosage compensation in male D. melanogaster with references 44–46 as follow-up commentaries (alternative model presented in ref. 64) [DOI] [PubMed] [Google Scholar]
- 32.Pferdehirt RR, Kruesi WS, Meyer BJ. An MLL/COMPASS subunit functions in the C. elegans dosage compensation complex to target X chromosomes for transcriptional regulation of gene expression. Genes Dev. 2011;25:499–515. doi: 10.1101/gad.2016011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Regnard C, et al. Global analysis of the relationship between JIL-1 kinase and transcription. PLoS Genet. 2011;7:e1001327. doi: 10.1371/journal.pgen.1001327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–1848. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Larschan E, et al. X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature. 2011;471:115–118. doi: 10.1038/nature09757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Khodor YL, et al. Nascent-seq indicates widespread cotranscriptional pre-mRNA splicing in Drosophila. Genes Dev. 2011;25:2502–2512. doi: 10.1101/gad.178962.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nechaev S, et al. Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–338. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dekker J, Marti-Renom MA, Mirny LA. Exploring the three-dimensional organization of genomes: interpreting chromatin interaction data. Nat Rev Genet. 2013;14:390–403. doi: 10.1038/nrg3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nora EP, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Splinter E, et al. The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Dev. 2011;25:1371–1383. doi: 10.1101/gad.633311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Simon MD, et al. High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 2013;504:465–469. doi: 10.1038/nature12719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Engreitz JM, et al. The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 2013;341:1237973. doi: 10.1126/science.1237973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vaquerizas JM, Cavalli FM, Conrad T, Akhtar A, Luscombe NM. Response to Comments on “Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters”. Science. 2013;340:273. doi: 10.1126/science.1232874. [DOI] [PubMed] [Google Scholar]
- 45.Straub T, Becker PB. Comment on “Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters”. Science. 2013;340:273. doi: 10.1126/science.1231895. [DOI] [PubMed] [Google Scholar]
- 46.Ferrari F, et al. Comment on “Drosophila dosage compensation involves enhanced Pol II recruitment to male X-linked promoters”. Science. 2013;340:273. doi: 10.1126/science.1231815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ehrensberger AH, Kelly GP, Svejstrup JQ. Mechanistic interpretation of promoter-proximal peaks and RNAPII density maps. Cell. 2013;154:713–715. doi: 10.1016/j.cell.2013.07.032. [DOI] [PubMed] [Google Scholar]
- 48.Bhadra MP, Bhadra U, Kundu J, Birchler JA. Gene expression analysis of the function of the male-specific lethal complex in Drosophila. Genetics. 2005;169:2061–2074. doi: 10.1534/genetics.104.036020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Birchler JA, Pal-Bhadra M, Bhadra U. Dosage dependent gene regulation and the compensation of the X chromosome in Drosophila males. Genetica. 2003;117:179–190. doi: 10.1023/a:1022935927763. [DOI] [PubMed] [Google Scholar]
- 50.Hamada FN, Park PJ, Gordadze PR, Kuroda MI. Global regulation of X chromosomal genes by the MSL complex in Drosophila melanogaster. Genes Dev. 2005;19:2289–2294. doi: 10.1101/gad.1343705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Straub T, Gilfillan GD, Maier VK, Becker PB. The Drosophila MSL complex activates the transcription of target genes. Genes Dev. 2005;19:2284–2288. doi: 10.1101/gad.1343105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Park SW, Oh H, Lin YR, Park Y. MSL cis-spreading from roX gene up-regulates the neighboring genes. Biochem Biophys Res Commun. 2010;399:227–231. doi: 10.1016/j.bbrc.2010.07.059. [DOI] [PubMed] [Google Scholar]
- 53.Alekseyenko AA, et al. A sequence motif within chromatin entry sites directs MSL establishment on the Drosophila X chromosome. Cell. 2008;134:599–609. doi: 10.1016/j.cell.2008.06.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Straub T, Grimaud C, Gilfillan GD, Mitterweger A, Becker PB. The chromosomal high-affinity binding sites for the Drosophila dosage compensation complex. PLoS Genet. 2008;4:e1000302. doi: 10.1371/journal.pgen.1000302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Smith ER, Allis CD, Lucchesi JC. Linking global histone acetylation to the transcription enhancement of X-chromosomal genes in Drosophila males. J Biol Chem. 2001;276:31483–31486. doi: 10.1074/jbc.C100351200. [DOI] [PubMed] [Google Scholar]
- 56.Alekseyenko AA, Larschan E, Lai WR, Park PJ, Kuroda MI. High-resolution ChIP-chip analysis reveals that the Drosophila MSL complex selectively identifies active genes on the male X chromosome. Genes Dev. 2006;20:848–857. doi: 10.1101/gad.1400206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gilfillan GD, et al. Chromosome-wide gene-specific targeting of the Drosophila dosage compensation complex. Genes Dev. 2006;20:858–870. doi: 10.1101/gad.1399406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Allahverdi A, et al. The effects of histone H4 tail acetylations on cation-induced chromatin folding and self-association. Nucleic Acids Res. 2011;39:1680–1691. doi: 10.1093/nar/gkq900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu Y, et al. Influence of histone tails and H4 tail acetylations on nucleosome-nucleosome interactions. J Mol Biol. 2011;414:749–764. doi: 10.1016/j.jmb.2011.10.031. [DOI] [PubMed] [Google Scholar]
- 60.Dunlap D, et al. Distinct contributions of MSL complex subunits to the transcriptional enhancement responsible for dosage compensation in Drosophila. Nucleic Acids Res. 2012;40:11281–11291. doi: 10.1093/nar/gks890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Cugusi S, et al. Topoisomerase II plays a role in dosage compensation in Drosophila. Transcription. 2013;4:32–44. doi: 10.4161/trns.26185. [DOI] [PubMed] [Google Scholar]
- 62.Johansson AM, Stenberg P, Allgardsson A, Larsson J. POF regulates the expression of genes on the fourth chromosome in Drosophila melanogaster by binding to nascent RNA. Mol Cell Biol. 2012;32:2121–2134. doi: 10.1128/MCB.06622-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Core LJ, et al. Defining the status of RNA polymerase at promoters. Cell Rep. 2012;2:1025–1035. doi: 10.1016/j.celrep.2012.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ferrari F, et al. “Jump start and gain” model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Rep. 2013;5:629–636. doi: 10.1016/j.celrep.2013.09.037. Provides evidence in favor of improved pausing release and transcriptional elongation as the molecular mechanisms underlying dosage compensation in male D. melanogaster (alternative model in ref. 31) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Prabhakaran M, Kelley RL. Mutations in the transcription elongation factor SPT5 disrupt a reporter for dosage compensation in Drosophila. PLoS Genet. 2012;8:e1003073. doi: 10.1371/journal.pgen.1003073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mets DG, Meyer BJ. Condensins regulate meiotic DNA break distribution, thus crossover frequency, by controlling chromosome structure. Cell. 2009;139:73–86. doi: 10.1016/j.cell.2009.07.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Csankovszki G, et al. Three distinct condensin complexes control C. elegans chromosome dynamics. Curr Biol. 2009;19:9–19. doi: 10.1016/j.cub.2008.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ercan S, et al. X chromosome repression by localization of the C. elegans dosage compensation machinery to sites of transcription initiation. Nat Genet. 2007;39:403–408. doi: 10.1038/ng1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ercan S, Dick LL, Lieb JD. The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Curr Biol. 2009;19:1777–1787. doi: 10.1016/j.cub.2009.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Jans J, et al. A condensin-like dosage compensation complex acts at a distance to control expression throughout the genome. Genes Dev. 2009;23:602–618. doi: 10.1101/gad.1751109. Provides evidence that C. elegans dosage compensation occurs through global rather than local control of gene expression. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McDonel P, Jans J, Peterson BK, Meyer BJ. Clustered DNA motifs mark X chromosomes for repression by a dosage compensation complex. Nature. 2006;444:614–618. doi: 10.1038/nature05338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerstein MB, et al. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010;330:1775–1787. doi: 10.1126/science.1196914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petty EL, Collette KS, Cohen AJ, Snyder MJ, Csankovszki G. Restricting dosage compensation complex binding to the X chromosomes by H2A.Z/HTZ-1. PLoS Genet. 2009;5:e1000699. doi: 10.1371/journal.pgen.1000699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Whittle CM, et al. The genomic distribution and function of histone variant HTZ-1 during C. elegans embryogenesis. PLoS Genet. 2008;4:e1000187. doi: 10.1371/journal.pgen.1000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Simon MD, et al. The genomic binding sites of a noncoding RNA. Proc Natl Acad Sci USA. 2011;108:20497–20502. doi: 10.1073/pnas.1113536108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chu C, Qu K, Zhong FL, Artandi SE, Chang HY. Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol Cell. 2011;44:667–678. doi: 10.1016/j.molcel.2011.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Plath K, et al. Role of histone H3 lysine 27 methylation in X inactivation. Science. 2003;300:131–135. doi: 10.1126/science.1084274. [DOI] [PubMed] [Google Scholar]
- 78.Zhao J, Sun BK, Erwin JA, Song JJ, Lee JT. Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 2008;322:750–756. doi: 10.1126/science.1163045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Maenner S, et al. 2-D structure of the A region of Xist RNA and its implication for PRC2 association. PLoS Biol. 2010;8:e1000276. doi: 10.1371/journal.pbio.1000276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jeon Y, Lee JT. YY1 tethers Xist RNA to the inactive X nucleation center. Cell. 2011;146:119–133. doi: 10.1016/j.cell.2011.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nozawa RS, et al. Human inactive X chromosome is compacted through a PRC2-independent SMCHD1-HBiX1 pathway. Nat Struct Mol Biol. 2013;20:566–573. doi: 10.1038/nsmb.2532. [DOI] [PubMed] [Google Scholar]
- 82.O’Sullivan JM, et al. Gene loops juxtapose promoters and terminators in yeast. Nat Genet. 2004;36:1014–1018. doi: 10.1038/ng1411. [DOI] [PubMed] [Google Scholar]
- 83.Tan-Wong SM, French JD, Proudfoot NJ, Brown MA. Dynamic interactions between the promoter and terminator regions of the mammalian BRCA1 gene. Proc Natl Acad Sci USA. 2008;105:5160–5165. doi: 10.1073/pnas.0801048105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tan-Wong SM, et al. Gene loops enhance transcriptional directionality. Science. 2012;338:671–675. doi: 10.1126/science.1224350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Grimaud C, Becker PB. The dosage compensation complex shapes the conformation of the X chromosome in Drosophila. Genes Dev. 2009;23:2490–2495. doi: 10.1101/gad.539509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ferrari F, et al. Jumpstart and gain model for dosage compensation in Drosophila based on direct sequencing of nascent transcripts. Cell Rep. 2013;5:629–636. doi: 10.1016/j.celrep.2013.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Churchman LS, Weissman JS. Nascent transcript sequencing visualizes transcription at nucleotide resolution. Nature. 2011;469:368–373. doi: 10.1038/nature09652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Dekker J, Rippe K, Dekker M, Kleckner N. Capturing chromosome conformation. Science. 2002;295:1306–1311. doi: 10.1126/science.1067799. [DOI] [PubMed] [Google Scholar]
- 89.Simonis M, et al. Nuclear organization of active and inactive chromatin domains uncovered by chromosome conformation capture-on-chip (4C) Nat Genet. 2006;38:1348–1354. doi: 10.1038/ng1896. [DOI] [PubMed] [Google Scholar]
- 90.Sexton T, et al. Sensitive detection of chromatin coassociations using enhanced chromosome conformation capture on chip. Nat Protoc. 2012;7:1335–1350. doi: 10.1038/nprot.2012.071. [DOI] [PubMed] [Google Scholar]
- 91.Dostie J, et al. Chromosome conformation capture carbon copy (5C): a massively parallel solution for mapping interactions between genomic elements. Genome Res. 2006;16:1299–1309. doi: 10.1101/gr.5571506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]