

Abstract
High tidal volume mechanical ventilation can cause inflammation and lung damage. Mechanical strain is also necessary for normal lung growth. The current work was performed to determine if mechanical ventilation with clinically utilized tidal volumes stimulates a proliferative response in the lung. Six to eight week old C57/Bl6 mice, anesthetized with ketamine/ xylozine, were ventilated for 6 hours with 10 ml/kg tidal volume, PEEP 3cm H2O. Pulmonary function testing demonstrated decreased compliance within 3 hours of ventilation. Assessment of bronchoalveolar lavage (BAL) demonstrated no significant increase in lactate dehydrogenase, total lavagable cell number or total protein after ventilation. There was evidence of inflammation in the lungs of ventilated mice, with an increased percentage of lymphocytes and neutrophils in BAL, and an increase in MIP-2 and IL-1β message in lung tissue.
Immunohistochemistry of inflation-fixed lungs demonstrated increased alveolar cell proliferation as measured by both proliferating cell nuclear antigen and Ki67 staining. Dual staining confirmed that proliferating cells labeled with proSP-B demonstrating that ventilation induces proliferation of alveolar type II cells. Ventilation did not increase apoptosis in alveolar type II cells, as measured by tunel staining. Ventilation at low tidal volumes leads to a mild inflammatory response and alveolar epithelial cell proliferation.
Keywords: lung, MIP-2, IL-1β, mechanical ventilation, proliferation
INTRODUCTION
Respiratory failure and severe hypoxemia unresponsive to supplemental oxygen may necessitate mechanical ventilation. This intervention, while life sustaining, can lead to significant lung injury and even death. Mechanical ventilation has been associated with ventilator induced lung injury (VILI) and multi-system organ failure resulting from initiation of an injurious inflammatory response in patients supported with high tidal volume ventilation [1]. Use of a protective low lung volume ventilator strategy, which minimizes lung distension, in patients with Acute Respiratory Distress Syndrome (ARDS) resulted in a 22% reduction in patient mortality [2, 3]. Mechanical ventilation is also used to support patients under general anesthesia for surgery and following intubation for surfactant administration. The pulmonary effects of such short-term ventilation are not well known.
While mechanical strain forces are potentially injurious to the lung, strain forces can also lead to lung growth. Mechanical strain results in post-pneumonectomy lung growth in rodents and humans [4-6]. The mechanisms by which mechanical strain exerts growth and injury responses remain poorly understood. Most murine studies to date have used supraphysiologic tidal volumes of 20-45 ml/kg to stimulate an injury response [7-13]. The degree to which more clinically relevant tidal volume ventilation affects pulmonary mechanics and inflammation is unknown, and the hypothesis that such ventilation can initiate a proliferative response in vivo untested. The current work uses an in vivo murine ventilation model with juvenile animals to investigate the proliferative response of the lung to mechanical ventilation with tidal volumes comparable to those used clinically. Alterations in pulmonary compliance are determined, the inflammatory response initiated by physiologic mechanical ventilation is assessed, and the hypothesis that this mode of ventilation initiates a proliferative response in pulmonary alveolar epithelial cells is tested. By identifying the early responses triggered by low tidal volume ventilation commonly used clinically, strategies can be developed to mitigate damaging responses while maintaining protective ones.
METHODS
Mouse ventilation and pulmonary function testing
6-8 week C57/Bl6 mice were anesthetized with ketamine/ xylazine. Six mice were used per condition per experiment. A total of 38 mice were used. A dose of 0.01 ml/g body weight of a solution of 100 mg/ml ketamine and 20 mg/ml xylazine was delivered intraperitoneally. Once anesthesia was induced, a tracheotomy was performed with a 16 gauge catheter. Mice were ventilated using a Harvard rodent ventilator with humidified room air at a tidal volume of 10 ml/kg, PEEP 3 cm H2O, rate 150 breaths per minute for 6 hours under continued anesthesia to a level resulting in no spontaneous respirations [14]. These settings were chosen based on published literature of physiologic murine tidal volume, respiratory rates and optimal PEEP, and confirmed as adequate to maintain adequate blood gases by blood gas determination [15]. Heart rate was determined every 30 min. Continuous plethysmography was performed using a Buxco plethysmography system (Buxco Research Systems, Wilmington, NC). After 6h of ventilation, a lethal dose of pentobarbitol was administered. Nonventilated animals were used for controls. The heart was exposed and a blood gas obtained by direct cardiac puncture immediately postmortem. The right lungs were removed and frozen for quantitation of steady-state cytokine mRNA levels using a multi-cytokine ribonuclease protection assay (RPA). The left lungs were inflation fixed with 10% neutral buffered formalin at 10 cm Hg for 24h followed by graded EtOH washes and paraffin embedding. In other animals, bronchoalveolar lavage (BAL) was performed (see subsequent methods for details). These lungs were not used for further analysis. Murine experiments were conducted in conformity with guiding principles in the care and use of animals using a protocol approved by the University of Rochester’s University Committee on Animal Resources.
Bronchoalveolar lavage (BAL)
Lungs were lavaged with 8-1ml aliquots of 37°C 0.9% NS. The first 2 lavages were spun at 150g × 5 min. Supernatant was analyzed for lactate dehydrogenase (LDH) per manufacturer’s recommendation (Sigma, St Louis, MO) and total protein measured by bicinchoninic acid analysis (Pierce Chemical Co, Rockford, IL). The remaining aliquots were spun, and cell pellets combined with aliquots 1 and 2 for total cell count and cytospin with Diff-Quick.
Ribonuclease Protection Assay (RPA)
Total RNA was isolated from frozen lung tissue using TRIzol Reagent (Life Technologies, Grand Island, NY) according to the manufacturer’s instructions. Each lung lobe (50-100 mg) was homogenized in 1 ml of TRIzol Reagent. Each final RNA pellet was resuspended in 50 μl of diethylpyrocarbonate-treated water. The RNA concentration and purity was quantified using the GeneQuant RNA/DNA Calculator (Pharmacia Biotech, Piscataway, NJ). RNase protection assays were performed as previously described [16] with a riboprobe template including interleukin (IL)-12 p35, IL-12 p40, IL-10, IL-1α, IL-1β, IL-1Ra, IL-18, Macrophage Migration Inhibitory Factor (MIF), interferon (IFN)-γ, L32, Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and a custom template (Lymphotactin (Ltn), tumor necrosis factor (TNF)β, IL-1β, Regulated upon Activation, Normal T-cell Expressed, and Secreted - Chemokine (RANTES), macrophage inflammatory protein (MIP)-1β, MIP-1α, MIP-2, monocyte chemotactic protein (MCP)-1, MIP-3α, IFN-γ, L32, GAPDH).
The protected radiolabeled RNA fragments were separated by electrophoresis on a 5% acrylamide/8 M urea sequencing gel and the dried gel exposed to X-AR film (Eastman Kodak, Rochester, NY) at −80°C with intensifying screens (Quanta III; Dupont, Wilmington, DE). For quantification, the dried gels were placed against PhosphoImager screens (Molecular Dynamics, Sunnyvale, CA). The intensity of each specific chemokine and cytokine band was measured using a computer-linked PhosphoImager with ImageQuant software (Molecular Dynamics). Each intensity score was normalized to the intensity of hybridization for the L32 band to correct for differences in loading.
Immunohistochemistry (IHC)
Tissue sections (4μm) were deparaffinized and rehydrated in graduated ethanol. Sections were stained with Hematoxylin and Eosin (H&E), or IHC for PCNA or Ki67 was performed as follows:
Proliferating cell nuclear antigen (PCNA) IHC
Endogenous peroxidase activity was blocked with hydrogen peroxide in MeOH. The sections were rinsed with PBS, incubated serially in biotinylated mouse anti-PCNA antibody, Streptavidin-Peroxidase, and 3, 3’-diaminobenzidine (DAB) (ZYMED, San Francisco CA). Cells were counterstained with methyl green, and staining quantified using Metamorph software (Universal Imaging, Downingtown, PA).
Ki67 IHC
Tissue sections (4μm) were deparaffinized, rehydrated in graduated ethanol, antigen retrieved in Tris pH 9.5 for 15 minutes, peroxidase activity blocked with 1% hydrogen peroxide in methanol, rinsed with TBS, and incubated serially in rat monoclonal anti-mouse Ki67 (DAKO, Carpinteria, CA) diluted 1:50 and biotinylated rabbit anti-rat IgG (Vector Labs, Burlingame, CA) diluted 1:200. Detection was with Vectastain ABC Elite (Vector Labs, Burlingame, CA) and DAB. Cells were counterstained with hematoxylin diluted 1:5 for 30s, and staining quantified using Metamorph software.
Ki67/proSP-B dual IHC
Tissue sections (4μm) were deparaffinized, rehydrated in graduated ethanol, antigen retrieved in Tris pH 9.5 for 15 minutes, peroxidase activity blocked with 1% hydrogen peroxide in methanol, rinsed with TBS, endogenous avidin and biotin activity blocked with Avidin/Biotin Blocking Kit (Vector Labs, Burlingame, CA). Slides were blocked with 5% normal rabbit serum in TBS-T then incubated in rat monoclonal anti-mouse Ki67 (DAKO, Carpinteria, CA) diluted 1:50 and biotinylated rabbit anti-rat IgG (Jackson ImmunoResearch, West Grove, PA) diluted 1:200. Detection was with Texas Red Avidin D (Vector Labs, Burlingame, CA), followed by treatment with Avidin/Biotin Blocking Kit, blocking with 5% normal goat serum and incubation with polyclonal rabbit anti-human prosurfactant protein B (proSP-B) (Chemicon/Millipore, Billerica, MA) diluted 1:500 and biotinylated goat anti-rabbit IgG (Jackson ImmunoResearch, West Grove, PA) diluted 1:500. Detection was with Fluorescein Avidin DCS (Vector Labs, Burlingame, CA). Cells were counterstained with 4’,6-diamidino-2-phenylindole (DAPI). Stained sections were visualized with a Nikon E800 fluorescence microscope. Ki67 fluorescence was visualized using a Texas Red filter and proSP-B using a FITC filter. Images were captured with a SPOT-RT digital camera.
Terminal transferase dUTP nick end-labeling (Tunel)/ SP-C dual IHC Staining
Apoptosis was assessed by tunel staining as previously described by O’Reilly et al [17]. Tissue sections were deparaffinized and rehydrated in graduated ethanol, washed in PBS. Using proprietary reagents from Chemicon Apoptag apoptosis detection kit (Chemicon, Temecula CA), and incubated with 20 ug/mL Proteinase.K in PBS for 15 min. Sections were equilibrated for 10 seconds and incubated in working strength of TdT enzyme for 1 hour at room temperature, then washed and stop buffer was applied with agitation for 10 minutes. Sections were again washed and incubated in anti-digoxigenin conjugate for 30 minutes in the dark at room temperature. After washing, the sections were counterstained with DAPI (1:400 in dH2O) for 3 minutes at room temperature, and washed.
SP-C Dual Staining
Antigen retrieval was performed in a Tris Buffer (pH 9.5) in a microwave. Nonspecific binding was blocked in a 3% normal donkey serum (NDS) in TBS-T for 30 min at room temperature and incubated at 1:100 in rabbit anti-SP-C (Santa Cruz Biotech, Santa Cruz CA) in TBS-T with 3% NDS overnight at 4C. Sections were then washed and incubated with Tx-red conjugate donkey anti-Rabbit secondary at a 1:200 dilution in TBS-T for 1 hour, followed by wash and counterstained with DAPI Nuclear Counterstain.
In each of the IHC conditions a negative control section using secondary antibody alone was used to demonstrate specificity of primary antibody.
Quantitative immunohistochemistry
Random, noncontiguous fields of parenchyma were acquired using the 40X objective of a Nikon E-800 microscope and a SPOT RT camera. Four random fields per lung were obtained from 6-8 separate animals at each condition. Fields that contained a large airway or blood vessel were rejected. Positively staining cells were identified and counted in a blinded fashion using Metamorph software.
Mean linear intercept
As a measure of interalveolar wall distance, mean linear intercept was calculated from the H&E stained lung sections of control and ventilated animals. Five random 40X fields were selected via light microscopy from each animal and the mean linear intercept was calculated by dividing the total length of a line (in micrometers) drawn across the entire field by the total number of alveolar intercepts encountered along the length of the line. Bars represent the mean of five random measurements.
Data Analysis
ANOVA with Scheffe’s post-hoc analysis was used for analysis of data quantified over time. An unpaired t-test was used when comparing two groups. A p value of < 0.05 was considered statistically significant. Values are shown as mean ± standard error of the mean (SEM).
RESULTS
Mice ventilated with humidified room air for 6 hours with 10 ml/kg tidal volume, PEEP 3 cm H2O, rate 150 breaths per minute (BPM) had no change in lung morphology by mean linear intercept quantification of alveolar size compared to nonventilated controls (Figure 1). The pH, pCO2 and pO2 of arterial blood obtained immediately postmortem did not differ significantly between ventilated and control animals (data not shown). Adverse effects were seen in dynamic pulmonary mechanics. Compliance decreased within 3 hours of ventilation, and peak pressure needed to maintain tidal volume was significantly increased by 5.5 hours of ventilation (Figure 2).
Figure 1.
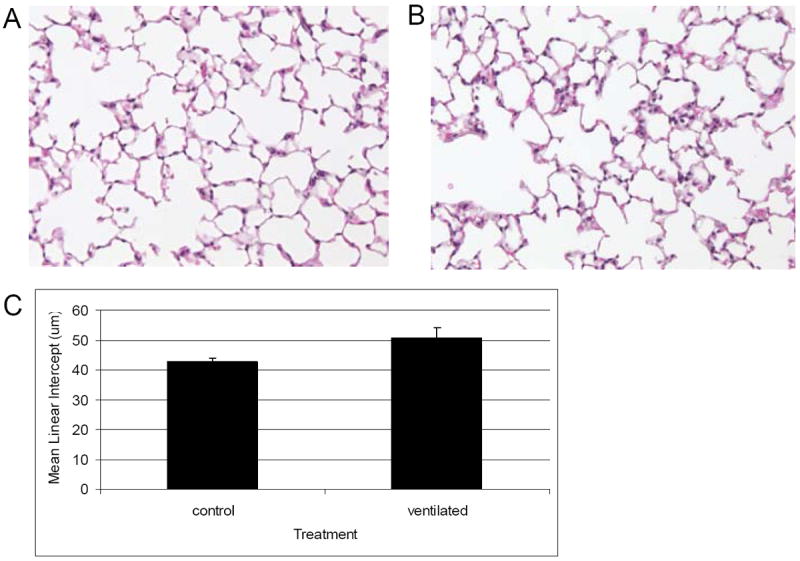
H and E staining of inflation fixed lungs from A: nonventilated and B: ventilated C57/Bl6 mice. C. Mean linear intercept quantification. There is no significant difference in mean linear intercept after 6 h ventilation. 20x magnification. N=6 per condition.
Figure 2.
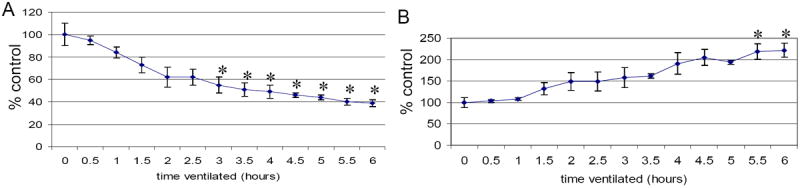
Compliance characteristics of mice assessed at 30 m intervals from 0-6 h ventilation. A: compliance B: peak pressure needed to maintain tidal volume. There was a significant difference in compliance and peak pressure needed to maintain tidal volume within 6 hours of mechanical ventilation. Values represent mean ± SEM (*p<0.05 compared to time 0 min compliance and peak pressure, respectively). N=6 per condition.
To assess if ventilation resulted in an influx of inflammatory cells or protein, BAL from control and ventilated animals was analyzed. There was an increase in number of cells and LDH activity, but these did not achieve statistical significance (Figure 3A, B). There was no change in BAL protein (Figure 3C). While the total number of inflammatory cells in BAL was not increased significantly after mechanical ventilation, the percent of lymphocytes and neutrophils were significantly increased (9 and 8 fold increase respectively, p<0.05) (Figure 3D, E).
Figure 3.
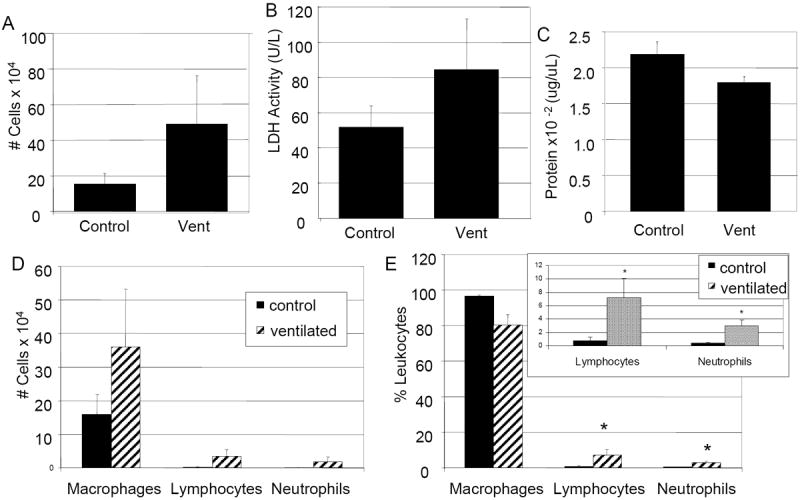
Differential cell counts of BAL from nonventilated and ventilated mice. A: Total number of cells. B: LDH activity. C: total protein. D: differential cell count. E: Percent leukocyte differential. Ventilated animals have a 9 fold increase in percent lymphocytes and an 8 fold increase in percent neutrophils compared to nonventilated control animals. E: Insert represents lymphocyte/ neutrophil percentages. Values represent mean ± SEM (*p<0.05). N=6 per condition.
To determine if ventilation results in increased mRNA for inflammatory mediators, RPA was performed. MIP-2 and IL-1β were significantly increased after 6 hours of ventilation (4.3 and 2.6 fold respectively, p<0.05) (Figure 4A, B). There were no changes in mRNA for IL-12 p35, IL-12 p40, IL-10, IL-1 β, IL-1Ra, IL-18/IGIF, MIF, IFN-γ, Ltn, TNFα, RANTES, MIP-1β, MIP-1α, MCP-1, or MIP-3 α (data not shown).
Figure 4.
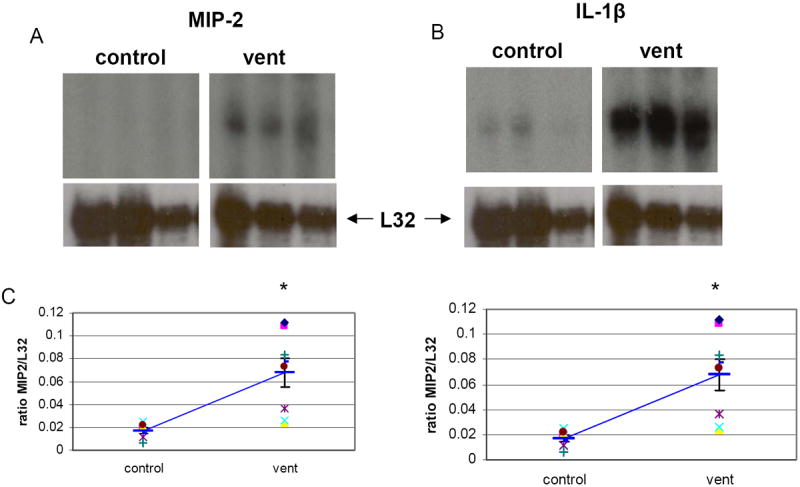
RPAs of RNA isolated from lungs of A: nonventilated controls (C) or B: ventilated (V, vent) mice demonstrate a significant increase in MIP-2 and IL1-β in ventilated mice. C: scatter graph of individual animals. There was no change in TNFa or RANTES (data not shown). Heavy blue bar = mean, error bars =SEM, *p<0.05. N=7 per condition.
To determine if ventilation initiates a proliferative response in alveolar epithelial cells, lungs from control and ventilated mice were analyzed. Lungs were inflation fixed and immunohistochemistry for proliferating cell nuclear antigen (PCNA) and Ki67 followed by morphometric analysis were performed. PCNA is an antigen expressed during the late G1 and early S phase of DNA synthesis as well as during DNA repair. Ki67 is an antigen that is expressed during active phases of the cell cycle but absent in resting G0 cells [18]. Alveolar epithelial cells were identified morphologically by their characteristic shape and location. Two and a half and 3 percent of pulmonary epithelial cells stained positive for PCNA and Ki67 respectively in control lungs, which is comparable to the published reports of basal proliferation in adult murine lungs [19, 20]. Mechanical ventilation resulted in a 3 fold increase in cells staining positive for PCNA, and a 6.5 fold increase in cells staining positive for Ki67 after 6 hours compared to control lungs (p<0.05 for both data sets) (Figures 5, 6). The cells staining positive for proliferating antigen had morphology similar to pulmonary type II alveolar epithelial cells. To confirm this, dual staining was performed with a type II cell marker, pro-SP-B. Control lungs had a very low number of cells that stained positive for Ki67, and those that did colocalized Ki67 and pro-SP-B (data not shown). Figure 7 demonstrates the dual staining of Ki67 with proSP-B in lungs after 6 hours of mechanical ventilation. Since proliferation can be a response to repopulate injured cells, tunel staining was used to assess apoptosis in control and ventilated lungs. There was minimal tunel staining in either control (0-1 cell per high power field (HPF)) or ventilated (1-4 cells per HPF) lungs. The rare tunel positive cells did not colocalize with the type II cell label SP-C (Figure 8).
Figure 5.
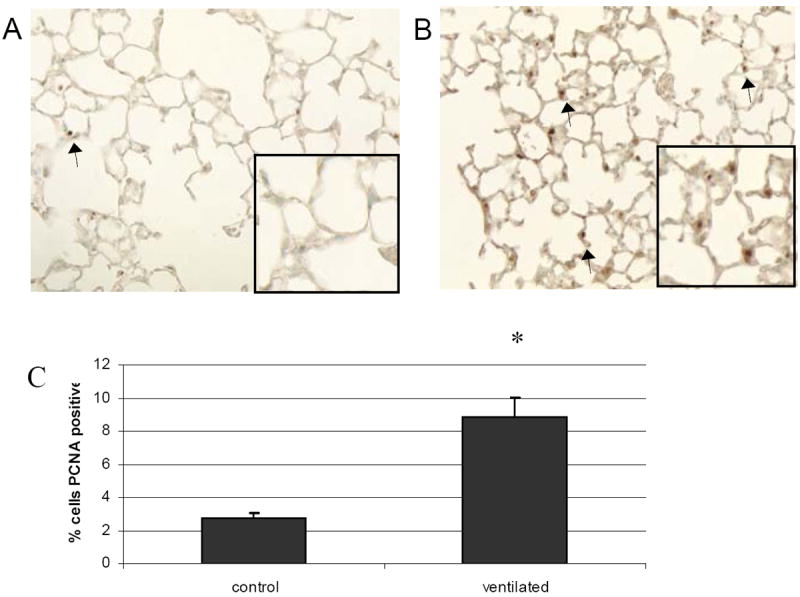
PCNA staining of A: nonventilated and B: ventilated mice. C: Quantification of IHC. Ventilated mice have a 3 fold increase in PCNA staining compared to nonventilated controls. Arrows note representative positively stained cells. 20x magnification. Values represent mean ± SEM (*p<0.05). N=6 per condition.
Figure 6.
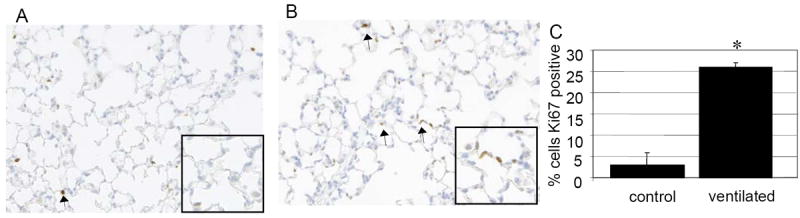
Ki67 staining of A: nonventilated and B: ventilated mice. C: quantification of IHC. Ventilated mice have a 6.5 fold increase in Ki67 staining compared to nonventilated controls. Arrows note representative positively stained cells. 20x magnification. Values represent mean ± SEM (*p<0.05). N=6 per condition.
Figure 7.
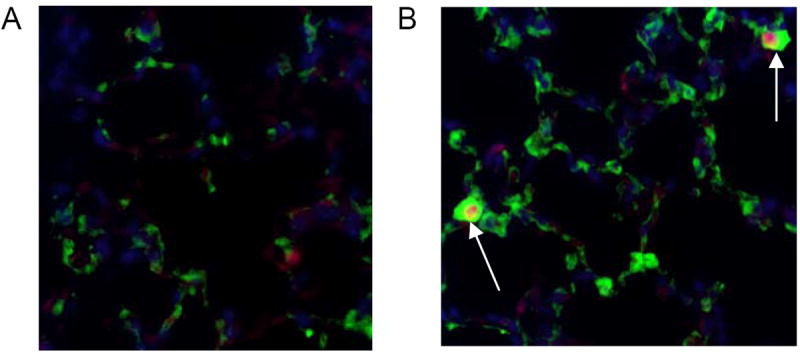
Dual IHC for Ki67 (red) and proSP-B (green). DAPI=blue. A= control B= ventilated. Ventilated mouse lungs demonstrate cells that colocalize staining for Ki67 and proSP-B (arrows). 40x magnification.
Figure 8.
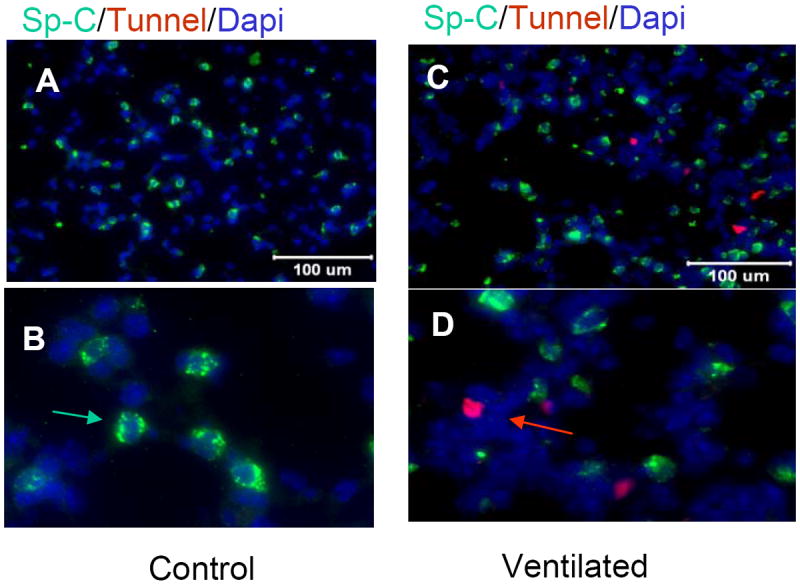
Tunel staining (red arrow) of control mice demonstrate 0-1 apoptotic cell per high power field (HPF) (A=20x, B=40X magnification). Ventilated mice demonstrate 1-4 tunel positive cells per HPF (C=20x, D=40X) Dual staining with SP-C (green arrow) demonstrates cells staining positively for tunel do not colocalize SP-C staining.
DISCUSSION
The lung is exposed to mechanical strain forces over baseline in humans during short-term ventilation for surgical procedures, longer term ventilation for respiratory failure, and subacutely following pneumonectomy for cancer resection or emphysema treatment. Little is known about biophysical changes in the lungs after short term ventilation for surgical procedures etc. While there is a sizable body of literature on the detrimental effects of high tidal volume ventilation as discussed below, less is lknown about the signaling cascades initiated early in the course of ventilation for respiratory failure when low tidal volumes are utilized. Continuous positive airway pressure (CPAP), such as that seen with patients using CPAP for sleep apnea or in post-pneumonectomy, has been demonstrated to initiate a proliferative response. CPAP induces lung growth in 6 week old ferrets [21] Postpneumonectomy lung growth is observed in humans and animal models, which may be stimulated by the strain forces experienced by the residual lung [22, 23]. The aim of this work was to assess the short term biophysical changes elicited by low tidal volume ventilation, using tidal volumes that are clinically utilized. The effect on pulmonary mechanics was assessed, and the hypothesis that ventilation induces a proliferative response in pulmonary epithelial cells with minimal inflammatory response tested. This work demonstrates that even relatively low tidal volume ventilation initiates an inflammatory response, and that mechanical strain-initiates proliferation of alveolar epithelial cells using a clinically relevant tidal volume in an in vivo model.
Lung distension during mechanical ventilation for acute respiratory failure, while life-sustaining, also transmits significant risk of morbidity and mortality [1, 3]. Infants born prematurely often receive ventilatory support. Many infants supported with mechanical ventilation develop lung injury that can be fatal [24]. The most common use of ventilation is to provide respiratory support during general anesthesia for surgery. While this is usually provided only for a few hours, the current work provides evidence that such short-term ventilation can result in significant alterations in pulmonary mechanics and molecular processes. Large animal models of ventilator-induced lung injury indicate that immature lungs are particularly prone to ventilator-induced lung injury [25, 26]. Children and adults with trauma, aspiration or pneumonia frequently progress to Acute Respiratory Distress Syndrome (ARDS) with a mortality rate of 40-70% [27]. Mechanical ventilation to support patients with respiratory failure in ARDS has been implicated in initiating systemic responses that contribute to morbidity and mortality [28-30]. Elucidating the mechanisms by which mechanical strain leads to lung growth or inflammation can allow therapeutic approaches to optimize strain-induced lung growth while mitigating strain-induced injury.
Large tidal volume ventilation is known to cause an inflammatory response in the lung. Rabbits and rats have been used to study mechanical strain effects with high tidal volume ventilation associated with inflammatory markers [8, 25, 31-37]. Mice ventilated with 20 to 43 ml/kg had an increase in inflammatory cells and MIP-2 protein in lung lavage and an increase in inflammatory mediator mRNA [9-13, 38]. Mice ventilated with PIP 45 cm H2O had increased MIP-2 protein and IL-6 in BAL [7]. Mice ventilated with 17 ml/kg tidal volumes for 4 hours had no change in IL-6, TNFα, VEGF or VEGF-R2 protein in lung tissue; however, VEGF increased in liver and kidney of ventilated animals. This ventilator strategy also exacerbated acid aspiration- induced increases in IL-6 and VEGF-R2 in the lungs of ventilated mice [39]. Mouse lungs ventilated ex vivo at 20 ml/kg for 2 hours had increased TNFα and IL-6 protein in BAL. Application of chest wall restriction in ventilated rabbits minimized the increased capillary permeability observed in response to ventilation at high peak inspiratory pressures, providing evidence that lung injury caused by mechanical ventilation is more closely related to mechanical strain (volutrauma) than to barotrauma [40]. Acute infection adds to mechanical ventilation-induced lung injury [41]. In vivo work demonstrating an inflammatory response from stretch is corroborated by in vitro experiments. A549 lung epithelial cells exposed to 30% elongation have increased IL-8 gene transcription, whereas cells exposed to 20% elongation or less do not have increased transcription [42]. While these data demonstrate an inflammatory response to mechanical strain, the tidal volumes used are excessive, and not comparable to typical tidal volumes used clinically, making extrapolation of results to clinical paradigms difficult. The present work assesses the inflammatory and proliferative response seen in response to mechanical ventilation with a lower, clinically relevant tidal volume of 10 ml/kg, PEEP 3 cm H2O. Although a relatively low tidal volume was used for ventilating the adolescent mice in the current study, an inflammatory response was initiated, as noted by an increase in MIP-2 and IL-1β mRNA in lung tissue and an increase percent lymphocytes and neutrophils in BAL of ventilated animals. At the same time, a mechanical strain-induced proliferative response was established. Neonatal mice ventilated for 24h with 40% oxygen with peak inspiratory pressure average of 11 mmHg and PEEP <1 to minimize circulatory compromise did not result in a change in alveolar cell proliferation assessed by PCNA. This may be due to changes in the different ventilator settings used, the age of the animals, or the oxygen exposure, since oxygen is known to alter cell proliferation [43, 44]. Proliferation is sometimes a response to replace injured cells. [45, 46]. To determine if cell proliferation in this model was in response to injury, apoptosis was assessed by tunel staining. Control lungs did not demonstrate tunnel staining. While there were rare tunnel stained cells in ventilated animals, there were no tunnel positive cells that costained for the type II cell marker. Since type II cells were the cells identified as proliferating in response to ventilation, proliferation does not appear to be a response to injury. They may be proliferating to replace damaged type I cells.
The current work demonstrates a significant change in compliance after brief ventilation. A number of factors could contribute to this, including atelectasis or surfactant inactivation from the mild inflammatory response. Injurious high tidal volume or zero peep ventilation is known to result in areas of atelectasis and overdistension.[47, 48]. While stretch has been demonstrated to stimulate surfactant release, ventilation-induced inflammation can inactivate surfactant function [49, 50]. Further studies to investigate the etiology of these changes in pulmonary function are underway.
The current work demonstrates an increase in inflammation in spite of a physiologic tidal volume ventilation strategy used for a relatively short period of time, such as that which is used during operations under general anesthesia, or for intubation for surfactant administration. This provides evidence that using low tidal volume ventilation is recommended, even in short- term ventilatory support. Additional novel findings include ventilation-induced increases of markers of proliferation in alveolar epithelial type II cells of ventilated animals. This finding provides support for the possibility of utilizing mechanical strain during ventilation to regulate lung growth, while highlighting the potentially injurious inflammatory response stimulated by a clinically utilized relatively low tidal volume ventilation strategy.
Acknowledgments
The authors would like to thank Min Yee, Libby Kimball, Rickard Watkins and Greg Roat for their technical assistance.
GRANTS: The current work was supported by HL03910
Footnotes
Declaration of interest: The authors have no disclosures related to this material
Contributor Information
Patricia Chess, University of Rochester, Pediatrics and Biomedical Engineering.
Randi Benson, University of Rochester, Environmental Medicine.
William Maniscalco, University of Rochester, Pediatrics.
Terry Wright, University of Rochester, Pediatrics.
Michael A O’Reilly, University of Rochester Department of Pediatrics and Environmental Medicine.
Carl Johnston, University of Rochester, Pediatrics.
References
- 1.Plotz FB, Slutsky AS, van Vught AJ, Heijnen CJ. Ventilator-induced lung injury and multiple system organ failure: a critical review of facts and hypotheses. Intensive Care Med. 2004;30(10):1865–72. doi: 10.1007/s00134-004-2363-9. [DOI] [PubMed] [Google Scholar]
- 2.Vlahakis NE, Hubmayr RD. Cellular stress failure in ventilator-injured lungs. Am J Respir Crit Care Med. 2005;171(12):1328–42. doi: 10.1164/rccm.200408-1036SO. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Brower RG, Matthay MA, Morris A, Schoenfeld D, Taylor Thompson B, Wheeler A. Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. New England Journal of Medicine. 2000;342(18):1301–1308. doi: 10.1056/NEJM200005043421801. [DOI] [PubMed] [Google Scholar]
- 4.Fernandez LG, Mehta CK, Kron IL, Laubach VE. Reinitiation of compensatory lung growth after subsequent lung resection. J Thorac Cardiovasc Surg. 2007;134(5):1300–5. doi: 10.1016/j.jtcvs.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Sekhon HS, Smith C, Thurlbeck WM. Effect of hypoxia and hyperoxia on postpneumonectomy compensatory lung growth. Experimental Lung Research. 1993;19(5):519–32. doi: 10.3109/01902149309031725. [DOI] [PubMed] [Google Scholar]
- 6.Kreisel D, Krupnick AS, Huddleston CB. Outcomes and late complications after pulmonary resections in the pediatric population. Semin Thorac Cardiovasc Surg. 2004;16(3):215–9. doi: 10.1053/j.semtcvs.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 7.Yoshikawa S, King JA, Lausch RN, Penton AM, Eyal FG, Parker JC. Acute ventilator-induced vascular permeability and cytokine responses in isolated and in situ mouse lungs. J Appl Physiol. 2004;97(6):2190–9. doi: 10.1152/japplphysiol.00324.2004. [DOI] [PubMed] [Google Scholar]
- 8.Uhlig U, Haitsma JJ, Goldmann T, Poelma DL, Lachmann B, Uhlig S. Ventilation-induced activation of the mitogen-activated protein kinase pathway. European Respiratory Journal. 2002;20(4):946–56. doi: 10.1183/09031936.02.01612001. [DOI] [PubMed] [Google Scholar]
- 9.Wilson MR, Choudhury S, Goddard ME, O’Dea KP, Nicholson AG, Takata M. High tidal volume upregulates intrapulmonary cytokines in an in vivo mouse model of ventilator-induced lung injury. Journal of Applied Physiology. 2003;95(4):1385–93. doi: 10.1152/japplphysiol.00213.2003. [DOI] [PubMed] [Google Scholar]
- 10.Wilson MR, Choudhury S, Takata M. Pulmonary inflammation induced by high-stretch ventilation is mediated by tumor necrosis factor signaling in mice. Am J Physiol Lung Cell Mol Physiol. 2005;288(4):L599–607. doi: 10.1152/ajplung.00304.2004. [DOI] [PubMed] [Google Scholar]
- 11.Ma SF, Grigoryev DN, Taylor AD, Nonas S, Sammani S, Ye SQ, et al. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;289(3):L468–77. doi: 10.1152/ajplung.00109.2005. [DOI] [PubMed] [Google Scholar]
- 12.Li LF, Yu L, Quinn DA. Ventilation-induced neutrophil infiltration depends on c-Jun N-terminal kinase. American Journal of Respiratory & Critical Care Medicine. 2004;169(4):518–24. doi: 10.1164/rccm.200305-660OC. [DOI] [PubMed] [Google Scholar]
- 13.Li LF, Liao SK, Ko YS, Lee CH, Quinn DA. Hyperoxia increases ventilator-induced lung injury via mitogen-activated protein kinases: a prospective, controlled animal experiment. Crit Care. 2007;11(1):R25. doi: 10.1186/cc5704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lundblad LK, Thompson-Figueroa J, Leclair T, Irvin CG, Bates JH. Thoracic gas volume measurements in paralyzed mice. Ann Biomed Eng. 2004;32(10):1420–7. doi: 10.1114/b:abme.0000042229.41098.6a. [DOI] [PubMed] [Google Scholar]
- 15.Lai-Fook SJ, Houtz PK, Lai YL. End-expiratory and tidal volumes measured in conscious mice using single projection x-ray images. J Appl Physiol. 2008;104(2):521–33. doi: 10.1152/japplphysiol.00729.2007. [DOI] [PubMed] [Google Scholar]
- 16.Johnston CJ, Finkelstein JN, Gelein R, Oberdorster G. Pulmonary cytokine and chemokine mRNA levels after inhalation of lipopolysaccharide in C57BL/6 mice. Toxicol Sci. 1998;46(2):300–7. doi: 10.1006/toxs.1998.2557. [DOI] [PubMed] [Google Scholar]
- 17.O’Reilly MA, Staversky RJ, Stripp BR, Finkelstein JN. Exposure to hyperoxia induces p53 expression in mouse lung epithelium. Am J Respir Cell Mol Biol. 1998;18(1):43–50. doi: 10.1165/ajrcmb.18.1.2950m. [DOI] [PubMed] [Google Scholar]
- 18.Zhang X, Barile G, Chang S, Hays A, Pachydaki S, Schiff W, et al. Apoptosis and cell proliferation in proliferative retinal disorders: PCNA, Ki-67, caspase-3, and PARP expression. Curr Eye Res. 2005;30(5):395–403. doi: 10.1080/02713680590956306. [DOI] [PubMed] [Google Scholar]
- 19.Lee J, Reddy R, Barsky L, Weinberg K, Driscoll B. Contribution of proliferation and DNA damage repair to alveolar epithelial type 2 cell recovery from hyperoxia. Am J Physiol Lung Cell Mol Physiol. 2006;290(4):L685–L694. doi: 10.1152/ajplung.00020.2005. [DOI] [PubMed] [Google Scholar]
- 20.Pagano A, Metrailler-Ruchonnet I, Aurrand-Lions M, Lucattelli M, Donati Y, Argiroffo CB. Poly(ADP-ribose) polymerase-1 (PARP-1) controls lung cell proliferation and repair after hyperoxia-induced lung damage. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L619–29. doi: 10.1152/ajplung.00037.2007. [DOI] [PubMed] [Google Scholar]
- 21.Zhang S, Garbutt V, McBride JT. Strain-induced growth of the immature lung. Journal of Applied Physiology. 1996;81(4):1471–6. doi: 10.1152/jappl.1996.81.4.1471. [DOI] [PubMed] [Google Scholar]
- 22.Brody JS. Time course of and stimuli to compensatory growth of the lung after pneumonectomy. Journal of Clinical Investigation. 1975;56(4):897–904. doi: 10.1172/JCI108169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cagle PT, Langston C, Thurlbeck WM. The effect of age on postpneumonectomy growth in rabbits. Pediatric Pulmonology. 1988;5(2):92–5. doi: 10.1002/ppul.1950050205. [DOI] [PubMed] [Google Scholar]
- 24.Overstreet DW, Jackson JC, van Belle G, Truog WE. Estimation of mortality risk in chronically ventilated infants with bronchopulmonary dysplasia. Pediatrics. 1991;88(6):1153–60. [PubMed] [Google Scholar]
- 25.Adkins WK, Hernandez LA, Coker PJ, Buchanan B, Parker JC. Age effects susceptibility to pulmonary barotrauma in rabbits. Critical Care Medicine. 1991;19(3):390–3. doi: 10.1097/00003246-199103000-00018. [DOI] [PubMed] [Google Scholar]
- 26.Gerstmann DR, deLemos RA, Coalson JJ, Clark RH, Wiswell TE, Winter DC, et al. Influence of ventilatory technique on pulmonary baroinjury in baboons with hyaline membrane disease. Pediatric Pulmonology. 1988;5(2):82–91. doi: 10.1002/ppul.1950050204. [DOI] [PubMed] [Google Scholar]
- 27.van Soeren MH, Diehl-Jones WL, Maykut RJ, Haddara WM. Pathophysiology and implications for treatment of acute respiratory distress syndrome. AACN Clinical Issues. 2000;11(2):179–97. doi: 10.1097/00044067-200005000-00004. [DOI] [PubMed] [Google Scholar]
- 28.Gillette MA, Hess DR. Ventilator-induced lung injury and the evolution of lung-protective strategies in acute respiratory distress syndrome. Respiratory Care. 2001;46(2):130–48. [PubMed] [Google Scholar]
- 29.dos Santos CC, Slutsky AS. Mechanotransduction, ventilator-induced lung injury and multiple organ dysfunction syndrome. Intensive Care Medicine. 2000;26(5):638–42. doi: 10.1007/s001340051217. [DOI] [PubMed] [Google Scholar]
- 30.Tsuno K, Prato P, Kolobow T. Acute lung injury from mechanical ventilation at moderately high airway pressures. Journal of Applied Physiology. 1990;69(3):956–61. doi: 10.1152/jappl.1990.69.3.956. [DOI] [PubMed] [Google Scholar]
- 31.Nin N, Lorente JA, Fernandez-Segoviano P, De Paula M, Ferruelo A, Esteban A. High-tidal volume ventilation aggravates sepsis-induced multiorgan dysfunction in a dexamethasone-inhibitable manner. Shock. 2009;31(4):429–34. doi: 10.1097/SHK.0b013e318188b720. [DOI] [PubMed] [Google Scholar]
- 32.Nin N, Lorente JA, de Paula M, El Assar M, Vallejo S, Penuelas O, et al. Rats surviving injurious mechanical ventilation show reversible pulmonary, vascular and inflammatory changes. Intensive Care Med. 2008;34(5):948–56. doi: 10.1007/s00134-007-0959-6. [DOI] [PubMed] [Google Scholar]
- 33.Welk B, Malloy JL, Joseph M, Yao LJ, Veldhuizen AW. Surfactant treatment for ventilation-induced lung injury in rats: effects on lung compliance and cytokines. Experimental Lung Research. 2001;27(6):505–20. doi: 10.1080/019021401750414038. [DOI] [PubMed] [Google Scholar]
- 34.Roth-Kleiner M, Ridsdale R, Cao L, Kuliszewski M, Tseu I, McKerlie C, et al. Lipopolysaccharide exposure modifies high tidal volume ventilation-induced proinflammatory mediator expression in newborn rat lungs. Pediatr Res. 2007;61(2):191–6. doi: 10.1203/01.pdr.0000252437.51779.21. [DOI] [PubMed] [Google Scholar]
- 35.Okutani D, Han B, Mura M, Waddell TK, Keshavjee S, Liu M. High-volume ventilation induces pentraxin 3 expression in multiple acute lung injury models in rats. Am J Physiol Lung Cell Mol Physiol. 2007;292(1):L144–53. doi: 10.1152/ajplung.00002.2006. [DOI] [PubMed] [Google Scholar]
- 36.Sinclair SE, Molthen RC, Haworth ST, Dawson CA, Waters CM. Airway strain during mechanical ventilation in an intact animal model. Am J Respir Crit Care Med. 2007;176(8):786–94. doi: 10.1164/rccm.200701-088OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Desai LP, Sinclair SE, Chapman KE, Hassid A, Waters CM. High tidal volume mechanical ventilation with hyperoxia alters alveolar type II cell adhesion. Am J Physiol Lung Cell Mol Physiol. 2007;293(3):L769–78. doi: 10.1152/ajplung.00127.2007. [DOI] [PubMed] [Google Scholar]
- 38.Sakashita A, Nishimura Y, Nishiuma T, Takenaka K, Kobayashi K, Kotani Y, et al. Neutrophil elastase inhibitor (sivelestat) attenuates subsequent ventilator-induced lung injury in mice. Eur J Pharmacol. 2007;571(1):62–71. doi: 10.1016/j.ejphar.2007.05.053. [DOI] [PubMed] [Google Scholar]
- 39.Gurkan OU, O’Donnell C, Brower R, Ruckdeschel E, Becker PM. Differential effects of mechanical ventilatory strategy on lung injury and systemic organ inflammation in mice. American Journal of Physiology - Lung Cellular & Molecular Physiology. 2003;285(3):L710–8. doi: 10.1152/ajplung.00044.2003. [DOI] [PubMed] [Google Scholar]
- 40.Hernandez LA, Peevy KJ, Moise AA, Parker JC. Chest wall restriction limits high airway pressure-induced lung injury in young rabbits. Journal of Applied Physiology. 1989;66(5):2364–8. doi: 10.1152/jappl.1989.66.5.2364. [DOI] [PubMed] [Google Scholar]
- 41.Dhanireddy S, Altemeier WA, Matute-Bello G, O’Mahony DS, Glenny RW, Martin TR, et al. Mechanical ventilation induces inflammation, lung injury, and extra-pulmonary organ dysfunction in experimental pneumonia. Lab Invest. 2006;86(8):790–9. doi: 10.1038/labinvest.3700440. [DOI] [PubMed] [Google Scholar]
- 42.Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. American Journal of Physiology. 1999;277(1 Pt 1):L167–73. doi: 10.1152/ajplung.1999.277.1.L167. [DOI] [PubMed] [Google Scholar]
- 43.Bland RD, Ertsey R, Mokres LM, Xu L, Jacobson BE, Jiang S, et al. Mechanical ventilation uncouples synthesis and assembly of elastin and increases apoptosis in lungs of newborn mice. Prelude to defective alveolar septation during lung development? Am J Physiol Lung Cell Mol Physiol. 2008;294(1):L3–14. doi: 10.1152/ajplung.00362.2007. [DOI] [PubMed] [Google Scholar]
- 44.Auten RL, Mason SN, Auten KM, Brahmajothi M. Hyperoxia impairs postnatal alveolar epithelial development via NADPH oxidase in newborn mice. Am J Physiol Lung Cell Mol Physiol. 2009;297(1):L134–42. doi: 10.1152/ajplung.00112.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.O’Reilly M, Hooper SB, Allison BJ, Flecknoe SJ, Snibson K, Harding R, et al. Persistent bronchiolar remodeling following brief ventilation of the very immature ovine lung. Am J Physiol Lung Cell Mol Physiol. 2009;297(5):L992–L1001. doi: 10.1152/ajplung.00099.2009. [DOI] [PubMed] [Google Scholar]
- 46.May M, Strobel P, Preisshofen T, Seidenspinner S, Marx A, Speer CP. Apoptosis and proliferation in lungs of ventilated and oxygen-treated preterm infants. European Respiratory Journal. 2004;23(1):113–21. doi: 10.1183/09031936.03.00038403. [DOI] [PubMed] [Google Scholar]
- 47.Kaditis AG, Motoyama EK, Zin W, Maekawa N, Nishio I, Imai T, et al. The effect of lung expansion and positive end-expiratory pressure on respiratory mechanics in anesthetized children. Anesth Analg. 2008;106(3):775–85. doi: 10.1213/ane.0b013e318162c20a. table of contents. [DOI] [PubMed] [Google Scholar]
- 48.Chu EK, Whitehead T, Slutsky AS. Effects of cyclic opening and closing at low- and high-volume ventilation on bronchoalveolar lavage cytokines. Crit Care Med. 2004;32(1):168–74. doi: 10.1097/01.CCM.0000104203.20830.AE. [DOI] [PubMed] [Google Scholar]
- 49.Wirtz HR, Dobbs LG. Calcium mobilization and exocytosis after one mechanical stretch of lung epithelial cells. Science. 1990;250(4985):1266–9. doi: 10.1126/science.2173861. [DOI] [PubMed] [Google Scholar]
- 50.Dixon DL, De Smet HR, Bersten AD. Lung mechanics are both dose and tidal volume dependant in LPS-induced lung injury. Respir Physiol Neurobiol. 2009;167(3):333–40. doi: 10.1016/j.resp.2009.06.008. [DOI] [PubMed] [Google Scholar]