

Abstract
Alzheimer’s disease (AD) is a protein misfolding disease characterized by a build-up of β-amyloid (Aβ) peptide as senile plaques, uncontrolled neurodegeneration, and memory loss. AD pathology is linked to the destabilization of cellular ionic homeostasis and involves Aβ peptide-plasma membrane interactions. In principle, there are two possible ways through which disturbance of the ionic homeostasis can take place: directly, where the Aβ peptide either inserts into the membrane and creates ion-conductive pores or destabilizes the membrane organization; or, indirectly, where the Aβ peptide interacts with existing cell membrane receptors. To distinguish between these two possible types of Aβ-membrane interactions, we took advantage of the biochemical tenet that ligand-receptor interactions are stereospecific; L-amino acid peptides, but not their D-counterparts, bind to cell membrane receptors. However, with respect to the ion channel-mediated mechanism, like L-amino acids, D-amino acid peptides will also form ion channel-like structures. Using atomic force microscopy (AFM) we imaged the structures of both D- and L-enantiomers of the full length Aβ1-42 when reconstituted in lipid bilayers. AFM imaging shows that both L- and D-Aβ isomers form similar channel-like structures. Molecular dynamics (MD) simulations support the AFM imaged 3D structures. Earlier we have shown that D-Aβ1-42 channels conduct ions similarly to their L-counter parts. Taken together, our results support the direct mechanism of Aβ ion channel-mediated destabilization of ionic homeostasis rather than the indirect mechanism through Aβ interaction with membrane receptors.
Keywords: Alzheimer, D-amino acids, Amyloid β, Aβ enantiomer, cellular stereospecificity, atomic force microscopy, molecular dynamics simulation, D-peptide, amyloid toxicity
INTRODUCTION
Alzheimer’s disease (AD) is characterized by an aberrant buildup of extracellular protein plaques, uncontrolled neurodegeneration, chronic dementia, and memory loss.1-4 AD plaques are predominantly composed of β-amyloid (Aβ1-39~42) peptides derived from the proteolytic cleavage of its precursor protein, amyloid precursor protein (APP),5,6 and form amyloid structures.7 The role of amyloidogenicity in cellular toxicity is unclear and mounting evidence supports the role of oligomeric Aβ.8,9 Further, the AD pathology is increasingly believed to be mediated by globular Aβ disrupting the ionic homeostasis through its interaction with cellular membranes.10-14 Understanding the interaction of Aβ with the cell membrane and its effects on cellular degeneration is crucial to the understanding of the pathological origin of AD and would eventually aid the prevention and treatment of the disease.
Two mechanisms have been proposed to explain Aβ-mediated toxicity. According to the first, the Aβ peptide forms oligomeric complexes organized as pores, similar to other cytotoxic peptides.15 Atomic force microscopy (AFM) images support the presence of Aβ channels in lipid bilayers.14,16 The activity and function of these channels have been supported by electrophysiological recordings, molecular dynamics (MD) simulations, and cell calcium and degeneration studies:17-22 amyloid channels conduct cations resulting in a gain-of-function type pathological response.15,23 According to this mechanism, Aβ inserts directly into the cell membrane and allows cellular Ca2+ uptake, thus unbalancing the cell ionic homeostasis which can lead to neurodegeneration.10,13,14,24,25 An alternative mechanism for Aβ-mediated destabilization of ionic homeostasis suggests that the Aβ peptide interacts with the membrane via stereospecific interactions involving membrane receptors.26-28 Several cellular stereospecificity studies related to Aβ membrane receptors have been reported, but with conflicting results.27-29 This may be due to variation in cell lines, sample preparation and handling, and most importantly, the lack of any definite single toxic mechanism.
To distinguish between the two mechanisms, we address the question of whether such specific peptide-receptor interaction is a requirement for Aβ-mediated toxicity. Stereospecificity can be studied through comparison of the biological activities of the L- and D-enantiomers. In a stereospecific receptor-ligand relationship only L- (and not D-) amino acids are known to interact with membrane receptors. Thus, the formation of pores in the presence of the D-enantiomer-only would suggest that pore formation can take place in the absence of stereospecific interactions and as such, any cellular effect(s) would result from Aβ toxic channels directly, without receptors.
To date, there has been no study focusing on the structural stereospecificity of Aβ ionic channels. We have reported that the all L- and D-amino acids Aβ1-42 isomers (L-Aβ1-42 and D-Aβ1-42, respectively) exhibit comparable channel conductivity in lipid bilayers.30 Here, using AFM imaging and MD simulations, we present direct structural evidence for the pore-like morphology of the natural L-Aβ1-42 peptide and its mirror image, the D-Aβ1-42 isomer. D-Aβ1-42 retains the properties of its L-analog: the oligomer and fibril formation of the D-Aβ1-42 are indistinguishable from the L-Aβ1-42.
EXPERIMENTAL METHODS
Materials
For storage, peptides were solubilized in ultrapure water at a concentration of 1 mg/mL, aliquoted, and stored at -80 °C. Aliquots were thawed once and used immediately. After thawing, an appropriate amount of 0.22 μm filtered NH4OH solution was added to the aliquot to reach a 1% solution. Molecular Biology Grade water from Fisher Scientific (Pittsburgh, PA) was used for sample preparations and Dulbecco’s phosphate buffered saline without Ca2+ and Mg2+ (Fisher Scientific) was used for AFM imaging in liquid. The phospholipid, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) was purchased from Avanti polar lipids (Alabaster, AL).
AFM Imaging
A multimode AFM equipped with a NanoscopeIIIa controller (Bruker, Santa Barbara, CA) was used. Oxide sharpened cantilevers with nominal spring constants (kn) of 0.02 N/m or 0.08 N/m were employed. For experiments performed in liquid, a fluid cell (Bruker) was utilized. Before each experiment, the fluid cell was washed with detergent (~5 min) and vigorously rinsed with constant stream of DI water. The fluid cell was then sonicated for 2 min in molecular grade water, dried with a Kim wipe and used immediately. Imaging in liquid was performed in phosphate-buffered saline (PBS) without Ca2+ and Mg2+. Images in liquid were acquired in tapping mode at scan frequencies of 0.5-3.0 Hz and drive amplitudes below 100 mV. The cantilever oscillation frequency was 5-10 kHz. Image analysis was performed using the Bruker Nanoscope software. Some of the AFM images were low-pass filtered to remove noise.
Since the widths of fibers are increased in the AFM images due to tip-sample convolution we used a simple geometrical deconvolution model to obtain a good estimation of the actual size of the fiber width, w, from the width, wO, observed in the AFM image,31-33
Here, R is the tip apex radius, and h is the height of the fiber. We assumed the tip radius to be 30 nm.31
Sample Preparation
Thawed from -80 °C, D- and L-Aβ1-42 aliquots were mixed with 1.01% NH4OH solution to bring the peptides to a 1% NH4OH solution. For time-lapse imaging of peptides, these were sonicated for 20 min in an ice bath. New cantilevers or used cantilevers cleaned for 5 minutes in a UV/ozone chamber (Bioforce Nanosciences, Ames, IA) were utilized for imaging. Freshly cleaved mica was imaged in PBS without Ca2+ and Mg2+ to check for contamination before adding the peptide. The peptide-containing solution was added through the channel of the fluid cell and mixed with a pipette several times. Additional PBS was added as needed to account for evaporation. Samples were kept inside the AFM over the entire span of the experiment. The AFM was maintained at room temperature and covered with parafilm in a semi-humid environment to minimize evaporation of the peptide solution.
DOPC bilayers were formed by drying 30 μL of DOPC (10 mg/mL) dissolved in chloroform in a rotovap and replacing vacuum with Ar. The dried lipid cake was hydrated with 300 μL (1 mg/mL) of an electrolyte solution containing 150 mM KCl and 1 mM MgCl2 buffered with 10 mM HEPES, pH 7.4 and vortexed gently. The liposomes formed through this procedure were sonicated for 5 minutes in an ice bath. Following thawing of the aliquoted peptide solutions, this was brought to a concentration of 1 mg/mL in 1% NH4OH and sonicated for ~30 s. To incorporate the peptides in the lipid bilayer DOPC liposomes and peptide were combined at 20:1 weight ratio and sonicated in an ice bath for 10 min. The liposome-peptide mixture was allowed to adhere to freshly cleaved mica for 30 s and washed 10 times with PBS without Ca2+ and Mg2+. Bilayers were imaged at room temperature.
For fibrils’ preparation and imaging the peptide solutions were thawed and brought to solution concentrations of 1% NH4OH (1 mg/mL). These were incubated in centrifuge tubes at 37 °C for up to 72 h without mixing. Incubated peptides (1 mg/mL) were allowed to adhere to freshly cleaved mica for 4 min, rinsed gently 3 times with Molecular Biology Grade Water and dried briefly under a stream of N2.
Molecular Dynamics Simulations
To simulate Aβ barrels, we used two U-shaped monomer conformations: one is Aβ1-42 as defined in the pentamer based on hydrogen/deuterium-exchange NMR data, side-chain packing constraints from pair-wise mutagenesis, solid-state NMR and EM (PDB code: 2BEG);34 the other is Aβ1-40 based on the solid-state NMR model of small protofibrils.35 However, both conformers miss the N-terminal coordinates due to conformational disorder. We used the N-terminal coordinates obtained from the solution NMR structure of Aβ1-16; however, removing the Zn2+ (PDB code: 1ZE7).36 This structure was used to fill in the missing N-terminal portion of the peptides. For each combination of the N-terminal structure with the U-shaped motifs, two Aβ1-42 conformers were generated. Conformer 1 has a turn at Ser26-Ile31, and conformer 2 has a turn at Asp23-Gly29.30 In the latter conformer, two C-terminal residues, Ile41 and Ala42 were added to create Aβ1-42.
The coordinates of all D-amino acids Aβ1-42 are mirror-imaged coordinates of all L-amino acids Aβ1-42 and can be obtained by reflecting the coordinates with respect to the reference plane. To simulate D-amino acids, a protein force field for asymmetric isomers is required. The standard CHARMM force field has been designed for L-amino acids. However, it can be directly used for D-amino acids, since a D-amino acid is a mirror-image of an L-amino acid. Thus, we adapted the same standard parameters to D-amino acids as used for the L-amino acids. However, the parameters include the dihedral angle cross term map (CMAP), which for D-amino acids needs to be corrected, since the map was constructed for L-amino acids.37 Thus, in our simulation, we corrected CMAP for D-amino acids by reflecting the phi-psi CMAP matrix for L-amino acids.
To construct the β-barrel structure, both D- and L-Aβ1-42 (each with two conformers) were inclined ~37° relative to the pore axis21 and then rotated 18 times with respect to the pore axis creating Aβ barrels (supplemental Fig. S1). The construction follows known structures of β-barrel membrane proteins such as those in the outer membranes of bacteria, mitochondria, and chloroplasts, which have 8 to 22 β-strands with shear numbers ranging from 8 to 24, yielding a β-strand tilt angle range of 36° to 44° relative to the barrel axis.38,39 The Aβ barrels were then embedded in an anionic lipid bilayer containing 1,2-dioleoyl-sn-glycero-3-phosphoserine (DOPS) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE). A unit cell containing two layers of lipids was constructed. An anionic lipid bilayer, composed of DOPS/POPE (mole ratio 1:2) containing a total of 420 lipids, constitutes the unit cell with TIP3P waters added at both sides. For a given number of lipid molecules, the optimal value of lateral cell dimensions can be determined. The bilayer system containing an Aβ barrel, lipids, salts, and waters has almost 190,000 atoms. For the bilayer construction, we closely follow previous β-sheet channel simulations.17-22,30
The CHARMM program40 using the revised CHARMM27 (C27r) force field for lipids41 and the modified TIP3P water model42 were used to construct the set of starting points and to relax the systems to a production-ready stage. A series of minimizations were performed for the initial configurations to remove overlaps of the alkane chains in the lipids and to gradually relax the solvents around the Aβ barrel, which was held rigid. The initial configurations were gradually relaxed through dynamic cycles with electrostatic cutoffs (12 Å). In the subsequent pre-equilibrium stages, a series of dynamic cycles were performed with the harmonically restrained peptides in the channels, and then the harmonic restraints were gradually diminished until gone with the full Ewald electrostatics calculation. The entire pre-equilibration cycle took 5 ns to yield the starting point. A Nosé-Hoover thermostat/barostat was used to maintain constant temperature of 303 K. The simulations for the preequilibrations and production runs were performed on the NPAT (constant number of atoms, pressure, surface area, and temperature) ensemble. Production runs of 100 ns for the starting points with the NAMD code43 on the Biowulf cluster (http://biowulf.nih.gov) at the NIH was used for the starting point with the same CHARMM27 force field.40 Averages were taken after 20 ns discarding initial transients.
Comparison with Other Models
Recently, the Aβ1-42 channel was also modeled into 36-mer β-barrel channels.44 These consisted of a hexamer-of-hexamers; that is, six β-barrels each consisting of six monomers, yielding a complex of exactly 36 monomers. The transmembrane pores were proposed to form between the hexamers’ barrels. The β-barrels of six hexamers span the bilayer, merging to form a 36-stranded β-barrel. In this model, two 12-stranded parallel β-barrels formed by the N-terminal domain of Aβ1-42 are located symmetrically at both extramembranous bilayer leaflets, and 6 parallel β-strands wrap around each β-barrel. In the membrane core, 24 β-strands containing the central residues (17-21) line an inner antiparallel β-barrel forming a solvated pore and the C-terminal domain forms an outer 36-stranded antiparallel β-barrel interacting with lipids. This 36-mer barrel complex differs from our Aβ barrel in size (being considerably larger than ours), complexity (it is more complex), Aβ monomer conformation, barrel organization with respect to the bilayer (as described above), and its size uniqueness (the channel always consists exactly of hexamer-of-hexamers, i.e. 36 monomers, whereas our channels are dynamic, in consideration of bilayer fluidity, thus varied channel sizes as the loosely-associated subunits associate/dissociate). However, both models share similar residues that are engaged in a lipid-contacting outer barrel and the solvated pore. We emphasize that as always, modeling only provides models; eventually, any modeling requires direct, high resolution experimental data. In the case of amyloids, given the heterogeneous landscape, we can expect a range of polymorphic channels.45
RESULTS
D-Aβ1-42 Forms Stable Globular Units
Both D- and L-Aβ1-42 form oligomeric structures and these structures remain stable during the imaging by the AFM for up to a 23-25 h period. Within the first 45 min of AFM imaging after the addition of the peptide we saw mostly monomers approximately 1-2 nm in height and small oligomeric clusters (Fig. 1A). The amount of oligomeric clusters increased successively over time though no structural changes were apparent over the 23-25 h time period. Significantly, we found this pattern for both D- and L-Aβ1-42 peptides.
FIGURE 1.
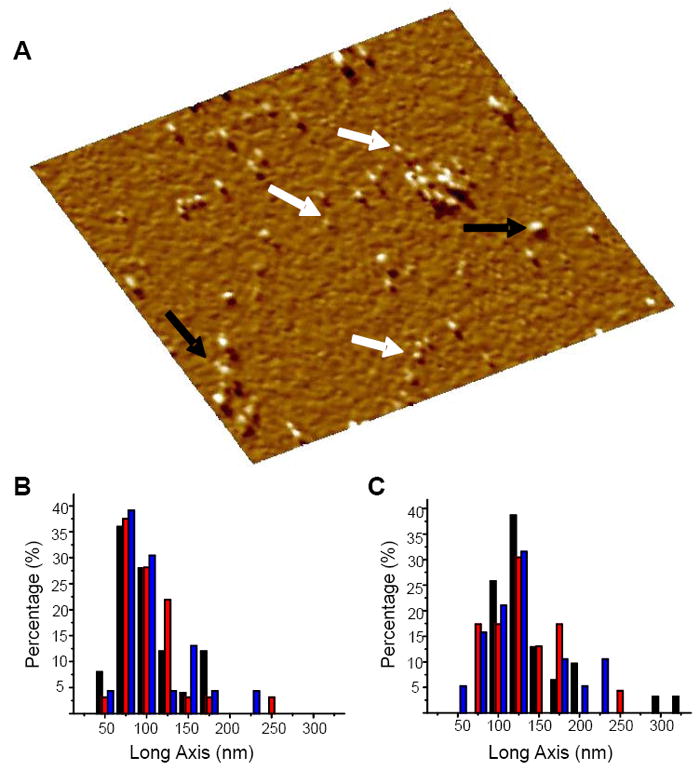
(A) AFM amplitude image (area 402 × 402 nm2) of freshly dissolved all D-amino acids Aβ1-42 in PBS without Ca2+ or Mg2+ at t = 0 of time lapsed imaging of the peptide. White arrows show peptides with heights of 1-2 nm, consistent with the size of monomers. Black arrows show higher order oligomers and clusters. The freshly cleaved mica surface was imaged in PBS solution alone to confirm zero contamination. The peptide was added to the PBS solution directly on the AFM stage to a final concentration of ≈ 35 μg/mL and the adsorbed peptide was imaged within 45 min. The length distribution of the long axis of the oligomers was measured for the (B) all D-amino acids Aβ1-42 and (C) all L-amino acids Aβ1-42 isomers. In B, the colors indicate: black for 1.5 h (n = 25, here n denotes the number of oligomers), red stripes for 5 h (n = 32), and blue for 23 h (n = 23). In C, the colors indicate: black for 1.5 h (n = 31), red stripes for 6 h (n = 23), and blue for 25 h (n = 19). There is little change in distribution for both isomers indicating stability of the oligomers.
We measured the long axis of the oligomers and globular species at each time point for both isomers. We found the distribution for the D-Aβ1-42 oligomers to remain similar throughout the observation period (Fig. 1B). Similarly, the length distribution of the L-Aβ1-42 oligomers measured along their long axis, remained stable from the initial time point, at 1.5 h, throughout the ~24 h observation period (Fig. 1C). It has been shown that many factors, including concentration, affect the fibril formation kinetics.46 In our time lapsed study different concentrations were used for the D-Aβ1-42 (≈35 μg/mL) and the L-Aβ1-42 (≈13 μg/mL) peptides, therefore, the total extent of oligomerization over the observation period cannot be strictly compared between isomers. We observed the stability of the D- and L-isomers to be similar over similar time periods under similar ambient conditions.
D-Aβ1-42 Forms Fibrils
Fibril formation of the Aβ1-42 peptide incubated in ultrapure water at 37 °C for 72 h was studied in vitro to evaluate the kinetics and hierarchy of fibril structure for L- and D-Aβ1-42.31,46,47 We found the fibril formations for both isomers to be indistinguishable after 72 h of incubation (Fig. 2, A and B). Both isomers form a complex network of fibers, protofibrils, and oligomers. In both images globular complexes can be identified as well as fiber-like complexes exhibiting varied widths and lengths.
FIGURE 2.
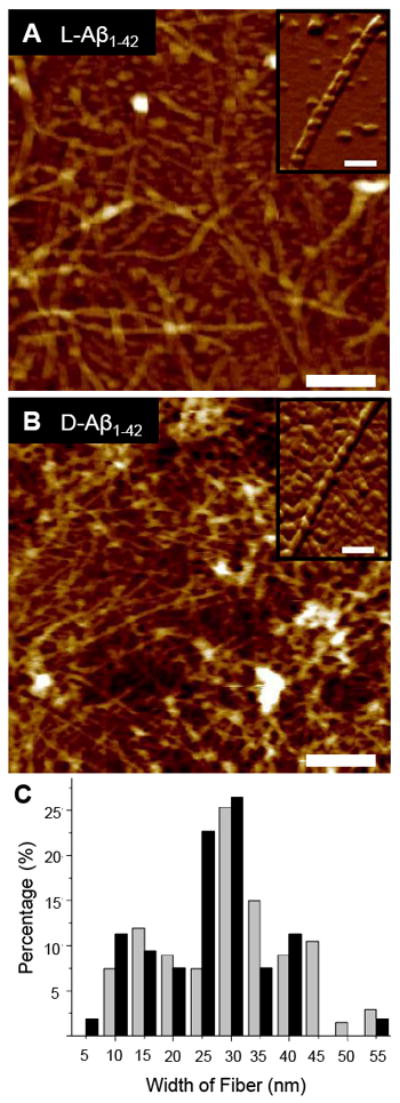
The (A) L- and (B) D-Aβ1-42 isomers induced towards fibril formation in the absence of a lipid membrane by incubation in 1% NH4OH at 37 °C for 72 h, dried overnight on fresh mica, and imaged with AFM in air. Both isomers form similar complex networks of globular units, oligomers, protofibrils, and fibrils. Insets show high resolution images of individual fibers (scale bars = 250 nm, height color scales = 25 nm, inset scale bars = 100 nm). (C) The distribution of randomly measured widths (n = 67, here n denotes the number of samples) of fibers of the D-Aβ1-42 fibers (gray bars) after incubation at 37 °C for 72 h shows a distribution with fiber widths most frequently between 25 nm and 30 nm. The distribution of the L-Aβ1-42 fibril widths (black bars), after similar incubation, also shows a distribution (n = 53) with fiber widths most frequently between 25 nm and 30nm. The overall range of values is comparable for both isomers.
Aliquots of both isomers after 72 h of incubation were diluted several times in order to identify and measure the width of individual fibrils (Fig. 2, A and B, insets). Numerous mature fibers had lengths greater than the maximum scan area (5 × 5 μm2) achievable with the scanner used for these experiments, and therefore could not be measured accurately. Random measurements of fibril widths were taken and deconvoluted.31-33 We calculated the histograms of the distribution of fibril widths for both isomers (Fig. 2C). The D-isomer (gray bars) and the L-isomer (black bars) show fibers with widths most frequently between 25 nm and 30 nm. The D-isomer shows a slight bias towards thicker fiber formation. 43.2% of the D-isomer width measurements were between 25 nm and 35 nm. The L-isomer showed a comparable but slightly lower distribution of width measurements with 49.1% of the measurements between 20 nm and 30 nm. The D-isomer and the L-isomer both show local minimum frequencies of width measurements just below these respective maximum ranges. The D-isomer local minimum frequency is between 20 nm and 25 nm and the L-isomer local minimum frequency is between 15 nm and 20 nm.
D-Aβ1-42 Forms Channels in Bilayers Characteristic of L-Aβ1-42
The channel formation of D-Aβ1-42was investigated by imaging the pores formed in supported lipid bilayers of DOPC. The DOPC lipids were chosen for their low transition temperature and consistency with previously reported evidence of Aβ1-42 pore formation by AFM studies and MD simulations.14,16-22,48 The bilayer sample preparation was optimized using the L-isomer to repeat and confirm previously reported results.14,16 The sample preparation process was repeated exactly for the D-isomer and channels were consistently and repeatedly observed. We imaged pore-like structures in bilayer membrane by first finding the edge of the bilayer (with usual height of >5 mn with respect to the mica substrate plane) and sequentially minimizing the scan area to obtain a high resolution image of the bilayer surface. This approach ensures that the features observed were characteristic of the bilayer and inserted peptide and not due to contamination of the mica surface. Control DOPC bilayers were imaged in high resolution in the absence of the peptide and no pore-like topography was observed.
The D-Aβ1-42 peptide forms channels in DOPC bilayers with a varying number of subunits (Fig. 3, A-C), structurally indistinguishable from the L-Aβ1-42 channels (Fig. 3D). The pores are heterogeneous, presenting trimers, tetramers, pentamers and hexamers, with tetramers being the most prevalent observed structures (Fig. 3E). Pores were identified by the presence of individual segments forming a circular group in the amplitude image, coinciding with a small height increase with respect to the bilayer membrane surface in the AFM height image. Because of the larger size of the AFM tip compared to the typical inner diameters of ion channels (~1 nm), the AFM images only provide information relating to depths which are in close proximity of the lipid bilayer/solution interface, and cannot discern whether the individual annular structures actually traverse the entire thickness of the lipid bilayer. However, functional evidence provided by electrophysiological experiments with lipid bilayers reconstituted with Aβ1-42 peptides suggests the presence of conductive pores, able to allow the selective passage of ions.14,16,30
FIGURE 3.
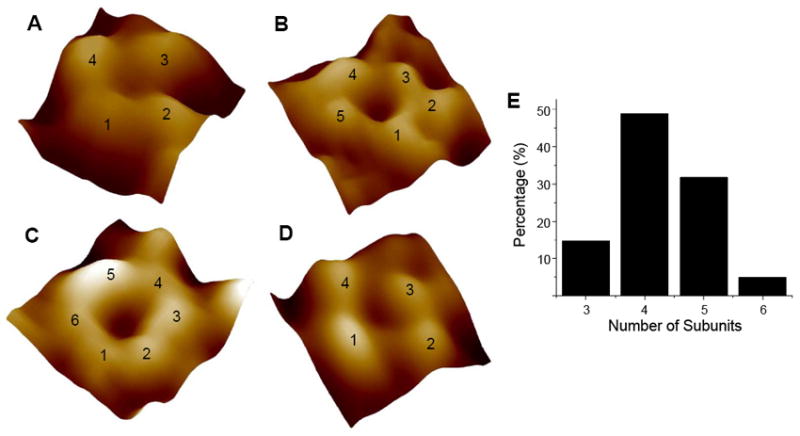
(A-C) AFM images of individual D-Aβ1-42 channels in high resolution. Channels are resolved in the error mode image, seen here. These channels are heterogeneous and were observed as trimeric (not shown), (A) tetrameric, (B) pentameric, and (C) hexameric subunits assemblies. The channels formed by D-Aβ1-42 are indistinguishable from the previously reported L-Aβ1-42 channels. (D) An example AFM image of the L-Aβ1-42 channels from the current study is shown. Channels are not observed in the absence Aβ1-42 peptide. Image sizes are 11.5 nm for (A), 18.1 nm for (B), 13.2 nm for (C), and 14.4 nm for (D). (E) The distribution of channels formed by a varying number of Aβ1-42 subunits. Channels with 3, 4, 5, or 6 subunits were observed. The most common structure is the channel with four subunits.
The inherent tendency of amyloid peptides to adopt the β-sheet conformation leads to formation of a complex β-barrel-like channel structure comprised of several β-sheet subunits in the membrane, where lipids promote the β-sheet formation.49-51 In our previous simulations, we modeled two truncated Aβ barrels, Aβ17-42 (p3) and Aβ9-42 (N9),21 using U-shaped peptides with the β-strand-turn-β-strand motif.34,35 To model the full sequence Aβ1-42 barrels, we again employ the U-shaped peptides as the membrane embedded portion and adopt the N-terminal structure from the Aβ1-16 (coordinates taken from PDB code: 1ZE7)36 as the extramembranous portion. Thus, two Aβ1-42 conformers define the turn: at Ser26-Ile31 (Conformer 1) and at Asp23-Gly29 (Conformer 2). Using these conformers, we constructed the D- and L-Aβ1-42 isomer barrels and performed 100 ns explicit MD simulations on the Aβ barrels embedded in an anionic lipid bilayer composed of DOPS/POPE (mole ratio 1:2). During the simulations, the Aβ barrels are gradually relaxed through the interaction with surrounding lipids (supplemental Fig. S2). While small fluctuations in the membrane embedded portions including the pore and C-terminal strands strongly preserve the U-shaped structure in the Aβ barrels, large fluctuations convert the N-terminal strands to disordered chains in the bulk water area (Fig. 4). In the Aβ barrels, the fluctuation of the individual peptide’s dynamics is inhomogeneous, although the dynamic motions for some adjacent peptides are correlated to each other (supplemental Fig. S3). We note that the MD simulations employed the anionic lipid bilayer composed of DOPS/POPE, and the AFM experiments used the zwitterionic lipid bilayer with DOPC; however, the results are expected to relate. In our modeling, the peptides are pre-inserted into the membrane core and assembled to form a channel. Under these circumstances, the hydrophobic interactions between lipid-facing residues in the channel and lipid tails should be an important factor to stabilize the channel conformation, and formation of the subunits in the channel structure may not completely rely on the interaction with the negatively charged PS lipid headgroups. Further, in our previous simulations we compared the subunits in the channel conformations for the p3 (Aβ17-42) and N9 (Aβ9-42) channels, when embedded in zwitterionic DOPC and in anionic POPC/POPG lipid bilayers, and observed no significant differences between the two.18,22 Therefore, our results with the anionic bilayer are expected to display the essential characteristics of Aβ1-42 channels in the zwitterionic bilayers.
FIGURE 4.
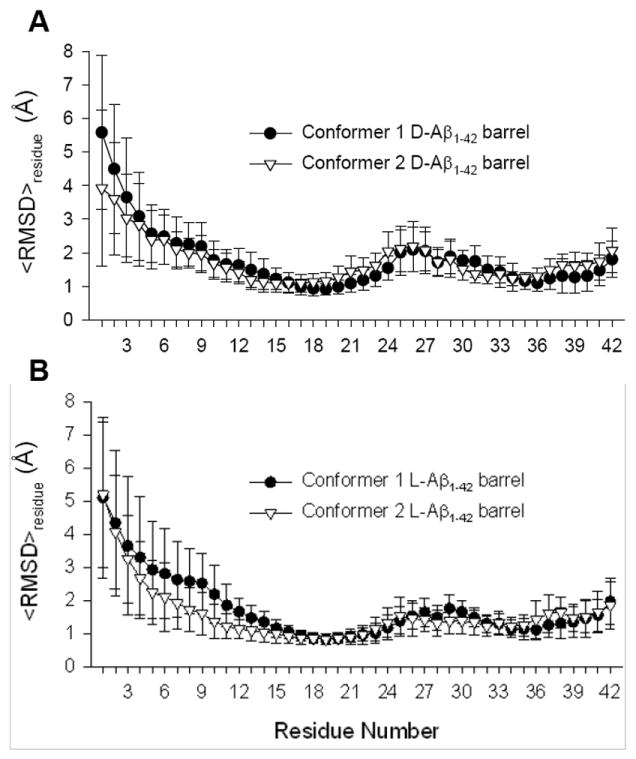
Residue averaged root-mean-squared deviation,<RMSD>residue, from the starting point for Cα atoms of the peptides for the (A) conformer 1 and 2 D-Aβ1-42 barrels, and the (B) conformer 1 and 2 L-Aβ1-42 barrels.
The molecular mass of our 18-mer Aβ barrel is ~81.2 kDa, which is in the intermediate range of the Aβ channels.18,22 The outer/pore diameters of our simulated barrels are ~7.8/~1.9 and ~8.2/~2.1 nm for the conformer 1 and 2 D-Aβ1-42 barrels, and ~8.3/~2.2 and ~8.1/~2.0 nm for the conformer 1 and 2 L-Aβ1-42 barrels, respectively. The sizes measured for the barrels largely depend on the number of peptides composing the Aβ barrels and partly on the location of the extramembranous N-terminal portions. The N-terminal strands containing several charged residues stretch toward the lipid headgroups. The strong electrostatic interactions can increase the channel size; alternatively they can interact with each other at the channel mouth blocking the entry into the pore. We speculate that the interactions of the N-terminal strands near the channel mouth may correlate with features of calcium selective amyloid channels and zinc blockage. The 18-mer simulations obtained three to five subunits in the anionic bilayer (Fig. 5). As we noted in our previous simulations, subunit formations result from the fluidic lipid bilayer dynamics, and even the same channel sizes may break into different number of subunits.17-22 To determine the subunits, we calculated the parameters, including the percentage of β-sheet content based on the intermolecular backbone hydrogen bonds (H-bonds), the β-strand order parameter, and the description of secondary structure, using our previous protocol (supplemental Fig. S4). Both D- and L-isomer Aβ barrels present a range of sizes and morphologies similar to the imaged AFM channels. No difference is found between D- and L-Aβ1-42 barrels, suggesting that both Aβ1-42 isomers form ion channels in the lipid membrane, and thus Aβ toxicity can take place in the absence of stereospecific interactions.
FIGURE 5.
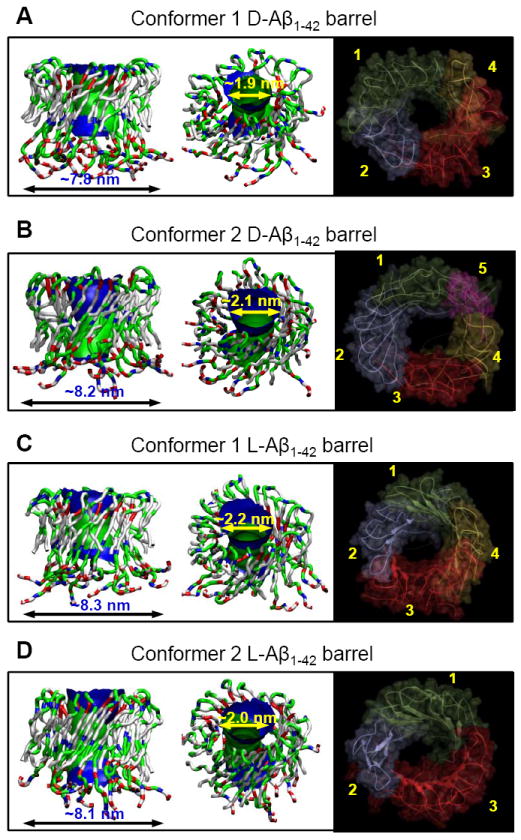
Averaged pore structures calculated by the HOLE program 55 embedded in the averaged barrel conformations during the simulations for the (A) conformer 1 and (B) 2 D-Aβ1-42 barrels, and the (C) conformer 1 and (D) 2 L-Aβ1-42 barrels. In the side (left) and angle (middle) views of the pore structure, whole barrel structures are shown with the ribbon representation. In the peptide, hydrophobic residues are shown in white, polar and Gly residues are shown in green, positively charged residues are shown in blue, and negatively charged residues are shown in red. For the pore structures in the surface representation, red denotes pore radius of r < 0.9 nm, green denotes pore radius in the range, 0.9 nm ≤ r ≤ 1.1 nm, and blue denotes pore radius of r > 1.3 nm. The simulated barrel structures (right) with highlighted subunits for the averaged barrels in the surface representation are shown in the view along the membrane normal.
DISCUSSION
We have studied the formation of channel-like structures using all D-amino acids Aβ1-42 and its chiral opposite, the all L-amino acids Aβ1-42. We imaged indistinguishable pore structures formed by both isomers. Since the imaging was carried out in a lipid bilayer composed of DOPC lipids and without any inserted membrane receptors, we infer that the channel formation by the peptides in the membrane does not depend on stereospecificity. Moreover, the formation of channels in the absence of the negatively charged phosphatidylserine (PS) phospholipids as used in previous studies,27 indicates that once inserted into the membrane, Aβ1-42 does not rely on a stereospecific interaction with PS to form the segmented channel structure.
Based on the structure-function relationship characteristics of peptides, we reasoned that the physicochemical behavior of the all D-amino acids enantiomer of Aβ1-42 will be identical to the naturally occurring all L-amino acids, if the structural and kinetic behavior of Aβ1-42 is independent of chirality. Time-lapse imaging of D-Aβ1-42 on mica showed that under appropriate physiological conditions, immediately after addition of freshly dissolved peptides, monomeric and oligomeric clusters are present and are stable over periods of time of ~23 h. Thus, these 3D structural units are characteristic of those present during the formation of pores in the presence of a lipid membrane. The physicochemical behavior of D-Aβ1-42 in the time lapsed study was similar to that of the previously reported behavior of L-Aβ1-42, which was also confirmed in this study.52 We observed the formation of Aβ1-42 fibers and larger oligomers after 72 h of incubation at 37 °C in 1% NH4OH. Both isomers exhibited a similar distribution of widths of fibers. The similar maximum frequency widths preceded by local minimum frequency widths is likely due to the amyloid fibers’ tendency to form hierarchical fibrillar structures.31,53 The D-enantiomer of Aβ1-42 showed comparable physicochemical behavior to the L-enantiomer, with monomeric and oligomeric forms stable over short periods of incubation at room temperature in physiological buffer and fiber formation over long time periods at 37 °C in ultrapure water. Therefore, the species available for channel formation in the preparation of the supported lipid membrane are monomers and oligomers, but not fibers.
Previous studies on the toxicity D- and L-Aβ1-42 enantiomers presented conflicting results. Ciccotosto et al reported Aβ1-42 toxicity through receptor interaction with the PS lipid flipped to the extracellular side of the lipid bilayer.27 Cribbs et al asserted that L- and D-Aβ1-42 show no difference in cellular toxicity.29 Both studies focused on the binding of Aβ1-42 to the cell membrane and the subsequent toxicity through fluorescence and viability assays. Neither study identified the mechanism of the toxicity, such as fibril adhesion, channel formation, oxidative stress, etc.27,29 In our study we focused on peptide insertion and channel formation with ion conductive pores in an effort to elucidate this path of toxicity and our results show that both L- and D-isomers form characteristic channels in supported lipid bilayer.
A number of cell membrane receptors which can bind to Aβ, whether in monomeric or fibrillar form, have been discussed and summarized by Verdier et al.54 Our current data as well as our previous experiments and MD simulations performed with model membranes reconstituted with Aβ peptides in a receptor free environment,14,16,20,30 show pore-like structural features and functional activity suggestive of an ion channel conductive mechanism that does not depend on the presence of cell receptors. However, due to the very nature of these model systems, they cannot rule out the action of cell receptor mechanisms in the complex cellular environment relevant to the disease.
CONCLUSSIONS
In summary, we have studied the structure of channels in the lipid membrane as well as fibril formation in solution by the naturally occurring L-Aβ1-42 peptide and its chiral opposite, the D-Aβ1-42 enantiomer. We show that the two isomers exhibit indistinguishable pore structures, consistent with the MD results, and similar fibril formation behavior and stability. Earlier we have shown that the D-Aβ1-42 analog conducts ions, also in a manner indistinguishable from the L-Aβ1-42 peptides.30 Therefore it is likely that insertion and toxic channel formation of Aβ1-42 does not occur through a stereospecific interaction but by a direct pathway – through an ion channel.
Supplementary Material
Acknowledgments
This research was supported by the National Institutes of Health (National Institute on Aging AG028709 to RL). This project has been funded in whole or in part with Federal funds from the National Cancer Institute, National Institutes of Health, under contract number HHSN261200800001E. This research was supported (in part) by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. All simulations had been performed using the high-performance computational facilities of the Biowulf PC/Linux cluster at the National Institutes of Health, Bethesda, MD (http://biowulf.nih.gov).
Footnotes
Supporting Information Available. Supplemental Figs. S1-S4. This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.Temussi PA, Masino L, Pastore A. The EMBO journal. 2003;22:355. doi: 10.1093/emboj/cdg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dobson CM. Nature. 2003;426:884. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Nature. 2003;426:900. doi: 10.1038/nature02264. [DOI] [PubMed] [Google Scholar]
- 4.Revesz T, Ghiso J, Lashley T, Plant G, Rostagno A, Frangione B, Holton JL. Journal of neuropathology and experimental neurology. 2003;62:885. doi: 10.1093/jnen/62.9.885. [DOI] [PubMed] [Google Scholar]
- 5.Neet KE, Thinakaran G. The Journal of biological chemistry. 2008;283:29613. doi: 10.1074/jbc.R800051200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Muller-Hill B. Nature. 1987;325:733. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Jr, Eckman C, Golde TE, Younkin SG. Science. 1994;264:1336. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 8.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Nature. 2002;416:535. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 9.Hardy J, Selkoe DJ. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 10.Arispe N, Rojas E, Pollard HB. Proc Natl Acad Sci U S A. 1993;90:567. doi: 10.1073/pnas.90.2.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pollard HB, Rojas E, Arispe N. Ann N Y Acad Sci. 1993;695:165. doi: 10.1111/j.1749-6632.1993.tb23046.x. [DOI] [PubMed] [Google Scholar]
- 12.Bhatia R, Lin H, Lal R. FASEB J. 2000;14:1233. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- 13.Rhee SK, Quist AP, Lal R. J Biol Chem. 1998;273:13379. doi: 10.1074/jbc.273.22.13379. [DOI] [PubMed] [Google Scholar]
- 14.Lin HAI, Bhatia R, Lal R. FASEB J. 2001;15:2433. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 15.Kourie JI, Shorthouse AA. American journal of physiology. Cell physiology. 2000;278:C1063. doi: 10.1152/ajpcell.2000.278.6.C1063. [DOI] [PubMed] [Google Scholar]
- 16.Quist A, Doudevski I, Lin H, Azimova R, Ng D, Frangione B, Kagan B, Ghiso J, Lal R. Proc Natl Acad Sci U S A. 2005;102:10427. doi: 10.1073/pnas.0502066102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jang H, Zheng J, Nussinov R. Biophys J. 2007;93:1938. doi: 10.1529/biophysj.107.110148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jang H, Arce FT, Capone R, Ramachandran S, Lal R, Nussinov R. Biophys J. 2009;97:3029. doi: 10.1016/j.bpj.2009.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jang H, Zheng J, Lal R, Nussinov R. Trends Biochem Sci. 2008;33:91. doi: 10.1016/j.tibs.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 20.Jang H, Arce FT, Ramachandran S, Capone R, Azimova R, Kagan BL, Nussinov R, Lal R. Proc Natl Acad Sci U S A. 2010:6538. doi: 10.1073/pnas.0914251107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. J Mol Biol. 2010;404:917. doi: 10.1016/j.jmb.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jang H, Arce FT, Ramachandran S, Capone R, Lal R, Nussinov R. J Phys Chem B. 2010;114:9445. doi: 10.1021/jp104073k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagan BL, Azimov R, Azimova R. J Membr Biol. 2004;202:1. doi: 10.1007/s00232-004-0709-4. [DOI] [PubMed] [Google Scholar]
- 24.Arispe N, Pollard HB, Rojas E. Proc Natl Acad Sci U S A. 1993;90:10573. doi: 10.1073/pnas.90.22.10573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demuro A, Smith M, Parker I. The Journal of cell biology. 2011;195:515. doi: 10.1083/jcb.201104133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Verdier Y, Penke B. Current protein & peptide science. 2004;5:19. doi: 10.2174/1389203043486937. [DOI] [PubMed] [Google Scholar]
- 27.Ciccotosto GD, Tew DJ, Drew SC, Smith DG, Johanssen T, Lal V, Lau T-L, Perez K, Curtain CC, Wade JD, Separovic F, Masters CL, Smith JP, Barnham KJ, Cappai R. Neurobiology of Aging. 2011;32:235. doi: 10.1016/j.neurobiolaging.2009.02.018. [DOI] [PubMed] [Google Scholar]
- 28.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2007;27:796. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cribbs DH, Pike CJ, Weinstein SL, Velazquez P, Cotman CW. J Biol Chem. 1997;272:7431. doi: 10.1074/jbc.272.11.7431. [DOI] [PubMed] [Google Scholar]
- 30.Capone R, Jang H, Kotler SA, Connelly L, Arce FT, Ramachandran S, Kagan BL, Nussinov R, Lal R. J Chem Theory Comput. 2011 doi: 10.1021/ct200885r. submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arimon M, Diez-Perez I, Kogan MJ, Durany N, Giralt E, Sanz F, Fernandez-Busquets X. The FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:1344. doi: 10.1096/fj.04-3137fje. [DOI] [PubMed] [Google Scholar]
- 32.Relini A, Torrassa S, Ferrando R, Rolandi R, Campioni S, Chiti F, Gliozzi A. Biophysical journal. 2010;98:1277. doi: 10.1016/j.bpj.2009.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jang H, Arce FT, Mustata M, Ramachandran S, Capone R, Nussinov R, Lal R. Biophys J. 2011;100:1775. doi: 10.1016/j.bpj.2011.01.072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lührs T, Ritter C, Adrian M, Riek-Loher D, Bohrmann B, Doeli H, Schubert D, Riek R. Proc Natl Acad Sci U S A. 2005;102:17342. doi: 10.1073/pnas.0506723102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Petkova AT, Yau WM, Tycko R. Biochemistry. 2006;45:498. doi: 10.1021/bi051952q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zirah S, Kozin SA, Mazur AK, Blond A, Cheminant M, Segalas-Milazzo I, Debey P, Rebuffat S. J Biol Chem. 2006;281:2151. doi: 10.1074/jbc.M504454200. [DOI] [PubMed] [Google Scholar]
- 37.Mackerell AD, Feig M, Brooks CL. J Comput Chem. 2004;25:1400. doi: 10.1002/jcc.20065. [DOI] [PubMed] [Google Scholar]
- 38.Schulz GE. Biochimica et biophysica acta. 2002;1565:308. doi: 10.1016/s0005-2736(02)00577-1. [DOI] [PubMed] [Google Scholar]
- 39.Marsh D, Pali T. Biophysical journal. 2001;80:305. doi: 10.1016/S0006-3495(01)76015-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comp Chem. 1983;4:187. [Google Scholar]
- 41.Klauda JB, Brooks BR, MacKerell AD, Jr, Venable RM, Pastor RW. J Phys Chem B. 2005;109:5300. doi: 10.1021/jp0468096. [DOI] [PubMed] [Google Scholar]
- 42.Durell SR, Brooks BR, Bennaim A. J Phys Chem. 1994;98:2198. [Google Scholar]
- 43.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, Chipot C, Skeel RD, Kale L, Schulten K. J Comp Chem. 2005;26:1781. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shafrir Y, Durell S, Arispe N, Guy HR. Proteins. 2010;78:3473. doi: 10.1002/prot.22853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Miller Y, Ma B, Nussinov R. Chem Rev. 2010;110:4820. doi: 10.1021/cr900377t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Antzutkin ON. Magnetic resonance in chemistry : MRC. 2004;42:231. doi: 10.1002/mrc.1341. [DOI] [PubMed] [Google Scholar]
- 47.Harper JD, Lieber CM, Lansbury PT., Jr Chem Biol. 1997;4:951. doi: 10.1016/s1074-5521(97)90303-3. [DOI] [PubMed] [Google Scholar]
- 48.Arce FT, Jang H, Ramachandran S, Landon PB, Nussinov R, Lal R. Soft Matter. 2011;7:5267. doi: 10.1039/C1SM05162H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Thundimadathil J, Roeske RW, Guo L. Biopolymers. 2006;84:317. doi: 10.1002/bip.20470. [DOI] [PubMed] [Google Scholar]
- 50.Kagan BL, Thundimadathil J. Advances in experimental medicine and biology. 2010;677:150. doi: 10.1007/978-1-4419-6327-7_13. [DOI] [PubMed] [Google Scholar]
- 51.Zhao LN, Chiu S-W, Benoit J, Chew LY, Mu Y. J Phys Chem B. 2011;115:12247. doi: 10.1021/jp2065985. [DOI] [PubMed] [Google Scholar]
- 52.Kowalewski T, Holtzman DM. Proc Natl Acad Sci U S A. 1999;96:3688. doi: 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ionescu-Zanetti C, Khurana R, Gillespie JR, Petrick JS, Trabachino LC, Minert LJ, Carter SA, Fink AL. Proc Natl Acad Sci U S A. 1999;96:13175. doi: 10.1073/pnas.96.23.13175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verdier Y, Zarandi M, Penke B. Journal of Peptide Science. 2004;10:229. doi: 10.1002/psc.573. [DOI] [PubMed] [Google Scholar]
- 55.Smart OS, Goodfellow JM, Wallace BA. Biophys J. 1993;65:2455. doi: 10.1016/S0006-3495(93)81293-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.