

Abstract
β-Cardiac myosin is a mechanoenzyme that converts the energy from ATP hydrolysis into a mechanical force that drives contractility in muscle. Thirty percent of the point mutations that result in hypertrophic cardiomyopathy are localized to MYH7, the gene encoding human β-cardiac myosin heavy chain (β-MyHC). Force generation by myosins requires a tight and highly conserved allosteric coupling between its different protein domains. Hence, the effects of single point mutations on the force generation and kinetics of β-cardiac myosin molecules cannot be predicted directly from their location within the protein structure. Great insight would be gained from understanding the link between the functional defect in the myosin protein and the clinical phenotypes of patients expressing them. Over the last decade, several single molecule techniques have been developed to understand in detail the chemomechanical cycle of different myosins. In this review, we highlight the single molecule techniques that can be used to assess the effect of point mutations on β-cardiac myosin function. Recent bioengineering advances have enabled the micromanipulation of single cardiomyocyte cells to characterize their force–length dynamics. Here, we briefly review single cell micromanipulation as an approach to determine the effect of β-MyHC mutations on cardiomyocyte function. Finally, we examine the technical challenges specific to studying β-cardiac myosin function both using single molecule and single cell approaches.
Keywords: Hypertrophic Cardiomyopathy, Single Molecule Analysis, Cardiac Myosin
Introduction
Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiovascular disease. HCM affects one in 500 people worldwide with outcomes ranging from reduced quality of life to early sudden cardiac death [1, 2]. To date, hundreds of different mutations in many of the sarcomeric genes have been identified [3], the majority of which affect proteins of the cardiomyocyte sarcomere. The clinical phenotype of HCM is variable, with the predominant morphologic change being abnormal hypertrophy of the left ventricle. This hypertrophy originates from improper function of sarcomeric contractile proteins [4].
Despite our understanding of the genetic underpinnings of cardiomyopathy, no specific pharmacological therapies currently exist to alleviate the source of this sarcomeric dysfunction, namely correcting the function of the sarcomeric proteins themselves. With 50 years of care and almost 20 years since the first gene mutation was identified, therapy for HCM remains palliative. Briefly, beta blockers, calcium channel blockers, and disopyramide are the mainstay of pharmacological management. Their aim is to reduce inotropy, encourage filling through bradycardia, and, perhaps, reduce the arrhythmogenicity of the heart. However, effects are modest and often limited by side effects. At the cellular level, beta blockers may in fact, impede relaxation via inhibition of PKA-mediated SER-CA2a activity. The advent of the implantable defibrillator was a significant opportunity to save lives in HCM. However, with more than 600,000 patients in the USA, most of whom are not at significant risk of sudden death, choosing patients in whom to implant is challenging and controversial. In addition, long-term effects of lead implantation in young patients are unknown.
From a therapeutic standpoint, targeting the potential signaling pathways upstream of muscle sarcomere function could suppress the myocardial hypertrophy but will not deal with the underlying abnormal function of the contractile apparatus. One way to address this problem would be to directly target the contractile apparatus with small molecule therapies. An example of this approach is a potential heart failure drug that binds specifically to β-cardiac myosin and activates its force producing capability, being developed by Cytokinetics, Inc.
The mutant proteins in HCM, although part of the sarcomere, perform very different functions, including enzymatic and force-generating roles (β-MyHC and associated light chains) and structural and regulatory roles (tropomyosin, the troponins, and myosin-binding protein C). Hence, a detailed understanding of the effects of HCM mutations on the function of proteins at the level of single molecules and as part of the cardiac muscle sarcomere is essential before appropriate therapies can be designed. This review will focus on mutations in MYH7, which encodes β-MyHC.
Although, more than 30% of all cases of HCM are caused by one of more than 200 described mutations in MYH7, very little is known about the effects of these mutations at either the single molecule or muscle sarcomere level. Human β-cardiac myosin is a mechanoenzyme that converts the energy from ATP hydrolysis into a mechanical force, which drives contractility in muscle. This process requires a tight and highly conserved allosteric coupling between different protein domains. As such, the effect of a single point mutation on the force generation and kinetics of a single β-cardiac myosin molecule cannot be predicted directly from its location within the protein. In recent years, significant advances have been made in understanding force generation by myosins at the level of single molecules and single cells. In this review, we discuss these single molecule and single cell techniques and the insights they have provided into β-MyHC mutation induced cardiomyopathies. From a clinical standpoint, mutations that reduce contractile force need to be treated differently than mutations that increase contractile force. For example, small molecule activators of β-cardiac myosin function could be used for the former and small molecule inhibitors for the latter. A combination of single molecule and single cell techniques can provide the vital information needed to assess the best therapeutic approach.
This review is structured as follows: We begin with a discussion on single molecule techniques. Specifically, we discuss in vitro motility assays, total internal reflection (TIRF) microscopy, gold nanoparticle tracking, and optical tweezers (optical traps). Each section begins with a brief description of the experimental setup, followed by a summary of studies carried out using the single molecule technique in relation to β-MyHC mutation-induced cardiomyopathy and ending with an overview of the advantages and challenges of using the particular single molecule approach. The second section discusses single cell approaches. We focus on the use of carbon nanofibers to attach the ends of cardiomyocytes for cell stretch. Following a brief description of the setup, we highlight studies using this approach. A quick comparison of the strengths and weaknesses of these different techniques can be found in Table 1. We end with a discussion on the technical challenges for the widespread use of these assays to study β-cardiac myosin function in cardiomyopathies and suggest a path forward.
Table 1.
Actomyosin techniques discussed in this review—a quick comparison
| Technique | Strengths | Weaknesses |
|---|---|---|
| In vitro motility assay | Gives quantitative measure of unloaded velocities | Does not quantify the effect of load on myosin function |
| Simple experimental set up | ||
| Small quantities of purified protein (<1 µg) | ||
| Good first screen to identify effect of mutation | ||
| Steady state ATPase assay | Characterizes the effect of mutation on the ATP cycle time and apparent affinities for actin and for ATP |
Larger quantities of protein needed (~1 mg) |
| Stopped flow techniques are needed to measure kinetic substeps in the ATPase cycle |
||
| Well established bulk assay | ||
| Optical tweezers | Directly quantify the effect of mechanical load on myosin function |
Sophisticated instrumentation for dual-beam optical trap set up |
| Quantitative measurement of effect of mutation on force generation of myosin |
||
| Single cell dynamic force–length measurements |
Measure the effect of the mutation on an intact beating cardiomyocyte |
Sophisticated instrumentation for force and length measurements |
| Cell attachment and measurements are trickly, requiring technical expertise for reproducible measurements |
||
| Characterize the effect of single mutations in myosin at the sarcomeric level |
Single Molecule Techniques
In Vitro Motility Assay
In vitro motility assays examine the movement of myosin relative to actin filaments. There have been several in vitro motility assays designed to examine actomyosin interaction [5–7]. In these assays, either F-actin or myosin is anchored to a stationary surface, and the movement of its counterpart is tracked temporally to obtain a velocity of movement (v, also called the sliding velocity). The velocity of movement can be expressed as v=d/ts, where d is the relative displacement between myosin and F-actin for each ATP hydrolyzed, and ts is the average strongly bound state time of the myosin on the actin. For processive motors like myosin V, where ts is the rate limiting step of the ATPase cycle, ts can be determined from an actin-activated ATPase assay [8]. Thus, combining ATPase assays and in vitro motility assays for some myosins gives us a good estimate of d, the displacement at the actomyosin interface, also termed the step size that the myosin molecule takes along the actin filament. This is not true for skeletal or cardiac myosin, which has an ATPase cycle that is not limited by the strongly bound state time, and ts must be determined by other methods [9].
The first in vitro motility assay to examine the movement of a myosin along actin filaments involved the use of rabbit skeletal myosin coated beads moving along polarized actin bundles in the algae Nitella axillaris [6]. In this model organism, the highly ordered actin bundles that are used for cytoplasmic streaming provided a polarized actin array along which myosin movement could be studied. This assay was subsequently modified to use purified reconstituted actin filaments that were aligned using flow [7] and provided the first demonstration that purified actin, myosin, and ATP alone are sufficient to bring about movement with rates that are consistent with those observed in muscle contraction. The in vitro motility assay was further modified by Kron and Spudich [5], with myosin anchored to the surface by adsorption and fluorescently labeled actin filaments in solution that land and move along the surface myosin, as depicted in Fig. 1. The speed of actin filament movement was used to estimate the stroke size of muscle myosin to be ~10 nm [9, 10], although higher values were reported in similar studies [11, 12]. Single-molecule laser trap measurements [13] clarified that the stroke size is indeed ~10 nm. The Kron in vitro motility assay, with myosin anchored to the surface and the movement of fluorescently labeled actin filaments tracked, has become the standard assay and has been used to characterize the function of multiple myosins, including myosins I [14], V [14], VI [15], IX [16], and X [17].
Fig. 1.
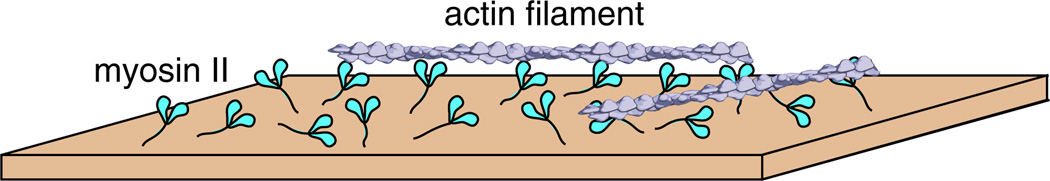
In vitro motility assay: cartoon schematic of the in vitro motility assay showing myosin II protein dimers anchored to a glass coverslip. Actin filaments are in a buffer solution with ATP (not shown). They land on the myosin coated surface and are moved by the myosin II heads. Actin filaments in this assay are labeled with fluorescent phalloidin so that their movement can be imaged on an epifluorescence microscope with a sensitive camera. The velocity of movement (v), also referred to as the “sliding velocity,” can be directly measured using this assay. The sliding velocity can be quantified as v=d/ts, where d is the stroke size of the myosin lever arm and ts is the length of time that the myosin head stays bound to the actin filament, also referred to as the dwell time
In vitro motility assays have yielded important insights into the function of normal and mutant cardiac myosins. Mammalian hearts express two isoforms of myosin: α and β and their relative proportions vary according to species, development, hormonal status, and disease. The healthy human heart expresses about 10% α-myosin and 90% β-myosin. Although they are 93% identical at the amino acid level, they are functionally distinct. Using the Nitella-based motility assay described above, Yamashita et al. [18] found that the sliding velocity for α-cardiac myosin was ~2-fold higher than β-cardiac myosin. Using the motility assay as a means to characterize the type of myosin (α vs β) involved, Yamashita el al. [19] found a redistribution of cardiac myosin isoforms in a hamster model of cardiomyopathy. These studies using in vitro motility assays demonstrated that the relative levels of α- and β-cardiac myosin isoform expression could directly regulate the contractility of the muscle, as inferred from the studies of Barany et al. [20], who showed that hearts with varying amounts of α and β myosin had different contractile velocities.
Cuda et al. [21] compared the in vitro motility-derived sliding velocities of cardiac myosins derived from HCM mutations to assess their impact on myosin function. β-Cardiac myosin in these studies was derived in limited amounts from patient biopsies. This was supplemented by skeletal myosin from soleus muscle, which is enriched in “slow” muscle fibers (containing the β isoform). To select for the β isoform in the in vitro motility assay, the surface was first coated with a β isoform-specific antibody rather than non-specifically sticking the myosin to the surface, as used in many previous motility assays. By comparing the in vitro motility sliding velocities from muscle and cardiac tissues, they were able to demonstrate the selection for the β isoform. Two β-MyHC mutations were studied, namely R403Q, which has an early onset and high frequency of deaths, and L908V, which has late onset and low frequency of deaths [22]. Despite the differences in the disease presentation, both of them showed a decreased velocity (3- and 5-fold, respectively), indicating decreased myosin function. This finding was extended to five more β-MyHC mutations using this assay, all of which showed decreased sliding velocities, to varying degrees [23]. The seven mutations in these studies lie in different structural domains, suggesting different modes of dysregulation of the myosin. The results of these two studies have since been contradicted by Palmiter et al. [24], who found a 30% increase in sliding velocity for β-MyHC isolated from cardiac biopsies of patients with R403Q and L908V mutations. Although the reason for discrepancies between the two sets of studies is unclear, Palmiter et al. [24] suggest that the higher sliding velocities found in their studies are likely a result of the improved quality of their myosin protein preparation. Cardiac biopsies from patients yield small amounts of β-MyHC protein, making it difficult to carry out additional purification steps to remove dead (enzymatically inactive) protein and also to ensure timely use of the protein sample, as both these steps further reduce protein available for the assay. Dead β-MyHC protein binds strongly to actin filaments without undergoing its ATPase cycle, and slows down movement. Therefore, any contamination with dead β-MyHC will reduce sliding velocities in in vitro motility assays, which Palmiter et al. [24] suggest could be falsely interpreted as compromised function arising from mutations. Overall, this is a limitation of using β-MyHC protein derived from cardiac biopsies. It must be noted that both of these studies involved tissue from patients who were heterozygous for the different mutations, leading to a mix of wild type and mutant protein used in the assays. An increase in sliding velocity with R403Q was also seen in an in vitro motility assay with α-cardiac myosin from heterozygous mutant mice [25]. Keller et al. [26] performed in vitro motility assays with β-cardiac myosin isolated from a transplant patient with a homozygous R403W mutation and found a 20% increase in sliding velocity. The increase in sliding velocity was much smaller than the ~2-fold increase in actin-activated ATPase activity of this mutant β-cardiac myosin, leading them to suggest that the inefficient ATP utilization and reduced mechanical efficiency resulting from this mutation are the source of the HCM phenotype.
Understanding the effect of cardiomyopathy mutations on β-cardiac myosin function using in vitro motility assays is limited by access to homogeneous and fully active protein samples with the appropriate mutations. As discussed above, β-cardiac myosin isolated from patient hearts is limited to the small amount of protein that can be derived from cardiac biopsies or from rare homozygous transplant patients. Recombinant techniques have thus far been unsuccessful, with the majority of recombinant protein (>95%) expressed using a baculovirus system rendered non-functional [27].
Researchers have therefore turned toward animal models using mouse, rat, rabbits, pigs, and hamsters [19, 25, 28]. In addition to HCM, there are a different set of β-MyHC mutations that result in dilated cardiomyopathy (DCM) [29]. The organ level phenotype is the reduced thickness of ventricular walls, finally precipitating in heart failure. DCM mutations S532P and F764L introduced into the α-MyHC of mice reproduce the organ level defects observed in human cardiomyopathy [30]. Those myosins have reduced sliding velocity in in vitro motility assays, suggesting reduced myosin function. The R403Q mutation in mouse α-MyHC on the other hand shows increased sliding velocity, whereas R453C does not alter the sliding velocity [25]. One drawback of these animal models is that the relative levels of α and β isoforms (also referred to as V1 and V3, respectively) of cardiac myosin is dependent on the specific animal used [28]. Specifically, adult mouse and rat hearts predominantly express α-MyHC, whereas rabbit, pig, and human hearts express predominantly (>90%) β-MyHC. A systematic comparison of α- and β-cardiac myosins from different species reveals that the ~2-fold ratio of sliding velocities of α- to β-cardiac myosins observed in humans is maintained in all species [28]. Although the amino acid sequences of α and β-MyHC isoforms are ~93% identical, the differences between α- and β-cardiac myosins are clustered around their actin-binding and ATPase domains, consistent with the different sliding velocities of these two isoforms [31]. It is therefore not surprising that the same mutation, namely R403Q introduced into mouse α-cardiac myosin and human β-cardiac myosin, have different phenotypes [32].
Although adult rodent hearts express predominantly α-MyHC, during embryogenesis and fetal development, they express β-MyHC [33]. Researchers have taken advantage of this finding to suppress the switch from β-cardiac myosin to α-cardiac myosin during development by manipulating thyroid function, such that adult rodent models express increasing levels of β-MyHC [34, 35]. However, the sliding velocity for mouse and rat α- and β-cardiac myosin are higher than their counterparts in rabbit, pig, and human, suggesting that both the isoform distribution and function is species specific [28]. These differences in sliding velocity for the same isoform, when comparing larger animals (rabbit, pig and human) to smaller rodents (mouse and rat), suggests that despite the high degree of homology (>90%) for the cardiac myosins across species, the differences are sufficient to alter sliding velocity and kinetics. Thus, it would be no surprise if the cardiomyopathy mutations in β-MyHC were to have different phenotypes in humans compared to animal models.
The in vitro motility assay discussed in this section requires very small quantities of protein (<1 µg) and can be set up and performed with relatively inexpensive equipment (epifluorescent microscope with sensitive camera). This assay, along with a steady-state ATPase assay, is therefore the best first screen to understand the effect of point mutations on myosin function. With the low concentrations of motor on the surface in the in vitro motility assay, there is very little load against which the myosin needs to stroke. Hence, the in vitro motility assay is best suited to study motor function in the absence of load. Nonetheless, modified in vitro motility assays have been used to estimate the force generation by cardiac myosins [28]. Accurate determination of force generated by myosins requires the use of optical tweezers, discussed further below. We first discuss briefly the steady-state ATPase assay and the complementary insight it provides into β-cardiac myosin function.
ATPase Assay
Steady-state ATPase assays and stop flow kinetic measurements use bulk solution measurements to dissect the kinetics of the different steps of the actomyosin kinetic cycle [36]. In vitro motility assays, in turn, examine relative movement of the actomyosin interface to infer myosin function. They provide complementary insights into myosin function. The extent of information that can be gained from these assays depends on the specific myosin being studied. Here, we contrast muscle myosin II with myosin V, where the latter has been extensively studied using ATPase and in vitro motility assays [37–41]. Studies on myosin V using these assays have yielded detailed insight into the chemomechanical cycle of myosins in general and have made myosin V the best characterized molecular motor [42].
We provide here a brief overview of ATPase assays. For a detailed description, we refer the reader to a recent review by De la Cruz and Ostap [36]. The most commonly performed ATPase assay involves a steady-state measurement of the maximum ATPase rate of a myosin (Vmax) and its apparent affinity for actin (KM) [26, 38, 43]. Vmax is a measure of the length of time the myosin takes to complete a single ATPase cycle in the presence of saturating actin concentrations, while KM measures the actin concentration at which one-half Vmax is achieved. In the steady-state ATPase assay, a known concentration of myosin is mixed with increasing concentration of actin filaments, in the presence of saturating levels of ATP.
The actomyosin hydrolyzes the ATP and releases free ADP and Pi into solution. There are several ways to monitor the kinetics of ATP hydrolysis by the actomyosin. One straight forward and very sensitive assay is to use γ-32P labeled ATP and follow the appearance of γ-phosphate with time [44]. Another assay couples ADP produced in the reaction to the conversion of NADH to NAD+, which is monitored in a spectrophotometer. The latter is widely used because it allows one to follow the reaction of single reaction mixtures kinetically [36]. For each molecule of ADP released, a single molecule of NADH is converted to NAD+. NADH absorbs 340 nm light but NAD+ does not. The time rate of decrease in absorbance of NADH yields the rate of ATP hydrolysis, at the specific actin concentration used. With increasing actin concentration, the ATPase rate saturates and yields Vmax (ATP hydrolyzed per second per myosin head). The concentration of actin that yields one half of Vmax is a measure of the apparent affinity (KM) of the myosin for the actin. Beyond this steady-state assay, stopped flow studies can be used to obtain rate constants for the different steps in the myosin kinetic cycle, such as rates of ATP association, ATP hydrolysis, phosphate (Pi) release, ADP release, and rates of conformational changes in the myosin head that alter its affinity for actin [36, 38, 43].
The specific rate constants that can be dissected depend on the time resolution of the instrumentation used in the stopped flow assays, relative to the characteristic time for each of the kinetic steps. For instance, in cardiac myosin, the time that a myosin head spends bound to an actin filament in each cycle is limited by the rate of ADP release [43]. This has been measured to be >150 s−1, a value limited by the time resolution of the instrument used. For myosin V, the corresponding value is ~12 s−1 [38].
Processivity and its Implications for Function
Myosin V is a native homo-dimer [45], similar to myosin II. However, unlike myosin II, myosin V dimers do not integrate to form thick filaments that enable sarcomeric contraction [46]. Detailed ATPase assays performed on the motor domain of myosin V showed that it spends ~70% of its ATPase cycle strongly bound to actin [38]. This contrasts with ~4% for cardiac myosin [43]. The fraction of time spent in the strongly bound state is also referred to as the duty ratio (dr). Thus, as dr increases, the length of time that at least one of the myosin heads in the dimer is bound to an actin filament increases. With dr >0.5, the two heads in a myosin dimer are likely to interact more than once with a single actin filament before both heads have dissociated from it. This ability of a single dimer to undergo multiple interactions with a single filament before detaching is referred to as processivity. A single myosin V molecule, with dr ~0.7, is processive. Each interaction is referred to as a step, as it results in a displacement at the actomyosin interface. Cardiac myosin II on the other hand, with a dr ~0.04, essentially never has both heads of the dimer bound simultaneously. A single head completes only a single step along an actin filament before both heads of the dimer are detached. Thus, myosin II is not processive.
Processivity has important implications for the physiological function of these motors. In the case of myosin V, it enables a few myosin V dimers to transport cargo over long distances [42], but the velocity is limited because of the long strongly bound state times. In contrast, the low dr values for skeletal and cardiac myosin II enables multiple myosins assembled into a thick filament to interact with actin filaments in a contracting sarcomere without significant interference between the heads [47], and much faster velocities can be achieved. Processivity of myosin V enables one to observe a single molecule of myosin V, as it steps along a single actin filament. Studies drawing upon this feature of myosin V have enhanced our understanding of all myosins and are discussed briefly below.
In vitro motility assays for myosins II and V measure the sliding velocity at the actomyosin interface. In the most commonly used in vitro motility assay, described earlier, myosins are bound to the surface of the coverslip, and actin filaments land from solution and are moved by the cooperative interaction of multiple myosins. A myosin on the surface binds and hydrolyzes ATP to ADP+Pi (see Fig. 2). This myosin then binds an actin filament and its lever arm swings and displaces the actin filament. The lever arm swing is accompanied by Pi release, followed by ADP release. An ATP molecule from solution then binds to the myosin head, which then releases from the actin filament. Following the lever arm swing, the myosin head needs to dissociate from the actin filament before another head can displace it further. The sliding velocity (vo) therefore depends on the distance the lever arm swings (also referred to as the stroke size d) and the time that the myosin spends bound strongly to actin (includes time to release Pi, ADP, rebind ATP, and transition to weak binding to actin, denoted as ts). The value of ts for myosin V (~83 ms [38]) is much much larger than that for myosin II (~10 ms for cardiac myosin [43]). Consequently, vo for cardiac myosin II (~1 µm/s) [43] is larger than that for myosin V (~300 nm/ s) [48]. The significantly smaller ts for cardiac myosin II implies that an actin filament at a surface that has very few cardiac myosin II dimers bound is likely to have nearly all of them bound weakly. When all myosin heads are weakly bound, the actin filament diffuses into solution before moving a significant distance. Increasing the number of myosin molecules bound to the surface (surface myosin concentration) or increasing the length of an actin filament both enhance the distance moved by the actin filament. This aspect of myosin II has been used in conjunction with sliding velocity to estimate the ts for skeletal myosin II9. The small ts also means that, in a contracting muscle sarcomere sliding at high velocity, at any given instant, a relatively small number of myosin heads are strongly bound to an actin filament. If the myosin heads were all simultaneously bound strongly, the muscle would be unable to function due to interference between stroking heads. Furthermore, for the most part, a small number of strongly bound myosin heads stroke and unbind from the actin filament before the next group of heads bind and stroke, minimizing interference among the heads (although mechanical strain is also likely involved that will cause heads to release faster when pulled on in the sliding direction by active heads). Thus, a simple treatment of the situation in the sarcomere is that each successive myosin II stroke is additive, and muscle speed is limited only by ts (~10 ms) and not by the Vmax ATPase rate for the myosin (~250 ms for cardiac myosin [43]) [49]. This synergy between heads is what enables the large speeds (~5 µm/s per sarcomere for skeletal muscle) in muscle contraction.
Fig. 2.

ATPase cycle of myosin in the presence of actin: cartoon schematic showing the different steps in the actin-activated ATPase cycle of myosin (starting from the top left and moving clockwise). A myosin molecule in solution (yellow) bound to ATP, ATP hydrolysis to ADP and phosphate (Pi). Catalytic head of myosin undergoes a conformational change which transitions it to a strongly bound actin state (red). Pi release is accompanied by a stroke in the lever arm. ADP from the head is released. ATP from solution rebinds to the head. The myosin head undergoes a conformational change which transitions it to a weakly bound actin state (yellow) and the ATPase cycle is repeated
Both ATPase and in vitro motility assays are easy to set up and implement and therefore will provide quick insights into changes in myosin function brought about by cardiomyopathy mutations. However, both of them study the function of myosin in the absence of load. Preload is an important physiological parameter in muscle function, and its effect on myosin can be understood using a dual beam optical trap. Before we proceed to discuss optical trapping, we provide an overview of two single molecule techniques which have been used to study the processive myosin V and have significantly enhanced our understanding of the functioning of all myosins.
Single-Molecule Microscopy Techniques Using Processive Myosins
This section discusses single-molecule microscopy techniques that are suited to the study of processive myosins. As discussed in the previous section, motors with dr<0.5, such as cardiac myosins, are non-processive. Unconventional dimeric myosins, such as myosins V and X, with a high duty ratio are processive and undergo many steps along an actin filament before detaching [42, 50]. Unconventional myosins that are monomeric but have a high duty ratio, such as myosin VI, can be artificially dimerized using a GCN4 leucine-zipper motif such that they are rendered processive [51]. The ability of a processive myosin to move (also referred to as “walk”) along actin filaments has been used in conjunction with single-molecule techniques to gain detailed insight into the function of myosins. Here, we briefly review two techniques that have significantly advanced our understanding of the myosin family of proteins.
TIRF Fluorescence Microscopy
A single, fluorescently labeled, processive myosin dimer walking along an actin filament (Fig. 3a) can be tracked with high accuracy (~1 nm) using TIRF [41, 52]. TIRF involves the use of a high numerical aperture objective lens such that the laser light is reflected back into the microscope objective at the water–glass interface of the coverslip [41]. The light reflection at the water–glass interface results in an evanescent wave that propagates several hundred nanometers into the coverslip, thereby illuminating a very small depth into solution. This prevents activation of fluorescently labeled molecules deep in solution (>1 µm) whose fluorescence would interfere with the detection of single processive molecules at the coverslip surface. Knowledge of the point spread function of a diffraction-limited fluorophore enables one to localize the fluorophore with high accuracy. Tracking the movement of myosins V and VI as they move along actin filaments confirms that they take large ~36 nm steps [41, 52]. Furthermore, labeling the two heads of myosin with two different fluorophores confirms that processive myosins walk along actin filaments in a hand-over-hand manner [37]. Although the TIRF assay requires more instrumentation (laser and TIRF objective) than the in vitro motility assay, it involves much less effort than the optical trap and therefore can be used to screen processive mutants or genetically engineered molecular motors. TIRF of green fluorescent protein labeled molecular motors transfected into living cells have also provided insights into their function in vivo [53].
Fig. 3.

Total internal reflection fluorescence (TIRF) and gold nanoparticle tracking of single myosin molecules: left half cartoon schematic of the TIRF assay showing the excitation beam (green line) entering a high numerical aperture TIRF objective and undergoing total internal reflection at the coverslip glass–water interface. An evanescent wave is set up by the excitation beam, and it illuminates fluorophores up to a few hundred nanometers into solution. Inset A myosin VI dimer moving processively (walking) along an actin filament anchored to the coverslip surface. A fluorophore is attached to the end of each myosin VI monomer tail (red circle). The evanescent wave excites the myosin VI fluorophore, and the resulting emission beam (red line) is imaged by a camera with appropriate optical path. Right half Cartoon schematic for gold nanoparticle tracking assay. Inset A processive myosin VI dimer is attached to a gold nanoparticle (20–40 nm in diameter) through a biotin–streptavidin linkage (yellow circle). A myosin VI dimer moving processively along an actin filament anchored to the glass surface is illuminated by a 542-nm laser beam (green line). A darkfield condenser lens obliquely illuminates the gold particle, which scatters the light beam. The scattered light (red line) is imaged on a camera using an appropriate optical path
High Temporal Resolution Gold Nanoparticle Tracking
TIRF microscopy of single molecules is limited by the intensity of a single fluorophore attached to a single processive motor. Accurate tracking requires the gathering of adequate photons by a sensitive camera, resulting in frame rates of at most 5 Hz using TIRF with single fluorophores [41, 52]. This time resolution is limiting, considering that multiple very fast steps in the actomyosin ATPase cycle is [42, 54]. When a myosin head is bound to ATP, it binds weakly to an actin filament. Hydrolysis of ATP results in a conformational change of the myosin head into a prestroke state that is still a weak binding state to actin. Associated with Pi release is the transition to a strongly bound state; both myosin VI bound to ADP or without nucleotide bind actin strongly [55]. The detailed coordination between the ATPase cycle (chemical) and the lever arm swing (mechanical) of the myosin is unclear, primarily due to the short time span (several microseconds) over which important substeps of this chemomechanical cycle occur. These include events that follow the binding of ATP to the myosin, which releases the myosin from the actin, ATP hydrolysis in the unbound head, the weak–strong transition in the unbound head, and its search for the next binding site [55]. The gold nanoparticle tracking assay provides significantly improved temporal resolution (~100 µs) to enable observation of some of these fast intermediate states, thereby shedding light on the chemomechanical cycle of myosins [39]. This assay involves attaching a gold nanoparticle (20–40 nm diameter) to a processive myosin through a biotin–streptavidin linkage. The movement of the gold nanoparticle is then tracked by virtue of the light scattered by the gold nanoparticle in a darkfield microscope (Fig. 3b). The light scattered by the gold nanoparticle is much more intense than the light emitted by a single fluorophore in the TIRF assay, and also, there is no photobleaching as you have with fluorescent tags. Thus, the tracking rate is only limited by the camera frame rate and camera noise, which currently are limited to ~100 µs. This gold nanoparticle assay helped resolve the release of the trailing head of a processive myosin V dimer and tracked its rebinding to the actin filament [39]. The results are consistent with a free swivel at the junction between the lever arms of the two myosin V heads, which would facilitate walking along branched actin meshworks.
Force Generation and Kinetics Studied Using Optical Tweezers
The assays we have discussed thus far measure the relative displacement of the actomyosin interface in the absence of significant load on the myosin. In the context of cardiac myosin, which generates a contractile force that propels blood throughout the circulatory system, it is necessary to understand myosin function under load. Specifically, we need to understand the effect of cardiomyopathy mutations on the contractility of cardiac myosins.
Early studies to measure the force exerted by cardiac myosins used glass microneedles to catch and pull on actin filaments, while a high density of myosin molecules on the surface were bound to the actin filament [56, 57]. The microneedle was displaced until the maximum steady-state force exerted by the bound myosins was balanced by the bending of the microneedle tip. Using this approach, it was found that α-cardiac myosin produces only one half the force exerted by β-cardiac myosin. Although this assay yields important insights into cardiac myosin function, it does not examine the force generation of single actomyosin interactions as is made possible by an optical trap [13]. Examining single actomyosin interactions enables one to quantify both force generation and kinetics of single myosin molecules and paves the way to a detailed understanding of the effects of cardiomyopathy mutations on myosin function.
The optical trap setup to examine single actomyosin interactions is depicted in Fig. 4. It consists of two focused infrared laser beams (referred to as “laser traps”), in each of which a polystyrene bead (~1 µm diameter) is trapped by the power of the laser beam [58]. The surface of the polystyrene beads is coated with streptavidin, which facilitates the binding of a biotinylated actin filament to each of the trapped beads. The two laser traps can be steered independently to pull the actin filament taut between the two trapped beads. The myosin is bound to “platform” beads, which are stuck to the surface of a glass coverslip, using an antibody and/or nitrocellulose coating on the platform bead surface. The concentration of the myosin is kept low such that only one in ten platform beads has a myosin that can interact with the actin filament. The actin filament is lowered onto the surface of the platform bead, resulting in the single myosin molecule binding to the actin filament. Using the energy derived by an ATP hydrolysis, the myosin strokes its lever arm, thereby displacing the optically trapped beads at the ends of the actin filament. The magnitude of this displacement is a measure of the stroke of the myosin lever arm. Following the stroke, the myosin head releases ADP and binds ATP, which causes it to release from the actin filament. The length of time for which the myosin is bound to the actin filament (also referred to as “dwell time”) is therefore a combination of ADP release and ATP-binding rates. The optical trap provides a linear restoring force that is proportional to the displacement of the trapped beads. The magnitude of this restoring force (quantified as trap stiffness) is proportional to the power of the laser beam and hence can be varied to alter the force experienced by the stroking myosin. In addition, a high-frequency feedback signal that moves the trap position in response to the myosin stroke can be used to quantify the maximum force generated by the myosin [13]. These properties have been used to gain detailed insight into myosin function.
Fig. 4.

Optical trapping of myosin molecules: cartoon schematic of a dualbeam laser optical trap for measuring the lever-arm stroke, force generation, and kinetics of single myosin molecules. Streptavidin-coated polystyrene beads are optically trapped by a focused infrared (IR) laser beam. An optically trapped bead is attached to each end of a single biotinylated actin filament (gray line). The filament is stabilized using fluorescently labeled phalloidin. The actin filament is stretched taut in the optical trap by independently steering the two laser beams. Inset A myosin VI monomeric construct with C-terminal GFP tags is attached to polystyrene “platform” beads coated with anti-GFP antibody. The position of each of the trap beads is determined accurately by two quadrant photo detectors (QPD) positioned above the optical trap. QPD readout can be calibrated to yield an accurate measure of myosin step size
The first optical trap quantified the stroke size of rabbit skeletal muscle myosin to be ~10 nm with forces ranging from 3 to 4 pN generated by the myosin [13]. The use of an optical trap, with its ability to detect single actomyosin interactions, provided extremely strong support for the lever arm theory of myosin motion, also referred to as the swinging cross-bridge hypothesis [59].
The dual-beam optical trap has since been applied to study cardiac myosin function. Palmiter et al. [60] used the dual-beam optical trap to observe differences between rabbit α- and β-cardiac myosin isoforms. They found that both α and β isoforms have similar stroke sizes and generate equal force in the optical trap. However, the β isoform has a significantly longer ts. The combination of myosin stroke size, strongly bound state time, and force generated, as measured by the optical trap, can be used to model the isometric contraction force generated by an ensemble of myosins, as in the muscle sarcomere [57].
The optical trap has also been used to characterize the effect of some cardiomyopathy mutations on myosin function. Tyska et al. [61] used an optical trap to show that the R403Q HCM mutation introduced into mouse α-MyHC does not affect single myosin force generation, stroke size, or ts despite a 1.6-fold increase in the sliding velocity in an in vitro motility assay and a 2.3-fold increase in ATPase activity. Tyska et al. [61] propose that the information provided by the optical trap suggests that the R403Q mutation in α-MyHC likely does not affect individual myosin function, but the coordination between the strokes of the α–α homodimers that exist in the myosin thick filament [61]. The R403Q mutation introduced into the equivalent site in chicken gizzard myosin (a smooth muscle myosin) does not alter force generation or stroke size but decreases the ts on the actin filament, suggesting a change in kinetics being responsible for the effect of this mutation in vivo [62]. The same effect of reduced ts was observed in this system for HCM mutations R719Q and D778G, suggesting a similar mode by which these three mutations alter myosin function in vivo [62]. The reduced strongly bound state times observed with chicken gizzard myosin is consistent with optical trapping measurements performed using R403Q and L908V mutant β-cardiac myosin obtained from cardiac biopsies of hypertrophic cardiomyopathic patients [24].
Two DCM mutations S532P and F764L introduced into mouse α-MyHC have also been examined using the optical trap [30]. While F764L measurements are similar to wild type, S532P myosin shows both a reduced stroke size and an increased ts, both of which combine to reduce myosin velocity [30]. A modified optical trap setup, which involved multiple motors on the platform bead surface that interact with the actin filament under constant load (using feedback as described earlier), showed that HCM mutations R403Q and R453C have increased maximum force production, whereas the DCM mutation S532P has reduced maximum force production and the DCM mutation F764L unaltered force production [63]. These findings led the authors to suggest that HCM mutations have increased myosin function, whereas DCM mutations have diminished function [30, 63]. However, none of these studies have analyzed these mutations in the context of human β-cardiac myosin. These findings, if extended to other HCM and DCM mutations, would lend itself to a systematic clinical regimen that targets myosin function differently for HCM and DCM.
Single Cell Approaches
Thus far, we have outlined studies of the interaction between purified myosin and actin. These studies have yielded detailed insight into myosin function, with regards to its kinetics and force generation. Cardiac muscle, however, is composed of highly organized arrays of actin and myosin filaments (Fig. 5) along with numerous structural and regulatory protein elements that are essential for its normal function [47]. In order to translate findings from the single molecule level to muscle function in vivo, intermediate model systems are needed that incorporate these protein elements. Several such model systems, including isolated skinned muscle trabeculae (fiber bundles) [64, 65], isolated cardiomyocytes [66, 67], and single myofibrils [68, 69], have been developed. To translate our knowledge of the force generated and kinetics of actomyosin measured in single-molecule assays to the forces and velocities generated by muscle, we need to understand how the interplay of all the actomyosin interactions occurring in muscle get combined into an organ-level contractile force/ velocity machine. Cardiac muscle myosin is a low duty ratio motor (each myosin head is bound to the actin filament less than 5% of the time). At any given instant, the number of myosin heads that interact with actin filaments and produce a force during isometric contraction is unknown [70]. Furthermore, the alignment of the individual sarcomeres within a muscle fiber will determine how the forces generated by them get combined into the force generated by the muscle fiber. Thus, the single myofibril, the single cardiomyocyte, and heart trabeculae are progressively more complex, where the contractile force generated by these systems depends on an increasing number of parameters. Below, we discuss recent advances in understanding the force–length changes in single cardiomyocytes and its potential for unraveling the effect of β-MyHC cardiomyopathy mutations on sarcomere function.
Fig. 5.

Single cardiomyocyte dynamic force–length measurements: cartoon schematic of a cardiomyocyte (light brown) suspended between two carbon nanofibers. The carbon nanofibers act as calibrated force transducers, whose displacement is a dynamic readout of the force generated by the cardiomyocyte. The carbon nanofibers can be coupled to a piezo-translator. This provides a high-frequency feedback that adjusts the position of the carbon nanofiber during cardiomyocyte contraction, thereby simulating physiological load conditions. Inset The contractile apparatus of a cardiomyocyte, namely, the muscle sarcomere, is shown, highlighting the spatial arrangement of actin filaments (gray) and myosin thick filaments (blue), which are integrated with numerous structural and regulatory proteins (not shown) to generate a contractile force
Force–Length Dynamic Measurements of Single Cardiomyocytes
The basic requirement to measure the contractile force generated by a single cardiomyocyte is to attach its two ends, along the longest cellular dimension (parallel to the length of the sarcomere), to calibrated force transducers whose displacement is a readout of the force being generated (Fig. 5). Cellular attachment to force transducers has been done using a variety of techniques, including twirling the cell ends onto calibrated glass micropipettes [71], suction micropipettes to hold the cell ends [72], and attachment of the ends of the cell to a carbon nanofiber [73]. Among these, the carbon nanofiber technique has seen extensive use in recent years [66, 74–79], as the carbon nanofiber attaches directly to the cell without adhesives and requires relatively less technical expertise compared to cell twirling or suction micropipettes. The mechanism of attachment of the cell to the nanofiber is unknown, though it is likely mediated by electrostatic interactions between the oxidized graphite (carboxyl groups) and amino groups in the cellular membrane [73]. The carbon nanofibers are inserted into glass micropipettes, which in turn are mounted on independently controlled piezo-translators [66, 79]. The cardiomyocyte is imaged using brightfield microscopy in real time, and feedback is provided to the piezo-translators such that the distance between the carbon nanofibers is maintained in order to simulate isometric contraction of the cardiomyocyte sarcomere. The technique can be implemented with feedback provided to either one [79] or both [66] of the carbon nanofibers. A detailed layout of the cell-stretching apparatus and the experimental protocol can be found elsewhere [67].
Carbon nanofiber-based stretch studies of single cardiomyocytes have provided important insights into their force–length dynamics while establishing a tractable system to study the effect of cardiomyopathy mutations at a cellular level. Nishimura et al. [79] showed that, for isolated cardiomyocytes used in this system, their shortening velocity, isometric force, and response to altered afterload (equivalent to varying end-systolic pressure) were similar to values for isolated trabeculae. Additionally, ionotropic stimulation (isoproterenol) altered the force–length loops in a manner observed at the organ level. These results show that the carbon nanofiber attachment did not alter the force–length dynamics of the cardiomyocytes. Cardiomyocytes isolated from cardiomyopathic hamsters were used to study the effect of cardiomyopathy mutations on cellular mechanics [78]. In the absence of preload, the shortening fraction (fractional change in length during the contraction cycle) and maximum shortening velocity were decreased. Similarly, the maximum force during isometric contraction was also decreased. These decreases were much more prominent in the presence of physiological load on the cardiomyocyte. Overall, the dysregulation at the organ level in cardiomyopathic hamsters was observed at the single cardiomyocyte level, validating this system as a means to evaluate the effect of specific cardiomyopathy mutations on cellular function. The technique has been progressively improved to keep the cardiomyocyte in focus during the contraction cycle [66]. This allows for monitoring changes in sarcomere length using brightfield techniques and fluorescence imaging of labeled sarcomeric proteins, both of which can be exploited to understand sarcomeric dysregulation under cardiomyopathy.
Gene Transfer as a Means of Inducing Cardiomyopathy Phenotype at the Single Cell Level
Herron et al. [80] have discussed emerging evidence that an increase in the of β-MyHC relative to α-MyHC is a marker of reduced sarcomeric function in cardiomyopathy. For instance, increasing β-MyHC content by reducing thyroid function decreases the maximal power output of isolated skinned rat cardiac myocytes [81]. Reduced thyroid function, which increases β-MyHC content, also results in heart failure with severe, progressive systolic dysfunction and thinning of the heart chamber walls [82]. In this context, Herron et al. [80] propose that restoration of the relative α-MyHC content by acute gene transfer might be a clinical approach to restoring sarcomeric function. This approach is supported by experiments on isolated rat cardiomyocytes that are induced to express MyHC proteins by adenoviral infection [83]. Herron et al. [83] were able to substantially increase β-MyHC protein expression, from trace amounts in wild type to ~20% of total myosin, using adenoviral infection in isolated rat cardiomyocytes. The expressed β-MyHC protein colocalizes to the endogenous sarcomeric α-MyHC. Maximal sarcomeric shortening, in single beating cardiomyocytes, was reduced in a β-MyHC expression dependent manner, suggesting that changes in the relative content of α and β-MyHC isoforms, induced by adenoviral infection, can be used to affect changes in sarcomeric function in a predictable manner.
Adenoviral transfection used in the context of another sarcomeric protein, troponin I, has shown results similar to that obtained using transgenic mouse models [84]. Yasuda et al. [84] used a combination of gene transfer and transgenic mice to examine the role of troponin I in the sarcomere relaxation times. A constitutively phosphorylated form of troponin I (S23/24D) showed significantly faster relaxation times relative to controls, when examined using single cell carbon nanofiber experiments. The isolated cardiomyocytes used in these experiments were obtained either from transgenic mice expressing S23/24D or from wild-type cardiomyocytes infected with adenovirus encoding S23/24D mutation in troponin I. The gene transfer experiments yielded comparable results to the transgenic experiments, suggesting that adenoviral infection can be effectively used to deliver the appropriate sarcomeric proteins to rescue normal function in cardiomyopathy.
Adenoviral expression of R403Q, R453C, and G584R mutations in MyHC has been used to study their effect on isolated cardiomyocytes or myofibrils. Marian et al. [85] infected feline cardiac myocytes with R403Q human β-MyHC adenovirus. Five days post-gene transfer with adenovirus, they saw significant disruption of normal sarcomeric organization, whereas control myocytes transduced with wild-type human β-MyHC adenovirus did not. This study suggests that the R403Q mutation can disrupt existing sarcomeres, thereby effecting organ level dysfunction in HCM. Another study by Wang et al. [86] used adenovirus to express chicken embryonic MyHC tagged with green fluorescent protein (GFP) in chicken embryonic cardiomyocytes. The R403Q mutation in this assay produced mild myofibril disarray, whereas R453C and G584R caused dramatic myofibril disarray. The spontaneous contractility of the chicken embryonic cardiomyocytes, however, was not affected by the introduction of these mutations. These two studies that introduced cardiomyopathy MyHC mutations into cells demonstrate the feasibility of using adenovirus transduction to understand the cardiomyopathy phenotype at the single cell level.
Summary and Conclusions
This review has focused on the need to understand at the most fundamental level the roots of cardiac dysfunction caused by β-MyHC mutations. Clearly, each single point mutation that causes this disease alters β-cardiac myosin in some fundamental way, either causing a loss of force producing capability of the motor or an unhealthy enhancement of its force producing capability. Knowing which of these two broad categories the mutations fall into would allow one to consider a potential chronic therapy for afflicted individuals since one could imagine the use of force-enhancing agents or force-inhibiting agents, respectively, that work directly on β-cardiac myosin or related sarcomeric proteins. The feasibility of this approach is demonstrated by a potential congestive heart drug being developed by Cytokinetics, Inc., which binds directly to β-cardiac myosin and activates its force producing capability. If one optimizes the force producing capability of the myosin at an early enough age, the downstream pleiotrophic effects of these single point mutations, which lead to hypertrophy and death, could be ameliorated.
Furthermore, understanding more precisely the effects of each mutation on every kinetic step in the actomyosin chemomechanical cycle could lead to more specific targeting of potential drug candidates. For example, mutations that enhance the ADP release rate will decrease the duty ratio and therefore decrease the average number of heads interacting with actin in the sarcomere during each contraction, resulting in a diminished force production by the sarcomere. Mutations that enhance Pi release, on the other hand, will increase the duty ratio, resulting in increased force production by the sarcomere. Screening for agents that more specifically reset the altered kinetic step may ultimately be the preferred therapeutic response.
Thus, it is clear that there is a great need to obtain a fundamental understanding of the effects of individual β-cardiac myosin mutations on the activities of the myosin molecule. The advent of in vitro motility assays, taken to the single molecule level, has added importantly to the variety of necessary tools for this to be accomplished. These assays, coupled with related assays at the single cardiomyocyte level, can yield the sought after information.
What is now limiting is the ability to obtain sufficient, homogeneous quantities of purified, homogeneous wild type and mutant human β-cardiac myosin. We need to focus on the human protein since animal models, such as the mouse, already have amino acid changes compared to the human protein that change the myosin function considerably. Furthermore, the mouse uses primarily α-cardiac myosin rather than β-cardiac myosin, and these isoforms are considerably different. Unfortunately, human β-MyHC expression using bacterial expression or the baculovirus system does not yield sufficient quality protein to study multiple mutations with a breadth of assays. Adenovirus-based transduction of muscle cells to produce purified human β-cardiac myosin points to a potential solution, and one can be optimistic that the much needed basic characterization of wild type and mutant forms of human β-cardiac myosin is near at hand.
Acknowledgments
We thank the Institute for Stem Cell Biology and Regenerative Medicine (inSTEM) and the National Center for Biological Sciences, Bangalore, India, for funding a symposium on Cardiac and Cardiovascular disorders which catalyzed this collaborative review.
Contributor Information
Sivaraj Sivaramakrishnan, Department of Biochemistry, Stanford University, Stanford, CA, USA.
Euan Ashley, Cardiovascular Medicine, Stanford University, Stanford, CA, USA.
Leslie Leinwand, Cardiovascular Institute, University of Colorado, Boulder, USA.
James A. Spudich, Email: jspudich@stanford.edu, Department of Biochemistry, Stanford University, Stanford, CA, USA.
References
- 1.Maron BJ, Gardin JM, Flack JM, Gidding SS, Kurosaki TT, Bild DE. Prevalence of hypertrophic cardiomyopathy in a general population of young adults. Echocardiographic analysis of 4111 subjects in the CARDIA Study. Coronary Artery Risk Development in (Young) Adults. Circulation. 1995;92(4):785–789. doi: 10.1161/01.cir.92.4.785. [DOI] [PubMed] [Google Scholar]
- 2.Maron BJ. Hypertrophic cardiomyopathy: A systematic review. JAMA. 2002;287(10):1308–1320. doi: 10.1001/jama.287.10.1308. [DOI] [PubMed] [Google Scholar]
- 3.Liew CC, Dzau VJ. Molecular genetics and genomics of heart failure. Nature Reviews. Genetics. 2004;5(11):811–825. doi: 10.1038/nrg1470. [DOI] [PubMed] [Google Scholar]
- 4.Ramaraj R. Hypertrophic cardiomyopathy: etiology, diagnosis, and treatment. Cardiology in Review. 2008;16(4):172–180. doi: 10.1097/CRD.0b013e318178e525. [DOI] [PubMed] [Google Scholar]
- 5.Kron SJ, Spudich JA. Fluorescent actin filaments move on myosin fixed to a glass surface. Proceedings of the National Academy of Sciences of the United States of America. 1986;83(17):6272–6276. doi: 10.1073/pnas.83.17.6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sheetz MP, Spudich JA. Movement of myosincoated fluorescent beads on actin cables in vitro. Nature. 1983;303(5912):31–35. doi: 10.1038/303031a0. [DOI] [PubMed] [Google Scholar]
- 7.Spudich JA, Kron SJ, Sheetz MP. Movement of myosin-coated beads on oriented filaments reconstituted from purified actin. Nature. 1985;315(6020):584–586. doi: 10.1038/315584a0. [DOI] [PubMed] [Google Scholar]
- 8.Lymn RW. Kinetic analysis of myosin and actomyosin atpase. Annual Review of Biophysics and Bioengineering. 1979;8:145–163. doi: 10.1146/annurev.bb.08.060179.001045. [DOI] [PubMed] [Google Scholar]
- 9.Uyeda TQ, Kron SJ, Spudich JA. Myosin step size. Estimation from slow sliding movement of actin over low densities of heavy meromyosin. Journal of Molecular Biology. 1990;214(3):699–710. doi: 10.1016/0022-2836(90)90287-V. [DOI] [PubMed] [Google Scholar]
- 10.Toyoshima YY, Kron SJ, Spudich JA. The myosin step size: Measurement of the unit displacement per ATP hydrolyzed in an in vitro assay. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(18):7130–7134. doi: 10.1073/pnas.87.18.7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ishijima A, Harada Y, Kojima H, Funatsu T, Higuchi H, Yanagida T. Single-molecule analysis of the actomyosin motor using nano-manipulation. Biochemical and Biophysical Research Communications. 1994;199(2):1057–1063. doi: 10.1006/bbrc.1994.1336. [DOI] [PubMed] [Google Scholar]
- 12.Yanagida T, Arata T, Oosawa F. Sliding distance of actin filament induced by a myosin crossbridge during one ATP hydrolysis cycle. Nature. 1985;316(6026):366–369. doi: 10.1038/316366a0. [DOI] [PubMed] [Google Scholar]
- 13.Finer JT, Simmons RM, Spudich JA. Single myosin molecule mechanics: Piconewton forces and nanometre steps. Nature. 1994;368(6467):113–119. doi: 10.1038/368113a0. [DOI] [PubMed] [Google Scholar]
- 14.Wolenski JS, Cheney RE, Forscher P, Mooseker MS. In vitro motilities of the unconventional myosins, brush border myosin-I, and chick brain myosin-V exhibit assay-dependent differences in velocity. Journal of Experimental Zoology. 1993;267(1):33–39. doi: 10.1002/jez.1402670106. [DOI] [PubMed] [Google Scholar]
- 15.Bryant Z, Altman D, Spudich JA. The power stroke of myosin VI and the basis of reverse directionality. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(3):772–777. doi: 10.1073/pnas.0610144104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Post PL, Tyska MJ, O'Connell CB, Johung K, Hayward A, Mooseker MS. Myosin-IXb is a single-headed and processive motor. Journal of Biological Chemistry. 2002;277(14):11679–11683. doi: 10.1074/jbc.M111173200. [DOI] [PubMed] [Google Scholar]
- 17.Homma K, Saito J, Ikebe R, Ikebe M. Motor function and regulation of myosin X. Journal of Biological Chemistry. 2001;276(36):34348–34354. doi: 10.1074/jbc.M104785200. [DOI] [PubMed] [Google Scholar]
- 18.Yamashita H, Sugiura S, Serizawa T, Sugimoto T, Iizuka M, Katayama E, et al. Sliding velocity of isolated rabbit cardiac myosin correlates with isozyme distribution. American Journal of Physiology. 1992;263(2 Pt 2):H464–H472. doi: 10.1152/ajpheart.1992.263.2.H464. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita H, Sugiura S, Sata M, Serizawa T, Iizuka M, Shimmen T, et al. Depressed sliding velocity of isolated cardiac myosin from cardiomyopathic hamsters: evidence for an alteration in mechanical interaction of actomyosin. Molecular and Cellular Biochemistry. 1993;119(1–2):79–88. doi: 10.1007/BF00926857. [DOI] [PubMed] [Google Scholar]
- 20.Barany M, Conover TE, Schliselfeld LH, Gaetjens E, Goffart M. Relation of properties of isolated myosin to those of intact muscles of the cat and sloth. European Journal of Biochemistry. 1967;2(2):156–164. doi: 10.1111/j.1432-1033.1967.tb00120.x. [DOI] [PubMed] [Google Scholar]
- 21.Cuda G, Fananapazir L, Zhu WS, Sellers JR, Epstein ND. Skeletal muscle expression and abnormal function of beta-myosin in hypertrophic cardiomyopathy. Journal of Clinical Investigation. 1993;91(6):2861–2865. doi: 10.1172/JCI116530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Epstein ND, Fananapazir L, Lin HJ, Mulvihill J, White R, Lalouel JM, et al. Evidence of genetic heterogeneity in five kindreds with familial hypertrophic cardiomyopathy. Circulation. 1992;85(2):635–647. doi: 10.1161/01.cir.85.2.635. [DOI] [PubMed] [Google Scholar]
- 23.Cuda G, Fananapazir L, Epstein ND, Sellers JR. The in vitro motility activity of beta-cardiac myosin depends on the nature of the beta-myosin heavy chain gene mutation in hypertrophic cardiomyopathy. Journal of Muscle Research and Cell Motility. 1997;18(3):275–283. doi: 10.1023/a:1018613907574. [DOI] [PubMed] [Google Scholar]
- 24.Palmiter KA, Tyska MJ, Haeberle JR, Alpert NR, Fananapazir L, Warshaw DM. R403Q and L908V mutant beta-cardiac myosin from patients with familial hypertrophic cardiomyopathy exhibit enhanced mechanical performance at the single molecule level. Journal of Muscle Research and Cell Motility. 2000;21(7):609–620. doi: 10.1023/a:1005678905119. [DOI] [PubMed] [Google Scholar]
- 25.Palmer BM, Fishbaugher DE, Schmitt JP, Wang Y, Alpert NR, Seidman CE, et al. Differential cross-bridge kinetics of FHC myosin mutations R403Q and R453C in heterozygous mouse myocardium. American Journal of Physiology. Heart and Circulatory Physiology. 2004;287(1):H91–H99. doi: 10.1152/ajpheart.01015.2003. [DOI] [PubMed] [Google Scholar]
- 26.Keller DI, Coirault C, Rau T, Cheav T, Weyand M, Amann K, et al. Human homozygous R403W mutant cardiac myosin presents disproportionate enhancement of mechanical and enzymatic properties. Journal of Molecular and Cellular Cardiology. 2004;36(3):355–362. doi: 10.1016/j.yjmcc.2003.12.006. [DOI] [PubMed] [Google Scholar]
- 27.Sweeney HL, Straceski AJ, Leinwand LA, Tikunov BA, Faust L. Heterologous expression of a cardiomyopathic myosin that is defective in its actin interaction. Journal of Biological Chemistry. 1994;269(3):1603–1605. [PubMed] [Google Scholar]
- 28.Malmqvist UP, Aronshtam A, Lowey S. Cardiac myosin isoforms from different species have unique enzymatic and mechanical properties. Biochemistry. 2004;43(47):15058–15065. doi: 10.1021/bi0495329. [DOI] [PubMed] [Google Scholar]
- 29.Shaw T, Elliott P, McKenna WJ. Dilated cardiomyopathy: a genetically heterogeneous disease. Lancet. 2002;360(9334):654–655. doi: 10.1016/S0140-6736(02)09879-3. [DOI] [PubMed] [Google Scholar]
- 30.Schmitt JP, Debold EP, Ahmad F, Armstrong A, Frederico A, Conner DA, et al. Cardiac myosin missense mutations cause dilated cardiomyopathy in mouse models and depress molecular motor function. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(39):14525–14530. doi: 10.1073/pnas.0606383103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kurabayashi M, Tsuchimochi H, Komuro I, Takaku F, Yazaki Y. Molecular cloning and characterization of human cardiac alpha- and beta-form myosin heavy chain complementary DNA clones. Regulation of expression during development and pressure overload in human atrium. Journal of Clinical Investigation. 1988;82(2):524–531. doi: 10.1172/JCI113627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lowey S, Lesko LM, Rovner AS, Hodges AR, White SL, Low RB, et al. Functional effects of the hypertrophic cardiomyopathy R403Q mutation are different in an alpha- or beta-myosin heavy chain backbone. Journal of Biological Chemistry. 2008;283(29):20579–20589. doi: 10.1074/jbc.M800554200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circulation Research. 1991;68(6):1742–1750. doi: 10.1161/01.res.68.6.1742. [DOI] [PubMed] [Google Scholar]
- 34.Umeda PK, Darling DS, Kennedy JM, Jakovcic S, Zak R. Control of myosin heavy chain expression in cardiac hypertrophy. American Journal of Cardiology. 1987;59(2):49A–55A. doi: 10.1016/0002-9149(87)90176-7. [DOI] [PubMed] [Google Scholar]
- 35.Morkin E. Control of cardiac myosin heavy chain gene expression. Microscopy Research and Technique. 2000;50(6):522–531. doi: 10.1002/1097-0029(20000915)50:6<522::AID-JEMT9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.De La Cruz EM, Ostap EM. Kinetic and equilibrium analysis of the myosin ATPase. Methods in Enzymology. 2009;455:157–192. doi: 10.1016/S0076-6879(08)04206-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Churchman LS, Okten Z, Rock RS, Dawson JF, Spudich JA. Single molecule high-resolution colocalization of Cy3 and Cy5 attached to macromolecules measures intramolecular distances through time. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(5):1419–1423. doi: 10.1073/pnas.0409487102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.De La Cruz EM, Wells AL, Rosenfeld SS, Ostap EM, Sweeney HL. The kinetic mechanism of myosin V. Proceedings of the National Academy of Sciences of the United States of America. 1999;96(24):13726–13731. doi: 10.1073/pnas.96.24.13726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dunn AR, Spudich JA. Dynamics of the unbound head during myosin V processive translocation. Nature Structural & Molecular Biology. 2007;14(3):246–248. doi: 10.1038/nsmb1206. [DOI] [PubMed] [Google Scholar]
- 40.Sakamoto T, Webb MR, Forgacs E, White HD, Sellers JR. Direct observation of the mechanochemical coupling in myosin Va during processive movement. Nature. 2008;455(7209):128–132. doi: 10.1038/nature07188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yildiz A, Selvin PR. Fluorescence imaging with one nanometer accuracy: Application to molecular motors. Accounts of Chemical Research. 2005;38(7):574–582. doi: 10.1021/ar040136s. [DOI] [PubMed] [Google Scholar]
- 42.Trybus KM. Myosin V from head to tail. Cellular and Molecular Life Sciences. 2008;65(9):1378–1389. doi: 10.1007/s00018-008-7507-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marston SB, Taylor EW. Comparison of the myosin and actomyosin ATPase mechanisms of the four types of vertebrate muscles. Journal of Molecular Biology. 1980;139(4):573–600. doi: 10.1016/0022-2836(80)90050-9. [DOI] [PubMed] [Google Scholar]
- 44.Spudich JA. Biochemical and structural studies of actomyosin-like proteins from non-muscle cells. II. Purification, properties, and membrane association of actin from amoebae of Dictyostelium discoideum. Journal of Biological Chemistry. 1974;249(18):6013–6020. [PubMed] [Google Scholar]
- 45.Cheney RE, O'Shea MK, Heuser JE, Coelho MV, Wolenski JS, Espreafico EM, et al. Brain myosin-V is a two-headed unconventional myosin with motor activity. Cell. 1993;75(1):13–23. doi: 10.1016/S0092-8674(05)80080-7. [DOI] [PubMed] [Google Scholar]
- 46.Huxley HE. Electron microscope studies on the structure of natural and synthetic protein filaments from striated muscle. Journal of Molecular Biology. 1963;7:281–308. doi: 10.1016/s0022-2836(63)80008-x. [DOI] [PubMed] [Google Scholar]
- 47.Bagshaw C. Muscle contraction. 2nd ed. London: Chapman & Hall; 1993. [Google Scholar]
- 48.Mehta AD, Rock RS, Rief M, Spudich JA, Mooseker MS, Cheney RE. Myosin-V is a processive actin-based motor. Nature. 1999;400(6744):590–593. doi: 10.1038/23072. [DOI] [PubMed] [Google Scholar]
- 49.Spudich JA. Optical trapping: motor molecules in motion. Nature. 1990;348(6299):284–285. doi: 10.1038/348284a0. [DOI] [PubMed] [Google Scholar]
- 50.Sousa AD, Cheney RE. Myosin-X: A molecular motor at the cell's fingertips. Trends in Cell Biology. 2005;15(10):533–539. doi: 10.1016/j.tcb.2005.08.006. [DOI] [PubMed] [Google Scholar]
- 51.Rock RS, Rice SE, Wells AL, Purcell TJ, Spudich JA, Sweeney HL. Myosin VI is a processive motor with a large step size. Proceedings of the National Academy of Sciences of the United States of America. 2001;98(24):13655–13659. doi: 10.1073/pnas.191512398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Okten Z, Churchman LS, Rock RS, Spudich JA. Myosin VI walks hand-over-hand along actin. Nature Structural & Molecular Biology. 2004;11(9):884–887. doi: 10.1038/nsmb815. [DOI] [PubMed] [Google Scholar]
- 53.Kerber ML, Jacobs DT, Campagnola L, Dunn BD, Yin T, Sousa AD, et al. A novel form of motility in filopodia revealed by imaging myosin-X at the single-molecule level. Current Biology. 2009;19(11):967–973. doi: 10.1016/j.cub.2009.03.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Altman D, Sweeney HL, Spudich JA. The mechanism of myosin VI translocation and its load-induced anchoring. Cell. 2004;116(5):737–749. doi: 10.1016/s0092-8674(04)00211-9. [DOI] [PubMed] [Google Scholar]
- 55.Sellers JR, Veigel C. Walking with myosin V. Current Opinion in Cell Biology. 2006;18(1):68–73. doi: 10.1016/j.ceb.2005.12.014. [DOI] [PubMed] [Google Scholar]
- 56.Kishino A, Yanagida T. Force measurements by micromanipulation of a single actin filament by glass needles. Nature. 1988;334(6177):74–76. doi: 10.1038/334074a0. [DOI] [PubMed] [Google Scholar]
- 57.VanBuren P, Harris DE, Alpert NR, Warshaw DM. Cardiac V1 and V3 myosins differ in their hydrolytic and mechanical activities in vitro. Circulation Research. 1995;77(2):439–444. doi: 10.1161/01.res.77.2.439. [DOI] [PubMed] [Google Scholar]
- 58.Rice SE, Purcell TJ, Spudich JA. Building and using optical traps to study properties of molecular motors. Methods in Enzymology. 2003;361:112–133. doi: 10.1016/s0076-6879(03)61008-6. [DOI] [PubMed] [Google Scholar]
- 59.Huxley HE. The mechanism of muscular contraction. Science. 1969;164(886):1356–1365. doi: 10.1126/science.164.3886.1356. [DOI] [PubMed] [Google Scholar]
- 60.Palmiter KA, Tyska MJ, Dupuis DE, Alpert NR, Warshaw DM. Kinetic differences at the single molecule level account for the functional diversity of rabbit cardiac myosin isoforms. Journal of Physiology. 1999;519(Pt 3):669–678. doi: 10.1111/j.1469-7793.1999.0669n.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tyska MJ, Hayes E, Giewat M, Seidman CE, Seidman JG, Warshaw DM. Single-molecule mechanics of R403Q cardiac myosin isolated from the mouse model of familial hypertrophic cardiomyopathy. Circulation Research. 2000;86(7):737–744. doi: 10.1161/01.res.86.7.737. [DOI] [PubMed] [Google Scholar]
- 62.Yamashita H, Tyska MJ, Warshaw DM, Lowey S, Trybus KM. Functional consequences of mutations in the smooth muscle myosin heavy chain at sites implicated in familial hypertrophic cardiomyopathy. Journal of Biological Chemistry. 2000;275(36):28045–28052. doi: 10.1074/jbc.M005485200. [DOI] [PubMed] [Google Scholar]
- 63.Debold EP, Schmitt JP, Patlak JB, Beck SE, Moore JR, Seidman JG, et al. Hypertrophic and dilated cardiomyopathy mutations differentially affect the molecular force generation of mouse alpha-cardiac myosin in the laser trap assay. American Journal of Physiology. Heart and Circulatory Physiology. 2007;293(1):H284–H291. doi: 10.1152/ajpheart.00128.2007. [DOI] [PubMed] [Google Scholar]
- 64.de Tombe PP, Stienen GJ. Protein kinase A does not alter economy of force maintenance in skinned rat cardiac trabeculae. Circulation Research. 1995;76(5):734–741. doi: 10.1161/01.res.76.5.734. [DOI] [PubMed] [Google Scholar]
- 65.Vahebi S, Ota A, Li M, Warren CM, de Tombe PP, Wang Y, et al. p38-MAPK induced dephosphorylation of alphatropomyosin is associated with depression of myocardial sarcomeric tension and ATPase activity. Circulation Research. 2007;100(3):408–415. doi: 10.1161/01.RES.0000258116.60404.ad. [DOI] [PubMed] [Google Scholar]
- 66.Iribe G, Helmes M, Kohl P. Force-length relations in isolated intact cardiomyocytes subjected to dynamic changes in mechanical load. American Journal of Physiology. Heart and Circulatory Physiology. 2007;292(3):H1487–H1497. doi: 10.1152/ajpheart.00909.2006. [DOI] [PubMed] [Google Scholar]
- 67.Sugiura S, Nishimura S, Yasuda S, Hosoya Y, Katoh K. Carbon fiber technique for the investigation of single-cell mechanics in intact cardiac myocytes. Natural Protocol. 2006;1(3):1453–1457. doi: 10.1038/nprot.2006.241. [DOI] [PubMed] [Google Scholar]
- 68.Lionne C, Iorga B, Candau R, Travers F. Why choose myofibrils to study muscle myosin ATPase? Journal of Muscle Research and Cell Motility. 2003;24(2–3):139–148. doi: 10.1023/a:1026045328949. [DOI] [PubMed] [Google Scholar]
- 69.Telley IA, Denoth J. Sarcomere dynamics during muscular contraction and their implications to muscle function. Journal of Muscle Research and Cell Motility. 2007;28(1):89–104. doi: 10.1007/s10974-007-9107-8. [DOI] [PubMed] [Google Scholar]
- 70.Cooke R. Actomyosin interaction in striated muscle. Physiological Reviews. 1997;77(3):671–697. doi: 10.1152/physrev.1997.77.3.671. [DOI] [PubMed] [Google Scholar]
- 71.Tarr M, Trank JW, Leiffer P, Shepherd N. Sarcomere length-resting tension relation in single frog atrial cardiac cells. Circulation Research. 1979;45(4):554–559. doi: 10.1161/01.res.45.4.554. [DOI] [PubMed] [Google Scholar]
- 72.Brady AJ, Tan ST, Ricchiuti NV. Contractile force measured in unskinned isolated adult rat heart fibres. Nature. 1979;282(5740):728–729. doi: 10.1038/282728a0. [DOI] [PubMed] [Google Scholar]
- 73.Le Guennec JY, Peineau N, Argibay JA, Mongo KG, Garnier D. A new method of attachment of isolated mammalian ventricular myocytes for tension recording: Length dependence of passive and active tension. Journal of Molecular and Cellular Cardiology. 1990;22(10):1083–1093. doi: 10.1016/0022-2828(90)90072-a. [DOI] [PubMed] [Google Scholar]
- 74.Nishimura S, Kawai Y, Nakajima T, Hosoya Y, Fujita H, Katoh M, et al. Membrane potential of rat ventricular myocytes responds to axial stretch in phase, amplitude and speed-dependent manners. Cardiovascular Research. 2006;72(3):403–411. doi: 10.1016/j.cardiores.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 75.Nishimura S, Nagai S, Katoh M, Yamashita H, Saeki Y, Okada J, et al. Microtubules modulate the stiffness of cardiomyocytes against shear stress. Circulation Research. 2006;98(1):81–87. doi: 10.1161/01.RES.0000197785.51819.e8. [DOI] [PubMed] [Google Scholar]
- 76.Nishimura S, Nagai S, Sata M, Katoh M, Yamashita H, Saeki Y, et al. Expression of green fluorescent protein impairs the force-generating ability of isolated rat ventricular cardiomyocytes. Molecular and Cellular Biochemistry. 2006;286(1–2):59–65. doi: 10.1007/s11010-005-9090-6. [DOI] [PubMed] [Google Scholar]
- 77.Nishimura S, Seo K, Nagasaki M, Hosoya Y, Yamashita H, Fujita H, et al. Responses of single-ventricular myocytes to dynamic axial stretching. Progress in Biophysics and Molecular Biology. 2008;97(2–3):282–297. doi: 10.1016/j.pbiomolbio.2008.02.011. [DOI] [PubMed] [Google Scholar]
- 78.Nishimura S, Yamashita H, Katoh M, Yamada KP, Sunagawa K, Saeki Y, et al. Contractile dysfunction of cardiomyopathic hamster myocytes is pronounced under high load conditions. Journal of Molecular and Cellular Cardiology. 2005;39(2):231–239. doi: 10.1016/j.yjmcc.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 79.Nishimura S, Yasuda S, Katoh M, Yamada KP, Yamashita H, Saeki Y, et al. Single cell mechanics of rat cardiomyocytes under isometric, unloaded, and physiologically loaded conditions. American Journal of Physiology. Heart and Circulatory Physiology. 2004;287(1):H196–H202. doi: 10.1152/ajpheart.00948.2003. [DOI] [PubMed] [Google Scholar]
- 80.Herron TJ, Devaney EJ, Metzger JM. Modulation of cardiac performance by motor protein gene transfer. Annals of the New York Academy of Sciences. 2008;1123:96–104. doi: 10.1196/annals.1420.011. [DOI] [PubMed] [Google Scholar]
- 81.Herron TJ, Korte FS, McDonald KS. Loaded shortening and power output in cardiac myocytes are dependent on myosin heavy chain isoform expression. American Journal of Physiology. Heart and Circulatory Physiology. 2001;281(3):H1217–H1222. doi: 10.1152/ajpheart.2001.281.3.H1217. [DOI] [PubMed] [Google Scholar]
- 82.Tang YD, Kuzman JA, Said S, Anderson BE, Wang X, Gerdes AM. Low thyroid function leads to cardiac atrophy with chamber dilatation, impaired myocardial blood flow, loss of arterioles, and severe systolic dysfunction. Circulation. 2005;112(20):3122–3130. doi: 10.1161/CIRCULATIONAHA.105.572883. [DOI] [PubMed] [Google Scholar]
- 83.Herron TJ, Vandenboom R, Fomicheva E, Mundada L, Edwards T, Metzger JM. Calcium-independent negative inotropy by beta-myosin heavy chain gene transfer in cardiac myocytes. Circulation Research. 2007;100(8):1182–1190. doi: 10.1161/01.RES.0000264102.00706.4e. [DOI] [PubMed] [Google Scholar]
- 84.Yasuda S, Coutu P, Sadayappan S, Robbins J, Metzger JM. Cardiac transgenic and gene transfer strategies converge to support an important role for troponin I in regulating relaxation in cardiac myocytes. Circulation Research. 2007;101(4):377–386. doi: 10.1161/CIRCRESAHA.106.145557. [DOI] [PubMed] [Google Scholar]
- 85.Marian AJ, Yu QT, Mann DL, Graham FL, Roberts R. Expression of a mutation causing hypertrophic cardiomyopathy disrupts sarcomere assembly in adult feline cardiac myocytes. Circulation Research. 1995;77(1):98–106. doi: 10.1161/01.res.77.1.98. [DOI] [PubMed] [Google Scholar]
- 86.Wang Q, Moncman CL, Winkelmann DA. Mutations in the motor domain modulate myosin activity and myofibril organization. Journal of Cell Science. 2003;116(Pt 20):4227–4238. doi: 10.1242/jcs.00709. [DOI] [PubMed] [Google Scholar]