

Abstract
We used human cardiomyocyte-derived cells to create an in vitro model to study lipid metabolism and explored the effects of PPARγ, ACSL1 and ATGL on fatty acid-induced ER stress. Compared to oleate, palmitate treatment resulted in less intracellular accumulation of lipid droplets and more ER stress, as measured by upregulation of CHOP, ATF6 and GRP78 gene expression and phosphorylation of eukaryotic initiation factor 2a (EIF2a). Both ACSL1 and PPARγ adenovirus-mediated expression augmented neutral lipid accumulation and reduced palmitate-induced upregulation of ER stress markers to levels similar to those in the oleate and control treatment groups. This suggests that increased channeling of non-esterified free fatty acids (NEFA) towards storage in the form of neutral lipids in lipid droplets protects against palmitate-induced ER stress. Overexpression of ATGL in cells incubated with oleate-containing medium increased NEFA release and stimulated expression of ER stress markers. Thus, inefficient creation of lipid droplets as well greater release of stored lipids induces ER stress.
Keywords: ER stress, heart, lipid, lipotoxicity, PPAR, ACSL1, triglycerides
1. Introduction
Long chain fatty acids (FAs), either associated with albumin or contained in lipoproteins, are the main energy source for the heart, accounting for about 70% of its energy needs. Under normal conditions, the heart metabolizes FAs rather immediately; it has little capacity for storage [1]. However, obese and/or diabetic conditions lead to an excess influx of lipid to the heart, resulting in increased cardiac lipid accumulation, which is associated with impaired contractility [2] and cardiac hypertrophy (reviewed in [3]). Induction of endoplasmic reticulum (ER) stress by lipid oversupply has been proposed as one of the underlying mechanisms explaining lipid-driven cardiac dysfunction [4, 5]. Induction of ER stress has been shown in vitro with conditions that mimic ischemia [6, 7] and in vivo with infarction [7] and pressure overload [8]. Saturated FAs increase the saturated lipid content of the ER, leading to changes in ER structure and integrity, and contributing to the unfolded protein response (ER stress) [9]. Consequences of ER stress include mitochondrial dysfunction and reduced energy expenditure, activation of inflammatory pathways, impaired protein synthesis and cell growth, and apoptosis (reviewed in [10–12]).
Lipotoxicity is the result of an imbalance between lipid uptake and utilization. Saturated fatty acids (FAs) cause considerably more aggravating effects than unsaturated FAs. One possible reason for this is that the saturated FA palmitate leads to greater ceramide synthesis [13], triggers reactive oxygen species (ROS) generation [14], induces fusion/fission events of ER membranes [9], and impairs the synthesis of the mitochondrial membrane phospholipid cardiolipin, which causes mitochondrial dysfunction [15]. In combination these processes lead to apoptotic cell death [16, 17]. Some of these effects are likely due to insufficient conversion of palmitate into triacylglycerol (TAG). Unsaturated FAs help prevent lipotoxic cell death via activation of cellular survival pathways and channeling of FAs towards storage as TAG in lipid droplets [5, 18]. Storage of lipids in the form of inert TAG is considered harmless [2, 18]. In contrast, accumulation of lipid intermediates like nonesterified FAs, —and signaling lipids such as ceramide and diacylglycerol (DAG) is associated with lipotoxicity [19–21].
In this report we describe studies of the effects of peroxisome proliferator-activated receptor γ (PPARγ) and acyl-CoA synthetase (ACSL1) on palmitate-induced ER stress in the human cardiomyocyte-like cell line AC16 [22], which was derived from adult ventricular heart tissue. PPARγ is a nuclear receptor involved in regulation of intracellular lipid storage, and ACSL1 catalyzes esterification of long chain FAs with co-enzyme A – the initial step in fatty acid metabolism. Although cardiomyocyte specific overexpression of either PPARγ or ACSL1 causes lipid accumulation and cardiac dysfunction, both PPARγ and ASCL1 inhibit inflammation in FA-treated macrophages [23]. Our study shows that PPARγ and ACSL1 can protect cardiomyocytes from ER stress. Moreover we found that oleate (OA), which is usually a non-toxic lipid, induces toxicity if its storage is disrupted by excess intracellular lipolysis.
2. Materials and Methods
2.1 Cells
The human cardiomyocyte cell line AC16, derived from primary cultures of adult ventricular heart tissue [22], was used for the experiments. Cells were grown in DMEM/F-12 medium (GIBCO Invitrogen Corporation, Carlsbad, California, USA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin in a 5% CO2 atmosphere at 37°C. Prior to the infection of the cells with recombinant adenovirus, the medium was changed to DMEM/F-12 medium supplemented with 2% heat inactivated horse serum and 1% penicillin-streptomycin. When cells were treated with FA, the medium was changed to DMEM/F-12 medium supplemented with 1% FBS and 1% penicillin-streptomycin.
2.2 Construction of recombinant adenoviruses
The plasmid that contained the cDNA of the human ACSL1 (pBS-hACS) was purchased from Open Biosystems. The hACSL1 cDNA was isolated with double digestion using BamHI and XbaI restriction enzymes. The 5′ and 3′ ends of hACSL1 were blunted with DNA polymerase I, Large (Klenow) Fragment. Accordingly, pAd-TrackCMV plasmid was digested with SalI restriction enzyme and ends were blunted with Klenow fragment. The cDNA of hACSL1 was then cloned in pAd-TrackCMV. The pAd-TrackCMV-hACSL1 plasmid was used to produce adenoviral particles as previously described [24] using the Ad-Easy-1 system [25]. The recombinant adenoviral vectors were linearized with PacI and used to infect human embryonic kidney 293 cells. The recombinant adenoviruses were purified by two consecutive cesium chloride ultracentrifugation steps, dialyzed, and titrated. Usually, titers of −5 × 1010 plaque-forming units (pfu)/ml were obtained. The adenovirus expressing the human PPARγ cDNA (Ad-PPARγ) was purchased from Vector Biolabs (Philadelphia, PA, USA). The adenovirus expressing ATGL was constructed as described previously [26]. Recombinant adenoviruses expressing green fluorescent protein (Ad-GFP) served as control.
2.3 Infection of cell cultures with recombinant adenoviruses and treatments with FA
At day 1, AC16 cells were plated in 6-well plates. The next day, cells were infected with either one of the recombinant adenoviruses, or solely GFP (control group) (multiplicity of infection - MOI: 10). Two days post-infection the cells were treated with FAs (palmitate or OA diluted in methanol, or solely methanol as a control) at a concentration of 0.1 mM or 0.4 mM. FA-free bovine serum albumin (BSA) fraction V (Sigma-Aldrich, St. Louis, Missouri, USA) was added (1%) to serve as a FA carrier. Cells were treated for 15h. Subsequently, cells were processed for RNA isolation, protein isolation, or Oil-red-O staining.
2.4 Western blots
Cellular protein was isolated using lysis buffer containing 20 mM Tris-HCL (pH 8.0), 2 mM EDTA, 2mM EGTA, 6 mM β-mercaptoethanol, 0.1 mM sodium vanadate, 50 mM NaF, and complete protease inhibitor cocktail (Roche Pharma, Nutley, New Jersey, USA). Protein concentration was determined using the Pierce® BCA protein assay kit (Thermo Scientific, Waltham, Massachusetts, USA) and equal amounts of protein were loaded per lane. GAPDH protein expression was used as loading control. Membranes were incubated with antibodies against ACSL1, PPARγ, eIF2a and phospho-eIF2a (Ser51) (all from Cell Signaling Technology, Danvers, Massachusetts, USA). Secondary antibodies (goat-anti-rabbit and goat-anti-mouse; Santa Cruz Biotechnology, Santa Cruz, California, USA) were horseradish peroxidase (HRP) conjugated and detected using an ECL Western blotting detection kit (GE Healthcare, Pittsburgh, Pennsylvania, USA).
2.5 RNA isolation, cDNA synthesis and gene expression analyses
Total RNA was purified using Trizol reagent and treated with DNase, following the manufacturer’s instructions (Invitrogen, Carlsbad, California, USA). cDNA was synthesized using the SuperScript cDNA synthesis system from Invitrogen (Carlsbad, California, USA) according to the supplier’s protocol. Quantitative real-time polymerase chain reactions (qPCR) were conducted with the Stratagene Mx3005 qPCR System (Stratagene, La Jolla, California, USA), using the Brilliant SYBR Green qPCR kit of Stratagene (La Jolla, California, USA). Primer sequences are listed in Supplemental Table 1. All samples were analyzed in duplicates and standardized to β-actin expression. Relative quantification of gene expression was performed with the comparative Ct method.
2.6 Oil-red-O staining
Neutral lipids were stained using Oil-red-O as previously described [2]. Microscopy was performed using a Nikon Eclipse E200 microscope (Nikon, Meltville, New York, USA) and digital images were obtained with a SPOT Insight Firewire digital camera (Diagnostic Instruments Inc., Sterling Heights, Michigan, USA) using SPOT Advanced software (Diagnostic Instruments Inc., Sterling Heights, Michigan, USA).
2.7 Cellular lipid analyses
Cellular lipids were extracted and TAG and free fatty acids (FFAs) measurements were performed as described previously [2].
2.8 Statistical analyses
The data were analyzed using the statistical program SPSS (SPSS 16.0 for Windows, Chicago, Illinois, USA). Differences between groups were evaluated with two-sided t-tests. Results were considered significant when P<0.05. Data are expressed as mean ± standard error of the mean (SEM).
3. Results
3.1 Palmitate induces ER stress in cultured human cardiomyocytes
AC16 cells were treated with 0.4 mM palmitate (PA; C16:0) or 0.4 mM monounsaturated fatty acid, oleate (OA; C18:1). Both treatments resulted in intracellular lipid accumulation (Fig. 1A). OA but not PA treatment significantly increases TAG accumulation (CTRL: 1.0-fold ± 0.19 vs PA: 1.46-fold ± 0.20, p=0.08 vs OA: 2.12-fold ± 0.20, p=0.008; Supplemental Fig. 1A). Treatment with PA induced the expression of genes encoding ER stress markers such as activating transcription factor 6 (ATF6) (1.4-fold, P=0.003) (Fig. 1B), DNA-damage-inducible transcript 3 (DDIT3/CHOP) (5.8-fold, P<0.001) (Fig 1C) and heat shock 70 protein GRP78 (1.4-fold, P=0.03) (Fig. 1D). This finding indicates that PA is a potent activator of ER stress. In contrast, treatment with OA did not modulate these ER stress markers (P=0.42 for ATF6, P=0.62 for CHOP) (Fig. 1). GRP94 was not altered in any of the treatment groups (Fig. 1E).
Fig. 1. Palmitate induces expression of ER stress markers.
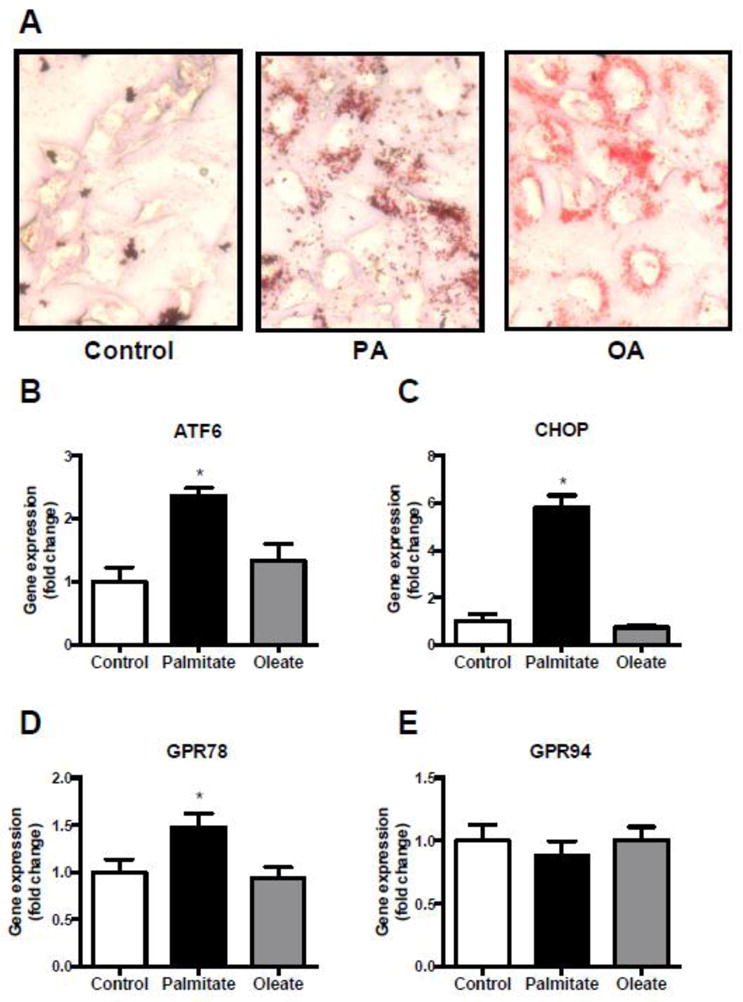
(A) Oil-red-O staining of cardiomyocytes after overnight incubation with 400μM BSA-coupled palmitate (PA), OA(OA) or BSA with methanol as a control (magnification x40). (B–E) Gene expression of the ER stress markers ATF6 (B), CHOP (C), GRP78 (D) and GRP94 (E) after overnight treatment with 400μM BSA-coupled palmitate (PA), oleate (OA) or solely BSA and methanol as a control. Data are presented as fold change in gene expression relative to the control group. *P<0.05, n=4, error bars represent SEM.
3.2 PPARγ-overexpression augments neutral lipid accumulation and inhibits PA-mediated induction of ER stress markers in cultured cardiomyocytes
TAG formation has been correlated with prevention of PA-mediated lipotoxicity [2, 18] and specifically ER-stress [27]. As PPARγ has been associated with cardiac TAG accumulation [28, 29] we performed adenovirus-mediated PPARγ overexpression in AC16 cells that were subsequently treated with PA. Recombinant adenovirus-mediated expression of PPARγ was verified by Western blot for PPARγ expression (Fig. 2A) and a dose-dependent induction of the target gene CD36 (Fig. 2B). Incubation with PA increased neutral lipid accumulation in cells transfected with Ad-PPARγ even when the PA concentration was 0.1 mM, which did not result in neutral lipid accumulation in control cells treated with Ad-GFP (Fig. 2C). Accordingly, Ad-PPARγ infection further increased neutral lipid accumulation in AC16 cells that were treated with a low concentration of OA (0.1 mM) (Fig. 2C). TAG levels upon palmitate treatment were increased in PPARγ-infected cells compared to GFP-controls (CTRL: 1.0-fold ± 0.19; PA: 1.46-fold ± 0.20, p=0.08 vs CTRL; PPARγ: 1.47-fold ± 0.071, p=0.05 vs CTRL; PPARγ + PA: 2.06 ± 0.072, p=0.034 vs PA; Supplemental Fig. 1A). Likewise, non-esterified fatty acid levels (NEFA) were reduced by PPARγ overexpression (Supplemental Fig. 1B).
Fig. 2. Ectopic expression of PPARγ increases neutral lipid accumulation in cardiomyocytes.
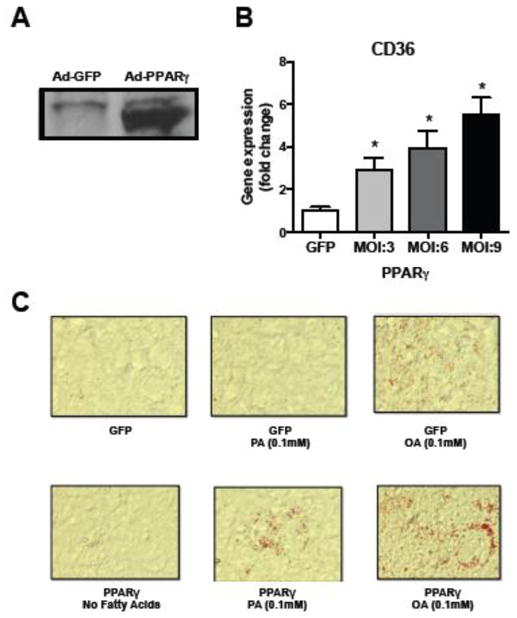
(A) Western Blot data demonstrating successful adenovirus-mediated overexpression of PPARγ in AC16 cells (MOI: 10). Ad: adenovirus. (B) Dose-dependent induction of the PPAR target gene CD36 upon PPARγ overexpression. MOI: Multiplicity of infection. *P<0.05 versus GFP-control, n=4–6 per condition. Error bars represent SEM. (C) PPARγ overexpression (MOI: 10) increased neutral lipid accumulation (Oil-red-O staining, magnification x10). PA: palmitate, OA: oleate.
AC16 cells were then treated with Ad-PPARγ and 0.4mM PA. As hypothesized, PPARγ expression increased neutral lipid storage and blunted PA-induced upregulation of the ER stress markers ATF6 (PA-treated PPARγ overexpressing cells versus Ad-GFP treated controls; P<0.001) (Fig. 3A), CHOP (P=0.02) (Fig. 3B) and GRP78 (P=0.03) (Fig. 3C). Phosphorylation of eukaryotic initiation factor 2a (EIF2a), which is known to be increased in ER stress conditions under the influence of PRKR-like ER kinase (PERK) activity [31], increased 174% after incubation with PA (P=0.03, Fig. 3D). Consistent with the blunted transcriptional induction of the ER stress markers ATF6 and CHOP, induction of EIF2a phosphorylation upon PA treatment was diminished in PPARγ overexpressing cells (Fig. 3B), while cells treated with Ad-PPARγ alone did not have altered pEIF2α/total EIF2α levels (Supplemental Fig. 2).
Fig. 3. PPARγ overexpression reduces palmitate-induced ER stress in cardiomyocytes.
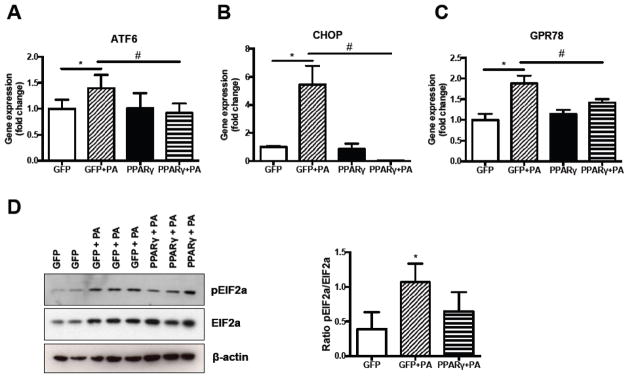
(A–C) Reduction in palmitate-induced induction of ER stress markers ATF6 (A), CHOP (B) and GRP78 (C) upon PPARγ overexpression. N=3–4. (B) Representative Western Blot and quantification of eIF2a phosphorylation. PA: palmitate. N=3. *P<0.05 BSA-control versus palmitate within groups, #P<0.05 PPARγ versus GFP, error bars represent SEM.
3.3 Acyl-CoA synthetase-mediated induction of intracellular neutral lipid accumulation protects against PA-mediated induction of ER stress markers
ASCL1 converts FA to fatty acyl-CoAs, and directs FAs to TAG synthesis [32]. To investigate whether TAG accumulation, which occurred with PPARγ overexpression, protects against PA-induced ER stress, we treated Ad-ACSL1 infected AC16 cells with PA. Infection of AC16 cells with Ad-ACSL1 followed by treatment with 0.1 mM PA (Fig. 4A) resulted in neutral lipid accumulation, as shown by Oil-red-O staining, while control Ad-GFP-treated cells did not accumulate TAG (Fig. 4B). Similar to combined Ad-PPARγ and OA treatments, Ad-ACSL1 infection further enhanced neutral lipid accumulation in AC16 cells that were treated with 0.1 mM OA (Fig. 4B). Accordingly, ACSL1 overexpression reduced the induction of ER stress markers in PA-treated cells (ATF6; P<0.001, CHOP; P=0.001) (Fig. 4C–D). However, in contrast to PPARγ, ACSL1 overexpression did not reduce PA-induced expression of the ER stress marker GRP78 nor did it significantly increase cellular TAG levels (CTRL: 1.0-fold ± 0.19; PA: 1.46-fold ± 0.20, p=0.08 vs CTRL; ACSL: 1.32-fold ± 0.17, p=0.14 vs CTRL; ACSL + PA: 1.69 ± 0.12, p=0.196 vs PA; Supplemental Fig. 1A) or reduce cellular NEFA levels (Supplemental Fig. 1B). These data indicate that PPARγ overexpression results in better protection against PA-induced ER stress than ACSL1 overexpression.
Fig. 4. Acyl-CoA synthetase (ACSL1) overexpression increases neutral lipid accumulation and relieves ER stress.
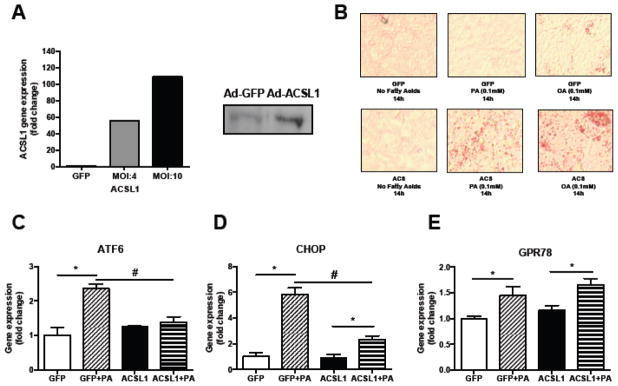
(A) qPCR and Western Blot data demonstrating successful adenovirus-mediated overexpression of ACSL1 in AC16 cells. (B) Representative images of Oil-red-O stained AdGFP or AdACSL1-infected cells treated with either palmitate (PA) or oleate (OA) (magnification x10). (C–E) ACSL1-mediated reduction in palmitate-induced expression of ER stress markers ATF6 (C), CHOP (D) and GRP78 (E). N=3–4 per condition. Data are presented as mean±SEM. *P<0.05 BSA-control versus palmitate within groups, #P<0.05 ACSL1+PA versus GFP+PA, n=4.
3.4 Increased NEFA release associated with reduced TAG storage due to overexpression of ATGL increases expression of ER stress markers
Our in vitro observations with reduced expression of ER stress markers in PA-treated cells that overexpress PPARγ contrast with previous in vivo findings showing that cardiomyocyte-specific overexpression of PPARγ (αMHC-PPARγ) increases CHOP mRNA levels [29]. However, whole genome microarray analysis of RNA obtained from hearts of αMHC-PPARγ mice show increased expression of ATGL and reduced ACSL1 (Supplemental Table 2). Thus, we tested whether ATGL-mediated release of FAs from stored TAGs might be the driving force that causes FA-induced ER stress. To this end, we treated AC16 cells with 0.4 mM of OA and adenovirus expressing ATGL (Ad-ATGL), which hydrolyzes TAG and releases NEFA. OA was chosen over PA for this experiment because OA is a better substrate for diacylglycerol acyl transferases (DGATs) and eventually leads to greater TAG synthesis compared to PA [33, 34]. Control cells were treated with Ad-GFP and OA (Fig. 5A). As anticipated, ATGL overexpression decreased intracellular neutral lipid accumulation in oleate-treated cells (Fig. 5B), reduced TAG levels (29% decrease, P=0.03) (Fig. 5C) and increased non-esterified FA (NEFA) levels (54% increase, P=0.01) (Fig. 5D). Although OA treatment alone does not induce ER stress, ectopic expression of ATGL increased mRNA levels of the ER stress markers ATF6 (4-fold, P=0.03) (Fig. 5E), CHOP (6-fold, P=0.05) (Fig. 5F), GRP78 (3-fold, P<0.01) (Fig. 5G), and GRP94 (3-fold, P<0.01) (Fig. 5H). These data suggest that NEFA released from the lipid droplet can be toxic.
Fig. 5. ATGL-mediated reduction in triglyceride (TAG) accumulation induces ER stress.

(A) qPCR data demonstrating successful adenovirus-mediated overexpression of ATGL in AC16 cells. Mean±SEM, n=7–8, *P<0.05. MOI: 10. (B) Oil-red-O staining of AdGFP or AdATGL-infected AC16 cells in the absence or presence of oleate (magnification x10). (C–D) ATGL overexpression reduces intracardiomyocellular TAG levels and increases FFA levels. Data are presented as fold change compared to the GFP control group. n=6, error bars represent SEM. *P<0.05 BSA-control versus palmitate within groups, #P<0.05 ATGL versus GFP. TAG: triacylglycerol, NEFA: non-esterified FA. (E–H) ATGL-mediated increase in ER stress markers ATF6 (E), CHOP (F), GRP78 (G) and GRP94 (H) in AC16 cells treated with oleate. *P<0.05 BSA-control versus palmitate within groups, #P<0.05 ATGL versus GFP, n=6, error bars represent SEM.
4. Discussion
Cardiac lipotoxicity is an important cause of heart failure [3, 15]. One of the mechanisms involved in the pathogenesis of heart failure is ER stress. Here we show that increased ACSL1 and PPARγ expression leads to increased neutral lipid storage in cardiomyocytes and protect against PA-induced ER stress. Increasing NEFA release by overexpression of ATGL increased ER stress. These data further support the hypothesis that neutral lipid storage capacity modulates lipotoxicity.
Although accumulation of lipids in non-adipose tissues correlates with markers for impaired cellular function [35–37], directing FAs to neutral lipid storage in lipid droplets, primarily in the form of TAG, has been shown to protect against lipotoxicity and lipid-induced insulin resistance in various tissues including skeletal muscle, liver and heart [2, 18, 38–40]. Storage of FAs in TAG limits accumulation of lipid species with greater lipotoxic potential such as DAGs and ceramides (reviewed in [41–43]). Our findings are similar to those found in vivo; cardiac lipid accumulation does not have lipotoxic consequences when due to exercise training-induced cardiac lipid accumulation [19] and DGAT1 overexpression [19, 44]. These conditions are associated with more TAG formation but less DAG and ceramide.
The use of an in vitro system allowed us to study the effects of treatment with PA or OA specifically on cells without the influences of tissue- and organ-interactions. We chose to study the quantitatively most important FAs in plasma [1] and the impact of overexpression of ACSL1 and PPARγ. Our findings are consistent with the observation made in various cell types showing that saturated FAs like PA induce lipotoxic responses, whereas unsaturated FA like OA do not. OA can even protect against PA-induced toxicity [16, 45–48]. PA, in contrast to OA, increased the expression of the ER stress markers CHOP and ATF6. OA is a better substrate for DGAT-mediated TAG synthesis [18]. Palmitoyl-CoA may accumulate in mitochondria [45, 49], trigger ROS generation [9], and function as a precursor for ceramide synthesis [13]. ACSL1 and PPARγ overexpression reduced PA-induced ER stress, as demonstrated by the elimination of the PA-induced expression of the ER stress markers CHOP and ATF6. Our findings support those by Miller et al., who showed that inhibition of ACSL1 by triacsin C resulted in a 2-fold increase in PA-mediated apoptosis [50]. Furthermore, Muoio et al., showed that overexpression of ACSL1 in hepatocytes increased incorporation of NEFA into TAGs [51]. Our results are also consistent with animal studies in which cardiac-specific increased expression of ACSL1 [32] or PPARγ [29] increased cardiac TAG levels. Although cardiac PPARγ and ACSL1 overexpression in mice caused toxicity [29, 32], our studies using an in vitro system do not allow us to assess the long-term results of these gene overexpressions. However, our studies do suggest that over the long-term ATGL hydrolysis of neutral stored lipids is likely to promote toxicity. The in vivo studies may also reflect toxic consequences of chronic TAG accumulation that is usually accompanied by accumulation of toxic lipids, such as DAG and ceramide [52].
The imbalance that renders greater lipid uptake toxic is unclear, but the comparison of our in vitro studies with those in mice illustrate the importance of TAG storage versus accumulation of less non-polar molecules. Protective effects of ACSL1 and PPARγ have been reported in other cell types. Particularly, PPARγ reduces inflammation in macrophages [23]. PPARγ overexpression was more effective in reducing palmitate-induced ER stress than ACSL1 overexpression. While ACSL1 merely catalyzes esterification of fatty acids with CoA, the initial step in intracellular fatty acid metabolism, PPARγ regulates key lipogenic pathways and regulates lipid droplet coat protein expression needed to facilitate TAG storage [53]. Our data do not exclude that the stronger protective effects of PPARγ versus ACSL1 overexpression are due to factors other than increased facilitation of TAG storage, such as increased utilization of NEFA for β-oxidation.
PPARγ actions in the heart have beneficial but sometimes also toxic actions. We recently showed that pharmacologic and genetic activation of cardiac PPARγ in mice treated with lipopolysaccharide (LPS) corrected cardiac dysfunction, further supporting the beneficial effects of increased PPARγ activity [54]. However, ectopic expression of ACSL1 or PPARγ in the heart eventually results in cardiac hypertrophy, myofibrillar disorganization, interstitial fibrosis, and left-ventricular dysfunction [32, 55, 56], indicating that prolonged overexpression of these proteins has negative consequences. Similarly, although treatment with some PPAR agonists alleviates lipid-induced toxicity by increasing uptake of circulating lipids by adipose tissue [57], more robust activation of PPARγ can cause cardiac hypertrophy [58]. Although increased cardiac ACSL1 and PPARγ levels may not be beneficial in the long-term, this study does suggest that short-term stimulation of ACSL1 and/or PPARγ may be beneficial in conditions of increased lipid supply.
To our surprise, we also found that release of FAs from stored TAGs led to ER stress. Besides free FAs other toxic lipids could be released from lipid droplets due to ATGL action, e.g. DAG. The aggravating effect of ATGL in ER stress may explain the increased CHOP mRNA levels observed in hearts of αMHC-PPARγ mice [29]; αMHC-PPARγ mice have increased cardiac ATGL expression [29]. Most studies of lipid-induced cellular and organ toxicity have focused on lipid entry leading to excess accumulation. In fact, others showed that release of lipids from lipid droplets by ATGL leads to the production of non-toxic molecules, specifically diacylglycerols that do not activate PKC [59]. Although we cannot be sure which ATGL-generated lipids lead to ER stress, our observations that ATGL activity causes non-toxic lipid treatments such as oleic acid to induce ER stress is novel and might explain some of the inflammatory and cachectic effects of cancers that are prevented in ATGL knockout mice [60]. The reduction in PPAR-mediated gene expression responsible for cardiac FA oxidation in ATGL knockout mice and the correction of cardiac dysfunction by PPAR agonists that induce FA oxidation [61] is strong evidence that accumulation of non-metabolized lipid species is responsible, at least in part, for heart dysfunction. Our data with ATGL overexpression in lipid-loaded cells show that toxic lipids can be released from stored TAG. Therefore, our data suggest that TAG storage without excess lipolysis is needed to maintain normal cardiomyocyte function.
In summary, both ACSL1 and PPARγ overexpression augmented neutral lipid accumulation in cardiomyocytes and reduced the PA-induced upregulation of ER stress markers. Reducing TAG storage by overexpression of ATGL increased ER stress. Our results indicate that channeling of FA to TAG storage in conditions of increased fat supply protects against cardiac lipid-induced ER stress. However, toxicity can occur upon accumulation of lipid intermediates that may be either due to reduced TAG synthesis or lipolysis of stored lipids.
Supplementary Material
Highlights.
Palmitate treatment of human cardiomyocyte cell line induces ER stress
ACSL1 and PPARγ augment intracellular neutral lipid accumulation
Increased neutral lipid formation ameliorates palmitate-induced ER stress
Increased lipolysis negates the protective effect of oleate and induces ER stress
Acknowledgments
This study was supported by National Heart, Lung, and Blood Institute (NHLBI) Grants HL45095, HL73029 (I.J.G) and HL112853 (K.D.). M.B. was supported by a Dr. E. Dekker Student Fellowship of the Netherlands Heart Foundation.
Abbreviations
- ACSL1
acyl-CoA synthase 1
- ATGL
adipose triglyceride lipase
- DAG
diacylglycerol
- ER
endoplasmic reticulum
- FA
fatty acid
- LD
lipid droplet
- OA
oleic acid
- PA
palmitic acid
- PPAR
peroxisome proliferator-activated receptor
- ROS
reactive oxygen species
- TAG
triacylglycerol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res. 2000;45:279–293. doi: 10.1016/s0008-6363(99)00263-1. [DOI] [PubMed] [Google Scholar]
- 2.Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, Agarwal R, Yu S, Jiang H, Steinberg SF, Blaner WS, Koch WJ, Goldberg IJ. Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metab. 2011;300:E489–499. doi: 10.1152/ajpendo.00569.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chess DJ, Stanley WC. Role of diet and fuel overabundance in the development and progression of heart failure. Cardiovasc Res. 2008;79:269–278. doi: 10.1093/cvr/cvn074. [DOI] [PubMed] [Google Scholar]
- 4.Perman JC, Bostrom P, Lindbom M, Lidberg U, StAhlman M, Hagg D, Lindskog H, Scharin Tang M, Omerovic E, Mattsson Hulten L, Jeppsson A, Petursson P, Herlitz J, Olivecrona G, Strickland DK, Ekroos K, Olofsson SO, Boren J. The VLDL receptor promotes lipotoxicity and increases mortality in mice following an acute myocardial infarction. J Clin Invest. 2011;121:2625–2640. doi: 10.1172/JCI43068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Borradaile NM, Schaffer JE. Lipotoxicity in the heart. Curr Hypertens Rep. 2005;7:412–417. doi: 10.1007/s11906-005-0035-y. [DOI] [PubMed] [Google Scholar]
- 6.Terai K, Hiramoto Y, Masaki M, Sugiyama S, Kuroda T, Hori M, Kawase I, Hirota H. AMP-activated protein kinase protects cardiomyocytes against hypoxic injury through attenuation of endoplasmic reticulum stress. Mol Cell Biol. 2005;25:9554–9575. doi: 10.1128/MCB.25.21.9554-9575.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC. Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes. Circ Res. 2006;99:275–282. doi: 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- 8.Okada K, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 9.Borradaile NM, Han X, Harp JD, Gale SE, Ory DS, Schaffer JE. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J Lipid Res. 2006;47:2726–2737. doi: 10.1194/jlr.M600299-JLR200. [DOI] [PubMed] [Google Scholar]
- 10.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: Programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91. doi: 10.1016/j.yjmcc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Groenendyk J, Sreenivasaiah PK, Kim DH, Agellon LB, Michalak M. Biology of endoplasmic reticulum stress in the heart. Circulation Res. 2010;107:1185–1197. doi: 10.1161/CIRCRESAHA.110.227033. [DOI] [PubMed] [Google Scholar]
- 12.Hotamisligil GS. Role of endoplasmic reticulum stress and c-Jun NH2-terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54:S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 13.Ostrander DB, Sparagna GC, Amoscato AA, McMillin JB, Dowhan W. Decreased cardiolipin synthesis corresponds with cytochrome c release in palmitate-induced cardiomyocyte apoptosis. J Biol Chem. 2001;276:38061–38067. doi: 10.1074/jbc.M107067200. [DOI] [PubMed] [Google Scholar]
- 14.Borradaile NM, Buhman KK, Listenberger LL, Magee CJ, Morimoto ET, Ory DS, Schaffer JE. A critical role for eukaryotic elongation factor 1A-1 in lipotoxic cell death. Mol Biol Cell. 2006;17:770–778. doi: 10.1091/mbc.E05-08-0742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Park TS, Yamashita H, Blaner WS, Goldberg IJ. Lipids in the heart: a source of fuel and a source of toxins. Curr Opin Lipidol. 2007;18:277–282. doi: 10.1097/MOL.0b013e32814a57db. [DOI] [PubMed] [Google Scholar]
- 16.De Vries JE, Vork MM, Roemen TH, de Jong YF, Cleutjens JP, van der Vusse GJ, van Bilsen M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J Lipid Res. 1997;38:1384–1394. [PubMed] [Google Scholar]
- 17.Dyntar D, Eppenberger-Eberhardt M, Maedler K, Pruschy M, Eppenberger HM, Spinas GA, Donath MY. Glucose and palmitic acid induce degeneration of myofibrils and modulate apoptosis in rat adult cardiomyocytes. Diabetes. 2001;50:2105–2113. doi: 10.2337/diabetes.50.9.2105. [DOI] [PubMed] [Google Scholar]
- 18.Listenberger LL, Han X, Lewis SE, Cases S, Farese RV, Jr, Ory DS, Schaffer JE. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci U S A. 2003;100:3077–3082. doi: 10.1073/pnas.0630588100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Shi X, Bharadwaj KG, Ikeda S, Yamashita H, Yagyu H, Schaffer JE, Yu YH, Goldberg IJ. DGAT1 expression increases heart triglyceride content but ameliorates lipotoxicity. J Biol Chem. 2009;284:36312–36323. doi: 10.1074/jbc.M109.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Aurich AC, Niemann B, Pan R, Gruenler S, Issa H, Silber RE, Rohrbach S. Age-dependent effects of high fat-diet on murine left ventricles: role of palmitate. Basic Res Cardiol. 2013;108:369. doi: 10.1007/s00395-013-0369-6. [DOI] [PubMed] [Google Scholar]
- 21.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knoll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–2853. doi: 10.1161/CIRCULATIONAHA.111.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol. 2005;39:133–147. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 23.Straus DS, Glass CK. Anti-inflammatory actions of PPAR ligands: new insights on cellular and molecular mechanisms. Trends Immunol. 2007;28:551–558. doi: 10.1016/j.it.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 24.Drosatos K, Sanoudou D, Kypreos KE, Kardassis D, Zannis VI. A dominant negative form of the transcription factor c-Jun affects genes that have opposing effects on lipid homeostasis in mice. J Biol Chem. 2007;282:19556–19564. doi: 10.1074/jbc.M700986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He TC, Zhou S, da Costa LT, Yu J, Kinzler KW, Vogelstein B. A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A. 1998;95:2509–2514. doi: 10.1073/pnas.95.5.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reid BN, Ables GP, Otlivanchik OA, Schoiswohl G, Zechner R, Blaner WS, Goldberg IJ, Schwabe RF, Chua SC, Jr, Huang LS. Hepatic overexpression of hormone-sensitive lipase and adipose triglyceride lipase promotes fatty acid oxidation, stimulates direct release of free fatty acids, and ameliorates steatosis. J Biol Chem. 2008;283:13087–13099. doi: 10.1074/jbc.M800533200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Peter A, Weigert C, Staiger H, Machicao F, Schick F, Machann J, Stefan N, Thamer C, Häring H, Schleicher E. Individual stearoyl-CoA desaturase 1 expression modulates endoplasmic reticulum stress and inflammation in human myotubes and is sssociated with skeletal muscle lipid storage and insulin sensitivity in vivo. Diabetes. 2009;58:1757–1765. doi: 10.2337/db09-0188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Son NH, Yu S, Tuinei J, Arai K, Hamai H, Homma S, Shulman GI, Abel ED, Goldberg IJ. PPARy-induced cardiolipotoxicity in mice is ameliorated by PPARa deficiency despite increases in fatty acid oxidation. J Clin Invest. 2010;120:3443–3454. doi: 10.1172/JCI40905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–2801. doi: 10.1172/JCI30335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiAugustine RP, Schaefer JM, Fouts JR. Hepatic lipid droplets. Isolation, morphology and composition. Biochem J. 1973;132:323–327. doi: 10.1042/bj1320323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kuloglu T, Aydin S, Eren MN, Yilmaz M, Sahin I, Kalayci M, Sarman E, Kaya N, Yilmaz OF, Turk A, Aydin Y, Yalcin MH, Uras N, Gurel A, Ilhan S, Gul E, Aydin S. Irisin: a potentially candidate marker for myocardial infarction. Peptides. 2014 doi: 10.1016/j.peptides.2014.02.008. [DOI] [PubMed] [Google Scholar]
- 32.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–822. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bell RM, Coleman RA. Enzymes of glycerolipid synthesis in eukaryotes. Annu Rev Biochem. 1980;49:459–487. doi: 10.1146/annurev.bi.49.070180.002331. [DOI] [PubMed] [Google Scholar]
- 34.Mayorek N, Grinstein I, Bar-Tana J. Triacylglycerol synthesis in cultured rat hepatocytes. The rate-limiting role of diacylglycerol acyltransferase. Eur J Biochem. 1989;182:395–400. doi: 10.1111/j.1432-1033.1989.tb14844.x. [DOI] [PubMed] [Google Scholar]
- 35.Goodpaster BH, Theriault R, Watkins SC, Kelley DE. Intramuscular lipid content is increased in obesity and decreased by weight loss. Metabolism. 2000;49:467–472. doi: 10.1016/s0026-0495(00)80010-4. [DOI] [PubMed] [Google Scholar]
- 36.Pan DA, Lillioja S, Kriketos AD, Milner MR, Baur LA, Bogardus C, Jenkins AB, Storlien LH. Skeletal muscle triglyceride levels are inversely related to insulin action. Diabetes. 1997;46:983–988. doi: 10.2337/diab.46.6.983. [DOI] [PubMed] [Google Scholar]
- 37.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. The FASEB Journal. 2004;18:1692–1700. doi: 10.1096/fj.04-2263com. [DOI] [PubMed] [Google Scholar]
- 38.Bosma M, Hesselink MKC, Sparks LM, Timmers S, Ferraz MJ, Mattijssen F, van Beurden D, Schaart G, de Baets MH, Verheyen FK, Kersten S, Schrauwen P. Perilipin 2 improves insulin sensitivity in skeletal muscle despite elevated intramuscular lipid levels. Diabetes. 2012;61:2679–2690. doi: 10.2337/db11-1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schenk S, Horowitz JF. Acute exercise increases triglyceride synthesis in skeletal muscle and prevents fatty acid-induced insulin resistance. J Clin Invest. 2007;117:1690–1698. doi: 10.1172/JCI30566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brown JM, Betters JL, Lord C, Ma Y, Han X, Yang K, Alger HM, Melchior J, Sawyer J, Shah R, Wilson MD, Liu X, Graham MJ, Lee R, Crooke R, Shulman GI, Xue B, Shi H, Yu L. CGI-58 knockdown in mice causes hepatic steatosis but prevents diet-induced obesity and glucose intolerance. J Lipid Res. 2010;51:3306–3315. doi: 10.1194/jlr.M010256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bosma M, Kersten S, Hesselink MKC, Schrauwen P. Re-evaluating lipotoxic triggers in skeletal muscle: Relating intramyocellular lipid metabolism to insulin sensitivity. Prog Lipid Res. 2012;51:36–49. doi: 10.1016/j.plipres.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 42.Summers SA. Ceramides in insulin resistance and lipotoxicity. Prog Lipid Res. 2006;45:42–72. doi: 10.1016/j.plipres.2005.11.002. [DOI] [PubMed] [Google Scholar]
- 43.Erion DM, Shulman GI. Diacylglycerol-mediated insulin resistance. Nat Med. 2010;16:400–402. doi: 10.1038/nm0410-400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu L, Yu S, Khan RS, Homma S, Schulze PC, Blaner WS, Yin Y, Goldberg IJ. Diacylglycerol acyl transferase 1 overexpression detoxifies cardiac lipids in PPARγ transgenic mice. J Lipid Res. 2012;53:1482–1492. doi: 10.1194/jlr.M024208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lockridge JB, Sailors ML, Durgan DJ, Egbejimi O, Jeong WJ, Bray MS, Stanley WC, Young ME. Bioinformatic profiling of the transcriptional response of adult rat cardiomyocytes to distinct fatty acids. J Lipid Res. 2008;49:1395–1408. doi: 10.1194/jlr.M700517-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab. 2006;291:E275–281. doi: 10.1152/ajpendo.00644.2005. [DOI] [PubMed] [Google Scholar]
- 47.Hickson-Bick DL, Buja LM, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol. 2000;32:511–519. doi: 10.1006/jmcc.1999.1098. [DOI] [PubMed] [Google Scholar]
- 48.Okere IC, Chandler MP, McElfresh TA, Rennison JH, Sharov V, Sabbah HN, Tserng KY, Hoit BD, Ernsberger P, Young ME, Stanley WC. Differential effects of saturated and unsaturated fatty acid diets on cardiomyocyte apoptosis, adipose distribution, and serum leptin. Am J Physiol Heart Circ Physiol. 2006;291:H38–44. doi: 10.1152/ajpheart.01295.2005. [DOI] [PubMed] [Google Scholar]
- 49.Duncan JG. Lipotoxicity: what is the fate of fatty acids? J Lipid Res. 2008;49:1375–1376. doi: 10.1194/jlr.E800010-JLR200. [DOI] [PubMed] [Google Scholar]
- 50.Miller TA, LeBrasseur NK, Cote GM, Trucillo MP, Pimentel DR, Ido Y, Ruderman NB, Sawyer DB. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem Biophys Res Commun. 2005;336:309–315. doi: 10.1016/j.bbrc.2005.08.088. [DOI] [PubMed] [Google Scholar]
- 51.Muoio DM, Lewin TM, Wiedmer P, Coleman RA. Acyl-CoAs are functionally channeled in liver: potential role of acyl-CoA synthetase. Am J Physiol Endocrinol Metab. 2000;279:E1366–E1373. doi: 10.1152/ajpendo.2000.279.6.E1366. [DOI] [PubMed] [Google Scholar]
- 52.Goldberg IJ, Trent CM, Schulze PC. Lipid metabolism and toxicity in the heart. Cell Metab. 2012;15:805–812. doi: 10.1016/j.cmet.2012.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nakachi Y, Yagi K, Nikaido I, Bono H, Tonouchi M, Schonbach C, Okazaki Y. Identification of novel PPARgamma target genes by integrated analysis of ChIP-on-chip and microarray expression data during adipocyte differentiation. Biochem Biophys Res Commun. 2008;372:362–366. doi: 10.1016/j.bbrc.2008.05.037. [DOI] [PubMed] [Google Scholar]
- 54.Drosatos K, Khan RS, Trent CM, Jiang H, Son NH, Blaner WS, Homma S, Schulze PC, Goldberg IJ. Peroxisome proliferator-activated receptor-gamma activation prevents sepsis-related cardiac dysfunction and mortality in mice. Circ Heart Fail. 2013;6:550–562. doi: 10.1161/CIRCHEARTFAILURE.112.000177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morrow JP, Katchman A, Son NH, Trent CM, Khan R, Shiomi T, Huang H, Amin V, Lader JM, Vasquez C, Morley GE, D’Armiento J, Homma S, Goldberg IJ, Marx SO. Mice with cardiac overexpression of peroxisome proliferator-activated receptor gamma have impaired repolarization and spontaneous fatal ventricular arrhythmias. Circulation. 2011;124:2812–2821. doi: 10.1161/CIRCULATIONAHA.111.056309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee Y, Naseem RH, Duplomb L, Park BH, Garry DJ, Richardson JA, Schaffer JE, Unger RH. Hyperleptinemia prevents lipotoxic cardiomyopathy in acyl CoA synthase transgenic mice. Proc Natl Acad Sci U S A. 2004;101:13624–13629. doi: 10.1073/pnas.0405499101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vikramadithyan RK, Hirata K, Yagyu H, Hu Y, Augustus A, Homma S, Goldberg IJ. Peroxisome proliferator-activated receptor agonists modulate heart function in transgenic mice with lipotoxic cardiomyopathy. J Pharmacol Exp Ther. 2005;313:586–593. doi: 10.1124/jpet.104.080259. [DOI] [PubMed] [Google Scholar]
- 58.Bilan VP, Salah EM, Bastacky S, Jones HB, Mayers RM, Zinker B, Poucher SM, Tofovic SP. Diabetic nephropathy and long-term treatment effects of rosiglitazone and enalapril in obese ZSF1 rats. J Endocrinol. 2011;210:293–308. doi: 10.1530/JOE-11-0122. [DOI] [PubMed] [Google Scholar]
- 59.Eichmann TO, Kumari M, Haas JT, Farese RV, Zimmermann R, Lass A, Zechner R. Studies on the substrate and stereo/regioselectivity of adipose triglyceride lipase, hormone-sensitive lipase, and diacylglycerol-O-acyltransferases. J Biol Chem. 2012 doi: 10.1074/jbc.M112.400416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Das SK, Eder S, Schauer S, Diwoky C, Temmel H, Guertl B, Gorkiewicz G, Tamilarasan KP, Kumari P, Trauner M, Zimmermann R, Vesely P, Haemmerle G, Zechner R, Hoefler G. Adipose triglyceride lipase contributes to cancer-associated cachexia. Science. 2011;333:233–238. doi: 10.1126/science.1198973. [DOI] [PubMed] [Google Scholar]
- 61.Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FPW, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat Med. 2011;17:1076–1085. doi: 10.1038/nm.2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.