

Abstract
The specific contribution of risk or candidate gene variants to the complex phenotype of schizophrenia is largely unknown. Studying the effects of such variants on brain function can provide insight into disease-associated mechanisms on a neural systems level. Previous studies found common variants in the complexin2 (CPLX2) gene to be highly associated with cognitive dysfunction in schizophrenia patients. Similarly, cognitive functioning was found to be impaired in Cplx2 gene-deficient mice if they were subjected to maternal deprivation or mild brain trauma during puberty. Here, we aimed to study seven common CPLX2 single nucleotide polymorphisms (SNPs) and their neurogenetic risk mechanisms by investigating their relationship to a schizophrenia-related functional neuroimaging intermediate phenotype.
We examined functional MRI and genotype data collected from 104 patients with DSM-IV diagnosed schizophrenia and 122 healthy controls who participated in the Mind Clinical Imaging Consortium study of schizophrenia. Seven SNPs distributed over the whole CPLX2 gene were tested for association with working memory-elicited neural activity in a frontoparietal neural network.
Three CPLX2 SNPs were significantly associated with increased neural activity in the dorsolateral prefrontal cortex (DLPFC) and intraparietal sulcus in the schizophrenia sample, but showed no association in healthy controls. Since increased working memory-related neural activity in individuals with or at risk for schizophrenia has been interpreted as ‘neural inefficiency’, these findings suggest that certain variants of CPLX2 may contribute to impaired brain function in schizophrenia, possibly combined with other deleterious genetic variants, adverse environmental events or developmental insults.
Keywords: Complexin2, Imaging Genetics, Intermediate Phenotype, Working Memory, Frontoparietal Circuit, Schizophrenia
1 Introduction
The neurobiological underpinnings of severe mental disorders such as schizophrenia are still poorly understood. Twin studies suggest that certain genes predispose individuals to schizophrenia, but a concordance rate of 40 – 65% in monozygotic twins [1, 2] indicates the importance of epigenetic or environmental risk factors. The widely accepted diathesis-stress model [3–5] proposes that susceptibility to psychological disorders is due to a pre-existing vulnerability that emerges after exposure to deleterious psychological states and environmental events. The diathesis-stress conceptual model is the basis for the ‘two (or second) hit’ hypothesis of schizophrenia. First described by Bayer et al. [6], the two hit hypothesis states that a genetic defect leads to deficiencies in neuronal networks (first hit), and a second hit (e.g., an environmental hazard) further perturbs the functioning of the network, perhaps through modulation of gene activity, which then leads to the emergence of an ongoing psychotic illness.
Since there is considerable evidence for synaptic dysfunction playing a central role in the etiology of schizophrenia [7, 8], synaptic molecules such as synapsins and complexins might be of great importance for the disease process. The complexin family of presynaptic regulatory proteins consists of 4 members, of which only complexin1 and complexin2 (CPLX2) are strongly expressed in the central nervous system [9]. Complexins regulate synaptic neurotransmitter release and thereby influence synaptic signaling [10], synaptic plasticity [11], and neuronal network function [12]. Dysregulation of complexin expression occurs in neurodegenerative disorders such as Huntington’s, Parkinson’s and Alzheimer’s disease [13–15], as well as in psychiatric disorders such as schizophrenia [16, 17]. Although results from case-control genetic association studies have been inconsistent [18, 19], Eastwood and Harrison [20] found markedly reduced CPLX2 expression in the dorsolateral prefrontal cortex (DLPFC) and the superior temporal cortex (STC) in schizophrenia patients.
Recent animal models allow us to better understand how genetic and environmental risk factors of schizophrenia interact [21]. Yamauchi et al. [22] examined the physiological characteristics of Cplx2 gene-deficient mice subjected to maternal deprivation stress and found a significant decrease in post-tetanic potentiation and LTP induction in the knockout mice compared to the wild type, suggesting that in the setting of an environmental insult (maternal deprivation), variation in the Cplx2 gene becomes a critical determinant of synaptic functioning. Radyushkin et al. [23] designed a similar experimental approach for a schizophrenia-like phenotype in mice to specifically test the aforementioned ‘second hit’ hypothesis. They studied behavior and cognitive functioning (which is typically impaired in schizophrenia) in Cplx2-null mutant (Cplx2−/−) mice jointly with a mild parietal neurotrauma, applied during puberty. Consistent with the ‘second hit’ hypothesis, they found reduced pre-pulse inhibition and deficits of spatial learning as well as decreased hippocampal volume in Cplx2−/− mice after the neurotrauma had been applied, but not in lesioned wild-type mice or non-lesioned Cplx2−/− mice.
Subsequently, Begemann et al. [24] tested for associations between CPLX2 single nucleotide polymorphisms (SNPs) and several domains of cognitive functioning (executive functioning, reasoning, and verbal learning/memory) in a large sample of patients with schizophrenia (NSZ = 1,071). They found associations between six CPLX2 SNPs and impaired neurocognitive performance in patients. Assuming that schizophrenia patients have been exposed to additional environmental risk, they interpreted CPLX2 as a modifier of cognitive functioning in individuals which were subject to adverse environmental events or developmental insults (‘second hit’). Although CPLX2 has not been shown to be a risk gene for a diagnosis of schizophrenia in a recent GWA study/meta-analysis [25, 26], the results presented by Begemann et al. illustrate the impact of CPLX2 risk variants on the severity of cognitive dysfunction in patients with schizophrenia. However, the precise roles of these variants in disease-associated mechanisms on a neural systems level are unknown. Impaired working memory processing and functional abnormalities of a lateral frontoparietal network, which mediates working memory functions, are established intermediate phenotypes of schizophrenia [27–29]. Therefore, the aim of our study was to investigate the neurogenetic risk mechanisms of CPLX2 SNPs by examining their associations with frontoparietal network efficiency during working memory. Based on the above mentioned studies in rodents and in patients with schizophrenia [24], we hypothesized the presence of such associations in patients with schizophrenia, assuming that they have been exposed to additional environmental risk factors.
2 Methods and Material
2.1 Participants
Imaging, genetic and behavioral data from participants of the multisite Mind Clinical Imaging Consortium (MCIC) study of schizophrenia [30, 31] were used for analyses. All subjects gave written informed consent prior to study enrollment. The human subjects research committees at each of the four sites (Massachusetts General Hospital (MGH) and the Universities of Iowa (UI), Minnesota (UMN), and New Mexico (UNM)) approved the study protocol. The patient group (SZ) consisted of subjects with a DSM-IV diagnosis of schizophrenia established using structured clinical interviews and review of case files by trained clinicians (see also Online Resource 1.1). Healthy controls (HC) were included if they had no history of a medical or Axis I psychiatric diagnosis. All participants were required to be at least 18 years of age and no older than 60, and had to be fluent in English. Participants were excluded if they had a history of neurologic disease, or psychiatric disease other than schizophrenia, history of a head injury with loss of consciousness, history of substance abuse or dependence within the past month, severe or disabling medical conditions, any contraindication to MR scanning or an IQ less than 70 (based on the reading subtest from the Wide Range Achievement Test (WRAT3)). For details on clinical measures see Online Resource 1.1.
2.2 Sternberg Item Recognition Paradigm
The Sternberg Item Recognition Paradigm (SIRP) is a working memory (WM) task, previously shown to activate the DLPFC in healthy controls and schizophrenia patients [32]. The SIRP was administered during six 46 second blocks per run for three 360 second runs. In each block a memory set, composed of one (load1), three (load3), or five (load5) digits, was presented (two blocks per load condition). The Encode phase was followed by a presentation of 14 digits, one at a time (the Probe phase) and participants responded to each probe to indicate whether or not the probe digit was in the memory set. For additional details about the paradigm, see Roffman et al. [33] and Online Resource 1.2. The stimuli and responses were presented and collected using E-prime software (EPrime v1.1, Psychology Software Tools, Inc., Pittsburg, PA) during fMRI scanning. Participants were excluded from further analysis if they completed a block with less than a 80% accuracy rate and/or with more than 6 probes not answered within a block.
2.3 Image Acquisition and Processing
Structural MRI data were acquired with either a 1.5T Siemens Sonata (UNM, MGH, UI) or a 3T Siemens Trio (UMN) and analyzed in an automated manner with atlas-based FreeSurfer parcellation and segmentation software, version 4.0.1 (http://surfer.nmr.mgh.harvard.edu) to generate cortical and subcortical volumetric measures of regions of interest (ROIs) using each participant’s individual anatomy [34]. We focused on two ROIs, the intraparietal sulcus (IPS), which is a standard output of FreeSurfer parcellation, and the DLPFC. DLPFC ROIs were derived from a combination of several FreeSurfer cortical parcellations as described previously [35, 36]. For details see Online Resource 1.3.
Functional MRI data were acquired with either a 1.5T Siemens Sonata (UNM) or a 3T Siemens Trio (UMN, MGH, UI) and analyzed using the Function Biomedical Informatics Research Network (FBIRN) Image Processing Stream (FIPS), a pipeline using the Functional MRI of the Brain (FMRIB) Software Library of FSL [37]. Variability due to different scanners and acquisition protocols was analyzed using ten MCIC subjects who were scanned at all four sites [36]. Variability of neural activity due to site differences was negligible small compared to variability due to subject differences. For additional information about data acquisition and processing, see Online Resource 1.3.
A Functional Imaging Linear Model [38] was fit to model the Probe phases of each subject’s preprocessed functional time series. We used the linear Contrasts Of Parameter Estimate (COPE) Probe-load 5 versus Probe-load 1and refer to responses to this condition as ‘load-dependent’ activation [33]. We obtained indices of activation for the left and right IPS and left and right DLPFC ROIs using the COPE obtained from the second-level fixed-effects analysis for each participant. We applied an additional functional mask, based on the COPE of all loads (average of Probe-load 1, Probe-load 3 and Probe-load 5) versus fixation exceeding a threshold of Z = 2.3, and extracted the mean percentage signal change. The use of a functional mask (within anatomical ROIs) from all working memory loads protected against biases in signal change calculations derived from individual conditions [39].
2.4 Genotyping and Imputation
Blood samples were obtained from 255 MCIC participants and sent to the Harvard Partners Center for Genetics and Genomics for DNA extraction. All DNA extraction and genotyping was done blind to group assignment. Genotyping was performed at the Mind Research Network Neurogenetics Core Lab using the IlluminaHumanOmni-Quad BeadChip. Quality control steps included common standard procedures [40] using PLINK, 1.07 [41]. SNPs on the X or Y chromosome, or those with a genotyping rate of less than 90% or a minor allele frequency of less than 5% were excluded from the analysis. We removed seven participants with extreme heterozygosity values (+/− 3SD). Another 22 subjects were excluded because of low quality or missing fMRI data (for imaging control steps see Online Resource 1.3). The final sample with complete and high-quality structural and functional MRI and genetic data comprised of 122 HC and 104 SZ.
Additional SNPs were imputed based on the Hapmap3 dataset. Imputation was done using IMPUTE2 [42] with a probability threshold of 0.95. The imputed data set was then again filtered for a minor allele frequency of 5%, a genotyping rate of less than 90% and a Hardy-Weinberg equilibrium in controls with a threshold of 10−6. Using this data set, seven CPLX2 SNPs (rs2443541, rs2243404, rs4242187, rs10072860, rs4868539, rs3892909, and rs3822674) were extracted, five of those being significantly associated with current cognition in patients with schizophrenia in a recent candidate gene study [24]. General information and quality control measures including details about SNP location, an LD plot and allele frequencies according to diagnostic group for all seven SNPs are given in Online Resource Table 1 and 2 and Online Resource Figure 1. Because of the low frequency of T/T genotypes for both rs2243404 (N = 6) and rs2443541 (N = 6), T/T and C/T participants were combined into a T allele carrier group (N = 37 and N = 38, respectively).
2.5 Statistics & Principal Component Analysis (PCA)
Given our strong hypothesis, we primarily analyzed the patient sample (N = 104). Additional (separate) analyses were carried out for the control group (N = 122). Basic demographic characteristics were compared across genotype group and across sites with different scanner field strengths using one way ANOVA and subsequent Bonferroni post-hoc tests where necessary. Chi-square statistics were used to examine differences in categorical variables. Alpha was set to 0.05 for all analyses.
Each of the seven CPLX2 SNPs was tested for association with neural activity in the a priori defined working memory-related brain regions using a linear mixed model and controlling for age, gender and scanner field strength. We assumed a compound symmetry covariance structure for the load-dependent change of neural activity in the four brain regions (DLPFC and IPS mean activity in the left and right hemisphere). Population stratification was addressed using ten principal components as additional covariates calculated by EIGENSTRAT of the EIGENSOFT 3.0 software package [43, 44]. Details are given in Online Resource 1.6. We ran separate mixed models for the two diagnostic groups (SZ and HC). Mixed models for rs2243404 and rs2443541 were performed contrasting C allele homozygotes and T allele carriers, respectively, because of the small number of participants in the TT group. Multiple testing was addressed by Bonferroni correction (P < 0.007; corrected for the number of tested SNPs). All statistical analyses were carried out in IBM SPSS Statistics 19.
To verify our results in an ethnically homogeneous sample we re-ran the analyses in a subsample defined by stringent genetic criteria, including individuals of European descent only (NSZ = 68; NHC = 95; see Online Resource 1.7 for further details). Additional analyses were carried out on a whole-brain level using FSL (FMRIB Software Library; version 4.1.7; Smith et al. [37]). Please refer to Online Resource 1.8 for methodological details.
3 Results
3.1 Sample characteristics
In the patient group, there were no differences among genotype groups (seven CPLX2 SNPs) with respect to gender, age, parental SES, handedness, length of illness, and negative or positive symptoms. Rs3892909 T allele homozygotes and rs4868539 A allele homozygotes showed significantly higher WRAT3-RT scores, rs3892909 C allele homozygotes had higher lifetime antipsychotic exposure and rs4242187 C/T heterozygotes showed a higher working memory performance than C/C or T/T homozygotes (see Online Resource 2.1, Online Resource Table 3). Also, patients from acquisition sites with different scanner field strengths did not differ in any demographic or clinical variables. In the control group, there were no differences among genotype groups for any of the tested variables (data not shown). An overview about the demographic (and clinical) variables of patients with schizophrenia and healthy controls, respectively, is displayed in Table 1. As expected, schizophrenia patients had a significantly lower performance on the Sternberg Paradigm when compared to healthy controls (F = 44.26; p < 0.001).
Table 1.
Demographic and clinical variables
| Scanner field strength |
Sex (female) |
Age (years) |
WRAT3-RT | Parental SES | Handedness | WM Performance |
Length of Illness (years) |
Negative Symptoms |
Positive Symptoms |
Medication (cum. dose years) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||||||||||||||
| N | N | % | mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | mean | SD | |
|
Schizophrenia patients
| |||||||||||||||||||||
| 1.5T | 19 | 5 | 26.3 | 36.26 | 13.82 | 45.28 | 6.92 | 2.88 | 1.11 | 1.32 | 3.06 | 95.47 | 3.98 | 11.71 | 12.53 | 8.58 | 3.58 | 4.74 | 2.66 | 26.66 | 32.99 |
| 3T | 85 | 21 | 24.7 | 34.19 | 10.26 | 46.65 | 6.63 | 2.81 | 0.98 | 1.13 | 2.99 | 95.58 | 4.50 | 11.56 | 9.78 | 7.84 | 3.83 | 4.82 | 2.84 | 63.85 | 134.65 |
| Total | 104 | 26 | 25.0 | 34.57 | 10.95 | 46.40 | 6.67 | 2.82 | 0.99 | 1.17 | 2.99 | 95.56 | 4.39 | 11.58 | 10.27 | 7.97 | 3.78 | 4.81 | 2.79 | 56.85 | 122.86 |
|
| |||||||||||||||||||||
|
Healthy controls
| |||||||||||||||||||||
| 1.5T | 22 | 4a | 18.2 | 30.27 | 12.84 | 51.50 | 3.79 | 2.14b | 0.77 | 1.05 | 2.42 | 98.90 | 0.70 | - | - | - | - | - | - | - | - |
| 3T | 100 | 42a | 42.0 | 33.06 | 10.87 | 50.75 | 4.08 | 2.80b | 0.68 | 0.85 | 2.62 | 98.32 | 1.72 | - | - | - | - | - | - | - | - |
| Total | 122 | 46 | 37.7 | 32.56 | 11.25 | 50.89 | 4.03 | 2.68 | 0.74 | 0.89 | 2.58 | 98.42 | 1.61 | - | - | - | - | - | - | - | - |
Means and standard deviations (SD) are given. SZ = patient with schizophrenia; HC = healthy control. WRAT3-RT = Wide Range Achievement Test 3 - Reading Test. Parental SES (socioeconomic status) was classified according to Hollingshead, and handedness determined using the Annett Scale of Hand Preference. Working memory (WM) performance represents the mean accuracy during SIRP across all loads (1, 3, 5). Severity of positive and negative symptoms was rated using the Scale for the Assessment of Positive Symptoms (SAPS) and the Scale for the Assessment of Negative Symptoms (SANS). Cumulative antipsychotic exposure was calculated using the chlorpromazine (CPZ) conversion factors of Woods et al. [62].
significantly different between 1.5T and 3T sites on basis of Chi-Square (p < 0.05).
significantly different between 1.5T and 3T sites on basis of Welch (p < 0.05).
The patient group comprised more females (X2(1) = 4.17, p = 0.041), showed lower WRAT3-RT scores (F(1,220) = 38.23, p < 0.001) and a lower performance in the WM task (F(1,221) = 44.26, p < 0.001).
3.2 Functional MRI
Three CPLX2 SNPs (rs2443541, rs2243404, and rs4868539) were associated with load-dependent neural activity in working memory-related brain regions (DLPFC and IPS; see Fig. 1) in the schizophrenia sample. For both, rs2443541 and rs2243404, C allele homozygotes showed an increased neural activity when compared to T allele carriers (F(df=1,104) = 12.387, p = 0.001 and F(df=1,104) = 12.972, p < 0.001, respectively). The same was found for rs4868539 G allele carriers compared to A allele homozygotes (F(df=2,104) = 5.530, p = 0.005; see Table 2). According to Bonferroni-corrected post-hoc tests, the estimated residuals of the neural activity differed significantly between G allele homozygotes and A allele homozygotes (p = 0.024), as well as between heterozygotes and A allele homozygotes (p = 0.005), but not between G allele homozygotes and heterozygotes. We found no evidence for associations between working memory-related neural activity and any of the seven CPLX2 SNPs in healthy participants. Table 3 displays the test statistics and p-values for the linear mixed models of each SNP. For CPLX2 rs2443541 and rs2243404 the aforementioned effects were also determinable on a whole-brain level (see Online Resource 2.2).
Fig 1. FreeSurfer cortical parcellation of the DLPFC and IPS in the schizophrenia sample.
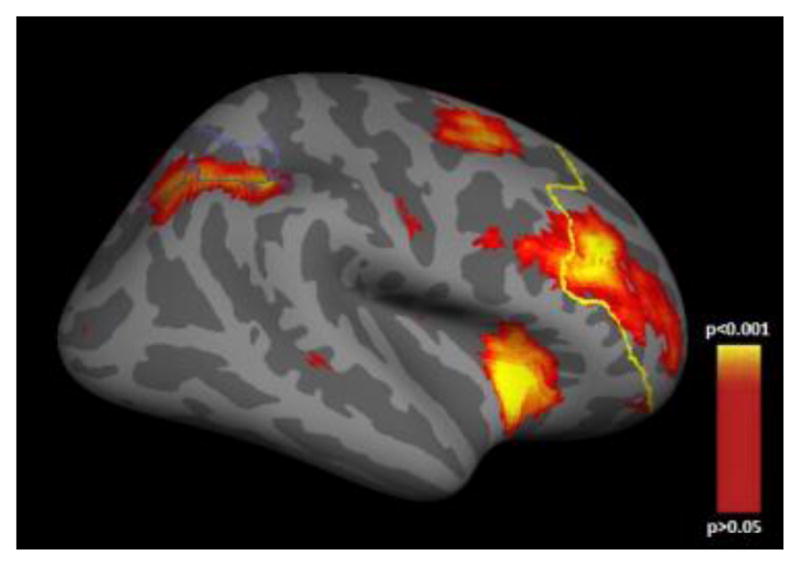
Cortical statistical map illustrating regions of working memory-related load-dependent neural activity in the dorsolateral prefrontal cortex (anterior outline) and the intraparietal sulcus (posterior outline) in the schizophrenia sample. This map is shown on the inflated surface of the standard average subject allowing visualization of data across the entire cortical surface without interference from cortical folding.
Table 2.
Associations between working memory-related load-dependent neural activity and CPLX2 SNPs in schizophrenia patients
| rs2443541 | rs2243404 | rs4242187 | rs10072860 | rs4868539 | rs3892909 | rs3822674 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CC | T carrier | CC | T carrier | CC | CT | TT | GG | GT | TT | GG | AG | AA | CC | CT | TT | CC | CT | TT | |
|
| |||||||||||||||||||
| N | 67 | 37 | 67 | 37 | 43 | 47 | 14 | 30 | 55 | 19 | 20 | 44 | 40 | 19 | 44 | 41 | 25 | 45 | 34 |
| left DLPFC | 0.072 | 0.043 | 0.072 | 0.042 | 0.062 | 0.063 | 0.055 | 0.084 | 0.053 | 0.049 | 0.085 | 0.068 | −0.003 | 0.072 | 0.070 | 0.047 | 0.056 | 0.050 | 0.080 |
| right DLPFC | 0.069 | −0.024 | 0.069 | −0.028 | 0.063 | 0.033 | −0.047 | 0.027 | 0.024 | 0.078 | 0.044 | 0.068 | −0.054 | 0.025 | 0.068 | 0.003 | 0.004 | 0.049 | 0.039 |
| left IPS | 0.087 | 0.044 | 0.087 | 0.043 | 0.077 | 0.077 | 0.035 | 0.076 | 0.068 | 0.072 | 0.076 | 0.076 | 0.053 | 0.067 | 0.083 | 0.061 | 0.086 | 0.077 | 0.053 |
| right IPS | 0.058 | 0.030 | 0.057 | 0.030 | 0.049 | 0.050 | 0.035 | 0.044 | 0.046 | 0.059 | 0.044 | 0.058 | 0.035 | 0.049 | 0.063 | 0.033 | 0.051 | 0.053 | 0.038 |
Working memory-related mean neural activity (% BOLD change) for corresponding genotype groups of each CPLX2 SNP in the schizophrenia sample; significant differences according to the linear mixed models (p < 0.007; Bonferroni-corrected for number of tested SNPs) are printed in bold face. DLPFC = dorsolateral prefrontal cortex; IPS = intraparietal sulcus. N = number of schizophrenia patients carrying the particular genotype.
Table 3.
Test statistics and p-values for the mixed models testing associations between CPLX2 variants and load-dependent working memory related neural activity in schizophrenia patients and healthy controls
| SNP | SZ | SZ (Europ. descent) | HC | HC (Europ. Descent) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | df | P | F | df | P | F | df | P | F | df | P | |
| rs2443541 | 12.387* | 1, 104 | 0.001 | 5.278 | 1, 68 | 0.025 | 1.233 | 1, 122 | 0.269 | 3.127 | 1, 95 | 0.080 |
| rs2243404 | 12.972* | 1, 104 | < 0.001 | 5.349 | 1, 68 | 0.024 | 1.454 | 1, 122 | 0.230 | 3.274 | 1, 95 | 0.074 |
| rs4242187 | 2.327 | 2, 104 | 0.103 | 0.535 | 2, 68 | 0.588 | 0.426 | 2, 122 | 0.654 | 1.380 | 2, 95 | 0.257 |
| rs10072860 | 0.849 | 2, 104 | 0.431 | 0.802 | 2, 68 | 0.453 | 1.095 | 2, 122 | 0.338 | 2.212 | 2, 95 | 0.115 |
| rs4868539 | 5.530* | 2, 104 | 0.005 | 1.638 | 2, 68 | 0.202 | 1.241 | 2, 122 | 0.293 | 1.981 | 2, 95 | 0.144 |
| rs3892909 | 1.890 | 2, 103 | 0.156 | 2.388 | 2, 67 | 0.100 | 0.007 | 2, 122 | 0.993 | 0.201 | 2, 95 | 0.819 |
| rs3822674 | 0.410 | 2, 104 | 0.665 | 0.012 | 2, 68 | 0.988 | 0.024 | 2, 122 | 0.976 | 0.661 | 2, 95 | 0.519 |
Single locus association analyses of working memory-related load-dependent neural activity (left and right DLPFC and left and right IPS) using mixed models and covarying for age, gender, scanner field strength and population stratification. Significant test statistics and p-values are displayed in bold face (p < 0.05) indicating differences between the genotype groups of each SNP.
p-value survives Bonferroni correction (p < 0.007; corrected for number of tested SNPs).
Additional models were carried out in healthy controls and in a subset of each participants group of European descent. SNP = single nucleotide polymorphism; SZ = patient with schizophrenia; HC = healthy control.
In a subsample of participants of European descent we found similar results for rs2443541 and rs2243404 in schizophrenia patients. Again, none of the tested polymorphisms showed any association with neural activity in the DLPFC or IPS in the subsample of healthy participants with European ancestry (Table 3). In subsequent models, we also covaried for the amount of lifetime antipsychotic exposure, working memory performance and WRAT3-RT scores. All main effects remained robust (Online Resource 2.3, Online Resource Table 4).
4 Discussion
Our analysis revealed an association betweenCPLX2 polymorphisms and working memory load-dependent neural activity in a frontoparietal brain network in schizophrenia patients but not in healthy controls. Results were strengthened by subsequent analyses including potentially confounding variables such as lifetime antipsychotic exposure and working memory performance. Our findings are in line with the results from Cplx2−/− rodent models and consistent with the ‘second hit’ hypothesis of schizophrenia, i.e. CPLX2 risk variants increase neural inefficiency if the individual has experienced an additional risk factor. Second hit risk factors such as prenatal infections, perinatal complications, early insults to the brain, stressful life events or drug abuse are more prevalent in people affected by schizophrenia than demographically similar control samples (for a review see Vilain et al. [45]).
The risk alleles of two (rs2443541 and rs2243404; C allele for both SNPs) out of the three genetic variants which were associated with working memory-related neural activity in our study had previously been related to cognitive functioning specifically in schizophrenia patients[24]. As described in the Supplementary Online content of Begemann et al. (eFigure 1A), for both SNPs T homozygotes performed significantly better compared to C allele carriers regarding executive functioning and verbal learning/memory, which is in line with our results. Increased load-dependent neural activity in a frontoparietal network in individuals with CPLX2 risk variants can be interpreted as ‘neural inefficiency’, i.e. the additional recruitment of neural resources to achieve a comparable task performance as non-risk carriers or healthy controls [46–48]. Based on similar findings in unaffected siblings, prodromal, medication-naïve and first-episode patients with schizophrenia, ‘neural inefficiency’ during working memory has been proposed to reflect an expression of genetic liability to schizophrenia [49–54]. A meta-analysis showed that working memory-related neural networks are consistently ‘disrupted’ in schizophrenia [55]. It has been hypothesized that this putative neural signature of schizophrenia may arise from altered signal transduction and synaptic functioning and plasticity [28, 56]. Synaptic proteins that are involved in the presynaptic secretory machinery such as complexions appear possible molecular candidates.
According to the Expression Atlas of EMBL-EBI (http://www.ebi.ac.uk/gxa/; last accessed on 2013-03-15), CPLX2 is primarily expressed in the human brain, but in the DLPFC and STC of schizophrenia patients CPLX2 expression is abnormally reduced [20]. The altered expression profile of CPLX2 and other synaptic proteins in schizophrenia suggests that there are aberrations in the precise organization and functioning of neural circuits, indicating that CPLX2 polymorphisms may contribute susceptibility to schizophrenia [19, 20]. Results of the current study provide a link between frontoparietal cortical function in schizophrenia and variation in the CPLX2 gene in schizophrenia.
The effects of CPLX2 are different from common risk genes of schizophrenia since CPLX2 seems to moderate the expression of the disease but not the general risk for schizophrenia. Other possible explanations for the case-only effect described here include gene X gene and gene X genome interactions. A variety of environmental and other factors can alter the influence of mutant alleles on organismal phenotypes, and the impact of these factors can vary with genetic background [57]. All explanations have in common, that CPLX2 is not an independent risk factor for schizophrenia.
The findings of this study should be interpreted in light of the following limitations. First, we focused on seven CPLX2 SNPs that had been previously investigated by other researchers. These SNPs may be in high linkage disequilibrium with other functional variants that are responsible for the effects described above. Second, we had an only medium-sized and rather heterogeneous sample, but subsequent models in European ancestry subsamples of patients and controls confirmed our initial results. Furthermore, results of additional whole-brain analyses support our main findings and the assumption that the observed SNP effects are rather specific to working memory-related brain regions. Third, we did not measure environmental factors in this study; we based our hypothesis on previous studies that have consistently shown a significantly higher prevalence of a variety of pre-, peri- and postnatal environmental risk factors in patients with schizophrenia compared to healthy subjects [58–60], which may represent a ‘second hit’. Finally, although we covaried for the amount of lifetime antipsychotic exposure in a secondary model and found our results to be essentially the same, we cannot completely exclude a potential effect of antipsychotic medication on our results in patients with schizophrenia. Sawada et al. [61] studied the effect of antipsychotic drug treatment on the expression of complexin and other presynaptic proteins in rats and observed no differences between treated and control animals for either Cplx1 or Cplx2 immunostaining in frontal cortex.
Taken together, our results indicate that CPLX2 may be involved in the etiology of schizophrenia and we identified one of its potential biological mechanisms by studying the effects of CPLX2 risk variants on working memory load-dependent neural activity in a frontoparietal network. Since the reported associations were restricted to the patient group, we interpret them in the framework of the ‘second hit’ hypothesis and assume that certain CPLX2 variants only confer risk for schizophrenia if other risk factors are also present. Although not directly related to the risk of the disorder, CPLX2 variants may moderate its expression. Our findings may help to understand the heterogeneity of schizophrenia and its underlying complex genetic architecture. However, additional studies are needed to replicate the findings and to fully untangle the effects of such genetic variants on brain function.
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (NIH/NCRR P41RR14075), Department of Energy (DE-FG02-99ER62764), MIND Research Network, Morphometry BIRN (1U24, RR021382A), Function BIRN (U24RR021992-01, NIH.NCRR MO1 RR025758-01), NARSAD Young Investigator Grant (SE), and the Deutsche Forschungsgemeinschaft (Research Fellowship to SE).
Footnotes
Conflict of interest
Veit Roessner has received lecture fees from Eli Lilly, Janssen-Cilag, Medice, Novartis and was a member of advisory boards of Eli Lilly and Novartis. All other authors declare that they have no conflicts of interest.
References
- 1.Cardno AG, Gottesman II. Twin studies of schizophrenia: from bow-and-arrow concordances to star wars Mx and functional genomics. Am J Med Genet. 2000;97:12–17. [PubMed] [Google Scholar]
- 2.Gottesman II, Shields J. A critical review of recent adoption, twin, and family studies of schizophrenia: behavioral genetics perspectives. Schizophr Bull. 1976;2:360–401. doi: 10.1093/schbul/2.3.360. [DOI] [PubMed] [Google Scholar]
- 3.Morley S. The stress-diathesis model of illness. J Psychosom Res. 1983;27:86–87. doi: 10.1016/0022-3999(83)90115-0. [DOI] [PubMed] [Google Scholar]
- 4.Walker EF, Diforio D. Schizophrenia: a neural diathesis-stress model. Psychol Rev. 1997;104:667–685. doi: 10.1037/0033-295x.104.4.667. [DOI] [PubMed] [Google Scholar]
- 5.Hankin BL, Abela JRZ. Development of Psychopathology: A Vulnerability-Stress Perspective. 1. SAGE Publications, Inc; 2005. [Google Scholar]
- 6.Bayer TA, Falkai P, Maier W. Genetic and non-genetic vulnerability factors in schizophrenia: the basis of the “two hit hypothesis. J Psychiatr Res. 1999;33:543–548. doi: 10.1016/s0022-3956(99)00039-4. [DOI] [PubMed] [Google Scholar]
- 7.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. image 5. [DOI] [PubMed] [Google Scholar]
- 8.Yin D-M, Chen Y-J, Sathyamurthy A, et al. Synaptic dysfunction in schizophrenia. Adv Exp Med Biol. 2012;970:493–516. doi: 10.1007/978-3-7091-0932-8_22. [DOI] [PubMed] [Google Scholar]
- 9.Reim K, Wegmeyer H, Brandstätter JH, et al. Structurally and functionally unique complexins at retinal ribbon synapses. J Cell Biol. 2005;169:669–680. doi: 10.1083/jcb.200502115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brose N. For better or for worse: complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–1413. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- 11.Takahashi S, Ujihara H, Huang GZ, et al. Reduced hippocampal LTP in mice lacking a presynaptic protein: complexin II. Eur J Neurosci. 1999;11:2359–2366. doi: 10.1046/j.1460-9568.1999.00652.x. [DOI] [PubMed] [Google Scholar]
- 12.Strenzke N, Chanda S, Kopp-Scheinpflug C, et al. Complexin-I is required for high-fidelity transmission at the endbulb of Held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basso M, Giraudo S, Corpillo D, et al. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteomics. 2004;4:3943–3952. doi: 10.1002/pmic.200400848. [DOI] [PubMed] [Google Scholar]
- 14.DiProspero NA, Chen E-Y, Charles V, et al. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–533. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- 15.Tannenberg RK, Scott HL, Tannenberg AEG, Dodd PR. Selective loss of synaptic proteins in Alzheimer’s disease: evidence for an increased severity with APOE varepsilon4. Neurochem Int. 2006;49:631–639. doi: 10.1016/j.neuint.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 16.Eastwood SL, Harrison PJ. Hippocampal synaptic pathology in schizophrenia, bipolar disorder and major depression: a study of complexin mRNAs. Mol Psychiatry. 2000;5:425–432. doi: 10.1038/sj.mp.4000741. [DOI] [PubMed] [Google Scholar]
- 17.Sawada K, Barr AM, Nakamura M, et al. Hippocampal complexin proteins and cognitive dysfunction in schizophrenia. Arch Gen Psychiatry. 2005;62:263–272. doi: 10.1001/archpsyc.62.3.263. [DOI] [PubMed] [Google Scholar]
- 18.Kishi T, Ikeda M, Suzuki T, et al. No association of complexin1 and complexin2 genes with schizophrenia in a Japanese population. Schizophr Res. 2006;82:185–189. doi: 10.1016/j.schres.2005.12.842. [DOI] [PubMed] [Google Scholar]
- 19.Lee HJ, Song JY, Kim JW, et al. Association study of polymorphisms in synaptic vesicle-associated genes, SYN2 and CPLX2, with schizophrenia. Behav Brain Funct. 2005;1:15. doi: 10.1186/1744-9081-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eastwood SL, Harrison PJ. Decreased expression of vesicular glutamate transporter 1 and complexin II mRNAs in schizophrenia: further evidence for a synaptic pathology affecting glutamate neurons. Schizophr Res. 2005;73:159–172. doi: 10.1016/j.schres.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 21.Ayhan Y, Sawa A, Ross CA, Pletnikov MV. Animal models of gene-environment interactions in schizophrenia. Behav Brain Res. 2009;204:274–281. doi: 10.1016/j.bbr.2009.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yamauchi Y, Qin L-H, Nishihara M, et al. Vulnerability of synaptic plasticity in the complexin II knockout mouse to maternal deprivation stress. Brain Res. 2005;1056:59–67. doi: 10.1016/j.brainres.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 23.Radyushkin K, El-Kordi A, Boretius S, et al. Complexin2 null mutation requires a “second hit” for induction of phenotypic changes relevant to schizophrenia. Genes Brain Behav. 2010;9:592–602. doi: 10.1111/j.1601-183X.2010.00590.x. [DOI] [PubMed] [Google Scholar]
- 24.Begemann M, Grube S, Papiol S, et al. Modification of cognitive performance in schizophrenia by complexin 2 gene polymorphisms. Arch Gen Psychiatry. 2010;67:879–888. doi: 10.1001/archgenpsychiatry.2010.107. [DOI] [PubMed] [Google Scholar]
- 25.Ripke S, O’Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013 doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Singh S, Kumar A, Agarwal S, et al. Genetic insight of schizophrenia: past and future perspectives. Gene. 2014;535:97–100. doi: 10.1016/j.gene.2013.09.110. [DOI] [PubMed] [Google Scholar]
- 27.Burns J, Job D, Bastin ME, et al. Structural disconnectivity in schizophrenia: a diffusion tensor magnetic resonance imaging study. BJP. 2003;182:439–443. doi: 10.1192/bjp.02.396. [DOI] [PubMed] [Google Scholar]
- 28.Deserno L, Sterzer P, Wüstenberg T, et al. Reduced prefrontal-parietal effective connectivity and working memory deficits in schizophrenia. J Neurosci. 2012;32:12–20. doi: 10.1523/JNEUROSCI.3405-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hall M-H, Smoller JW. A new role for endophenotypes in the GWAS era: functional characterization of risk variants. Harv Rev Psychiatry. 2010;18:67–74. doi: 10.3109/10673220903523532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ehrlich S, Yendiki A, Greve DN, et al. Striatal function in relation to negative symptoms in schizophrenia. Psychol Med. 2011:1–16. doi: 10.1017/S003329171100119X. [DOI] [PubMed] [Google Scholar]
- 31.Gollub RL, Shoemaker JM, King MD, et al. The MCIC Collection: A Shared Repository of Multi-Modal, Multi-Site Brain Image Data from a Clinical Investigation of Schizophrenia. Neuroinformatics. 2013 doi: 10.1007/s12021-013-9184-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Manoach DS, Press DZ, Thangaraj V, et al. Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task, as measured by fMRI. Biol Psychiatry. 1999;45:1128–37. doi: 10.1016/s0006-3223(98)00318-7. [DOI] [PubMed] [Google Scholar]
- 33.Roffman JL, Gollub RL, Calhoun VD, et al. MTHFR 677C → T genotype disrupts prefrontal function in schizophrenia through an interaction with COMT 158Val → Met. Proc Natl Acad Sci USA. 2008;105:17573–17578. doi: 10.1073/pnas.0803727105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fischl B, Salat DH, Busa E, et al. Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron. 2002;33:341–355. doi: 10.1016/s0896-6273(02)00569-x. [DOI] [PubMed] [Google Scholar]
- 35.Ehrlich S, Morrow EM, Roffman JL, et al. The COMT Val108/158Met polymorphism and medial temporal lobe volumetry in patients with schizophrenia and healthy adults. Neuroimage. 2010;53:992–1000. doi: 10.1016/j.neuroimage.2009.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yendiki A, Greve DN, Wallace S, et al. Multi-site characterization of an fMRI working memory paradigm: reliability of activation indices. Neuroimage. 2010;53:119–131. doi: 10.1016/j.neuroimage.2010.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Smith SM, Jenkinson M, Woolrich MW, et al. Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage. 2004;23(Suppl 1):S208–19. doi: 10.1016/j.neuroimage.2004.07.051. [DOI] [PubMed] [Google Scholar]
- 38.Woolrich MW, Ripley BD, Brady M, Smith SM. Temporal autocorrelation in univariate linear modeling of FMRI data. Neuroimage. 2001;14:1370–1386. doi: 10.1006/nimg.2001.0931. [DOI] [PubMed] [Google Scholar]
- 39.Mitsis GD, Iannetti GD, Smart TS, et al. Regions of interest analysis in pharmacological fMRI: how do the definition criteria influence the inferred result? Neuroimage. 2008;40:121–32. doi: 10.1016/j.neuroimage.2007.11.026. [DOI] [PubMed] [Google Scholar]
- 40.Anderson CA, Pettersson FH, Clarke GM, et al. Data quality control in genetic case-control association studies. Nat Protoc. 2010;5:1564–1573. doi: 10.1038/nprot.2010.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Howie BN, Donnelly P, Marchini J. A Flexible and Accurate Genotype Imputation Method for the Next Generation of Genome-Wide Association Studies. PLoS Genet. 2009;5:e1000529. doi: 10.1371/journal.pgen.1000529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genet. 2006;2:e190. doi: 10.1371/journal.pgen.0020190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Price AL, Patterson NJ, Plenge RM, et al. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 45.Vilain J, Galliot A-M, Durand-Roger J, et al. Environmental risk factors for schizophrenia: a review. Encephale. 2013;39:19–28. doi: 10.1016/j.encep.2011.12.007. [DOI] [PubMed] [Google Scholar]
- 46.Callicott JH, Egan MF, Mattay VS, et al. Abnormal fMRI response of the dorsolateral prefrontal cortex in cognitively intact siblings of patients with schizophrenia. Am J Psychiatry. 2003;160:709–19. doi: 10.1176/appi.ajp.160.4.709. [DOI] [PubMed] [Google Scholar]
- 47.Manoach DS, Press DZ, Thangaraj V, et al. Schizophrenic subjects activate dorsolateral prefrontal cortex during a working memory task, as measured by fMRI. Biol Psychiatry. 1999;45:1128–1137. doi: 10.1016/s0006-3223(98)00318-7. [DOI] [PubMed] [Google Scholar]
- 48.Potkin SG, Turner JA, Brown GG, et al. Working memory and DLPFC inefficiency in schizophrenia: the FBIRN study. Schizophr Bull. 2009;35:19–31. doi: 10.1093/schbul/sbn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fusar-Poli P, Howes OD, Allen P, et al. Abnormal frontostriatal interactions in people with prodromal signs of psychosis: a multimodal imaging study. Arch Gen Psychiatry. 2010;67:683–691. doi: 10.1001/archgenpsychiatry.2010.77. [DOI] [PubMed] [Google Scholar]
- 50.He H, Sui J, Yu Q, et al. Altered small-world brain networks in schizophrenia patients during working memory performance. PLoS ONE. 2012;7:e38195. doi: 10.1371/journal.pone.0038195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.MacDonald AW, 3rd, Thermenos HW, Barch DM, Seidman LJ. Imaging genetic liability to schizophrenia: systematic review of FMRI studies of patients’ nonpsychotic relatives. Schizophr Bull. 2009;35:1142–62. doi: 10.1093/schbul/sbn053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Meda SA, Bhattarai M, Morris NA, et al. An fMRI study of working memory in first-degree unaffected relatives of schizophrenia patients. Schizophr Res. 2008;104:85–95. doi: 10.1016/j.schres.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Van Veelen NMJ, Vink M, Ramsey NF, Kahn RS. Left dorsolateral prefrontal cortex dysfunction in medication-naive schizophrenia. Schizophr Res. 2010;123:22–29. doi: 10.1016/j.schres.2010.07.004. [DOI] [PubMed] [Google Scholar]
- 54.Whitfield-Gabrieli S, Thermenos HW, Milanovic S, et al. Hyperactivity and hyperconnectivity of the default network in schizophrenia and in first-degree relatives of persons with schizophrenia. Proc Natl Acad Sci USA. 2009;106:1279–84. doi: 10.1073/pnas.0809141106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Glahn DC, Ragland JD, Abramoff A, et al. Beyond hypofrontality: a quantitative meta-analysis of functional neuroimaging studies of working memory in schizophrenia. Hum Brain Mapp. 2005;25:60–9. doi: 10.1002/hbm.20138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stephan KE, Baldeweg T, Friston KJ. Synaptic plasticity and dysconnection in schizophrenia. Biol Psychiatry. 2006;59:929–939. doi: 10.1016/j.biopsych.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 57.Chandler CH, Chari S, Dworkin I. Does your gene need a background check? How genetic background impacts the analysis of mutations, genes, and evolution. Trends Genet. 2013;29:358–366. doi: 10.1016/j.tig.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hultman CM, Sparén P, Takei N, et al. Prenatal and perinatal risk factors for schizophrenia, affective psychosis, and reactive psychosis of early onset: case-control study. BMJ. 1999;318:421–426. doi: 10.1136/bmj.318.7181.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Khandaker GM, Zimbron J, Lewis G, Jones PB. Prenatal maternal infection, neurodevelopment and adult schizophrenia: a systematic review of population-based studies. Psychol Med. 2013;43:239–257. doi: 10.1017/S0033291712000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rehn AE, Rees SM. Investigating the neurodevelopmental hypothesis of schizophrenia. Clin Exp Pharmacol Physiol. 2005;32:687–696. doi: 10.1111/j.1440-1681.2005.04257.x. [DOI] [PubMed] [Google Scholar]
- 61.Sawada K, Young CE, Barr AM, et al. Altered immunoreactivity of complexin protein in prefrontal cortex in severe mental illness. Mol Psychiatry. 2002;7:484–492. doi: 10.1038/sj.mp.4000978. [DOI] [PubMed] [Google Scholar]
- 62.Woods SW. Chlorpromazine equivalent doses for the newer atypical antipsychotics. J Clin Psychiatry. 2003;64:663–7. doi: 10.4088/jcp.v64n0607. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.