

Abstract
Coibamide A is a highly potent antiproliferative cyclic depsipeptide, which was originally isolated from a Panamanian marine cyanobacterium. In this study, the synthesis of coibamide A has been investigated using Fmoc-based solid-phase peptide synthesis followed by the cleavage of the resulting linear peptide from the resin and its subsequent macrolactonization. The peptide sequence of the linear coibamide A precursor was constructed on a solid-support following the optimization of the coupling conditions, where numerous coupling agents were evaluated. The macrocyclization of the resulting linear peptide provided the [d-MeAla11]-epimer of coibamide A, which exhibited nanomolar cytotoxic activity towards a number of human cancer cell lines.
Keywords: Antiproliferative peptide, coibamide A, depsipeptide, N-methylamino acid
Coibamide A (1) is a cyclic depsipeptide, which was originally isolated from a Panamanian marine cyanobacterium (Figure 1).1 This compound has been reported to exhibit potent antiproliferative activity against a number of human cell lines, where it operates according to a unique mechanism of action. For this reason, there has been considerable interest in the use of coibamide A as a potential lead in cancer drug discovery. The recent results also revealed that coibamide A induces macroautophagy and can trigger apoptosis and non-apoptotic forms of cell death in human cancer cells.2 The production and isolation of coibamide A via the cultivation of coibamide A-producing cyanobacterium is a laborious process, and the development of new methods for the chemical synthesis of coibamide A and its derivatives is therefore highly desired to allow for further pharmaceutical research and development.1 The proposed structure of coibamide A was recently synthesized by He et al.,3 but the analytical and biological activity data for the synthetic sample were inconsistent with those reported for natural coibamide A.
Figure 1.
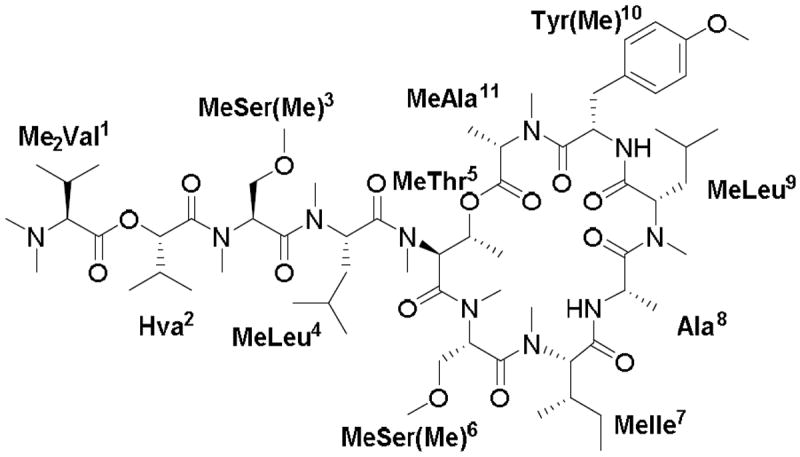
Structure of coibamide A.
Coibamide A has eight N-methylamino acids and two ester bonds. The coupling of an amino acid onto the N-methylamino terminus of a peptidyl resin proceeds slowly because of steric hindrance, and the synthesis of N-methyl-rich peptides can be made difficult by the sluggish nature of this reaction.4 The synthesis of N-methyl-rich peptides on a solid-support therefore requires the optimization of the reagent used for each coupling step, as exemplified by the syntheses of [MeLeu1]-cyclosporin A,5 the potent nematicide omphalotin A,6 the antitumor agent IB-012127 and the immunosuppressive antifungal compound petriellin A.8 The coupling conditions developed for the synthesis of [MeLeu1]-cyclosporin A5 involved the use a highly reactive DIC/1-hydroxy-7-azabenzotriazole (HOAt) system9 and O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl-uronium hexafluorophosphate (HATU),10 which led to significant improvements in the yields of the coupling products. Triphosgene (BTC) has also been used to improve the coupling efficiency of amidation reactions by converting Fmoc-amino acids into highly reactive and sterically unhindered acid chlorides in situ.6
It was envisaged in the current study that coibamide A could be synthesized via the Fmoc-based solid-phase peptide synthesis (SPPS) of a linear peptidyl resin, followed by the cleavage of the resulting linear peptide from the resin and a subsequent macrocyclization reaction between its MeThr5 β-hydroxy group and its C-terminal carboxylic acid moiety (Scheme 1). A series of different coupling protocols were investigated in the current study, including the use of DIC/HOAt, HATU and BTC systems to optimize the coupling conditions required for all seven of the N-methylamino acid-termini on the solid-support. The progress of each coupling reaction was monitored by HPLC analysis following the cleavage of the peptide product from the peptidyl resin with a 5:95 (v/v) mixture of TFA/DCM.
Scheme 1.
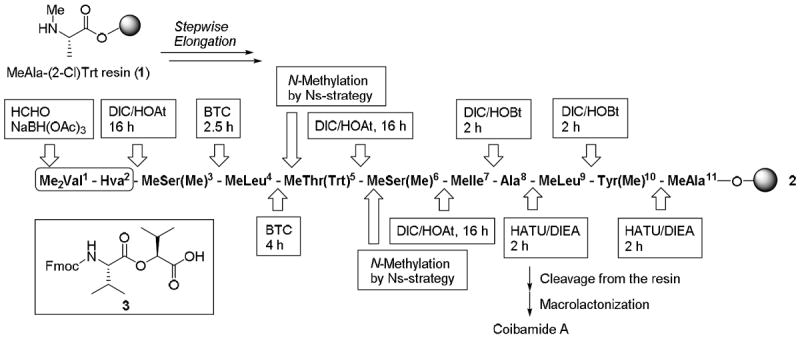
Overview of the synthetic strategy used for the construction of coibamide A.
Tyr(Me)10 and Ala8 were efficiently coupled to N-methylamino acids using a HATU/(i-Pr)2NEt system. In contrast, MeLeu9 and MeIle7 were successfully coupled onto N-methylamino acids using the standard protocol for Fmoc-based SPPS (i.e., DIC/HOBt). The coupling of MeSer(Me)6 onto the N-methylamino group of MeIle7 in the presence of HATU/(i-Pr)2NEt led to the partial epimerization of the product (dr = 72:28), which was attributed to slow reaction of the two coupling partners under the basic conditions. When the same coupling reaction was conducted with DIC/HOAt, which produces an HOAt ester as the active species without the addition of base, the reaction did not proceed to completion, although no epimerization was observed. In contrast, the coupling of Fmoc-Ser(Me)-OH proceeded smoothly to completion using the same DIC/HOAt reagents. Subsequent deprotection of the Fmoc group followed by an on-resin N-methylation protocol using an o-nitrobenzenesulfonyl (Ns)-strategy (Scheme 2)11 provided the desired hexapeptide containing MeSer(Me)6 in sufficient purity. Overall, this N-methylation protocol involved the nosylation of the Ser(Me) α-amino group, N-methylation of the resulting nosylated amine using a Mitsunobu reaction and the subsequent deprotection of the Ns-group. The coupling of Fmoc-Thr(Trt)-OH was also followed by an on-resin N-methylation process using the Ns-strategy described above to give the MeThr(Trt)5 residue.
Scheme 2.
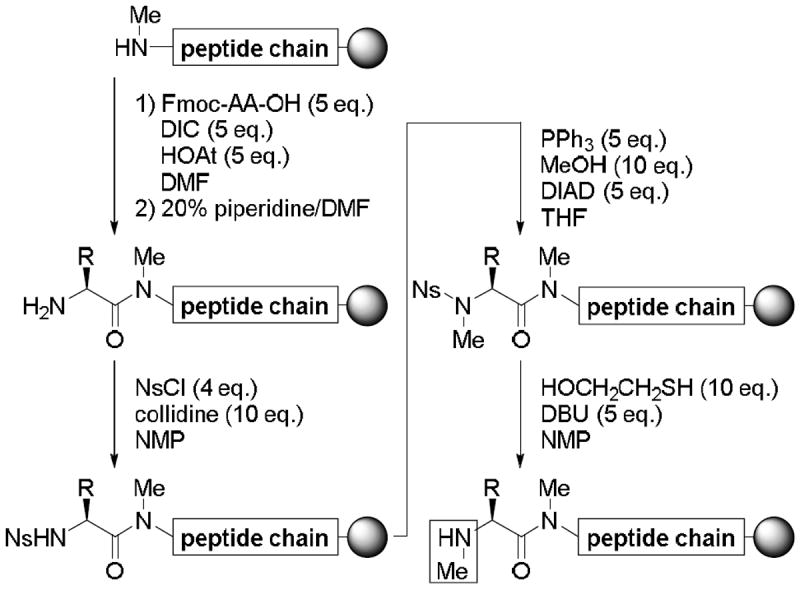
Preparation of N-methylamino acids [i.e., MeSer(Me)6 and MeThr(Trt)5] using the Ns-strategy on a solid support.
As was the case for MeSer(Me)6, the HATU-mediated coupling of MeLeu4 did not proceed to completion (61% conversion). The failure of this reaction to reach completion was most likely due to steric hindrance of the N-methylamino group and the protected bulky side chain of MeThr(Trt)5. Among the various coupling conditions investigated, BTC was found to be the only coupling agent capable of driving the MeLeu4 coupling to completion to obtain the desired octapeptide in good purity (83% purity). The second MeSer(Me)3 residue was also incorporated into the linear peptide by the BTC-mediated coupling of Fmoc-MeSer(Me)-OH, which was synthesized according to the Freidinger method.12 Finally, to allow for the incorporation of the N-terminal esterified dimer (Me2Val1-Hva2) into the linear peptide resin, the resulting nonapeptide was conjugated to ester 3 using DIC/HOAt; ester 3 was synthesized in advance of this particular step using solution phase chemistry. The resulting peptide was then N,N-dimethylated under reductive amination conditions with formalin and NaBH(OAc)3 to afford the desired linear undecapeptidyl resin 2 in 52% purity.
The final deprotection and cleavage of peptidyl resin 2 was achieved by the treatment of the resin-bound peptide with a 5:95 (v/v) mixture of TFA/DCM, which led to the formation of two products with identical molecular weights (Figure 2). The treatment of the minor product with Et3N provided only the major product, while the major product underwent partial conversion to the minor product by treatment with TFA/DCM (5:95) (accompanied by the unchanged major product). These suggested that the major product was the desired linear peptide 4, and that the minor product was the O-acyl peptide 5, which was most likely formed by the rearrangement of the N-terminal tetrapeptide from the MeThr5 α-amino group to the side-chain hydroxyl group. The desired product 4 could be recovered by the treatment of the mixture of 4 and 5 with Et3N followed by purification by reversed-phase HPLC. The overall isolated yield of 4 from resin 1 was 29%.
Figure 2.
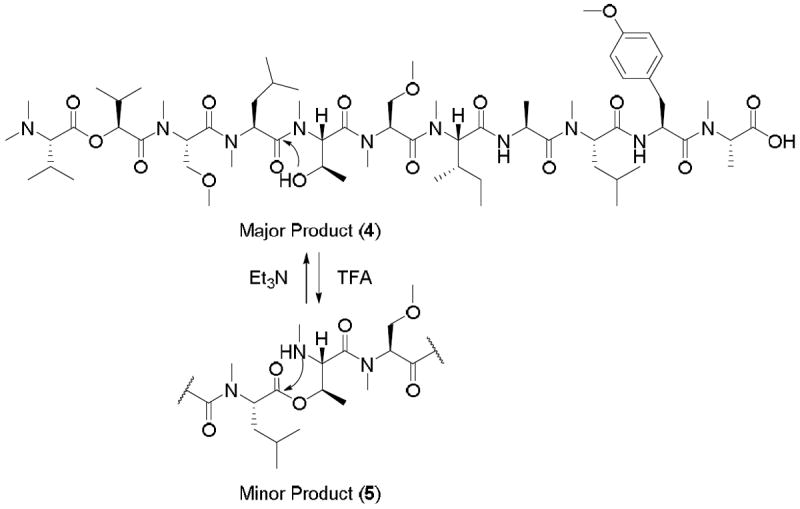
Possible N- to O-acyl shift at the MeThr5 residue following the treatment of linear undecapeptide 4 with a 5:95 (v/v) mixture of TFA/DCM.
With the linear precursor 4 in hand, we proceeded to investigate the macrolactonization using LC-MS analysis to monitor the reaction progress. Although various macrolactonization conditions have been reported in the literature,13-17 the application of these conditions to peptide 4 failed to provide any of the desired cyclic depsipeptide. Pleasingly, however, the use of MSNT18/(i-Pr)2NEt provided access to the desired cyclization product, albeit in a low yield of 3.8%.
The cyclic product was characterized by ESI-MS, MALDI-TOF-MS and ESI-TOF-MS/MS analyses (Figure 3). The expected molecular ions were detected by ESI-MS ([M+2H]2+, [M+H]+ and [M+Na]+) and MALDI-TOF-MS ([M+H]+ and [M+Na]+). Analysis of the cyclic peptide by ESI-TOF-MS/MS revealed that its fragmentation pattern was identical to that of the natural product, except that an MS/MS fragmentation peak with an m/z value of 880 was not observed for the synthetic material. The 13C NMR spectrum of the synthetic product was almost identical to that of the natural product (see supplementary data). Although the 1H NMR spectrum of the synthetic product was similar to that of natural coibamide A (Figure S1), significant differences were observed in the regions spanning δ 1.5–2.0 and 4.5–6.2 ppm (Figure 4). In particular, there were considerable differences between the 1H signals attributed to MeLeu4-β-CH2, MeSer(Me)3-α-CH and Hva2-α-CH. A comparison of these two compounds by HPLC analysis revealed that the synthetic product eluted slightly ahead of the natural product, which indicated that the synthetic product could be a diastereomer of the natural product. It is entirely possible that the MeAla11 residue may have been epimerized during the macrolactonization step and that only the epimerized precursor underwent the cyclization reaction to provide the corresponding cyclic product, which had an identical molecular weight. The configurations of the hydroxy and amino acids moieties of the synthetic product were therefore investigated using Marfey’s analysis (by LC-MS) and chiral GC-MS using similar methods to those published previously for the analysis of natural coibamide A.1 Based on the results of this analysis (see supplementary data), the MeAla residue of the synthetic precursor was assigned a d configuration, which confirmed our suspicions that the extended reaction time required for the macrolactonization process had resulted in the racemization and selective cyclization of the linear [d-MeAla11]-undecapeptide.
Figure 3.
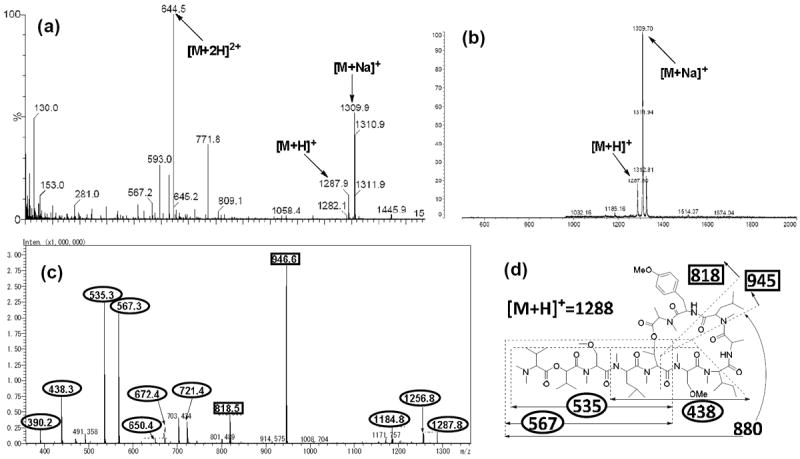
Mass spectrometric analysis of synthetic [d-MeAla11]-coibamide A. (a) ESI-MS spectrum, (b) MALDI-TOF-MS spectrum, (c) ESI-TOF-MS/MS spectrum, and (d) the reported MS/MS fragmentation pattern of natural coibamide A.
Figure 4.
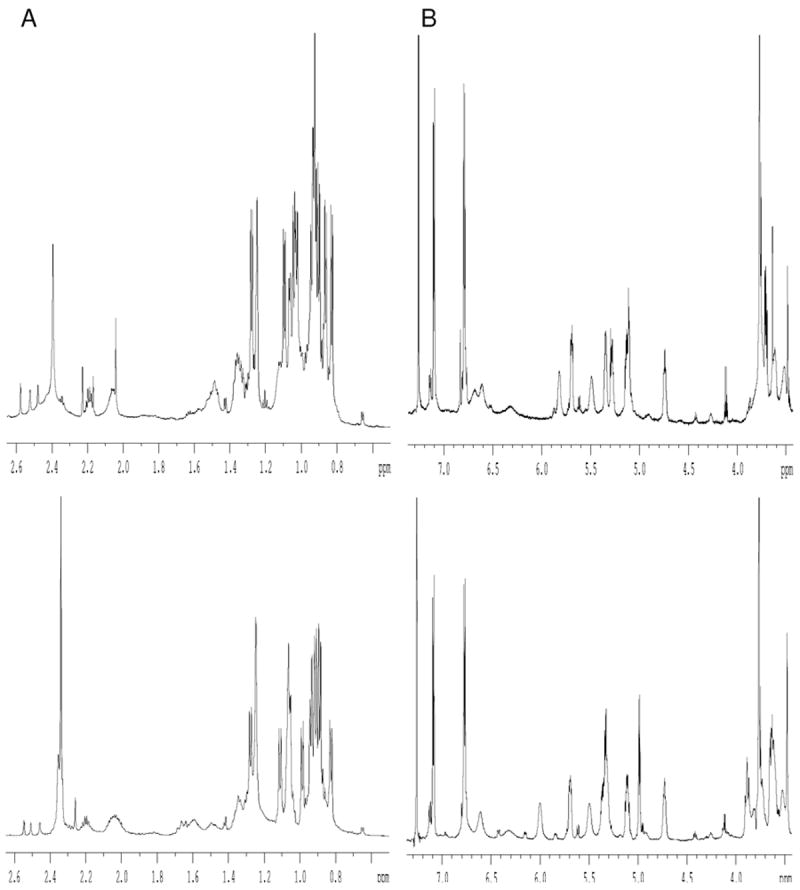
Comparison of A) the alkyl- and upper N-methyl and B) the α-proton and aromatic regions of the 1H NMR spectra for synthetic [d-MeAla11]-coibamide A (upper) and natural coibamide A (lower).
Initial biological testing of this [D-MeAla11]-epimer of coibamide A revealed that it was potently cytotoxic against A549 (non-small cell lung cancer), HCT116 (colon cancer), MCF-7 (breast cancer) and B16 (murine melanoma) cells at nanomolar concentrations (Table 1). The cytotoxicity of the [d-MeAla11]-epimer of coibamide A was also directly compared with that of natural coibamide A against a panel of four additional human cancer cell lines including H292 (lung carcinoma), MDA-MB-231 (breast cancer), PC-3 (prostate cancer) and SF-295 (glioblastoma) cells. The [d-MeAla11]-epimer of coibamide A showed potent cytotoxic activity towards all four of these cell lines, although the potency exhibited by this material was 3.7- to 8.3-fold less than that of natural coibamide A (Table 2).
Table 1.
Cytotoxicities of [d-MeAla11]-coibamide A against human non-small cell lung (A549), colon (HCT116), breast (MCF-7) and murine melanoma (B16) cancer cell lines.a
| cell line | IC50 (nM) |
|---|---|
| A549 | 19.0 |
| HCT116 | 44.6 |
| MCF-7 | 48.6 |
| B16 | 54.4 |
Concentration-response relationships were analyzed using Graphpad Prism Software (Graphpad Software Inc., San Diego, CA), and IC50 values were derived using nonlinear regression analysis fit to a logistic equation.
Table 2.
Comparative cytotoxicities for natural coibamide A and the [d-MeAla11]-epimer of coibamide A against human lung carcinoma (H292), breast (MDA-MB-231), prostate (PC-3) and glioblastoma (SF-295) cancer cell lines.a
| cell line | IC50 (nM)
|
|
|---|---|---|
| coibamide A (natural product) | [d-MeAla11]-coibamide A (synthetic product) | |
| H292 | 124 | 610 |
| MDA-MB-231 | 66 | 545 |
| PC-3 | 80 | 424 |
| SF-295 | 219 | 816 |
Concentration-response relationships were analyzed using Graphpad Prism Software, and IC50 values were derived using nonlinear regression analysis fit to a logistic equation.
In conclusion, we have investigated the synthesis of coibamide A using Fmoc-SPPS followed by the macrolactonization of the resulting linear peptide. The coupling conditions used for the solid-phase synthesis were optimized to obtain the linear precursor in high yield. During the macrocyclization process, only the d-MeAla11-epimer was obtained, most likely because of the epimerization of the MeAla11 residue during the slow ester bond formation. The [d-MeAla11]-epimer of coibamide A was found to be slightly less potent than the natural coibamide A, but still exhibited nanomolar cytotoxicity towards a number of different cancer cell lines, and is the first in a series of coibamide A epimers that could be obtained for structure-activity relationship studies.
Supplementary Material
Acknowledgments
This work was supported by Grants-in-Aid for Scientific Research (24659004, 26·7872) from JSPS, Japan; Platform for Drug Discovery, Informatics and Structural Life Science from MEXT, Japan; and research grants from the Uehara Memorial Foundation, Takeda Science Foundation and Teijin Pharm. Ltd. K.L.M. and J.E.I. gratefully acknowledge the USA NIH Fogarty International Center (ICBG grant TW006634-06). R.N. and R.M. are grateful for JSPS Research Fellowships for Young Scientists.
Abbreviations
- Hva
α-Hydroxyisovaleric acid
- HATU
O-(7-azabenzotriazol-1-yl)-1,1,3,3-tetra-methyl-uronium hexafluorophosphate
- BTC
Bis(trichloromethyl) carbonate
- DIC
1,3-diisopropylcarbodiimide
- HOBt
1-hydroxybenzotriazole
- HOAt
1-hydroxy-7-azabenzotriazole
- TFA
trifluoroacetic acid
- MSNT
1-(mesitylene-2-sulfonyl)-3-nitro-1,2,4-triazole
- NMI
N-methylimidazole
- DCM
dichloromethane
Footnotes
Supplementary data
Supplementary data associated with this article can be found in the online version at ########.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Medina RA, Goeger DE, Hills P, Mooberry SL, Huang N, Romero LI, Ortega-Barría E, Gerwick WH, McPhail KL. J Am Chem Soc. 2008;130:6324. doi: 10.1021/ja801383f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hau AM, Greenwood JA, Löhr CV, Serrill JD, Proteau PJ, Ganley IG, McPhail KL, Ishmael JE. PLoS One. 2013;8:e65250. doi: 10.1371/journal.pone.0065250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He W, Qui H–B, Chen Y–J, Xi J, Yao Z–J. Tetrahedron Lett. 2014;55:6109. [Google Scholar]
- 4.Teixidó M, Albericio F, Giralt E. J Pept Res. 2005;65:153. doi: 10.1111/j.1399-3011.2004.00213.x. [DOI] [PubMed] [Google Scholar]
- 5.Angell YM, Thomas TL, Flentke GR, Rich DH. J Am Chem Soc. 1995;117:7279. [Google Scholar]
- 6.Thern B, Rudolph J, Jung G. Angew Chem Int Ed. 2002;41:2307. doi: 10.1002/1521-3773(20020703)41:13<2307::AID-ANIE2307>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 7.Cruz LJ, Cuevas C, Giralt E, Cañedo LM, Albericio F. J Org Chem. 2006;71:3339. doi: 10.1021/jo051601h. [DOI] [PubMed] [Google Scholar]
- 8.Sleebs MM, Scanlon D, Karas J, Maharani R, Hughes AB. J Org Chem. 2011;76:6686. doi: 10.1021/jo201017w. [DOI] [PubMed] [Google Scholar]
- 9.Carpino LA. J Am Chem Soc. 1993;115:4397. [Google Scholar]
- 10.Carpino LA, El-Faham A, Minor CA, Albericio F. J Chem Soc Chem Commun. 1994:201. [Google Scholar]
- 11.Miller SC, Scanlan TS. J Am Chem Soc. 1997;119:2301. [Google Scholar]
- 12.Freidinger RM, Hinkle JS, Perlow DS, Arison BH. J Org Chem. 1983;48:77. [Google Scholar]
- 13.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. Bull Chem Soc Jpn. 1979;52:1989. [Google Scholar]
- 14.Shiina I, Ibuka R, Kubota M. Chem Lett. 2002:286. [Google Scholar]
- 15.Mukaiyama T, Araki M, Takei HJ. J Am Chem Soc. 1973;95:4763. [Google Scholar]
- 16.Corey EJ, Nicolaou KC. J Am Chem Soc. 1974;96:5614. [Google Scholar]
- 17.Boden EP, Keck GE. J Org Chem. 1985;50:2394. [Google Scholar]
- 18.Reese CB, Titmas RC, Yau L. Tetrahedron Lett. 1978;19:2727. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.